
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-templated synthesis of a switchable fluorescent [2]catenane with sulfate sensing capability†
Krzysztof M.
Bąk
 a,
Bartosz
Trzaskowski
b and
Michał J.
Chmielewski
a
a,
Bartosz
Trzaskowski
b and
Michał J.
Chmielewski
a
aFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, 02-089, Warsaw, Poland. E-mail: mchmielewski@chem.uw.edu.pl
bCentre of New Technologies, University of Warsaw, Banacha 2c, 02-097, Warsaw, Poland
First published on 18th December 2023
Abstract
Anion templation strategies have facilitated the synthesis of various catenane and rotaxane hosts capable of strong and selective binding of anions in competitive solvents. However, this approach has primarily relied on positively charged precursors, limiting the structural diversity and the range of potential applications of the anion-templated mechanically interlocked molecules. Here we demonstrate the synthesis of a rare electroneutral [2]catenane using a powerful, doubly charged sulfate template and a complementary diamidocarbazole-based hydrogen bonding precursor. Owing to the unique three-dimensional hydrogen bonding cavity and the embedded carbazole fluorophores, the resulting catenane receptor functions as a sensitive fluorescent turn-ON sensor for the highly hydrophilic sulfate, even in the presence of a large excess of water. Importantly, the [2]catenane exhibits enhanced binding affinity and selectivity for sulfate over its parent macrocycle and other acyclic diamidocarbazole-based receptors. We demonstrate also, for the first time, that the co-conformation of the catenane may be controlled by reversible acid/base induced protonation and deprotonation of the anionic template, SO42−. This approach pioneers a new strategy to induce molecular motion of interlocked components using switchable anionic templates.
Introduction
Mechanically interlocked molecules (MIMs), such as catenanes, rotaxanes, or knots, have emerged as captivating hosts for molecular recognition,1–5 with ever-expanding applications in sensing,6–12 catalysis,9,13–18 and the construction of molecular machines.18–28 They are particularly appealing for their three-dimensional cavities, which can serve as distinctive, solvent-shielded binding sites,29,30 and for the unique dynamic properties of their interlocked components (e.g. shuttling or pirouetting motions). Anion templation strategies,31,32 pioneered by Beer and co-workers, have facilitated the synthesis of numerous hydrogen bonding33–37 (HB) and halogen bonding38–42 (XB) interlocked hosts capable of strong and selective binding of anions in competitive solvents.43–46 Importantly, such receptors often outperform their non-interlocked analogues in terms of both affinity and selectivity, positioning them as highly promising candidates for molecular sensing applications.47–51To date, however, anion templation strategies have predominantly relied on positively charged precursors, leveraging their capacity for effective preorganization through strong interactions with anionic templates. This significantly limits both the structural diversity and potential applications of anion templated MIMs. For example, anion transport across lipid bilayers requires highly lipophilic, and hence typically uncharged, hosts,52,53 and therefore only very recently has the first [2]catenane anion transporter been described.54 Moreover, the ever-present counterions compete for binding sites of charged receptors, diminishing their selectivity. Thus, further development and future applications of the anion templation methodology depend on the availability of charge neutral and synthetically versatile building blocks, capable of forming orthogonal complexes with strongly interacting anionic templates.
Tetrahedral sulfate with its dinegative charge, strong HB-accepting capabilities, and presumed orthogonal coordination preference, is a particularly attractive anionic template (Fig. 1).55–57 However, there are only two examples of its use in the synthesis of MIMs thus far, both described by Beer and co-workers. The first is a doubly charged [2]catenane, which binds sulfate strongly but less selectively than its acyclic precursor, presumably due to its higher charge.58 The other is an electroneutral triply interlocked capsule, which sequesters sulfate during synthesis, but cannot release it, even in the presence of water.59 In both cases the interlocked cavity proved to be a promising binding site for sulfate, yet with limited functionality and no sensing capabilities.
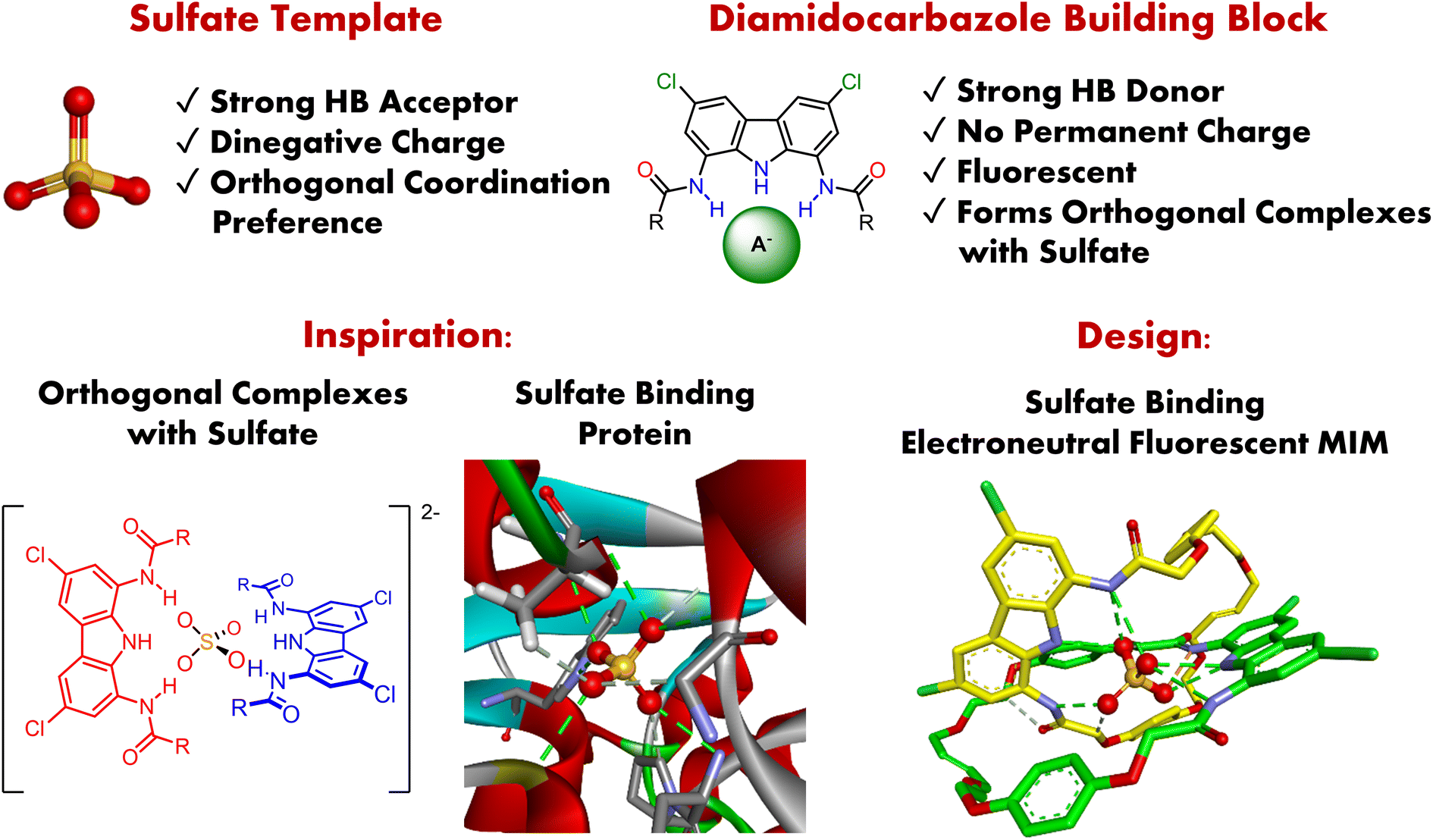 | ||
| Fig. 1 Sulfate template and high affinity diamidocarbazole building blocks enable the synthesis of the electroneutral fluorescent [2]catenane, capable of binding sulfate inside an interlocked hydrogen bonding cavity reminiscent of the binding site of Sulfate Binding Protein (SBP). | ||
In a previous report, we showed that simple and readily available diamidocarbazoles exhibit exceptionally high affinity for sulfate.60 Despite the lack of permanent charge and the presence of only three hydrogen bond donors, they form robust 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ligand:anion) complexes with this anion, even in highly competitive DMSO–water mixtures (Fig. 1). Most importantly, these complexes adopt orthogonal geometry, which is a prerequisite for the successful assembly of interlocked structures. Together with the outstanding fluorescent sensing properties of diamidocarbazoles, this encouraged us to strategically use these building blocks in the anion templated synthesis of electroneutral fluorescent MIMs capable of binding and sensing sulfate.
:1 (ligand:anion) complexes with this anion, even in highly competitive DMSO–water mixtures (Fig. 1). Most importantly, these complexes adopt orthogonal geometry, which is a prerequisite for the successful assembly of interlocked structures. Together with the outstanding fluorescent sensing properties of diamidocarbazoles, this encouraged us to strategically use these building blocks in the anion templated synthesis of electroneutral fluorescent MIMs capable of binding and sensing sulfate.
Sulfate recognition in aqueous media draws significant scientific and technological interest due to its important role in biology, medicine, and industry.61–83 Sulfate is the fourth most abundant anion in human plasma (serum levels 0.3–0.5 mM) and has important functions in human physiology.84,85 For example, it is used in the sulfonation of various endo- and exogenous compounds, which, inter alia, regulate functionality of secreted proteins, hormones, and signaling molecules. Sulfonation is involved in the control of various biological processes, including blood clotting and the formation of connective tissues.86–88 Unusually low levels of sulfate have been found in the plasma of patients with rheumatoid arthritis and irritable bowel disease.89–92 In industry, sulfate is known, for example, to interfere with the remediation of nuclear waste.62 Hence, sulfate-selective sensors could be used to elucidate the diverse roles of sulfate in living organisms, allow improved diagnostics, and facilitate monitoring of environmental samples.93–108
Despite the growing understanding of the biological, environmental, and technological importance of sulfate, examples of its fluorescent sensing in aqueous media are very limited.99–107 In fact, sulfate remains one of the most challenging anionic targets for molecular recognition in water, primarily due to its extremely high hydration energy (ΔG = −1090 kJ mol−1). Therefore, inspired by the sulfate binding protein, which strongly and selectively binds sulfate in water by encapsulating the anion using a network of hydrogen bonds,109–111 we envisaged that strategic incorporation of strong hydrogen bond donors inside a three-dimensional cavity of an electroneutral fluorescent catenane could mimic these characteristics and allow strong binding and selective sensing of sulfate in aqueous media (Fig. 1).
Herein we show that by using powerful, doubly charged anionic template, SO42−, and complementary HB precursors, it is possible to synthesize charge-neutral [2]catenane A (Scheme 1), which is a rare example of an electroneutral MIM achieved by the anion templation strategy.38,54,59,112,113 Importantly, catenane A binds sulfate more strongly and more selectively than its parent macrocycle, its acyclic precursor, and a model receptor containing two diamidocarbazole units linked covalently. Catenane A serves as a sensitive turn-ON fluorescent sensor for the extremely hydrophilic sulfate, even in highly competitive aqueous-organic medium. Additionally, we demonstrate, for the first time, that the relative disposition of the macrocyclic components of the catenane (its co-conformation) can be controlled by reversible acid/base induced protonation–deprotonation of SO42−. This approach pioneers a new strategy to induce reversible molecular motions using switchable anionic templates.
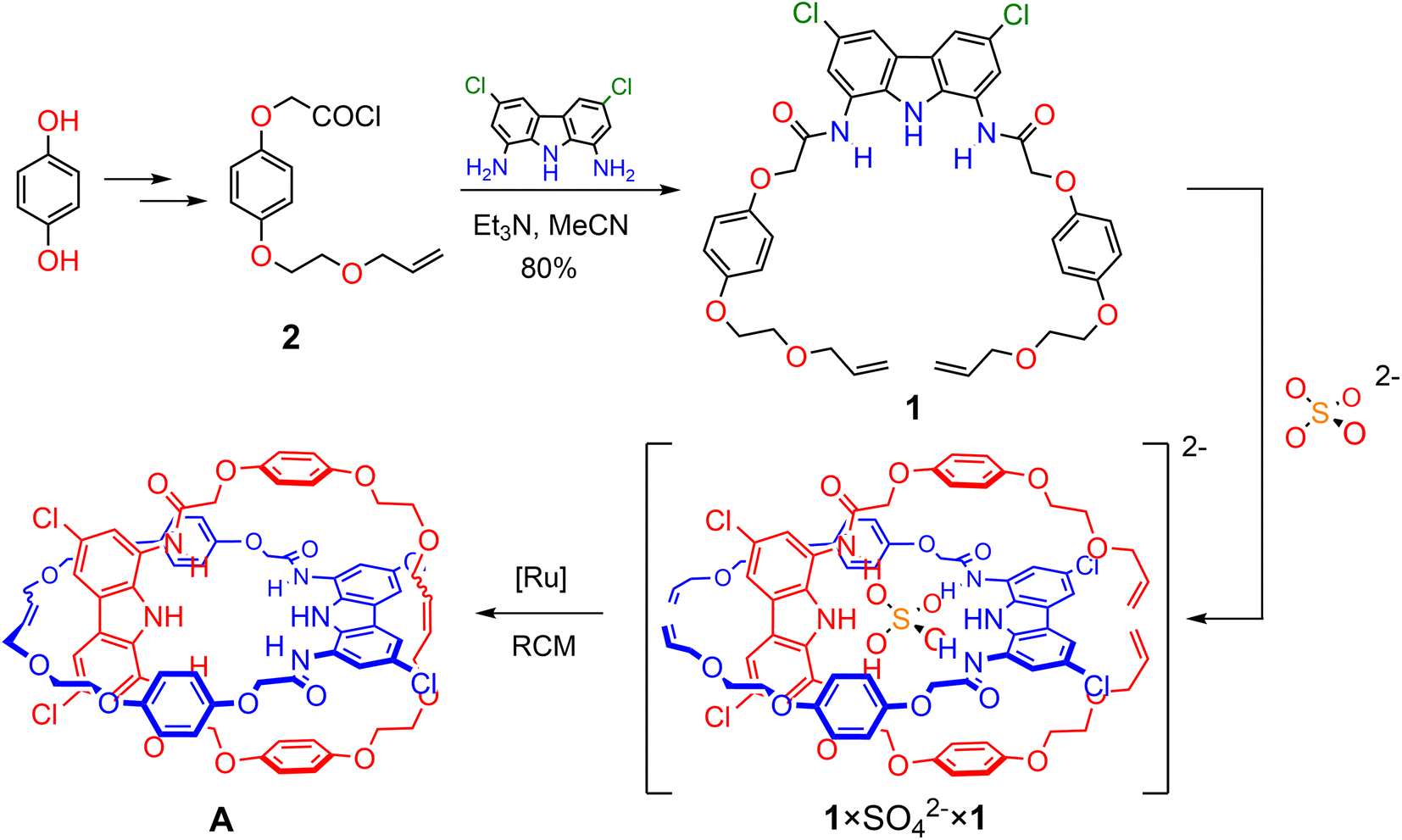 | ||
| Scheme 1 Synthesis of [2]catenane Avia RCM of the orthogonal assembly of 1 × SO42− × 1. | ||
Results and discussion
Synthesis of the [2]catenane precursor and its sulfate-directed self-assembly
Based on our previous studies,60 we envisaged that sulfate directed orthogonal assembly of two appropriately designed diamidocarbazole ligands 1 would enable the synthesis of homo[2]catenane A in a one-pot, double ring-closing metathesis (RCM) reaction (Scheme 1). Alkene-terminated precursor 1 was obtained in a convergent synthesis, ending with the reaction of acyl chloride 2 with 1,8-diamino-3,6-dichlorocarbazole (Scheme 1). The alkene-terminated arm 2 was synthesized in 4 steps from 1,4-hydroquinone (Scheme S1†), and 1,8-diamino-3,6-dichlorocarbazole was obtained in 3 steps from carbazole, according to the previously reported procedure.114,115The sulfate-directed self-assembly of precursor 1 was investigated by 1H NMR titrations. The addition of tetrabutylammonium (TBA) sulfate to the solution of 1 in DMSO-d6 induced large downfield perturbations of the carbazole (α, Δδ ≈ 2.5 ppm) and amide (β, Δδ ≈ 2.0 ppm) NH proton signals, indicating that the anion forms strong hydrogen bonds with these donors (Fig. 2). Interestingly, the signals of the hydroquinone protons B and C shifted significantly upfield (B, Δδ ≈ 0.2 ppm; C, Δδ ≈ 0.4 ppm) until 0.5 equivalent of TBA2SO4 was added, whereas further addition of sulfate shifted them in the opposite direction. At around 1:1 ligand-to-sulfate ratio these signals returned almost to their starting positions and at a host:guest ratio > 1.2 no further changes could be observed (Fig. 2c). A similar perturbation pattern was observed for signals D–F, corresponding to the –OCH2– protons, but the magnitude of these changes decreased with increasing distance from the hydroquinone ring. This characteristic behavior has been previously observed during titrations of simple diamidocarbazoles with TBA sulfate, and attributed to the consecutive formation of 2:1 and 1:1 ligand:anion complexes.60 In the orthogonal 2:1 complex, protons positioned close to the carbazole core of the other ligand experience substantial shielding. However, as the concentration of sulfate increases, the orthogonal 2:1 complex gives way to the 1:1 complex, in which these close contacts are very unlikely.
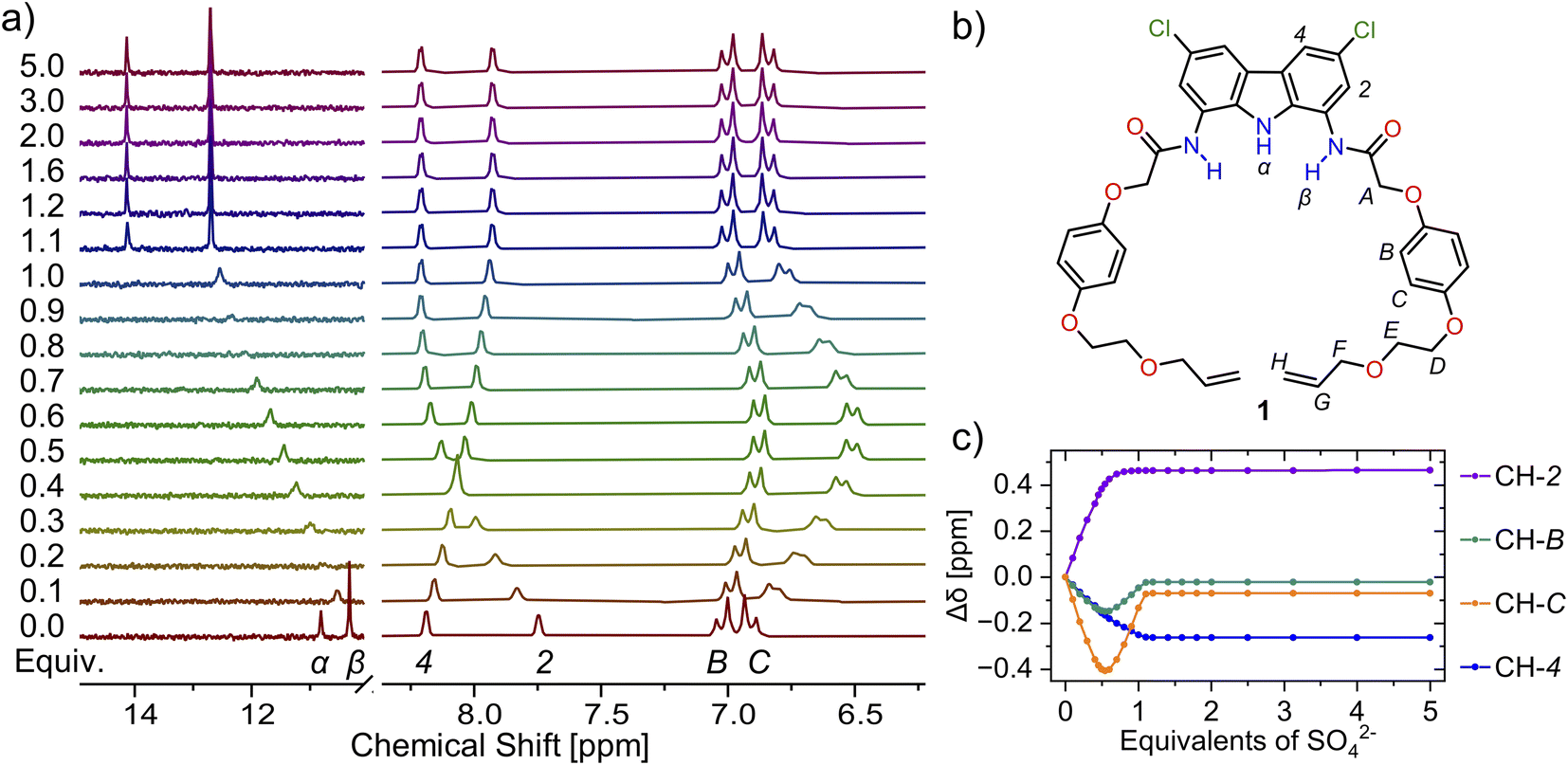 | ||
| Fig. 2 (a) Truncated 1H NMR spectra of precursor 1 (b) with increasing amount of TBA2SO4 in DMSO-d6 + 0.5% H2O. (c) Corresponding binding isotherms. The solid lines are visual aid only. | ||
The lack of strong auxiliary interactions (e.g. π–π stacking of electron-deficient and electron-rich aromatic rings) suggests that the orthogonal assembly depends mostly on the strong hydrogen bonding between the two ligands and the tetrahedral sulfate. Indeed, despite the highly competitive solvent – DMSO-d6, the association constant of electrically neutral receptor 1 with SO42− turned out to be too high to be reliably measured by 1H NMR titrations (K1:1 > 105 M−1, K2:1 > 103 M−1). As DMSO is not compatible with RCM, the analogous binding studies were also conducted in CDCl3. The results confirmed that strong orthogonal assembly also takes place in this medium (Fig. S42 and S43;† K1:1 > 105 M−1, K2:1 > 103 M−1), boding well for the sulfate-templated synthesis of [2]catenane via ring-closing metathesis.
Sulfate-templated synthesis of [2]catenane A
Catenane precursor 1 is poorly soluble in typical solvents used for RCM, such as DCM or toluene. However, it dissolves well in the presence of 0.5 equivalents of TBA sulfate, demonstrating again the strong affinity of diamidocarbazoles to the anionic template. 2nd Generation Grubbs catalyst, which is commonly used in the anion templated synthesis of interlocked structures, turned out to be inactive in the RCM of 1 × SO42− × 1. We suspect that the high basicity of sulfate in anhydrous and aprotic medium is responsible for the rapid deactivation of the catalyst. Screening of other commercially available catalysts showed that only the relatively fast-initiating Hoveyda-type complexes, particularly nitro-Grela (SIPr), allowed for the complete conversion of the substrate. To further limit the exposure of the catalyst to sulfate, the catalyst was slowly added to the reaction mixture using a syringe pump. Also, tetrafluoro-1,4-benzoquinone was added to the reaction mixture to prevent isomerization of the terminal allyloxy groups to unreactive vinyl ethers. The latter unwanted reaction often accompanies olefin metathesis catalysed by Ru complexes.112 Ultimately, two major products were isolated and separated from SO42− template by flash chromatography: [2]catenane A and macrocycle 3 (Fig. 3).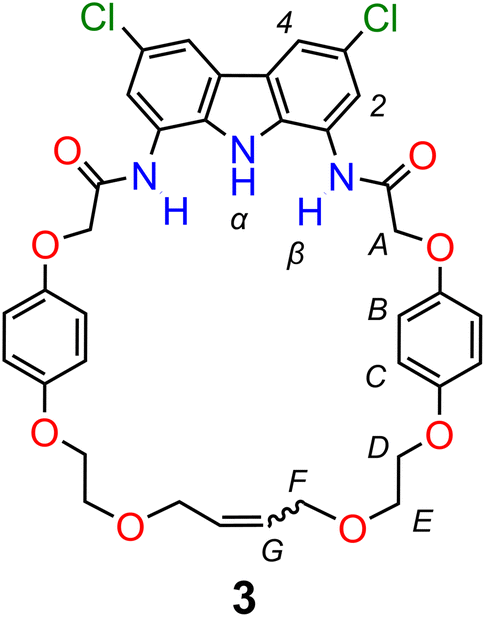 | ||
| Fig. 3 Macrocycle 3 – a side product of the synthesis of [2]catenane A. | ||
The product ratio was found to strongly depend on substrate concentration. While the high concentration of both the precursor and the template favors the formation of the orthogonal 2:1 complex, it also lowers the overall macrocyclization yield due to the formation of insoluble oligomers. On the other hand, very high dilution also decreased the yield of catenane A, likely due to the dissociation of the 2:1 complex. Under optimized conditions (1 mM of 1 in DCM, RT), catenane A was synthesized in 26% yield and macrocycle 3 in 14% yield.
Sulfate templation was found to be indispensable for obtaining catenane A in considerable yield (Table 1). Attempts to synthesize A in the absence of any anionic template failed due to the poor solubility of precursor 1 in DCM. However, the solubility of 1 in chloroform was sufficient to carry out RCM without the addition of any anion. The main product under these conditions was macrocycle 3 (40%) with only trace amounts of catenane A (2%). Chloride, which has been the most popular anionic template for the synthesis of catenanes and rotaxanes,43 did not significantly improve the solubility of 1 in DCM. In chloroform, however, RCM in the presence of 0.5 equivalent of chloride afforded similar product distribution as in the absence of any anion: 43% of macrocycle 3 and 5% of catenane A.
|
|
||||
|---|---|---|---|---|
| Entry | Concentration of 1 | Template | Solvent | Yield of A |
| a Isolated yields of a single run. | ||||
| 1 | 1 mM | 0.5 equiv. SO42− | DCM | 26% |
| 2 | 0.5 mM | 0.5 equiv. SO42− | DCM | 23% |
| 3 | 20 mM | 0.5 equiv. SO42− | DCM | 0% |
| 4 | 1 mM | 1.0 equiv. SO42− | DCM | 4% |
| 5 | 1 mM | — | CHCl3 | 2% |
| 6 | 1 mM | 0.5 equiv. Cl− | CHCl3 | 5% |
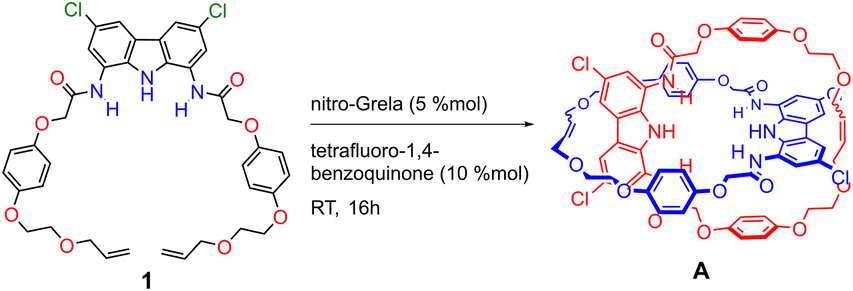
Catenane A and macrocycle 3 were characterized by high-resolution mass spectrometry (HR-MS) and a series of NMR experiments. The HR-MS of A revealed an intense signal at m/z = 1435.54, corresponding to a single-charged ion with an isotopic profile matching [A × Na]+. The 1H NMR spectra of catenane A and macrocycle 3 in DMSO were very similar (Fig. S11†). The only differences were minor upfield perturbations (Δδ ≈ 0.05 ppm) of aromatic CH signals of carbazole and hydroquinone moieties in catenane A, when compared to its parent macrocycle 3. This small shift may be attributed to the proximity of aromatic rings in some conformations of the mechanically interlocked structure. Nevertheless, the interlocked nature of A was further confirmed by 2D 1H–1H ROESY NMR and titration experiments, as described below.
The ROESY NMR spectra of A and 3 in DMSO-d6 did not reveal any specific through-space contacts, which would be unequivocally attributed to the interlocked structure (Fig. S10†). This is likely due to rapid (co-)conformational changes of A on the NMR time scale. However, in the presence of 1 equivalent of TBA2SO4, distinct cross-peaks appeared in the ROESY spectrum of A, due to close contacts between hydroquinone protons B and C, as well as ether protons D–F, with carbazole protons CH-2 and CH-4 (Fig. 4 and S12†). The interlocked nature of A allows the hydroquinone rings of one macrocycle to be positioned above/below the carbazole ring of the other, what becomes clearly visible when sulfate binding rigidifies the whole structure.
 | ||
| Fig. 4 (a) Fragment of the 2D ROESY spectra of A × SO42−. (b) Contacts between protons of the carbazole ring and the hydroquinone ring, which are possible due to the interlocked nature of the receptor. | ||
Anion binding studies of [2]catenane A and macrocycle 3
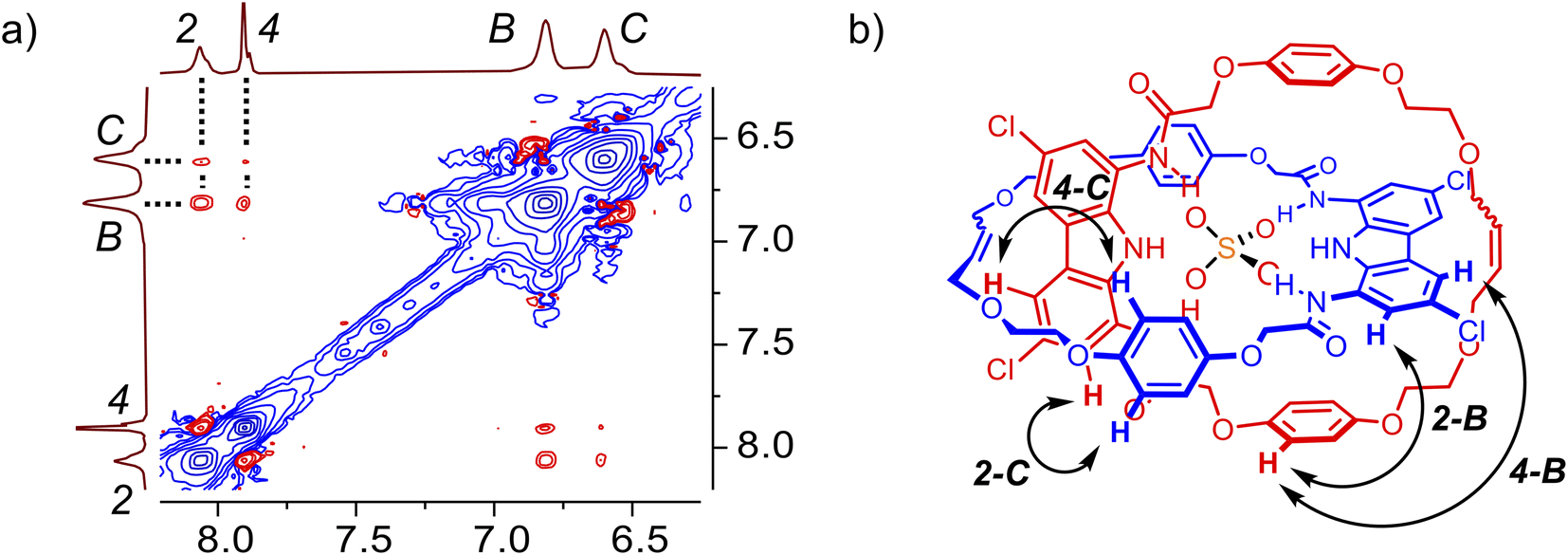
:1 complex with sulfate, in which the anion is bound by two carbazole moieties. Slow exchange on the NMR timescale suggests that sulfate is particularly tightly bound in the interlocked cavity, in contrast to the complexes formed by macrocycle 3 (vide infra). The 1:2 (receptor:anion) complex, which dominates in the presence of excess of sulfate, appears to have a distinct co-conformation, in which the two carbazole subunits individually bind different sulfate ions and presumably move away from each other due to electrostatic repulsion between the anions (Fig. 5a). Another proof of this notable co-conformational change was provided by the ROESY spectrum of catenane A in the presence of 2.5 equivalents of sulfate (Fig. S13†). Specifically, no cross-peaks were found between hydroquinone and carbazole CH signals, in stark contrast to the results obtained in the presence of 1 equivalent of sulfate (vide supra).
 | ||
| Fig. 5 (a) Graphical representation of the complexes formed during the titration of [2]catenane A with TBA2SO4. (b) Truncated 1H NMR spectra of [2]catenane A with increasing amount of TBA2SO4 in DMSO-d6 + 0.5% H2O. | ||
The co-conformational change can also be tracked by diffusion-ordered NMR spectroscopy (DOSY). The addition of 1 equiv. of TBA2SO4 to the solution of free catenane A in DMSO-d6 results in an increase of the diffusion coefficient by 8%, from 1.03 × 10−6 cm2 s−1 to 1.11 × 10−6 cm2 s−1. This increase in mobility despite increasing mass may be attributed to locking of the flexible free catenane in a more compact, contracted co-conformation, characteristic of the 1:1 complex with sulfate. Further addition of sulfate (to 2.5 equiv.) leads to a significant decrease of the diffusion coefficient by 20% (to 0.89 × 10−6 cm2 s−1), much higher than might be expected based on the increasing molecular mass. This is in line, however, with the adoption of the expanded 1:2 complex with a much larger hydrodynamic radius (Fig. S14–S17†).116
Increase of water content significantly simplifies the sulfate binding equilibria of A. In DMSO-d6/H2O 9:1, only the 1:1 complex is formed (Section S5.1.2†), even in the presence of excess of sulfate. This is presumably due to stronger stacking of the catenane's aromatic surfaces in the 1:1 A × SO42− complex as a consequence of the hydrophobic effect.117,118 Moreover, the interlocked cavity efficiently shields the bound guest from the surrounding solvent molecules, further increasing the stability of the complex.119 This result clearly illustrates the potential of interlocked microenvironments in the design of molecular receptors aimed at binding in aqueous media.
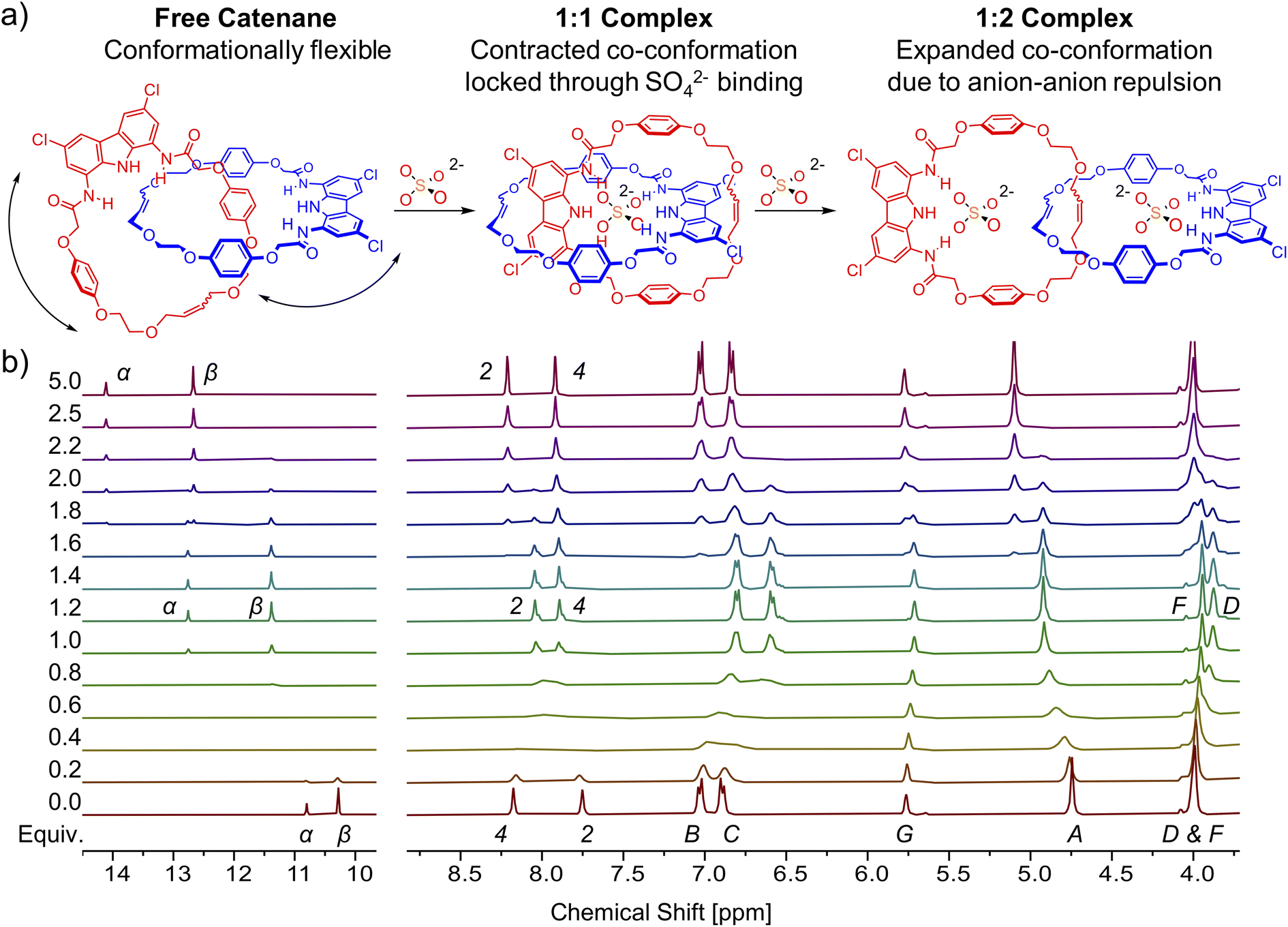
Similar to catenane A, titration of TBA2SO4 to the solution of macrocycle 3 in DMSO-d6 + 0.5% H2O broadens both the NH signals and shifts them significantly downfield (α, Δδ ≈ 3 ppm; β, Δδ ≈ 2.5 ppm). In case of 3, however, no further changes were observed after the addition of 1.2 equivalents of sulfate (Fig. 6). Hydroquinone signals B and C shifted only marginally during the course of the titration, but close inspection of the binding isotherm for signal B revealed a shallow minimum at around 0.5 equivalents of sulfate (Fig. 6c), likely due to its weak shielding by an aromatic ring. Apparently, alongside the 1:1 complex, the macrocycle 3 forms also a 2:1 (receptor:anion) complex. Molecular modeling of the 2:1 complexes using Conformer–Rotamer Ensemble Sampling Tool with the GFN2-xTB approach reveled multiple low energy arrangements,120,121 including orthogonal and sandwich type structures, suggesting that several binding modes could contribute to the overall association (Section S6.3†).
 | ||
| Fig. 6 (a) Graphical representation of the complexes formed during the titration of macrocycle 3 with TBA2SO4. (b) Truncated 1H NMR spectra of macrocycle 3 with increasing amount of TBA2SO4 in DMSO-d6 + 0.5% H2O. (c) Corresponding binding isotherms. The solid lines are visual aid only. | ||
:1. The addition of oxoanions such as sulfate (SO42−), dihydrogen phosphate (H2PO4−) and benzoate (PhCOO−), as TBA salts, elicited a sharp increase of absorbance in the 340–390 nm region of the UV-Vis spectra of catenane A (Fig. 7, S50–S53†), macrocycle 3 (Fig. S56–S61†) and acyclic precursor 1 (Fig. S62–S65†). In contrast, no significant changes were observed upon the addition of chloride (Cl−), indicating that it is not appreciably bound by these receptors in this competitive medium.
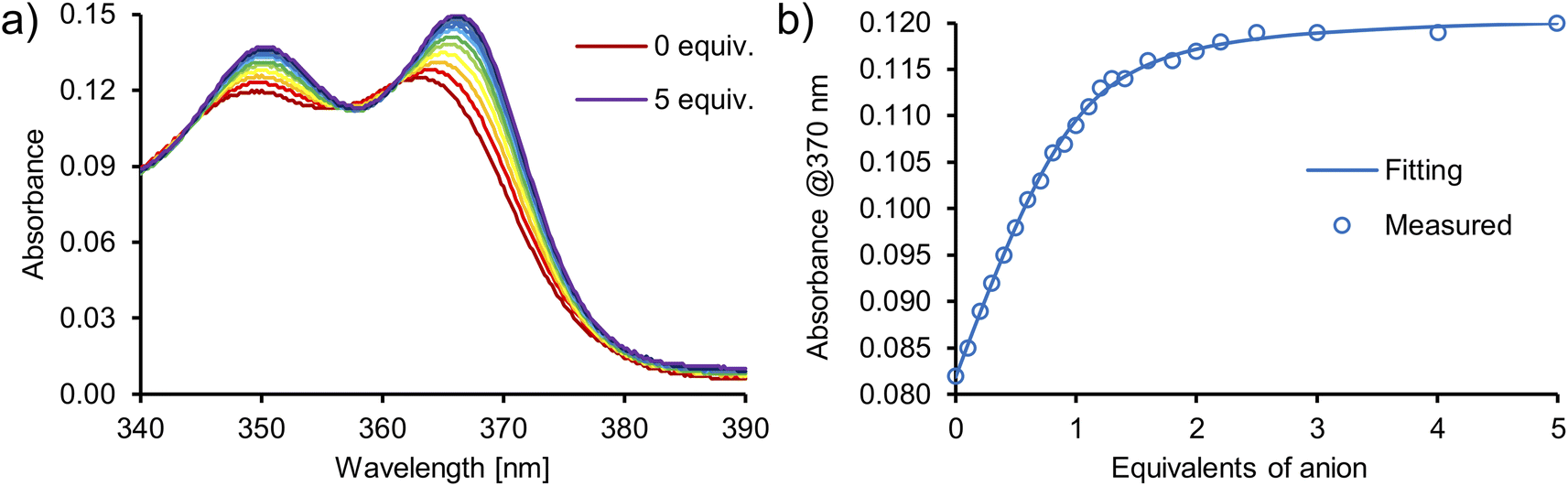 | ||
| Fig. 7 (a) UV-Vis spectra recorded during the titration of [2]catenane A with TBA2SO4 in DMSO/H2O 9:1. (b) 1:1 host–guest binding curve fitted to the results obtained during the titration of [2]catenane A with TBA2SO4 in DMSO/H2O 9:1. | ||
Numerical analysis performed using the HypSpec software suite revealed that catenane A binds sulfate stronger than other oxoanions with a high binding constant of K = 776000 M−1 for the 1:1 complex. This value is almost 10 times higher than that of acyclic precursor 1 and more than 20 times higher than that of macrocycle 3 (Table 2). Surprisingly, macrocycle 3 binds sulfate more weakly than its acyclic precursor 1, presumably due to its increased rigidity and energetic cost associated with conformational changes and solvent rearrangement upon binding. This supposition was supported by the results of DFT calculations (Section S6.4†). Notably, catenane A binds sulfate 2.5 times stronger than the previously reported receptor 4 (Fig. 8), which binds sulfate with two diamidocarbazole moieties connected covalently, rather than by a mechanical bond.103 The observed selectivity trend: SO42− > H2PO4− > PhCOO− > Cl− follows the series of HB acceptor parameters for anions.122 However, the 1:1 association constant of catenane A with sulfate is almost 100 times higher than that of dihydrogen phosphate and over 3000 times higher than that of benzoate, revealing excellent selectivity, which is also significantly enhanced in comparison to acyclic precursor 1, macrocycle 3, and bis(diamidocarbazole) receptor 4. The high affinity and selectivity of catenane A for sulfate could be attributed to exceptionally good size and shape complementarity between the interlocked binding cavity of A and tetrahedral SO42−.
| Receptor | SO42−a | H2PO4−a | PhCOO−a | Selectivity for SO42−vs. H2PO4−b | Selectivity for SO42−vs. PhCOO−b |
|---|---|---|---|---|---|
|
a Measured by UV-Vis titrations in DMSO/H2O 9:1 at 298 K. Error < 15%.
b Calculated as a ratio of K1:1(SO42−) and K1:1(H2PO4−) or K1:1(PhCOO−) for a given receptor. Error < 20%.
|
|||||
| Catenane A | K1:1 = 776000 |
K1:1 = 8130 | K1:1 = 230 | 95 | 3400 |
| K1:2 = 3240 | |||||
| Macrocycle 3 | K1:1 = 32400 |
K1:1 = 760 | K1:1 = 300 | 43 | 110 |
| K2:1 = 2290 | K2:1 = 4900 | ||||
| Acyclic precursor 1 | K1:1 = 83200 |
K1:1 = 2750 | K1:1 = 250 | 30 | 330 |
| K2:1 = 1170 | |||||
| Bis(diamidocarbazole) receptor 4 | K1:1 = 295000 |
K1:1 = 10700 |
K1:1 = 810 | 28 | 360 |
| K1:2 = 1230 | |||||
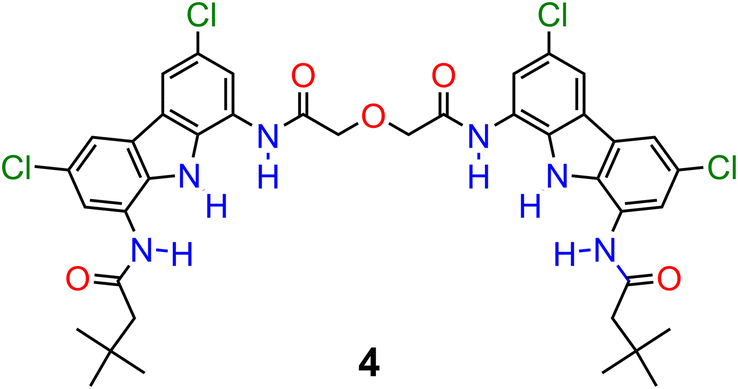 | ||
| Fig. 8 Bis(diamidocarbazole) receptor 4. | ||
Fluorescent sensing of anions by [2]catenane A
The carbazole moiety directly incorporated into the interlocked cavity serves as a convenient fluorescent binding probe. Addition of TBA2SO4 to a 10 μM solution of catenane A in DMSO/H2O 9:1 elicited a vivid increase of fluorescence emission intensity in the 360–440 nm range (up to 60%, Fig. 9). Strong sulfate binding resulted in saturation of the response after only 1.5 equivalents of SO42− (10 μM) were added. In comparison, benzoate did not noticeably affect the fluorescence emission of the catenane in a similar concentration range, and led to only a 15% increase of emission intensity after the addition of 1000 equivalents (Fig. S68†). The turn-ON sensing response is likely due to the restriction of vibrational and rotational modes of the receptor upon anion binding. Such rigidification limits nonradiative internal conversion, which competes with the fluorescence emission. Interestingly, sensing selectivity for sulfate vs. dihydrogen phosphate is even more pronounced, owing to a distinct optical response elicited by phosphate binding. More specifically, the addition of TBAH2PO4 caused a decrease of emission intensity in the 360–400 nm range and the appearance of a new band between 400 and 500 nm, which was saturated after the addition of 125 equivalents of the anion.123 Accordingly, catenane A is a rare example of a turn-ON fluorescent sensor with high sensitivity and selectivity for sulfate (Fig. 9c).
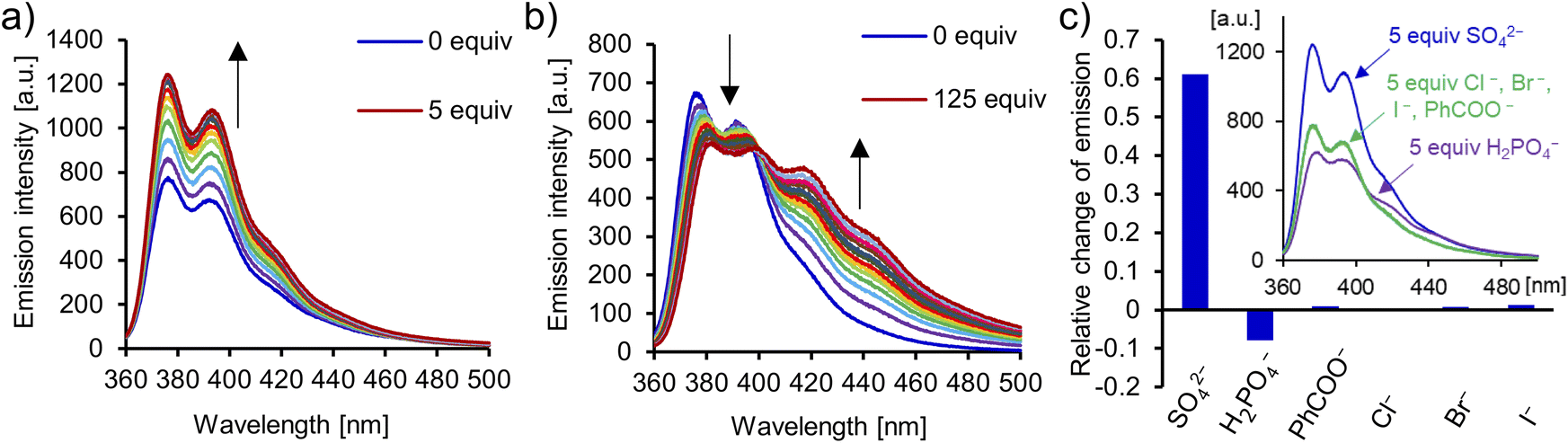 | ||
| Fig. 9 Emission spectra recorded during titration of [2]catenane A with TBA2SO4 (a) and TBAH2PO4 (b) in DMSO/H2O 9:1 (ex: 350 nm). (c) Relative change of emission intensity: (I–I0)/I0 of catenane A at 370 nm in the presence of 5 equiv. of TBA salts. Inset: comparison of emission spectra of [2]catenane A in the presence of 5 equiv. of TBA salts of SO42−, H2PO4−, PhCOO−, and halides (Cl−, Br−, and I−) in DMSO/H2O 9:1 (ex: 350 nm). | ||
Co-conformational switching of [2]catenane A
Although catenane A binds sulfate (SO42−) strongly, it shows virtually no affinity towards bisulfate (HSO4−) in DMSO-d6 (Section S5.1.6†). Since co-conformations of the 1:1 and 1:2 sulfate complexes differ significantly and can be easily distinguished by 1H NMR, we decided to investigate switching between these two well-defined states using acid and base.124 In DMSO, SO42− is much more basic than in water, and therefore can be protonated even by rather weak acids, such as benzoic or acetic acid. However, to simplify NMR spectra, we decided to use strong, NMR-silent trifluoromethanesulfonic (triflic) acid.
In a model experiment, the addition of 2 equivalents of sulfate to the 2 mM solution of catenane A in DMSO-d6 led to the formation of the 1:2 (receptor:anion) complex, in which each carbazole moiety binds its own sulfate. Then, the addition of 1 equivalent of triflic acid protonated one of the sulfates to the poorly bound HSO4−, enforcing rotation of the interlocked subunits to form the 1:1 complex, in which both carbazole units bind the same anion (Fig. 10). The change of co-conformation manifested as the upfield shift of hydroquinone proton signals in 1H NMR. Next, 1 equivalent of TBAOH was added to regenerate sulfate and trigger rotation of the subunits back to the 1:2 complex. This switching cycle was repeated eight times with excellent reproducibility. The switching between SO42− with strong HB accepting capabilities and HSO4− with poor HB accepting capabilities122 is reminiscent of the much better established switching of cationic templates, such as Cu2+/Cu+ switching elicited by redox reactions.125
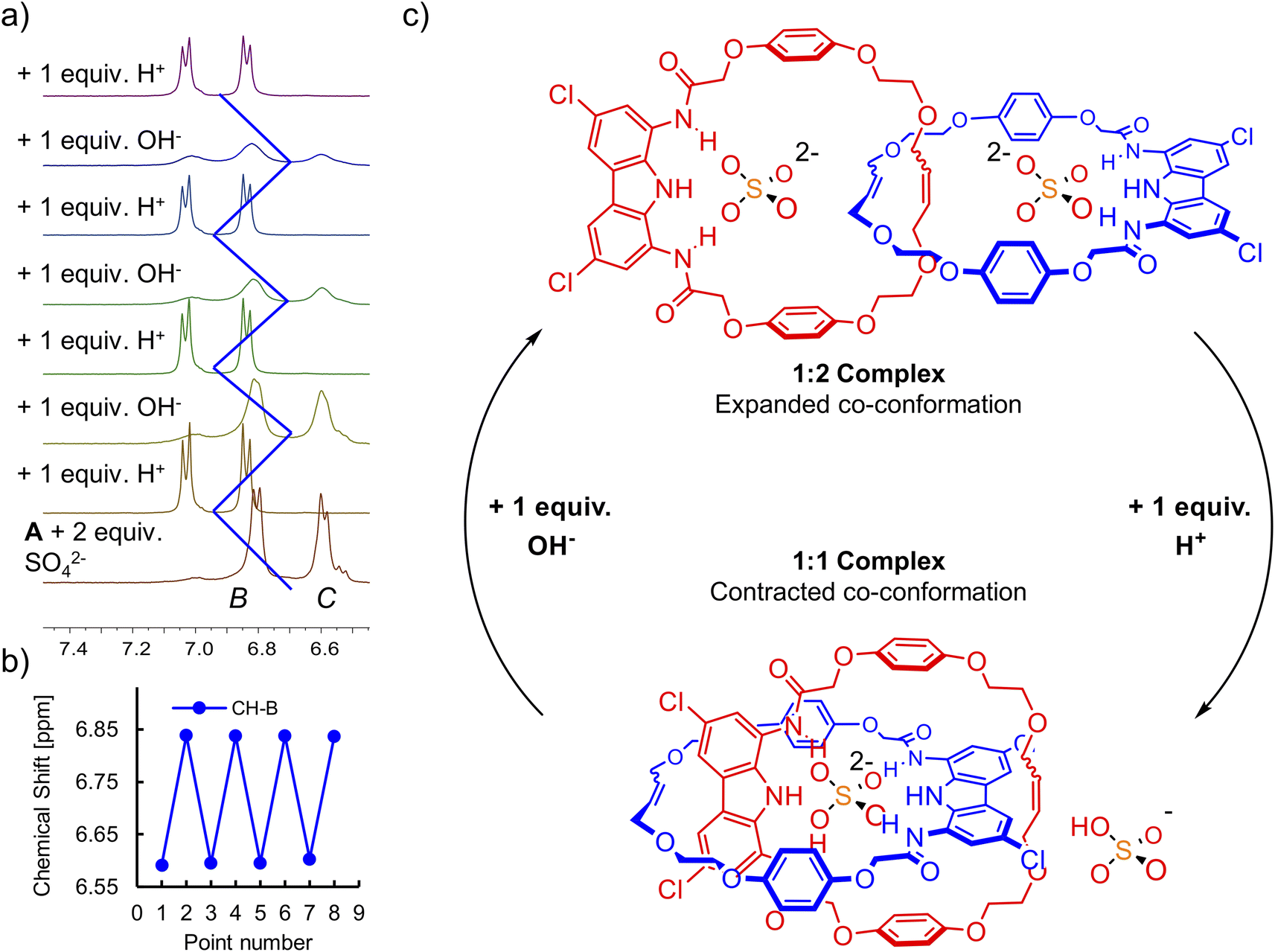 | ||
| Fig. 10 (a) Acid-base triggered switching between the 1:1 and 1:2 complexes of [2]catenane A and sulfate tracked by 1H NMR. (b) Chemical shift of signal CH–B during the 1H NMR switching experiment. (c) Graphical representation of the switching cycle. | ||
Computational studies of [2]catenane A
Further insights into sulfate complexation and anion-induced rotary motions of catenane A were obtained through conformational analysis and molecular dynamics (MD) simulations undertaken with the Macromodel suite, using OPLS 2005/2015 and Charmm General v. 3.0.1 force fields (Sections S6.1 and S6.2†). The initial conformational search performed for catenane A revealed three major conformations (Fig. 11). In the lowest-energy conformation both diamidocarbazole moieties are situated orthogonally (Fig. 11a). One of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of each diamidocarbazole moiety is pointing towards the center of the cavity, forming an intramolecular hydrogen bond with the carbazole NH. Additionally, it also forms hydrogen bonds with two NHs of the other diamidocarbazole, which leads to the formation of an array of 6 intramolecular hydrogen bonds. The conformation is additionally stabilized by π-stacking interactions between the carbazole moiety of one macrocycle and the hydroquinone moieties of the other.
O groups of each diamidocarbazole moiety is pointing towards the center of the cavity, forming an intramolecular hydrogen bond with the carbazole NH. Additionally, it also forms hydrogen bonds with two NHs of the other diamidocarbazole, which leads to the formation of an array of 6 intramolecular hydrogen bonds. The conformation is additionally stabilized by π-stacking interactions between the carbazole moiety of one macrocycle and the hydroquinone moieties of the other.
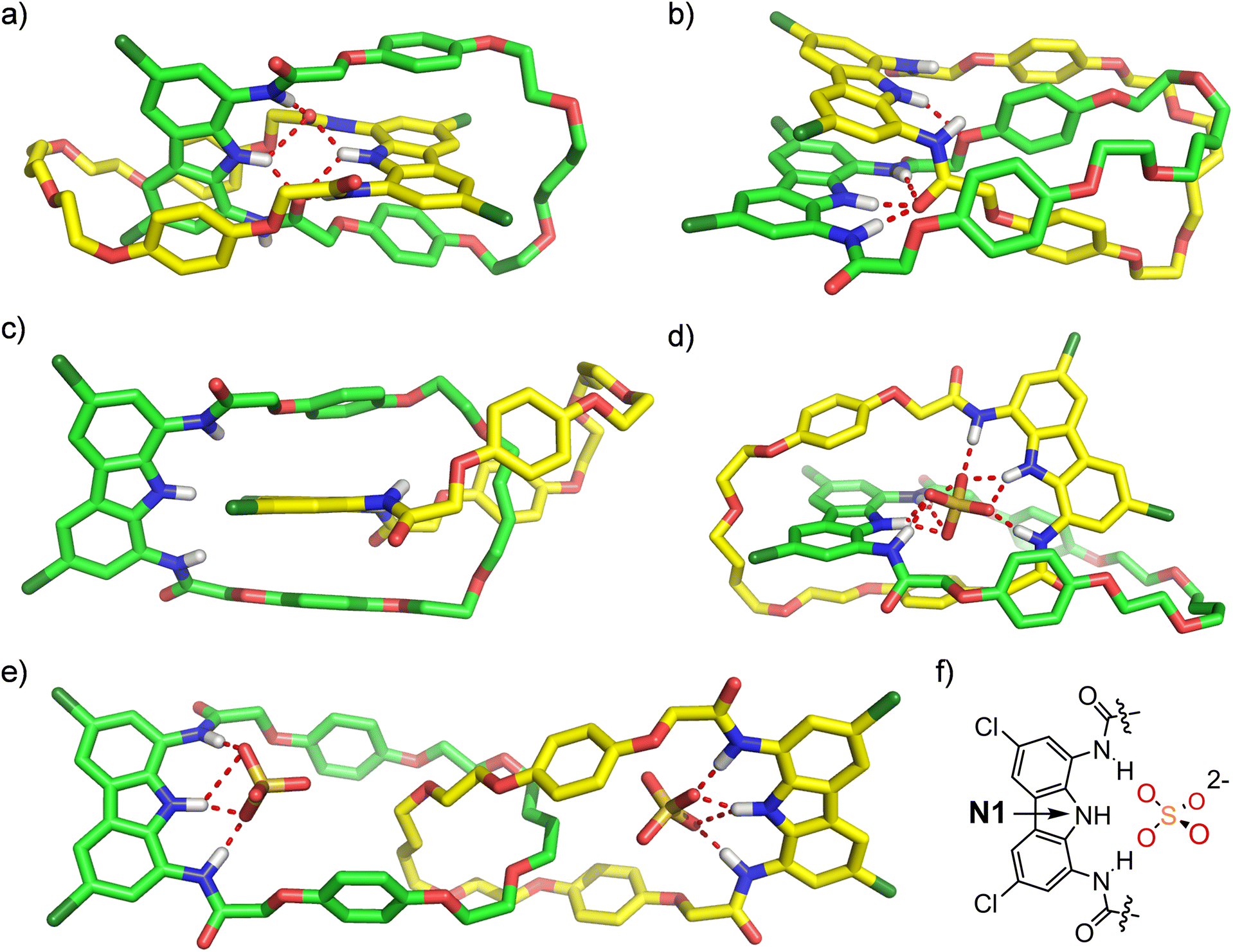 | ||
| Fig. 11 Low-energy conformations (a–c) of free catenane A obtained from conformational search. Snapshots from MD simulations showing stable conformations of (d) A × SO42− and (e) SO42− × A × SO42−. (f) Atom labels used in the discussion of MD simulations. | ||
In the second most stable conformation the two diamidocarbazoles are situated in a parallel manner and are held together by π–π stacking interactions and four HBs (Fig. 11b). Three of these HBs are formed between the three NH groups of one diamidocarbazole and the CO HB acceptor of the other, and the fourth HB binds together the central carbazole NH and the hydroquinone oxygen atom of the other macrocycle. This conformation is additionally stabilized by π–π stacking interactions of three hydroquinone moieties, where two hydroquinones of one macrocycle sandwich the hydroquinone ring of the other. The conformation was found to be 6.3 kcal mol−1 energetically less stable than the first conformation, apparently due to the lower number of HBs.
The third conformation is not stabilized by any HBs, and is therefore 15.5 kcal mol−1 higher in energy than the first one. It is only stabilized by π–π stacking interactions between two hydroquinone moieties of one macrocycle and the carbazole moiety of the other (Fig. 11c).
No other significantly different conformations were found in the 25 kcal mol−1 window. However, MD simulations of the three conformations revealed that they are very dynamic, with multiple low-energy states observed even during 100 ns simulations at 25 °C. While the hydrogen-bonded cores of the two most stable conformations are largely preserved during the MD runs, the π–π stacking interactions are easily broken. This might be responsible for the lack of specific contacts in 1H ROESY spectrum of free catenane A (vide supra).
A similar MD analysis was performed for the 1:1 and 1:2 sulfate complexes. In the 1:1 complex, sulfate is bound inside the interlocked cavity of the catenane by nine strong hydrogen bonds, formed by all six NH donors from both diamidocarbazole moieties (Fig. 11d). It is worth noting that while sulfate binding disrupts the array of hydrogen bonds observed in the lowest-energy conformation of A, it does not induce any major changes in the mutual orientation of the macrocyclic components. Also, despite the breaking of 6 intramolecular hydrogen bonds, sulfate binding is still energetically favorable by approximately 79.9 kcal mol−1. The complex is very stable over the simulation time and unlikely to undergo any major conformational changes. During the entire 100 ns run the two N1(diamidocarbazole)-S(sulfate) distances vary only between 3.2 and 3.5 Å. During this time however, the sulfate rotates in the cavity quickly (Fig. S72b†).
The co-conformation of the 1:2 complex is significantly different. Each sulfate anion is bound by a single diamidocarbazole moiety, forming 4 hydrogen bonds in total, with either two (edge binding) or three (plane binding) oxygen atoms of the anion (Fig. 11e). Due to the electrostatic repulsion between the anions, both diamidocarbazoles are pushed away from each other to an ‘expanded’ co-conformation, which is in agreement with the ROESY and DOSY spectra of the complex. During the 100 ns simulation run, diamidocarbazole moieties maintain the relatively long distance of 15 to 22 Å (measured between the nitrogen atoms of the carbazole rings), as the two macrocycles move to a certain extent with respect to each other. This means that the 1:2 complex is significantly more flexible than the 1:1 complex. However, both the N1(diamidocarbazole)-S(sulfate) distances are very stable during the entire 100 ns simulation and remain in the 3.0–3.7 Å range. This means that both sulfates are tightly bound in their respective binding sites, although rotate even faster than in the 1:1 complex (Fig. S68b†).
To further investigate the conformational change between the 1:1 and 1:2 sulfate complexes, we performed MD runs for the 1:2 complex from which one sulfate anion and two random TBA cations were removed, keeping the system electrically neutral. The exemplary MD run resulted in rapid change of the catenane co-conformation, which converged to the low-energy 1:1 complex in less than 1 ns (Fig. 12). This change from the ‘expanded’ to the ‘contracted’ co-conformation, driven by chelation of the sulfate by the second diamidocarbazole moiety, leads to a drastic decrease (from ca. 20 Å to ca. 9 Å) of the distance between the N1 nitrogen atoms of the two carbazoles, as well as of the distance between one of the carbazole's N1 atoms and the sulfate's sulfur atom (from 17 Å to 3.5 Å).
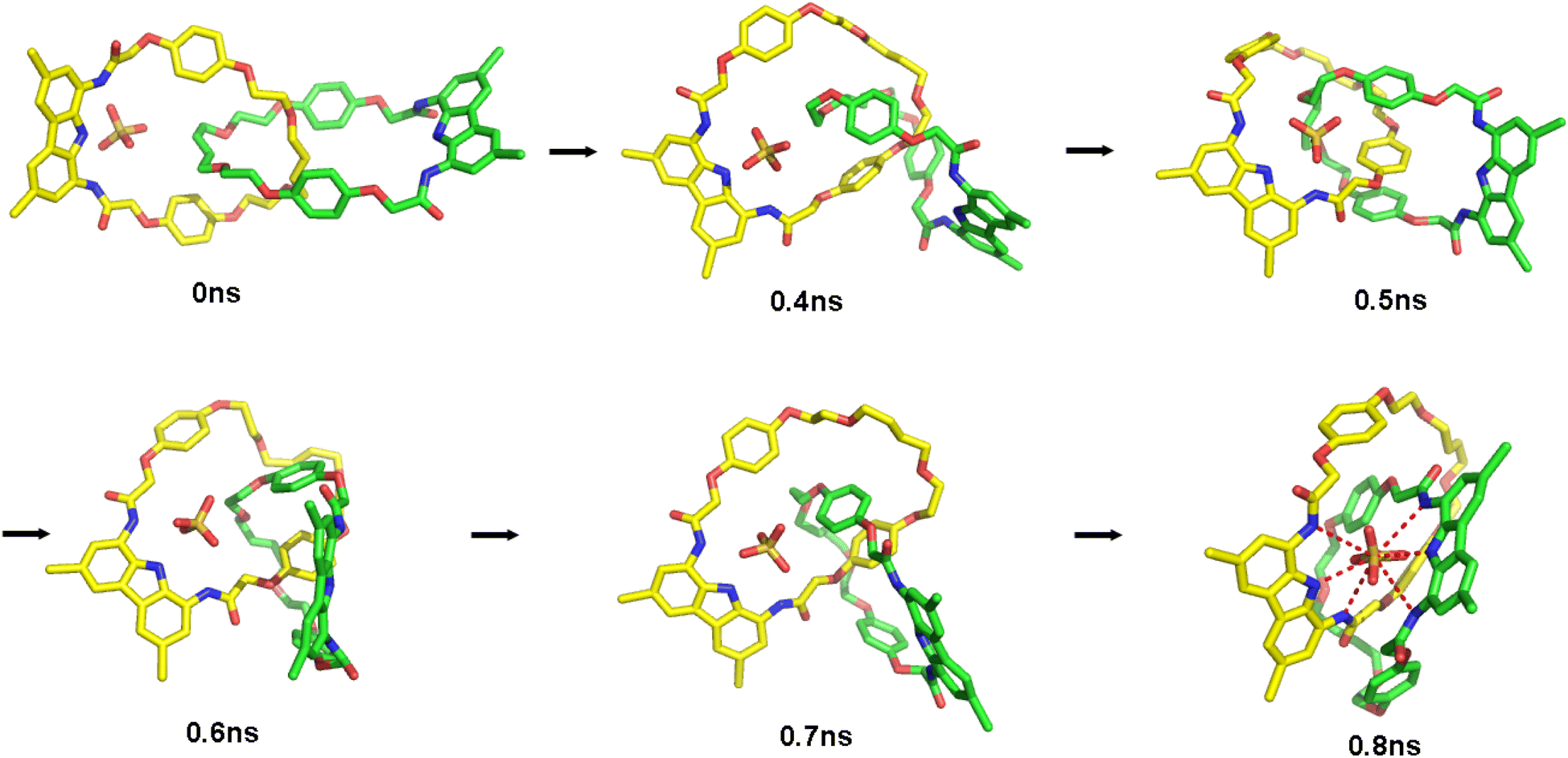 | ||
| Fig. 12 Snapshots from MD runs showing switch from the ‘expanded’ to the ‘contracted’ co-conformation of catenane A. | ||
Upon the transition to the ‘contracted’ co-conformation, the system becomes energetically and geometrically stable. In some MD runs, however, removal of one sulfate from the 1:2 complex resulted in a conformation, in which one of the macrocycles binds sulfate using all three NH groups, while the other uses only a single amide NH (Fig. S77†). As a result, the sulfate is bound more weakly than in the ‘contracted’ co-conformation described above, and the distance between sulfur and nitrogen of one carbazole ring remains relatively large: on average 6.5 Å. Energy data suggest that this conformation is energetically less favorable than the most stable one by approximately 30–40 kcal mol−1, but might be quite stable kinetically, since no major conformational/energy changes were observed during the entire 50 ns MD run. This is because the metastable conformation is stabilized by the intramolecular HB between amide CO and carbazole NH, and this HB has to be broken to allow transition to the most stable arrangement. It is likely therefore, that following sulfate removal from the 1:2 complex the resulting 1:1 complex becomes temporarily trapped in this high-energy, metastable state, although ultimately converges to the lowest-energy conformation. This computational result highlights challenges associated with the development of fast molecular switches based on hydrogen bonding interactions.
Conclusions
In this work, we have developed an anion-templated synthesis of electroneutral [2]catenane with remarkable affinity, selectivity, and a strong turn-ON fluorescence response towards sulfate anion, SO42−. Anion binding studies revealed that the catenane binds the extremely hydrophilic sulfate strongly even in the presence of large excess of water, and with high selectively over other oxoanions, notably over dihydrogen phosphate H2PO4−. Sulfate is bound inside the interlocked cavity by two diamidocarbazole moieties, forming strong, hydrogen bonded 1:1 complex. The excellent size and shape complementarity between the interlocked host and sulfate guest resulted in a significant increase of binding strength and selectivity when compared to the acyclic precursor, the parent macrocycle, and the acyclic receptor containing two similar diamidocarbazole moieties connected covalently by a flexible linker.
Our results indicate that by using a powerful anionic template and complementary high-affinity building blocks, it is possible to expand the scope of anion templated synthesis, which is therefore no longer limited to positively charged building blocks. Our findings showcase sulfate as a highly effective anionic template for the synthesis of MIMs and potentially also other complex supramolecular structures, such as helicates, foldamers and capsules, from electroneutral components. Moreover, diamidocarbazole has emerged from this study as a privileged building block for the anion templated synthesis, programmed self-assembly and the construction of highly sensitive fluorescent receptors.
Furthermore, sulfate may be used as a unique switchable template, able to enforce large amplitude molecular motions within an interlocked structure. Therefore, SO42− fills the gap of missing switchable anionic templates and further expands the applications of anion templation strategies.
The unique properties of catenane A illustrate the utility of electroneutral ion binding MIMs as receptors, sensors and molecular switches, with promising future applications in extraction, transmembrane transport, and the development of responsive devices and materials.
Data availability
General information, experimental procedures, characterization data for all new compounds, raw data and fitting procedures for 1H NMR, UV-Vis and fluorescence titration, detailed MD analysis and additional figures are available in the ESI.†Author contributions
Krzysztof M. Bąk: conceptualization, data curation, formal analysis, investigation (synthesis, binding studies), developing methodology, validation of results, visualization, writing – original draft, writing – reviewing & editing. Bartosz Trzaskowski: formal analysis (MD simulations), investigation (MD simulations), visualization (MD simulations), writing – review & editing. Michał J. Chmielewski: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – reviewing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland (OPUS grant 2018/31/B/ST5/02085 to M. J. C.). This study was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within a project co-financed by the European Union through the European Regional Development Fund under the Operational Programme Innovative Economy 2007–2013.Notes and references
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond, John Wiley & Sons, Hoboken, New Jersey, 2017 Search PubMed.
- D. Sluysmans and J. F. Stoddart, Trends Chem., 2019, 1, 185–197 CrossRef CAS.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef.
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- S. J. Nicholson, S. R. Barlow and N. H. Evans, Chemistry, 2023, 5, 106–118 CrossRef CAS.
- H. M. Tay and P. Beer, Org. Biomol. Chem., 2021, 19, 4652–4677 RSC.
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC.
- N. H. Evans, Chem.–Eur. J., 2018, 24, 3101–3112 CrossRef CAS.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434–2455 CrossRef CAS.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 RSC.
- A. W. Heard, J. M. Suárez and S. M. Goldup, Nat. Rev. Chem, 2022, 6, 182–196 CrossRef.
- C. Kwamen and J. Niemeyer, Chem.–Eur. J., 2021, 27, 175–186 CrossRef CAS PubMed.
- A. Martinez-Cuezva, A. Saura-Sanmartin, M. Alajarin and J. Berna, ACS Catal., 2020, 10, 7719–7733 CrossRef CAS.
- N. Pairault and J. Niemeyer, Synlett, 2018, 29, 689–698 CrossRef CAS.
- D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- M. N. Tasbas, E. Sahin and S. Erbas-Cakmak, Coord. Chem. Rev., 2021, 443, 214039 CrossRef CAS.
- A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- I. Aprahamian, ACS Cent. Sci., 2020, 6, 347–358 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS.
- C. Cheng and J. F. Stoddart, ChemPhysChem, 2016, 17, 1780–1793 CrossRef CAS.
- M. A. Watson and S. L. Cockroft, Chem. Soc. Rev., 2016, 45, 6118–6129 RSC.
- E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- Y. Liu, F. C. Parks, E. G. Sheetz, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2021, 143, 3191–3204 CrossRef CAS PubMed.
- K. M. Bąk, S. C. Patrick, X. Li, P. D. Beer and J. J. Davis, Angew. Chem., Int. Ed., 2023, 62, e202300867 CrossRef PubMed.
- R. Vilar, Eur. J. Inorg. Chem., 2008, 357–367 CrossRef CAS.
- R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460–1477 CrossRef CAS.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- T. A. Barendt, L. Ferreira, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 9026–9037 CrossRef CAS PubMed.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463–11466 CrossRef CAS PubMed.
- M. J. Langton and P. D. Beer, Chem. Commun., 2014, 50, 8124–8127 RSC.
- M. J. Langton, L. C. Duckworth and P. D. Beer, Chem. Commun., 2013, 49, 8608 RSC.
- A. Docker, Y. C. Tse, H. M. Tay, A. J. Taylor, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2022, 61, e202214523 CrossRef CAS.
- G. Turner, A. Docker and P. D. Beer, Dalton Trans., 2021, 50, 12800–12805 RSC.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584–588 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657–668 CrossRef CAS.
- M. D. Lankshear and P. D. Beer, Coord. Chem. Rev., 2006, 250, 3142–3160 CrossRef CAS.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS PubMed.
- D. A. McNaughton, M. Fares, G. Picci, P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2021, 427, 213573 CrossRef CAS.
- S. E. Bodman and S. J. Butler, Chem. Sci., 2021, 12, 2716–2734 RSC.
- R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS.
- P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS.
- N. Akhtar, O. Biswas and D. Manna, Chem. Commun., 2020, 56, 14137–14153 RSC.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
- H. Min Tay, T. G. Johnson, A. Docker, M. J. Langton and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202312745 CrossRef CAS PubMed.
- K. M. Mullen and P. D. Beer, Chem. Soc. Rev., 2009, 38, 1701–1713 RSC.
- L. Liang, W. Zhao, X.-J. Yang and B. Wu, Acc. Chem. Res., 2022, 55, 3218–3229 CrossRef CAS PubMed.
- M. J. Chmielewski, L. Zhao, A. Brown, D. Curiel, M. R. Sambrook, A. L. Thompson, S. M. Santos, V. Félix, J. J. Davis and P. D. Beer, Chem. Commun., 2008, 3154–3156 RSC.
- B. Huang, S. M. Santos, V. Félix and P. D. Beer, Chem. Commun., 2008, 4610–4612 RSC.
- Y. Li, K. M. Mullen, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, Chem. Commun., 2009, 7134–7136 RSC.
- K. M. Bąk and M. J. Chmielewski, Chem. Commun., 2014, 50, 1305–1308 RSC.
- I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077–3098 RSC.
- B. A. Moyer, R. Custelcean, B. P. Hay, J. L. Sessler, K. Bowman-James, V. W. Day and S.-O. Kang, Inorg. Chem., 2013, 52, 3473–3490 CrossRef CAS PubMed.
- C. Jia, W. Zuo, D. Zhang, X.-J. Yang and B. Wu, Chem. Commun., 2016, 52, 9614–9627 RSC.
- Y.-C. He, H.-B. Tong, C.-G. Xu, Z.-X. Ren, Y.-Z. Wang, Y.-M. Yan and M.-L. Wang, Org. Lett., 2023, 25, 1737–1741 CrossRef CAS.
- S.-Q. Chen, W. Zhao and B. Wu, Front. Chem., 2022, 10, 905563 CrossRef CAS.
- N. A. Tzioumis, D. A. Cullen, K. A. Jolliffe and N. G. White, Angew. Chem., Int. Ed., 2023, 62, e202218360 CrossRef CAS PubMed.
- W. Zhang, Y. Feng, B. Li, D. Yang, L. Hou, W. Zhao, X. Yang and B. Wu, Chem.–Eur. J., 2022, 28, e202103671 CrossRef CAS PubMed.
- S.-Q. Chen, S.-N. Yu, W. Zhao, L. Liang, Y. Gong, L. Yuan, J. Tang, X.-J. Yang and B. Wu, Inorg. Chem. Front., 2022, 9, 6091–6101 RSC.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- L. Qin, J. R. Wright, J. D. E. Lane, S. N. Berry, R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2019, 55, 12312–12315 RSC.
- L. Qin, A. Hartley, P. Turner, R. B. P. Elmes and K. A. Jolliffe, Chem. Sci., 2016, 7, 4563–4572 RSC.
- V. J. Dungan, H. T. Ngo, P. G. Young and K. A. Jolliffe, Chem. Commun., 2013, 49, 264–266 RSC.
- H. Xie, T. J. Finnegan, V. W. Liyana Gunawardana, R. Z. Pavlović, C. E. Moore and J. D. Badjić, J. Am. Chem. Soc., 2021, 143, 3874–3880 CrossRef CAS PubMed.
- S. Xiong and Q. He, Chem. Commun., 2021, 57, 13514–13517 RSC.
- Z. Huang, C. Jia, B. Wu, S. Jansone-Popova, C. A. Seipp and R. Custelcean, Chem. Commun., 2019, 55, 1714–1717 RSC.
- N. J. Williams, C. A. Seipp, K. A. Garrabrant, R. Custelcean, E. Holguin, J. K. Keum, R. J. Ellis and B. A. Moyer, Chem. Commun., 2018, 54, 10048–10051 RSC.
- R. Custelcean, N. J. Williams, C. A. Seipp, A. S. Ivanov and V. S. Bryantsev, Chem.–Eur. J., 2016, 22, 1997–2003 CrossRef CAS PubMed.
- R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525–10529 CrossRef CAS PubMed.
- W. A. Al Isawi, A. Z. Salome, B. M. Ahmed, M. Zeller and G. Mezei, Org. Biomol. Chem., 2021, 19, 7641–7654 RSC.
- M. Zaleskaya, M. Karbarz, M. Wilczek, Ł. Dobrzycki and J. Romański, Inorg. Chem., 2020, 59, 13749–13759 CrossRef CAS PubMed.
- D. Jagleniec, Ł. Dobrzycki, M. Karbarz and J. Romański, Chem. Sci., 2019, 10, 9542–9547 RSC.
- A. Schaly, R. Belda, E. García-España and S. Kubik, Org. Lett., 2013, 15, 6238–6241 CrossRef CAS PubMed.
- C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS PubMed.
- P. A. Dawson, S. Petersen, R. Rodwell, P. Johnson, K. Gibbons, A. McWhinney, F. G. Bowling and H. D. McIntyre, BMC Pregnancy Childbirth, 2015, 15, 96 CrossRef PubMed.
- D. E. C. Cole and J. Evrovski, Crit. Rev. Clin. Lab. Sci., 2000, 37, 299–344 CrossRef CAS PubMed.
- A. W. Y. Leung, I. Backstrom and M. B. Bally, Oncotarget, 2016, 7, 55811–55827 CrossRef PubMed.
- S. Lee, P. A. Dawson, A. K. Hewavitharana, P. N. Shaw and D. Markovich, Hepatology, 2006, 43, 1241–1247 CrossRef CAS PubMed.
- S. Wu, W. L. Green, W. Huang, M. T. Hays and I. J. Chopra, Thyroid, 2005, 15, 943–958 CrossRef CAS PubMed.
- P. A. Dawson, C. S. Russell, S. Lee, S. C. McLeay, J. M. Van Dongen, D. M. Cowley, L. A. Clarke and D. Markovich, J. Clin. Invest., 2010, 120, 706–712 CrossRef CAS PubMed.
- A. V. Nieuw Amerongen, J. G. M. Bolscher, E. Bloemena and E. C. I. Veerman, Biol. Chem., 1998, 379, 1–18 CAS.
- S. H. Murch, T. T. MacDonald, J. A. Walker-Smith, P. Lionetti, M. Levin and N. J. Klein, Lancet, 1993, 341, 711–714 CrossRef CAS PubMed.
- A. B. Olomu, C. R. Vickers, R. H. Waring, D. Clements, C. Babbs, T. W. Warnes and E. Elias, N. Engl. J. Med., 1988, 318, 1089–1092 CrossRef CAS PubMed.
- P. Piotrowski, J. Bukowska, B. Pałys, R. Pomorski and M. J. Chmielewski, Sens. Actuators, B, 2019, 283, 172–181 CrossRef CAS.
- P. Gołębiewski, B. Puciłowski, F. Sommer, S. Kubik, M. Daniels, W. Dehaen, U. Sivasankaran, K. G. Kumar, H. Radecka and J. Radecki, Sens. Actuators, B, 2019, 285, 536–545 CrossRef.
- P. Gołębiewski, F. Sommer, S. Kubik, H. Radecka and J. Radecki, J. Electroanal. Chem., 2018, 812, 249–257 CrossRef.
- M. Zaleskaya, D. Jagleniec and J. Romański, Dalton Trans., 2021, 50, 3904–3915 RSC.
- J. Bartl, L. Reinke, M. Koch and S. Kubik, Chem. Commun., 2020, 56, 10457–10460 RSC.
- K. T. Hamorsky, C. M. Ensor, P. Pasini and S. Daunert, Anal. Biochem., 2012, 421, 172–180 CrossRef CAS PubMed.
- P. J. Pacheco-Liñán, C. Alonso-Moreno, F. Carrillo-Hermosilla, A. Garzón-Ruiz, C. Martín, C. Sáez, J. Albaladejo and I. Bravo, ACS Sens., 2021, 6, 3224–3233 CrossRef PubMed.
- L.-X. Huang, H.-Y. Bai, H. Tao, G. Cheng and Q.-Y. Cao, Dyes Pigm., 2020, 181, 108553 CrossRef CAS.
- A. M. Agafontsev, T. A. Shumilova, P. A. Panchenko, S. Janz, O. A. Fedorova and E. A. Kataev, Chem.–Eur. J., 2016, 22, 15069–15074 CrossRef CAS PubMed.
- T. A. Shumilova, T. Rüffer, H. Lang and E. A. Kataev, Chem.–Eur. J., 2018, 24, 1500–1504 CrossRef CAS PubMed.
- K. M. Bąk, K. Masłowska and M. J. Chmielewski, Org. Biomol. Chem., 2017, 15, 5968–5975 RSC.
- C. Reyheller and S. Kubik, Org. Lett., 2007, 9, 5271–5274 CrossRef CAS PubMed.
- H. Zhou, Y. Zhao, G. Gao, S. Li, J. Lan and J. You, J. Am. Chem. Soc., 2013, 135, 14908–14911 CrossRef CAS PubMed.
- R. Saini and S. Kumar, RSC Adv., 2013, 3, 21856–21862 RSC.
- M. J. Langton and P. D. Beer, Chem.–Eur. J., 2012, 18, 14406–14412 CrossRef CAS PubMed.
- N. H. Evans, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 8775–8777 RSC.
- J. W. Pflugrath and F. A. Quiocho, Nature, 1985, 314, 257–260 CrossRef CAS PubMed.
- J. J. He and F. A. Quiocho, Science, 1991, 251, 1479–1481 CrossRef CAS PubMed.
- J. J. He and F. A. Quiocho, Protein Sci., 1993, 2, 1643–1647 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, Y. Li, H. Zheng, X. Yin and H. Liu, Chem. Commun., 2010, 46, 5698–5700 RSC.
- M. K. Chae, J. Suk and K.-S. Jeong, Tetrahedron Lett., 2010, 51, 4240–4242 CrossRef CAS.
- K. M. Bąk, K. Chabuda, H. Montes, R. Quesada and M. J. Chmielewski, Org. Biomol. Chem., 2018, 16, 5188–5196 RSC.
- M. J. Chmielewski, M. Charon and J. Jurczak, Org. Lett., 2004, 6, 3501–3504 CrossRef CAS PubMed.
- C. D. Do, D. Pál, A. Belyaev, M. Pupier, A. Kiesilä, E. Kalenius, B. Galmés, A. Frontera, A. Poblador-Bahamonde and F. B. L. Cougnon, Chem. Commun., 2023, 59, 13010–13013 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- C. J. Serpell, A. Y. Park, C. V. Robinson and P. D. Beer, Chem. Commun., 2021, 57, 101–104 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. J. Pike, J. J. Hutchinson and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6700–6706 CrossRef CAS PubMed.
- D. Zhang, J. R. Cochrane, A. Martinez and G. Gao, RSC Adv., 2014, 4, 29735–29749 RSC.
- K. M. Bąk and M. J. Chmielewski, Eur. J. Org Chem., 2015, 2015, 4077–4080 CrossRef.
- S. Durot, F. Reviriego and J.-P. Sauvage, Dalton Trans., 2010, 39, 10557–10570 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05086f |
| This journal is © The Royal Society of Chemistry 2024 |