
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of C2-indolyl-quaternary centers by branch-selective allylation: enabling concise total synthesis of the (±)-mersicarpine alkaloid†
Minakshi
Ghosh
,
Samrat
Sahu
 ,
Shuvendu
Saha
and
Modhu Sudan
Maji
*
,
Shuvendu
Saha
and
Modhu Sudan
Maji
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, WB, India. E-mail: msm@chem.iitkgp.ac.in
First published on 18th December 2023
Abstract
Herein we report a branch-selective allylation strategy for accessing C2-indolyl-all-carbon quaternary centers using allylboronic acids. This approach boasts broad functional group tolerance, scalability, and relies on easily accessible allyl alcohol precursors. Importantly, the C3-position of the indole remains free, offering a handle for further synthetic refinement. Mechanistic pathways, corroborated by density functional theory (DFT), suggest the involvement of an indolenine intermediate and a Zimmerman–Traxler-like transition state during allylboration. Demonstrating its efficacy, the method was applied to the total synthesis of the (±)-mersicarpine alkaloid and enabled formal synthesis of additional alkaloids, such as (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine.
Introduction
The construction of all-carbon quaternary stereocenters remains a formidable challenge in organic synthesis due to issues of steric hindrance, regioselectivity, and the frequent incompatibility with existing functional groups. Quaternary centers possessing one 2-indolyl group and a two-carbon moiety serve as a linchpin for several alkaloids, particularly indole terpenoids (Scheme 1a).1 This key structural moiety (1) can ideally be accessed by the branch-selective indole C2-allylation. Despite numerous applications and future prospects in total synthesis, this reaction is still underdeveloped, and to our knowledge, Danishefsky's reverse prenylation is the only method known to date (Scheme 1b).2a This method has played a key role in the late-stage reverse prenylation to access several C2-reverse-prenylated indole alkaloids of diverse chemical structures displaying a broad spectrum of biological activities.3 Extension of this method to other functionalized γ,γ-disubstituted-allyl-9-BBN reagents to access such all-carbon quaternary centers did not progress further which might stem from the fact that these are prepared via hydroboration of allenes or borylation of allyl-lithium reagents.4 These precursors are either difficult to access or key allylic C–B bond formation reactions are incompatible with common functional groups. This long-standing problem can be addressed by developing a unique and general reverse-allylation strategy by employing readily accessible allylating agents.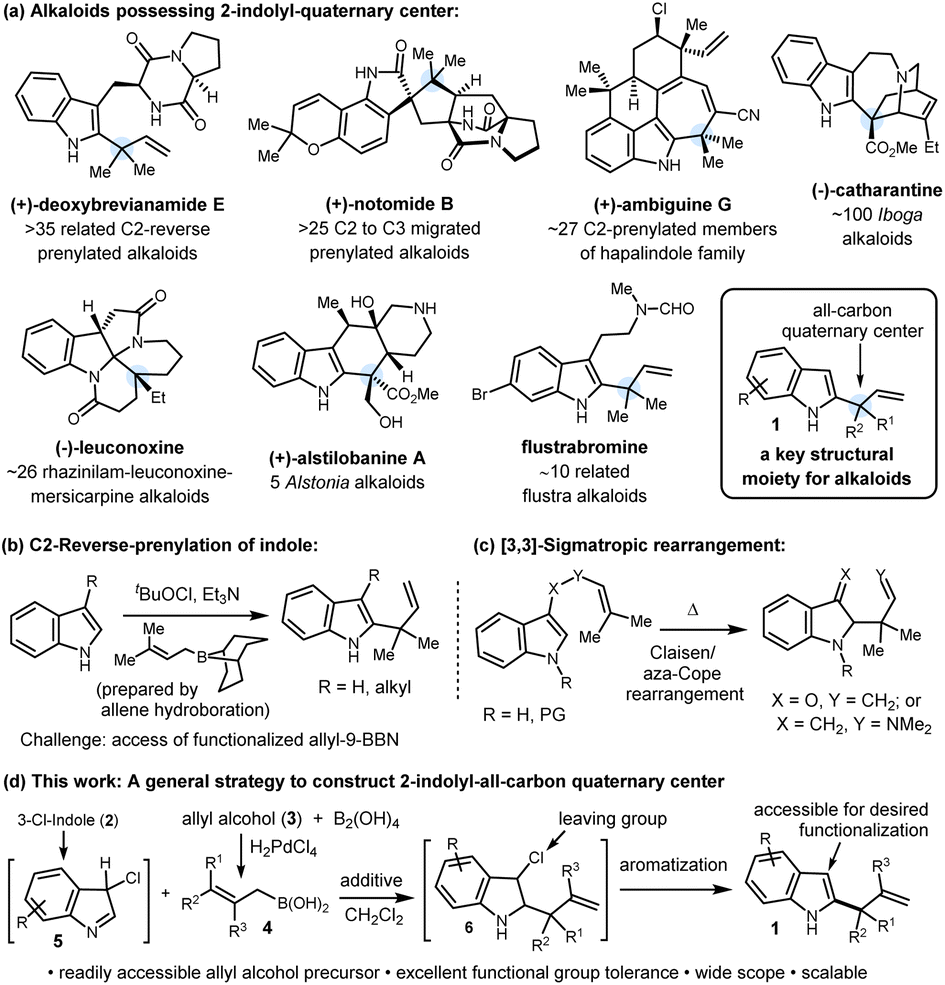 | ||
| Scheme 1 (a) Representative alkaloids possessing structural moiety 1. (b and c) Literature reports for the construction of a 2-indolyl quaternary center. (d) Reverse allylation using allylboronic acids. | ||
Although indole allylation chemistry has drawn considerable attention over the past few decades, the studies mostly focused on linear allylation. Transition-metal-catalyzed C–H allylation,5a–c acid catalyzed or mediated allylation,5d,e and lithiation of indole followed by allylation5f are some representative methods of linear allylation where indole plays the role of a nucleophile. In sharp contrast, branch-selective allylation using an Umpolung strategy where indole plays an electrophilic part is highly challenging and elusive in the literature. Besides Danishefsky's reverse prenylation reaction,2a [3,3]-sigmatropic rearrangement has been utilized to access reverse allylated products, and as a part of the method, the products contain specific substitution at the C3-position of indole (Scheme 1c).2b,c Thus a strategy which can provide direct access to several functionalized C2-indolyl-all-carbon quaternary centers 1 while the C3-position is free for further synthetic manipulation is challenging and highly desirable, particularly considering the total synthesis of indole alkaloids.
In recent years, allylboronic acid has emerged as a potent allylating agent of carbonyls and imines to generate adjacent quaternary stereocenters.6a,b Easy accessibility, geometrical stability, high reactivity, and functional group tolerance are the key aspects of allylboronic acids.6c Recently, indole C2-reverse allylation with allyl-trifluoroborate salts and allylboronic acids has been accomplished; with their own merits, these protocols led to indoline formation.7 In continuation of our interest in the synthesis of alkaloids and developing new reactions using allylboronic acids,8 herein, we report a mild, catalyst-free indole-C2-reverse-allylation to construct 1 employing easily accessible 3-chloroindoles as electrophiles and a wide range of functionalized γ,γ-disubstituted allylboronic acids as nucleophiles (Scheme 1d). The application of this newly developed method is also demonstrated in the total synthesis of (±)-mersicarpine and formal synthesis of (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine indole alkaloids.
Results and discussion
We hypothesize that, like indole, 3-chloroindole 2 can undergo protonation at the C3-position to eventually produce an imine intermediate 5 which upon in situ reaction with an allylboronic acid should furnish allylated indoline 6. Finally, aromatization should occur under redox-neutral conditions through the elimination of HCl leading to the formation of the desired C2-reverse allylated indoles 1 (Scheme 1d). To achieve our goal, we started investigating several reaction parameters by employing 3-chloroindole 2a as an electrophile and prenylboronic acid 4a as a nucleophilic allylating agent. As allylboronic acids are known to generate an indolenine intermediate from indoles,6b we initiated our study without employing any Brønsted acid additive. Among different screened solvents such as toluene, ethyl acetate, ether, and dichloromethane, the last one was found to be the most suitable providing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1aa and 7a in 67% yield (Table 1, entry 1). The formation of 7a can be explained by two mechanisms, firstly, 6 may be aromatized in the presence of molecular oxygen as an oxidant. In a second route, the product 1aa may undergo successive chlorination by the in situ generated 3-Cl-indolenine intermediate 5 which can potentially act as a chlorinating agent.9a
:1 mixture of 1aa and 7a in 67% yield (Table 1, entry 1). The formation of 7a can be explained by two mechanisms, firstly, 6 may be aromatized in the presence of molecular oxygen as an oxidant. In a second route, the product 1aa may undergo successive chlorination by the in situ generated 3-Cl-indolenine intermediate 5 which can potentially act as a chlorinating agent.9a
|
|
||||
|---|---|---|---|---|
| Entry | Additive/equiv of additive | Time (h) | Yieldb (%) | Ratio [1aa:7a]c |
| a Reaction conditions: 2a (0.2 mmol, 1.0 equiv), 4a (0.4 mmol, 2.0 equiv), additive, CH2Cl2 (2 mL), 80 °C, Ar. b Isolated yield. c Determined from the crude 1H NMR analysis. d At 60 °C. e At 100 °C. f 1.0 equiv of allylboronic acid was used. g Prenyl-BPin (0.4 mmol, 2.0 equiv) was used as an allylating agent. h 3-Bromoindole (0.2 mmol, 1.0 equiv) was used instead of 2a. i 3-Iodoindole (0.2 mmol, 1.0 equiv) was used instead of 2a. | ||||
| 1 | No additive/– | 10 | 67 | 1:1 |
| 2 | 2,6-Di-tBuC6H3OH/3.0 | 12 | 64 | 4:1 |
| 3 | 3-Methoxyphenol/3.0 | 12 | 62 | 1.7:1 |
| 4 | Resorcinol/3.0 | 12 | 57 | 1.66:1 |
| 5 | 1,3,5-(MeO)3C6H3/3.0 | 12 | 42 | 6:1 |
| 6 | N,N-dimethylaniline/3.0 | 12 | 78 | >20:1 |
| 7 | N,N-dimethylaniline/2.0 | 24 | 65 | >20:1 |
| 8 | N,N-dimethylaniline/1.0 | 24 | 83 |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 9 | N,N-dimethylaniline/0.5 | 48 | 53 | 3:1 |
| 10d | N,N-dimethylaniline/1.0 | 36 | 51 | >20:1 |
| 11e | N,N-dimethylaniline/1.0 | 24 | 79 | >20:1 |
| 12f | N,N-dimethylaniline/1.0 | 24 | 61 | >20:1 |
| 13g | N,N-dimethylaniline/1.0 | 24 | 0 | — |
| 14h | N,N-dimethylaniline/1.0 | 24 | 81 | >20:1 |
| 15i | N,N-dimethylaniline/1.0 | 24 | — | — |
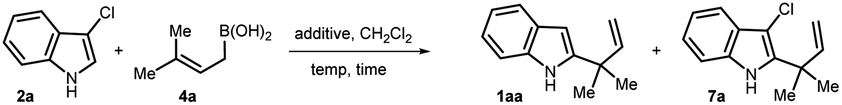
We postulated that, if the second route is operative, then formation of 7a can be suppressed by introducing an external electron-rich aromatic as a Cl+ scavenger9b or a base which can control the generation of 5 by neutralizing in situ generated HCl. Pleasingly, 2,6-di-tBuPhOH additive improved the product ratio to 4:1 (entry 2). Use of other additives, such as 3-methoxyphenol, resorcinol, and 1,3,5-trimethoxybenzene, was not very effective (entries 3–5). Interestingly, N,N-dimethylaniline was found to be a superior additive almost suppressing the formation of 7a and providing 1aa in 78% yield (entry 6). 1.0 equiv of this additive gave the best results (entry 8) as reducing the amount of additive further led to a decrease in yield and product selectivity (entries 7 to 9). A decrease, as well as an increase in the reaction temperature, diminished the yield of the reaction, although the product ratio remained unaltered (entries 10–11). Lowering the equivalency of allylboronic acid was not effective as yield decreased (entry 12). When the nucleophile was changed to prenyl-pinacol boronate ester, no allylated product was detected, highlighting the superior reactivity profile of allylboronic acid (entry 13). Next, the suitability of 3-bromo- and 3-iodoindole as electrophiles was tested in place of 2a. Pleasingly, 3-bromoindole was found to be highly effective for this reverse-prenylation reaction providing 1aa in 81% yield (entry 14). However, 3-iodoindole failed to provide 1aa (entry 15).
To probe the generality of our method, we first explored the scope of allylboronic acids of diverse structures and bearing different functional groups under the optimized conditions (Scheme 2). Allyl boronic acids having linear and branched alkyl substitutions at the γ,γ-position efficiently reacted to provide 1aa–1ad possessing the desired 2-indolyl-quaternary-center in good to excellent yields (38–83%). The decrease in yield with increased branching at the α-position of the newly generated quaternary center could be explained by the increased steric congestion. Nonetheless, even the tert-butyl group in 1ad can also be introduced as a substitution at the quaternary center albeit in low isolated yield. The rigid γ,γ-cyclic boronic acids of varied chain lengths smoothly reacted to furnish 1ae–1af in 73–91% yields. Both geranyl and farnesyl boronic acids underwent smooth conversion and gave the corresponding allylated products in excellent yields (1ag–1ah, 81–82%). Pleasingly, γ,γ-disubstituted allylboronic acids bearing ether (–OBn, –OPMB, –OMe), sulfonate (–OTs), thioether (–SPh) and ester (–CO2Me) functional groups were successfully tolerated under the reaction conditions to provide the desired products in good to excellent yields (1ai–1an, 53–92%). Allylboronic acids with substitutions at the β- and γ-positions were next tested, and to our delight, allylated products 1ao–1aq containing ester and cyanide functional groups were isolated in 42–85% yields. This proves that our developed method can also be applicable for accessing secondary as well as tertiary centers at the C2-position. To our delight, the secondary boronic acid also underwent smooth conversion and furnished the corresponding allylated product 1ar in 66% yield. The compatibility of key functional groups is another strength of this protocol as they could be utilized for further downstream modifications in organic synthesis.
 | ||
| Scheme 2 Scope of allylboronic acids and indolesa. aReaction conditions: 3-Cl-indole 2 (0.2 mmol, 1.0 equiv), allylboronic acid 4 (0.4 mmol, 2.0 equiv), N,N-dimethylaniline (0.2 mmol, 1.0 equiv), CH2Cl2 (2 mL), 80 °C, under Ar; isolated yield. | ||
We further investigated the scope of electrophiles by incorporating different substitutions on the benzene ring of indole, revealing pronounced trends in reactivity and selectivity. 4-Bromo and -cyano containing 3-chloroindoles underwent smooth conversion to furnish the desired allylated products 1ba–1ca in 69–77% yields (Scheme 2). Remarkably, the reaction appears largely insensitive to the electronic nature of the substituents at the C5-position, as both electron-donating and withdrawing groups such as methoxy, nitro, cyanide, and bromide furnished desired products 1da–1ga in comparable yields (75–86%). Notably, alanine coupled indole-5-carboxylic acid was also well tolerated, providing 1ha in 60% yield. Further probing revealed that 3-chloroindoles with bromo, cyano, ester, and nitro groups at the C6-position were amenable substrates, albeit with slightly diminished yields (1ia–1la, 35–65%). Being a reactive functional group toward allyl-boronic acid,6a to our joy, a ketone also survived under the reaction conditions to furnish 1ma in 64% yield. Indole containing a free-hydroxyl group also participated in this reaction providing 1na in 62% yield. Next, the effect of substitution closer to the coordinating imine nitrogen was tested which revealed that, due to sensitivity to spatial constraints, 3-chloro-7-methylindole failed to react under our optimized conditions. Next, the scalability of the developed protocol is demonstrated by carrying out the reaction on a 5.0 mmol scale by employing geranyl boronic acid 4g, and pleasingly 1.1 g of 1ag was isolated, recording a yield of 87%. In sum, our method offers a nuanced landscape of both opportunities and limitations for targeted molecule synthesis.
To access reverse-allylated products directly from indole, a one-pot two step strategy is adopted. Here, first, indole was subjected to chlorination by employing N-chlorosuccinimide, and then, subsequently in situ generated 2a was reacted with the allyl-boronic acids. Through this one-pot strategy, 1aa and 1ag were isolated in 62–74% yields from indole (Scheme 3a). Next, considering the applicability of the allylated products 1, the nucleophilicity of the indole nucleus was further exploited by in situ reacting with various electrophiles. This would lead to a formal one-pot indole 2,3-difunctionalization in an elegant way and can potentially furnish valuable intermediates for total synthesis. To this end, first 1aa was generated by reacting 2a with 4a, and then, in one pot sequentially treated with an appropriate electrophile to access 7. Through this strategy, chlorination, bromination, sulfenylation, and also the formation of bis(indolyl)methane were successfully achieved to furnish 7a–7d in 51–77% yields (Scheme 3b).
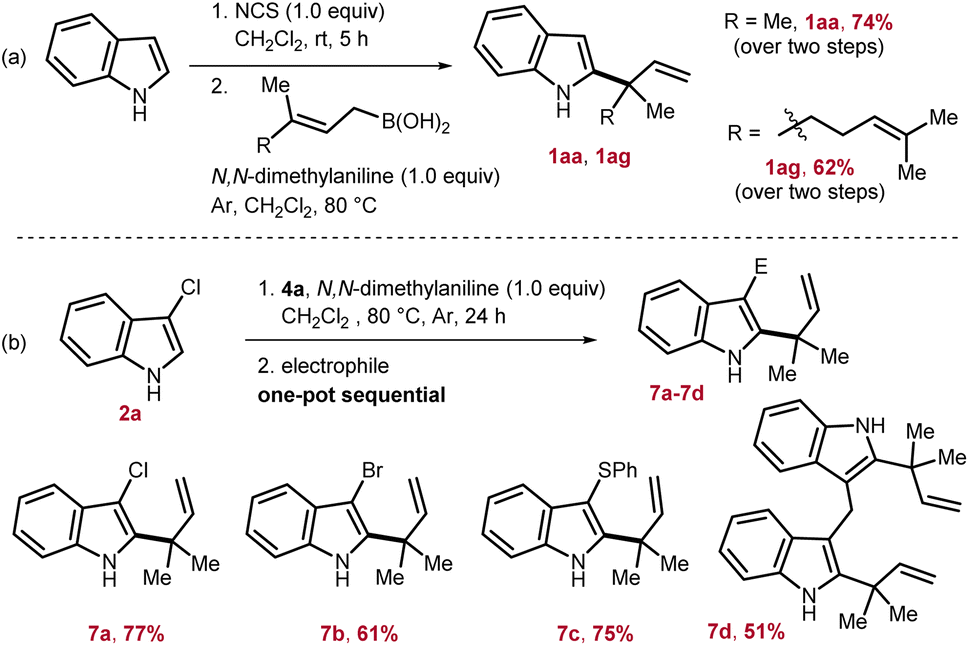 | ||
| Scheme 3 (a) One-pot allylation from indole. (b) One-pot sequential indole 2,3-difunctionalization. | ||
Based on the previous report7b we proposed that, initially, 3-chloroindole 2 underwent allylboronic acid induced tautomerization resulting in an indolenine intermediate 5 which coordinates with the electrophilic boron center of the allyl boronic acid to generate the adduct 8. Subsequently, intermediate 8 was transformed into an allylated indoline 6 through the Zimmerman–Traxler-like chair TS. Finally, aromatization provided the desired product 1aa. To support our hypothesis, the DFT calculations were performed using the B3LYP(SMD)/Def2-TZVP//B3LYP/TZVP level of theory. The allylboration proceeds via the chair-like transition state TS1 with a low activation barrier (10.0 kcal mol−1) compared to the corresponding boat-like transition state TS2 (14.9 kcal mol−1) providing intermediate 9 with a significant stabilization in energy. It is also well known that the boat form is unfavourable due to 1,4-diaxial strain. The bond distances in complex 8 [N–B, 1.68 Å & B–C, 1.64 Å], chair TS [N–B, 1.53 Å; B–C, 1.90 Å; & C–C, 2.23 Å] and complex 9 [N–B, 1.43 Å & C–C, 1.57 Å] suggested that B–C bond cleavage and C–C bond formation occurred through a concerted pathway and the product formation in this sequence was a thermodynamically downhill process (Scheme 4).
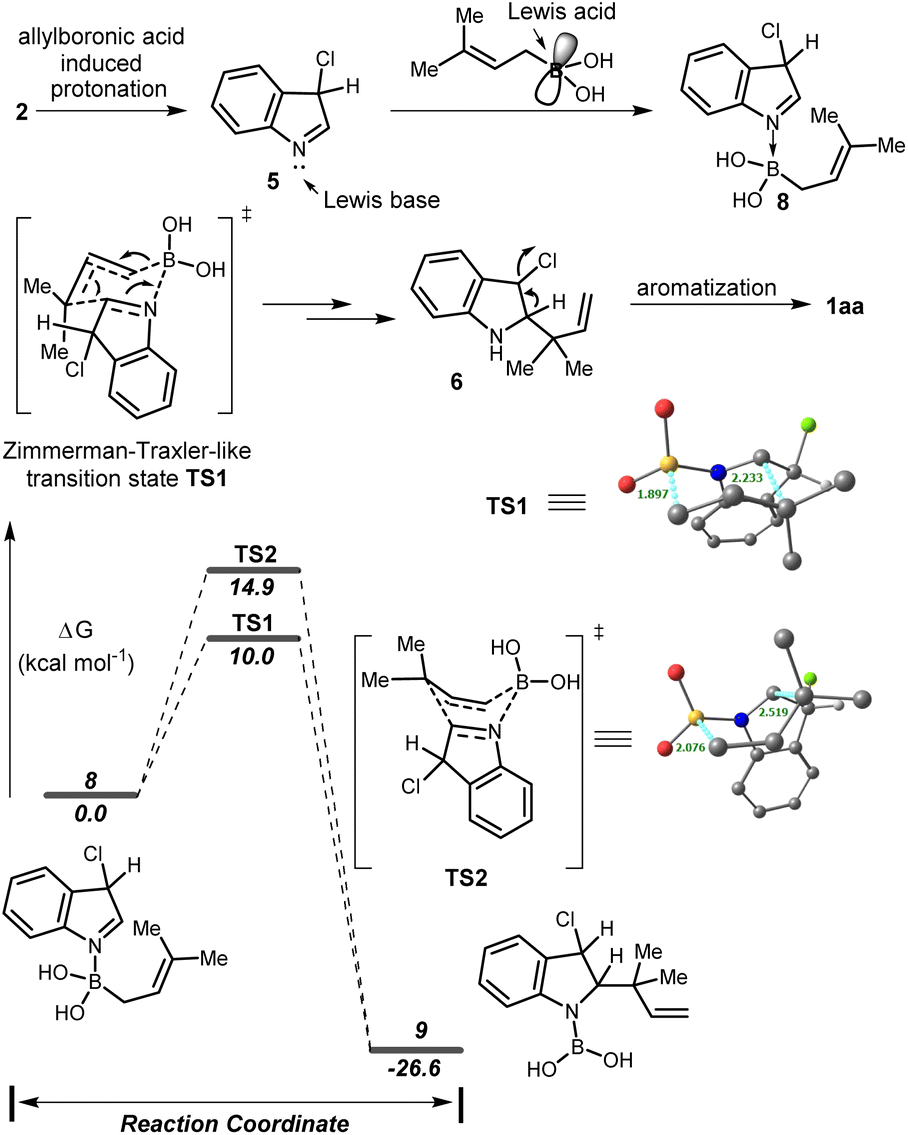 | ||
| Scheme 4 Plausible mechanism of reverse allylation and relative Gibbs free energy profile at the B3LYP(SMD)/Def2-TZVP//B3LYP/TZVP level. | ||
Facile generation of the indolyl-C2-quaternary center prompted us to apply this newly developed method to the total synthesis of indole-based alkaloids (±)-mersicarpine, (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine. These alkaloids belong to the leuconolam–leuconoxine–mersicarpine triads, a subfamily of aspidosperma alkaloids, among 2000 known monoterpene indole alkaloids.10 Their unprecedented structural features, significant biological activities, and interesting biosynthetic relationships made them an attractive target for total synthesis.1h,11,12 A detailed overview of these polycyclic alkaloids suggested that despite having different ring connectivity they share a common core having an all-carbon quaternary center at the C2-position of the indole ring. Starting from indole derivatives, this key quaternary center is generated mainly by intramolecular radical,12b–d Pd-catalyzed reductive Heck,12e and cationic cyclization12f of indole tethered alkenyl or alcohol moieties. In another strategy, the indole ring is constructed at a later stage on the properly assembled advanced precursors.11a–e,12g,h In our approach, we aim to generate 1 through a more challenging intermolecular amalgamation of the indole moiety and properly designed allyl unit possessing the required functional groups (Scheme 5).
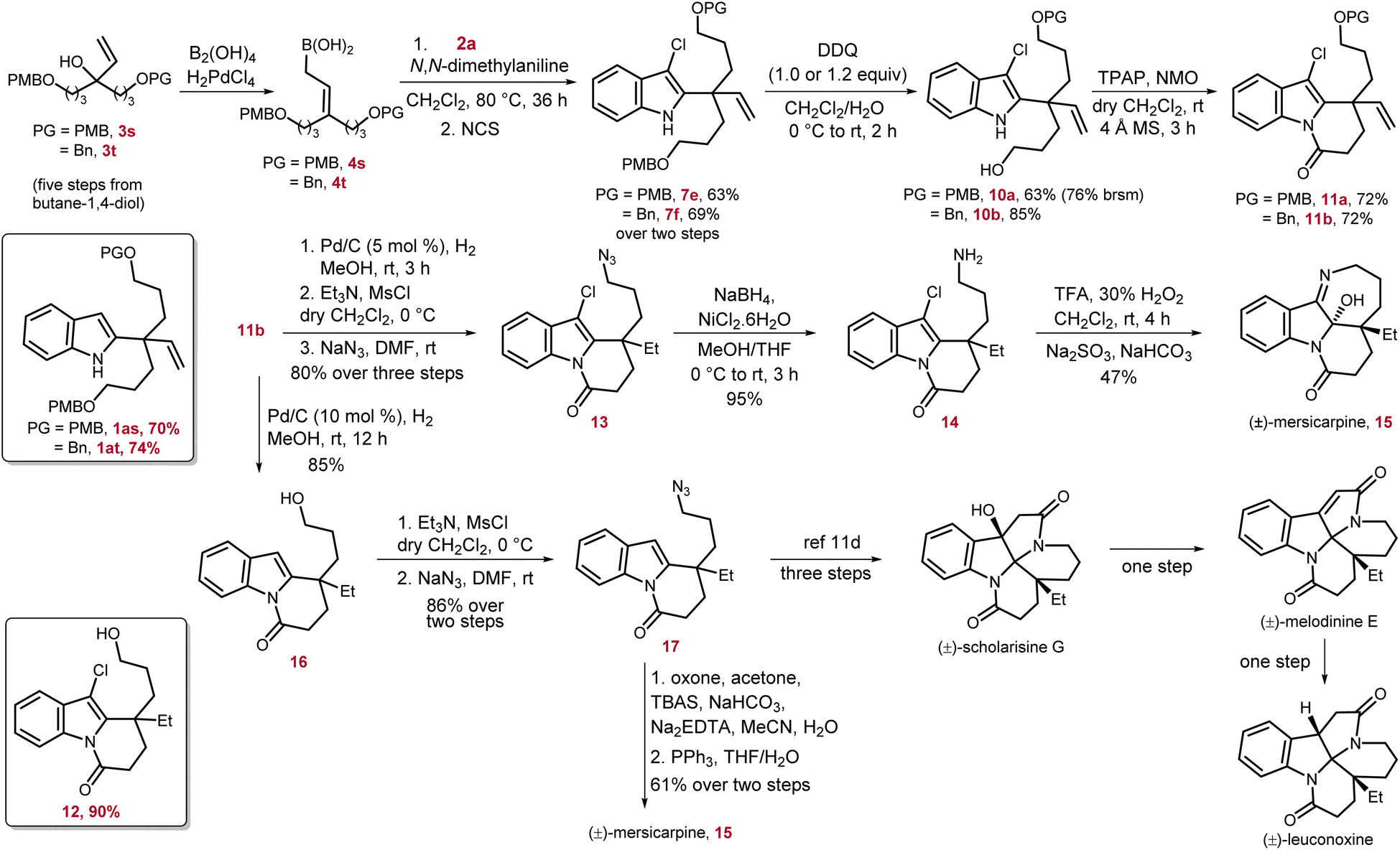 | ||
| Scheme 5 Total synthesis of (±)-mersicarpine and formal synthesis of (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine alkaloids. | ||
Mersicarpine was chosen as the first target for this venture owing to its intriguing structural features consisting of three heterocycles i.e., indoline, seven-membered cyclic imine, and δ-lactam, fused with each other around a hemiaminal group adjacent to the quaternary carbon center. In addition, in 2021, Usui and co-authors studied the biological activity of mersicarpine on the human leukemia cell line HL60, and the results suggested that it is a novel protein translation inhibitor.13 In our retrosynthetic analysis, along with vinyl substitution which will provide the ethyl group after hydrogenation, two more terminally functionalized three-carbon moieties were required to construct the cyclic imine and δ-lactam ring.
At the outset of the synthesis of mersicarpine, we devised a rapid synthetic route to access the key tertiary alcohol 3s, having two 4-methoxy-benzyl (PMB) protected three-carbon units, from commercially available 1,4-butanediol.14 The desired γ,γ-disubstituted allyl boronic acid 4s was readily prepared from 3s which upon treatment with 3-chloroindole 2a under the optimized conditions resulted in the desired indole derivative 1as possessing a quaternary center in 70% yield. Our attempts to selectively remove one of the PMB groups was unsuccessful as treatment of 1as with DDQ resulted in a complex reaction mixture. We speculated this issue might have arisen due to the reaction of electron-rich and highly nucleophilic 1as and oxidized side product of the PMB protecting group or DDQ itself. To prevent this, 1as was converted to 7e in one pot in 63% yield. To our delight, 7e underwent selective mono PMB-deprotection to furnish 10a in 63% overall yield, and 10% of diol by removal of both PMB groups was also isolated. The alcohol 10a was subjected to Ley oxidation conditions to construct the required δ-lactam ring by an oxidation–cyclization–oxidation sequence to obtain 11a in 72% yield. As the selective mono-deprotection of the PMB group in 7e was relatively low yielding and less selective, to increase the efficiency of our synthesis, one PMB group was replaced by a benzyl group in 3t. The same reaction sequence was then repeated with 3t to reach the intermediate 7f with a slight increase in overall yields. The selective mono-PMB deprotection of 7f proceeded smoothly and produced 10b with much higher yield of 85%, and no diol was detected. The δ-lactam ring formation on 10b also occurred efficiently to provide 11b in 72% yield. The intermediate 11b has four functional groups, namely, alkene, OBn group, C–Cl bond and indole nucleus, which can potentially be reduced under hydrogenation conditions. To achieve the total synthesis of mersicarpine, first alkene and OBn groups were selectively hydrogenated by 5 mol% Pd/C and running the reaction over 3 h to afford 12 in 90% yield. Interestingly, another key intermediate 16 was also obtained in 85% yield by increasing the Pd/C catalyst loading to 10 mol% and prolonging the reaction time to 12 h. Of note, increasing the catalyst loading further in order to decrease the reaction time led to over-reduction of 16 to the corresponding indoline product. Although the intermediate 12 was isolated and then subjected to mesylation followed by azidation, the same azide product 13 was also synthesized from 11b by a three step protocol in 80% overall yield. The reduction of azide to amine was extremely facile to produce 14 in 95% yield. Finally, the total synthesis of (±)-mersicarpine 15 was accomplished in 47% yield by oxidation of 14 with a mixture of trifluoroacetic acid and hydrogen peroxide followed by imine formation at an appropriate pH.12f
The total synthesis of mersicarpine was also achieved via a second strategy starting from intermediate 16. In this route, first the alcohol functional group was mesylated and converted to azide 17. Following Wang's route, treatment of 17 with dimethyl dioxirane, prepared in situ from acetone and oxone, followed by PPh3-mediated Staudinger–aza-Wittig reduction facilitated the intramolecular cyclization to furnish (±)-mersicarpine in 61% yield over two steps.11d The NMR spectrum of mersicarpine was recorded in base-washed CDCl3 due to the presence of an acid sensitive hemiaminal group. The intermediate 17 has been used as an advanced synthetic intermediate for the total synthesis of other leuconoxine family alkaloids by Wang et al. Thus, the formal synthesis of (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine was also achieved.
Conclusions
We developed an indole C2-reverse allylation strategy for the construction of an indolyl-C2-quaternary center employing allylboronic acids under mild conditions starting from readily available 3-chloroindole derivatives. The reaction shows excellent functional group tolerance as a wide range of allyl boronic acids and indoles took part in this reaction. As allylboronic acids can be accessed from simple allyl alcohol starting precursors, this method holds enormous potential for the total synthesis of indole-based alkaloids. According to our hypothesis, the nucleophilic addition of allylboronic acids occurred on the 3H-indole-imine tautomer. DFT calculations were carried out to support the proposed six-membered chair-like TS (Zimmerman–Traxler model) for the allylation step. The applicability of this strategy is demonstrated by total synthesis of the (±)-mersicarpine alkaloid via two routes in a divergent way from 3-chloroindole. Moreover, this strategy is also applied to the formal synthesis of other Aspidosperma alkaloids, such as (±)-scholarisine G, (±)-melodinine E, and (±)-leuconoxine, from a common intermediate.Data availability
The detailed experimental procedures, compound characterization data and related spectra are provided in the ESI.†Author contributions
M. G. and S. Sahu performed all the experiments in the laboratory. All the authors contributed to the design of the experiments, discussion, and preparation of the manuscript. S. Saha performed the DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. M. gratefully acknowledges the Council of Scientific & Industrial Research, India (Sanction No. 02(0433)/21/EMR-II) for funding. We thank DST (Sanction No. SR/FST/CSII-026/2013) for the 500 MHz NMR facility and acknowledge the Supercomputing Facility “PARAM Shakti” at IIT Kharagpur, established under the National Supercomputing Mission (NSM), Government of India. M. G. thanks IIT Kharagpur, S. Sahu thanks CSIR, India, and S. Saha thanks UGC, India for fellowships.Notes and references
- For selected alkaloids bearing a 2-indolyl-quaternary center, see: (a) S.-M. Li, Nat. Prod. Rep., 2010, 27, 57–78 RSC; (b) K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Nat. Prod. Rep., 2018, 35, 532–558 RSC; (c) J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS PubMed; (d) R. E. Johnson, H. Ree, M. Hartmann, L. Lang, S. Sawano and R. Sarpong, J. Am. Chem. Soc., 2019, 141, 2233–2237 CrossRef CAS PubMed; (e) J. Xu and V. H. Rawal, J. Am. Chem. Soc., 2019, 141, 4820–4823 CrossRef CAS PubMed; (f) C. Lavaud and G. Massiot, Prog. Chem. Org. Nat. Prod., 2017, 105, 89–136 CAS; (g) R. N. Iyer, D. Favela, G. Zhang and D. E. Olson, Nat. Prod. Rep., 2021, 38, 307–329 RSC; (h) M. Pfaffenbach and T. Gaich, Alkaloids-Chem. Biol., 2017, 77, 1–84 CAS; (i) K. Koyama, Y. Hirasawa, K. Zaima, T. C. Hoe, K.-L. Chan and H. Morita, Bioorg. Med. Chem., 2008, 16, 6483–6488 CrossRef CAS PubMed; (j) X. Di, S. Wang, J. T. Oskarsson, C. Rouger, D. Tasdemir, I. Hardardottir, J. Freysdottir, X. Wang, T. F. Molinski and S. Omarsdottir, J. Nat. Prod., 2020, 83, 2854–2866 CrossRef CAS PubMed.
- For related works to access the reverse prenyl moiety, see: (a) J. M. Schkeryantz, J. C. G. Woo, P. Siliphaivanh, K. M. Depew and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11964–11975 CrossRef CAS; (b) T. Kawasaki, K. Masuda, Y. Baba, K. Takada and M. Sakamoto, Chem. Pharm. Bull., 1994, 42, 1974–1976 CrossRef CAS; (c) X. Chen, H. Fan, S. Zhang, C. Yu and W. Wang, Chem.–Eur. J., 2016, 22, 716–723 CrossRef CAS PubMed; (d) Y.-C. Hu, D.-W. Ji, C.-Y. Zhao, H. Z. heng and Q.-A. Chen, Angew.Chem., Int. Ed., 2019, 58, 5438–5442 CrossRef CAS PubMed; (e) W.-S. Jiang, D.-W. Ji, W.-S. Zhang, G. Zhang, X.-T. Min, Y. C. Hu, X.-L. Jiang and Q.-A. Chen, Angew. Chem., Int. Ed., 2021, 60, 8321–8328 CrossRef CAS PubMed.
- For indole reverse-prenylation in alkaloid synthesis, see: (a) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS PubMed; (b) C. A. Kuttruff, H. Zipse and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 1402–1405 CrossRef CAS PubMed; (c) T. Lindel, N. Marsch and S. K. Adla, Top. Curr. Chem., 2012, 309, 67–130 CrossRef CAS PubMed; (d) R. C. Godfrey, H. E. Jones, N. J. Green and A. L. Lawrence, Chem. Sci., 2022, 13, 1313–1322 RSC.
- (a) G. W. Kramer and H. C. Brown, J. Organomet. Chem., 1977, 132, 9–27 CrossRef CAS; (b) Y. Nagashima, K. Sasaki, T. Suto, T. Sato and N. Chida, Chem.–Asian J., 2018, 13, 1024–1028 CrossRef CAS PubMed.
- For indole C2-linear allylation, see: (a) N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821–2847 CrossRef CAS; (b) M. R. Sk, S. S. Bera and M. S. Maji, Org. Lett., 2018, 20, 134–137 CrossRef CAS PubMed; (c) S. Basuli, S. Sahu, S. Saha and M. S. Maji, Adv. Synth. Catal., 2021, 363, 1–8 CrossRef; (d) A. S. P. Cardoso, M. M. B. Marques, N. Srinivasan, S. Prabhakar, A. M. Lobo and H. S. Rzepa, Org. Biomol. Chem., 2006, 4, 3966–3972 RSC; (e) Y.-C. Hu, Y. Li, D.-W. Ji, H. Liu, H. Zheng, G. Zhang and Q.-A. Chen, Chin. J. Catal., 2021, 42, 1593–1607 CrossRef CAS; (f) S. Zhao, T. Gan, P. Yu and J. M. Cook, Tetrahedron Lett., 1998, 39, 7009–7012 CrossRef CAS.
- For allylboronic acids in allylation reactions, see: (a) M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 51, 13050–13053 CrossRef CAS PubMed; (b) R. Alam, A. Das, G. Huang, L. Eriksson, F. Himo and K. J. Szabó, Chem. Sci., 2014, 5, 2732–2738 RSC; (c) C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2–14 CrossRef CAS PubMed.
- For the synthesis of indoline through indole C2-allylation, see: (a) F. Nowrouzi and R. A. Batey, Angew. Chem., Int. Ed., 2013, 52, 892–895 CrossRef CAS PubMed; (b) R. Alam, C. Diner, S. Jonker, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 14417–14421 CrossRef CAS PubMed.
- (a) S. Sahu, B. Das and M. S. Maji, Org. Lett., 2018, 20, 6485–6489 CrossRef CAS PubMed; (b) S. Saha, A. Banerjee and M. S. Maji, Org. Lett., 2018, 20, 6920–6924 CrossRef CAS PubMed; (c) G. Karan, S. Sahu and M. S. Maji, Chem. Commun., 2021, 57, 5274–5277 RSC; (d) S. Sahu, G. Karan, L. Roy and M. S. Maji, Chem. Sci., 2022, 13, 2355–2362 RSC; (e) A. Banerjee, S. Saha and M. S. Maji, J. Org. Chem., 2022, 87, 4343–4359 CrossRef CAS PubMed.
- (a) T. Chen, T. J. Y. Foo and Y.-Y. Yeung, ACS Catal., 2015, 5, 4751–4755 CrossRef CAS; (b) Y. Shi, Z. Ke and Y.-Y. Yeung, Green Chem., 2018, 20, 4448–4452 RSC.
- For reviews on monoterpenoid indole alkaloids, see: (a) J. Hájíček, Collect. Czech. Chem. Commun., 2011, 76, 2023–2083 CrossRef; (b) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC.
- For selected literature on Aspidosperma alkaloids, see: (a) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2013, 135, 19127–19130 CrossRef CAS PubMed; (b) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 6712–6724 CrossRef CAS PubMed; (c) Z. Li, Q. Geng, Z. Lv, B. P. Pritchett, K. Baba, Y. Numajiri, B. M. Stoltz and G. Liang, Org. Chem. Front., 2015, 2, 236–240 RSC; (d) Y. Liu and H. Wang, Chem. Commun., 2019, 55, 3544–3547 RSC; (e) R. Kim, A. J. Ferreira and C. M. Beaudry, Angew. Chem., Int. Ed., 2019, 58, 12595–12598 CrossRef CAS PubMed; (f) Y. Yang, Y. Bai, S. Sun and M. Dai, Org. Lett., 2014, 16, 6216–6219 CrossRef CAS PubMed.
- For the synthesis of the mersicarpine alkaloid, see: (a) T.-S. Kam, G. Subramaniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995–5998 CrossRef CAS; (b) J. Magolan, C. A. Carson and M. A. Kerr, Org. Lett., 2008, 10, 1437–1440 CrossRef CAS PubMed; (c) A. Biechy and S. Z. Zard, Org. Lett., 2009, 11, 2800–2803 CrossRef CAS PubMed; (d) W.-L. Peng, Y.-J. Jhang, C.-Y. Chang, P.-K. Peng, W.-T. Zhao and Y.-K. Wu, Org. Biomol. Chem., 2022, 20, 6193–6195 RSC; (e) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Chem. –Eur. J., 2013, 19, 9325–9334 CrossRef CAS PubMed; (f) Z. Lv, Z. Li and G. Liang, Org. Lett., 2014, 16, 1653–1655 CrossRef PubMed; (g) R. Nakajima, T. Ogino, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 1236–1237 CrossRef CAS PubMed; (h) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Org. Lett., 2012, 14, 2320–2322 CrossRef CAS PubMed; (i) X. Zhong, Y. Li and F.-S. Han, Chem.–Eur. J., 2012, 18, 9784–9788 CrossRef CAS PubMed; (j) M. Frahm, A. Voss and M. Brasholz, Chem. Commun., 2022, 58, 5467–5469 RSC.
- T. Shiobara, Y. Nagumo, R. Nakajima, T. Fukyama, S. Yokoshima and T. Usui, Biosci., Biotechnol., Biochem., 2021, 85, 92–96 CrossRef PubMed.
- See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04732f |
| This journal is © The Royal Society of Chemistry 2024 |