
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accessing a synthetic FeIIIMnIV core to model biological heterobimetallic active sites†
Justin L.
Lee
 a,
Saborni
Biswas
b,
Joseph W.
Ziller
a,
Emile L.
Bominaar
b,
Michael P.
Hendrich
b and
A. S.
Borovik
*a
a,
Saborni
Biswas
b,
Joseph W.
Ziller
a,
Emile L.
Bominaar
b,
Michael P.
Hendrich
b and
A. S.
Borovik
*a
aDepartment of Chemistry, University of California–Irvine, Irvine, CA 92697, USA. E-mail: aborovik@uci.edu
bDepartment of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213, USA
First published on 27th December 2023
Abstract
Metalloproteins with dinuclear cores are known to bind and activate dioxygen, with a subclass of these proteins having active sites containing FeMn cofactors and activities ranging from long-range proton-coupled electron transfer (PCET) to post-translational peptide modification. While mechanistic studies propose that these metallocofactors access FeIIIMnIV intermediates, there is a dearth of related synthetic analogs. Herein, the first well-characterized synthetic FeIII–(μ-O)–MnIV complex is reported; this complex shows similar spectroscopic features as the catalytically competent FeIIIMnIV intermediate X found in Class Ic ribonucleotide reductase and demonstrates PCET function towards phenolic substrates. This complex is prepared from the oxidation of the isolable FeIII–(μ-O)–MnIII species, whose stepwise assembly is facilitated by a tripodal ligand containing phosphinic amido groups. Structural and spectroscopic studies found proton movement involving the FeIIIMnIII core, whereby the initial bridging hydroxido ligand is converted to an oxido ligand with concomitant protonation of one phosphinic amido group. This series of FeMn complexes allowed us to address factors that may dictate the preference of an active site for a heterobimetallic cofactor over one that is homobimetallic: comparisons of the redox properties of our FeMn complexes with those of the di-Fe analogs suggested that the relative thermodynamic ease of accessing an FeIIIMnIV core can play an important role in determining the metal ion composition when the key catalytic steps do not require an overly potent oxidant. Moreover, these complexes allowed us to demonstrate the effect of the hyperfine interaction from non-Fe nuclei on 57Fe Mössbauer spectra which is relevant to MnFe intermediates in proteins.
Introduction
Many dioxygen-binding and/or activating non-heme metalloproteins contain a dinuclear active site, with the majority of them utilizing a diiron core, including the hydroxylase component of soluble methane monooxygenase (sMMO) and the R2 subunit of Class Ia ribonucleotide reductase (RNR).1–5 There is an increasing number of reports on dinuclear enzymes that employ a heterobimetallic FeMn active site to reduce O2, such as Class Ic RNR6–14 and R2-like ligand-binding oxidase (R2lox).15–19 The reproducible coordination of one Fe ion and one Mn ion in designated sites is unusual: Fe is more abundant than Mn under physiological conditions,20,21 and the Irving–Williams series supports stronger binding of FeII over MnII, so an FeIIFeII active site is thermodynamically more likely to form than an FeIIMnII one.22 It has been proposed that the tertiary structures of proteins enforce the site-specific binding of these metal ions,17 but the thermodynamic requirements for enzymatic functions (e.g., the relative accessibility of various oxidation levels of Fe vs. Mn) may also serve as a driving force for the metal selectivity, and these factors are underexplored. Synthetic model compounds are useful to address these fundamental considerations, but the preparation of heterobimetallic complexes is synthetically challenging: few examples of FeMn systems have been reported relative to their homobimetallic counterparts,23–30 and their oxidative chemistry has rarely been investigated.31–35 In this work, we describe a series of FeMn complexes in the same ligand architecture and explore their proton transfer, electron transfer, and proton-coupled electron transfer (PCET) properties. We examine a high-valent complex that contains an FeIII–(μ-O)–MnIV core that is relevant to key intermediates found for Class Ic RNR and R2lox.We have previously reported the usage of the multifunctional ligand [poat]3− (N,N′,N′′-[nitrilotris-(ethane-2,1-diyl)]tris(P,P-diphenylphosphinic amido))36–40 in which the C3 trianionic framework can support a metal center up to the 4+ oxidation level in high spin states. Our studies have shown that the phosphinic amido groups can form intramolecular hydrogen bonds (H-bonds),38 act as sites for proton storage, and be part of an auxiliary metal ion binding site, allowing us to construct discrete unsymmetrical bimetallic complexes. We have systematically varied the identity of the auxiliary metal center in a series of M/Fe dinuclear compounds (M = first row transition metals, group II alkali earth metals) to probe their effects on the electronic structures of FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units36 as well as magnetic and redox phenomena,37 and prepared a series of di-Fe complexes with the general formulation [(TMTACN)Fem–(μ-O(H))–Fen(H)poat]z+ (TMTACN = 1,4,7-trimethyl-1,4,7-triazacyclononane; m = II, III; n = II, III, IV; z = 0, 1, 2) that spans four oxidation states.39 We now report that replacing the Fe center in the [poat]3− site with a Mn ion provides a new heterobimetallic system that can be oxidized to the FeIIIMnIV level and serve as a synthetic model for FeMn-containing enzymatic active sites (Scheme 1).
O units36 as well as magnetic and redox phenomena,37 and prepared a series of di-Fe complexes with the general formulation [(TMTACN)Fem–(μ-O(H))–Fen(H)poat]z+ (TMTACN = 1,4,7-trimethyl-1,4,7-triazacyclononane; m = II, III; n = II, III, IV; z = 0, 1, 2) that spans four oxidation states.39 We now report that replacing the Fe center in the [poat]3− site with a Mn ion provides a new heterobimetallic system that can be oxidized to the FeIIIMnIV level and serve as a synthetic model for FeMn-containing enzymatic active sites (Scheme 1).
 | ||
| Scheme 1 Individual or coupled proton and electron transfers in the FeMn complexes described in this work. | ||
Results and discussion
Preparative routes and structure
The starting Mn synthon, K[MnIIpoat], was prepared by treating H3poat with three equivalents of KH and then subsequently adding MnII(OAc)2 (Scheme 2). The isolated K[MnIIpoat] salt, in the presence of 18-crown-6 and [FeII(TMTACN)(OTf)2], was allowed to react with isopropyl 2-iodoxybenzoate at −35 °C to yield [(TMTACN)FeIII–(μ-O)–MnIIIpoat](OTf) ([FeIII(O)MnIIIpoat]OTf); crystalline needles suitable for X-ray diffraction were obtained after multiple rounds of recrystallization at room temperature. The molecular structure revealed an Fe–O–Mn core with Mn–O1 and Fe–O1 bond lengths of 1.767(3) and 1.802(3) Å, respectively, and an Fe⋯Mn distance of 3.205(5) Å (Fig. 1A and Table 1); these values are comparable with those of the di-FeIII analog.39 The MnIII site in the [poat]3− framework adopts a trigonal bipyramidal geometry with an N4O primary coordination sphere comprising the N-atom donors of the [poat]3− ligand and a bridging oxido ligand; the FeIII site in the Fe-TMTACN adduct is 6-coordinated with an N3O3 primary coordination sphere comprising the TMTACN ligand, two O-atoms from the phosphinic amido groups from the [poat]3− ligand, and the bridging oxido ligand. This heterobimetallic complex, with its three-atom-bridge motif, a dynamic primary coordination sphere, and the ability to form an intramolecular H-bond network, incorporates crucial elements observed in the FeMn active sites of Class Ic RNR and R2lox.12,14,16,41 | ||
| Scheme 2 Preparative routes for K[MnIIpoat], [(TMTACN)FeIII–(μ-O)–MnIIIpoat]OTf, and [(TMTACN)FeIII–(μ-O)–MnIIIHpoat](OTf)2. | ||
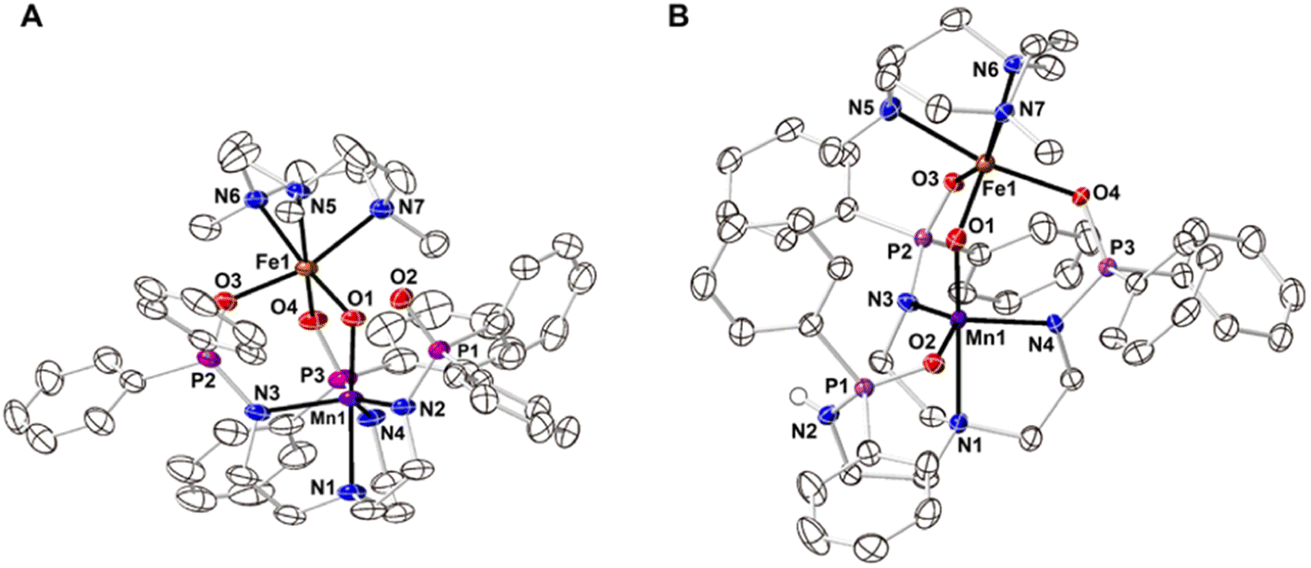 | ||
| Fig. 1 Thermal ellipsoid diagrams depicting the molecular structures of [FeIII(O)MnIIIpoat]+ (A) and [FeIII(O)MnIIIHpoat]2+ (B). Ellipsoids are drawn at the 50% probability level, and only the phosphinic amide H atom is shown for clarity. The triflate counterions are outer-sphere and are not interacting with the cation. | ||
| [FeIII(O)MnIIIpoat]+ | [FeIII(O)MnIIIHpoat]2+ | |
|---|---|---|
| a d[M–N/Ox] denotes the displacement of the metal atom from the 3-atom plane. N/Oeq represented the plane formed by N2, N3, N4 or O2, N3, N4. NTMTACN represents the plane formed by N5, N6, N7. b Trigonality structural parameter, τ5 = (β − α)/60°. β is the largest bond angle observed, and α is the second largest bond angle observed. | ||
| Bond lengths/distances (Å) | ||
| Mn1–N1 | 2.071(3) | 2.288(2) |
| Mn1–N2 | 2.008(3) | — |
| Mn1–O2 | — | 2.033(2) |
| Mn1–N3 | 2.079(4) | 2.007(2) |
| Mn1–N4 | 2.058(4) | 2.009(2) |
| Mn1–O1 | 1.767(3) | 1.793(2) |
| Fe1–O1 | 1.802(3) | 1.795(2) |
| Fe1–O3 | 2.026(3) | 2.000(1) |
| Fe1–O4 | 2.046(3) | 2.060(2) |
| Fe1–N5 | 2.219(3) | 2.234(2) |
| Fe1–N6 | 2.261(3) | 2.211(2) |
| Fe1–N7 | 2.225(4) | 2.212(2) |
| Mn1⋯Fe1 | 3.205(4) | 3.206(1) |
| av Mn1–N/Oeq | 2.048(3) | 2.016(2) |
| av Fe1–NTMTACN | 2.235(3) | 2.219(2) |
| d[Mn1–N/Oeq]a | 0.301 | 0.244 |
| d[Fe1–NTMTACN]a | 1.524 | 1.499 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Angles ( o ) | ||
| O1–Mn1–N1 | 177.85(14) | 178.25(7) |
| N2–Mn1–N3 | 120.53(15) | — |
| O2–Mn1–N3 | — | 133.29(7) |
| N3–Mn1–N4 | 103.63(15) | 107.64(7) |
| N2–Mn1–N4 | 129.26(15) | — |
| O2–Mn1–N4 | — | 114.56(7) |
| Mn1–O1–Fe1 | 127.80(15) | 126.62(8) |
| O3–Fe1–O4 | 96.08(12) | 98.29(6) |
| N5–Fe1–N6 | 78.54(12) | 79.89(7) |
| N5–Fe1–N7 | 79.00(13) | 78.84(7) |
| N6–Fe1–N7 | 78.24(13) | 79.29(7) |
|
||
| Calculated values | ||
| τ 5 | 0.810 | 0.749 |
The movement of protons is critical in many enzymatic processes,4,42–46 and so we explored the protonation/deprotonation steps within [FeIII(O)MnIIIpoat]+. The addition of one equivalent of 2,6-lutidinium triflate to [FeIII(O)MnIIIpoat]+ at −60 °C resulted in the immediate replacement of the λmax = 795 nm feature with a new band at 750 nm (Scheme 1 and Fig. S1A†), which was determined from electron paramagnetic resonance (EPR) spectroscopy (see below) to be the protonated species [(TMTACN)FeIII–(μ-OH)–MnIIIpoat]2+ ([FeIII(OH)MnIIIpoat]2+). This hydroxido-bridged complex was thermally unstable, even at −60 °C, and further converted to a species having a peak at λmax = 648 nm after one hour or upon warming to room temperature (Scheme 1 and Fig. S1B†). Isolation of the product at room temperature gave [(TMTACN)FeIII–(μ-O)–MnIIIHpoat](OTf)2 ([FeIII(O)MnIIIHpoat](OTf)2), whose structure revealed that the proton was transferred to the N-atom of one phosphinic amido group with its PO unit now coordinated to the MnIII center (Fig. 1B and Table 1).‡ The Mn–O1 and Fe–O1 bond lengths of 1.793(2) and 1.795(2) Å, respectively, and the Fe⋯Mn distance of 3.206(1) Å are similar to the measurements in the core of [FeIII(O)MnIIIpoat]+. The H-atom at the N-position could be identified using a difference-Fourier map and was also corroborated by a strong ν(N–H) feature at 3260 cm−1 in the FT-IR spectrum (Fig. S2†). The systematic blue-shifting of this optical transition upon protonation, as well as the intramolecular H+ transfer, were also observed for the di-Fe series.39
Magnetic properties of the MnIIIFeIII complexes
S- and X-band EPR spectroscopy (Fig. 2) revealed that each FeIIIMnIII complex has a distinct six-line hyperfine feature from its 55Mn center near g = 2. Although the spectral differences between the complexes are subtle, they are reproducible and could be simulated with distinctly different parameters for each species. The hyperfine pattern at g = 2 is indicative of an S = 1/2 spin-coupled system containing an S = 5/2 FeIII ion antiferromagnetically coupled to an S = 2 MnIII ion.6,17–19,32,33,47 Simultaneous least-squares fitting of the spectra for both frequencies provided the g- and A(55Mn)-tensors for the coupled system (Table 2). A rotation between the g- and A-tensors was required to simulate both the S- and X-band spectra with the same parameter set. The g-values are all larger than 2 because the g-values of the coupled S = 1/2 state are given by gc = 2 − (4/3)(gMn − 2) where the g-values for the MnIII centers are all less than 2 assuming gFe = 2. The deviations in the MnIIIg-values from 2 are at most 0.03, indicating that the spin–orbit contributions to the A-tensor are small. The spin-dipolar contribution to the A-tensor (ASD) was obtained by subtracting the isotropic value (Aiso) = trace(AMn)/3 from AMn for the uncoupled S = 2 MnIII site. The uncoupled values were determined from AMn = −3/4 ACMn, where ACMn is the magnetic hyperfine tensor for coupled spin S = 1/2. Only the magnitude of the hyperfine values can be determined from EPR spectroscopy, but the isotopic value is known to be negative. The Aiso values are near −200 MHz that are typical of MnIII complexes.6,18,19,32,33,48–50 The values of ASD are close to those obtained from density functional theory (DFT) calculations for the complexes and are within the uncertainty of the small spin–orbit contributions. The g- and ASD-tensors for the MnIII sites are rhombic, owing to a distorted empty dz2 orbital having mixtures with other d-orbitals as indicated by DFT. The distortion is influenced by the Fe–O–Mn angle being substantially less than 180°. As we have previously described, a rhombic ASD tensor was observed for the isoelectronic d4 FeIV site of [FeIII(O)FeIVpoat]2+, in which the di-Fe oxido bridge core is also bent; moreover, these findings contrast with those found for the related [FeIVpoat(O)]− complex that has an axial ASD tensor.36,39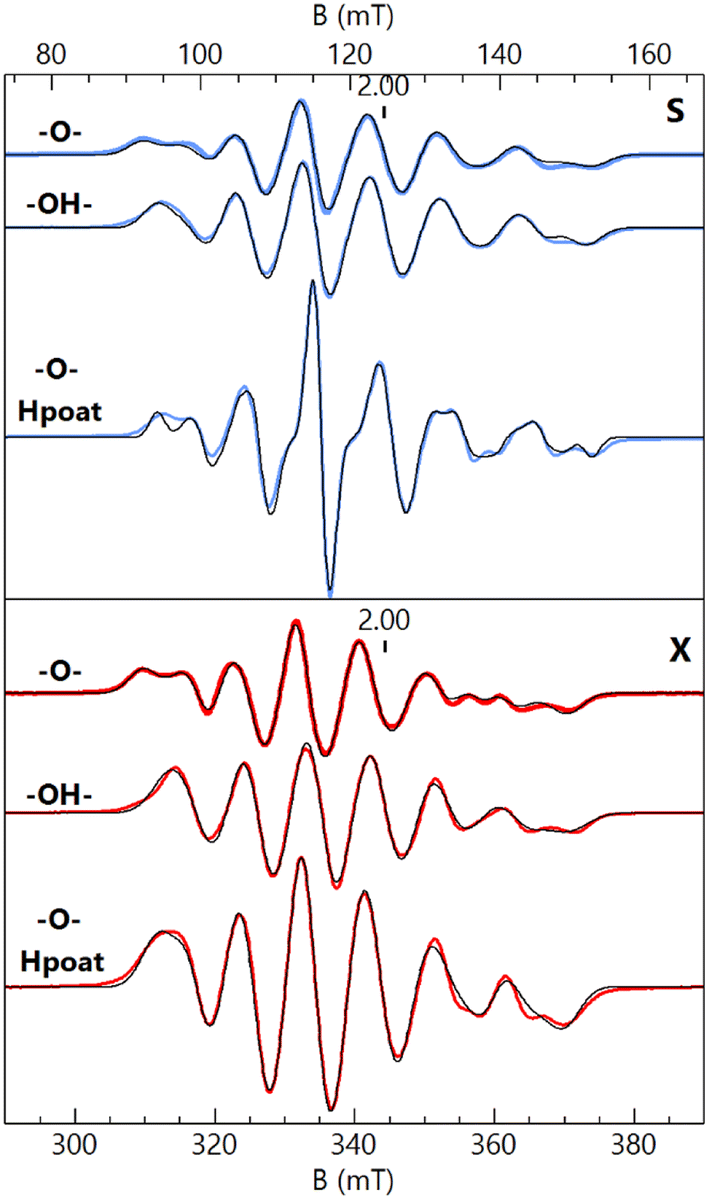 | ||
| Fig. 2 S- (blue traces, 3.485 GHz) and X-band (red traces, 9.636 GHz) EPR spectra of [FeIII(O)MnIIIpoat]+ (–O–), [FeIII(OH)MnIIIpoat]2+ (–OH–), and [FeIII(O)MnIIIHpoat]2+ (–O– Hpoat), 3 mM in CH2Cl2 (–O–, –OH–) or CH2Cl2:THF (–O– Hpoat). Measurement temperature, 20 K. The black traces are S = 1/2 simulations using the parameters given in Table 2. The g = 2 position is indicated. | ||
| Complex | g | A, S = 1/2 | g ∠A α, β, γb | AisoSMn = 2 | A SD, S Mn = 2 (DFT ASD) | J |
|---|---|---|---|---|---|---|
| a A-values are in MHz. b Euler angles α, β, γ indicate the rotation of A relative to g. c cm−1 (JSFe·SMn). | ||||||
| [FeIII(O)MnIIIpoat]+ | 2.042 | 215 | 73 | −200 | 39, 17, −57 (51, 11, −62) | 120 (170) |
| 2.031 | 243 | 54 | ||||
| 2.007 | 341 | 76 | ||||
| [FeIII(OH)MnIIIpoat]2+ | 2.026 | 209 | 90 | −197 | 41, 5, −46 (43, 10, −53) | 40 (40) |
| 2.024 | 255 | 27 | ||||
| 2.007 | 323 | 62 | ||||
| [FeIII(O)MnIIIHpoat]2+ | 2.037 | 202 | 77 | −196 | 44, 6, −50 (40, 25, −65) | 100 (125) |
| 2.025 | 254 | 53 | ||||
| 2.008 | 327 | 71 | ||||
The EPR features of [FeIII(OH)MnIIIpoat]2+ were further examined at higher temperatures to demonstrate the presence of the hydroxido bridging ligand. Upon warming above 20 K, a new signal appeared with features at g = 5.8, 4.9, and 3.0, which was absent from both [FeIII–(μ-O)–MnIII] complexes (Fig. 3). These g-values are expected for an S = 3/2 spin system with E/D = 0.16. When the spectra were plotted as intensity × temperature, an intensity increase was found at higher temperature as the S = 3/2 excited state became populated. The temperature dependence of the S = 3/2 signal (Fig. 3, inset) was fitted using the spin Hamiltonian JSFe·SMn with J = +40 cm−1, a value indicative of an antiferromagnetically coupled dinuclear system with a hydroxido bridging ligand.51 DFT calculations of the [FeIII(OH)MnIIIpoat]2+ complex also gave a J-value of +40 cm−1. The zero-field values D for the sites cannot be determined but are ∼0.5 cm−1 for FeIIITACN complexes and ∼2 cm−1 for MnIII complexes in a trianionic ligand scaffold similar to [poat]3−.49 These values are small relative to J = 40 cm−1 and have only a minor effect (only a few percent) on the observed A-tensor.
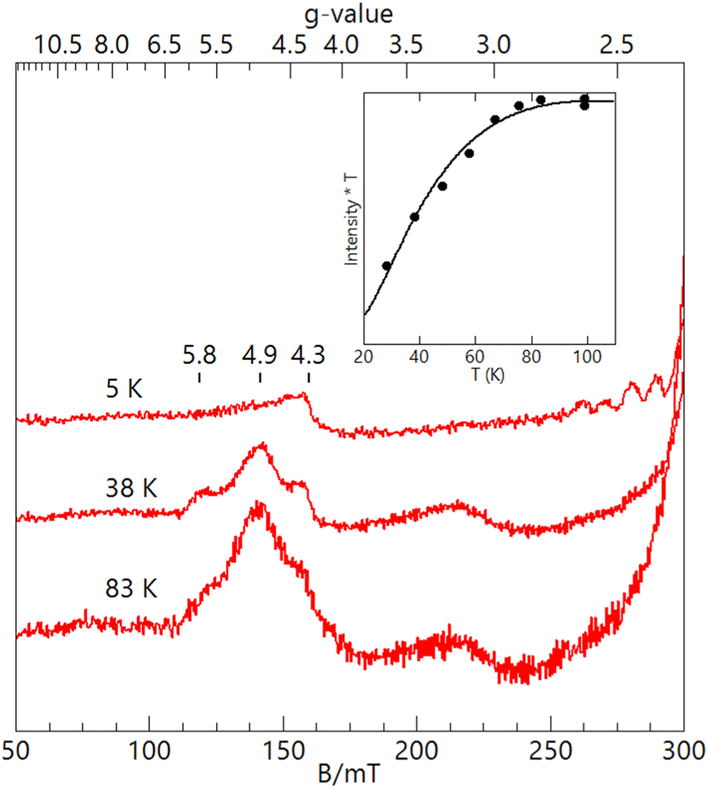 | ||
| Fig. 3 X-band (9.620 GHz) EPR spectra of [FeIII(OH)MnIIIpoat]2+ at 3 mM in CH2Cl2 at the measurement temperatures listed. The inset shows the temperature dependence of the S = 3/2 signal fitted with J = 40 cm−1 (JSFe·SMn). The signal at g = 4.3 is from a minor FeIII impurity. | ||
For [FeIII(O)MnIIIpoat]+ and [FeIII(O)MnIIIHpoat]2+, the temperature dependence of the microwave power required to half-saturate the EPR signal (P1/2) was measured over the temperature range 4–40 K (Fig. S3†). A fit of the data (Fig. S3†) to an Orbach relaxation curve and DFT gave the experimental and computed J-values, respectively (Table 2). The values are greater than 100 cm−1 and are consistent with an oxido bridging ligand.51 These values are similar to those of the oxido bridged FeIIIMnIII complexes with TACN and TMTACN ligands (J ∼ 130 cm−1) from magnetic susceptibility measurements.31
Variable-field 57Fe Mössbauer spectra and simulations of [FeIII(O)MnIIIpoat]+ (Fig. 4) are indicative of an S = 5/2 FeIII center exchange-coupled antiferromagnetically to an S = 2 MnIII center with J > 100 cm−1, thus producing a coupled S = 1/2 ground state. The FeIII site has an isomer shift (δ) of 0.53 mm s−1, quadrupole splitting (ΔEQ) of −1.84 mm s−1, and an isotropic A-tensor of −20 T. These parameters are indicative of a high-spin FeIII center.7,47 However, the experimental and simulated 57Fe Mössbauer spectra did not match at low magnetic field (45 mT, Fig. 4). The cause of this mismatch appears to result from the spin expectation of the Fe being reduced from its normal value to give an overall spectral line splitting that was smaller than expected. One possibility is that this lower spin expectation occurs for coupled metal spin systems with zero-field splitting (D) comparable to the exchange coupling (J). However, this possibility would cause the simulations at higher fields to not match the data, which they clearly do (Fig. 4), and was therefore ruled out.
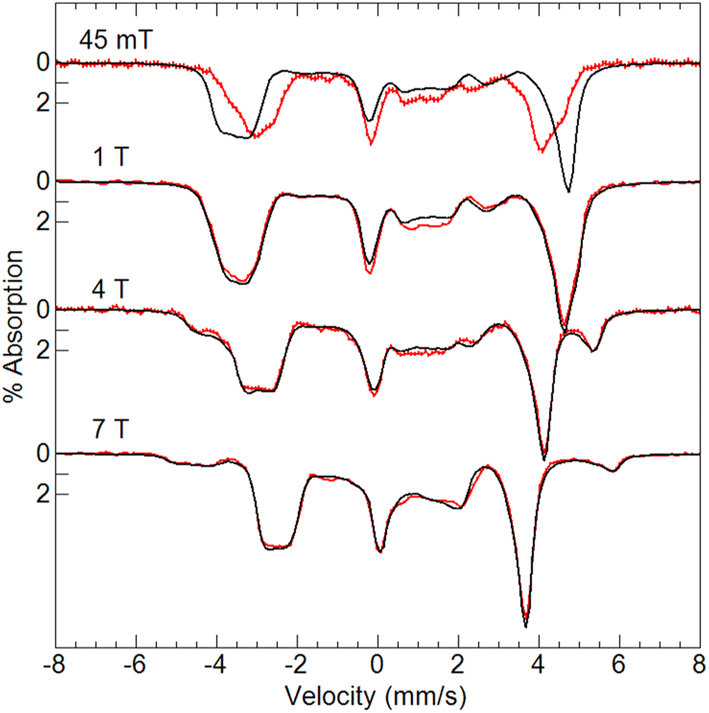 | ||
| Fig. 4 Mössbauer spectra (red traces) of [57FeIII(O)MnIIIpoat]+ in PrCN at 4.2 K and the magnetic fields listed. The simulations (black traces) are for S = 5/2 FeIII antiferromagnetically exchange-coupled to S = 2 MnIII (see text for parameters). | ||
The origin of the effect was discovered to be from the hyperfine interaction of the 55Mn nuclear spin (I = 5/2) with the electronic system spin (S = 1/2). For Fe complexes, the energy of the hyperfine interaction (S·AFe·IFe) is approximately 0.001 cm−1. For small magnetic fields produced by permanent magnets (∼50 mT) that are commonly used in Mössbauer spectrometers, the energy splitting between the electronic spin states is ∼0.04 cm−1, consequently, the electronic spin expectation at the Fe site is dominated by the magnetic field interaction (mBS·g·B). The EPR analysis of the S = 1/2 signal in [FeIII(O)MnIIIpoat]+ (Table 2) gave an isotropic hyperfine constant for 55Mn of Aiso = 260 MHz (0.0087 cm−1), which is more than 10% of the electronic energy splitting. Thus, the nuclear spin of Mn site significantly alters the electronic spin expectation observed at the Fe site.
To examine how this interaction affects the analysis of the Mössbauer data, we have incorporated the 55Mn hyperfine interaction (S·AMn·IMn) into our simulation software to allow diagonalization of the spin Hamiltonian with the electronic and Mn nuclear spin states. The new simulations of Mössbauer spectra collected at 7.5 and 45 mT show a dramatic change which closely matches the data (Fig. 5). The effect of this interaction is only observed at lower magnetic fields, while simulations at higher fields are unaffected by the Mn nuclear spin because the electronic spin at the Fe site is dominated by the magnetic field interaction.
 | ||
| Fig. 5 Mössbauer spectra (red traces) of [57FeIII(O)MnIIIpoat]+ in PrCN at 4.2 K and the magnetic fields listed. The simulations (black traces) are for S = 5/2 FeIII antiferromagnetically exchange-coupled to S = 2 MnIII with inclusion of the 55Mn hyperfine interaction using the A-tensor and rotation derived from EPR spectroscopy (Table 2). The dashed lines are without the 55Mn hyperfine interaction. | ||
Preparation and properties of an FeIII–(μ-O)–MnIV complex
Dioxygen-activating FeMn enzymes such as RNR Class Ic and R2lox have been proposed to access higher oxidation levels (e.g., FeIIIMnIV and FeIVMnIV) to achieve function;6–9,12,14,18 we therefore used cyclic voltammetry to evaluate the redox properties of our FeIIIMnIII complexes. The cyclic voltammogram of [FeIII(O)MnIIIpoat]+ revealed a nearly reversible, one-electron redox event at +0.20 V vs. [FeCp2]+/0, which was assigned to the FeIIIMnIV/III process (Fig. 6A). This potential is significantly more negative than the FeIIIFeIV/III potential of +0.55 V vs. [FeCp2]+/0 observed in the di-Fe analog,39 suggesting that the redox change occurs at the Mn site. Treatment of [FeIII(O)MnIIIpoat]+ with [FeCp(C5H4C(O)Me)]OTf or [N(p-C6H4Me)3]OTf at −60 °C resulted in the loss of the diagnostic peak at λmax = 795 nm and the concurrent appearance of shoulders at 420, 490, and 610 nm (Fig. 6B). While these features are poorly resolved, they resemble those reported for the catalytic FeIIIMnIV intermediate in RNR Ic.12 This new species did not show an EPR signal, suggesting an integer spin complex (Fig. S4†). This redox process was reversible: the addition of FeCp2 to this new compound regenerated the optical and EPR features of [FeIII(O)MnIIIpoat]+ and [FeCp2]+, supporting the hypothesis that the reaction was an outer-sphere electron transfer.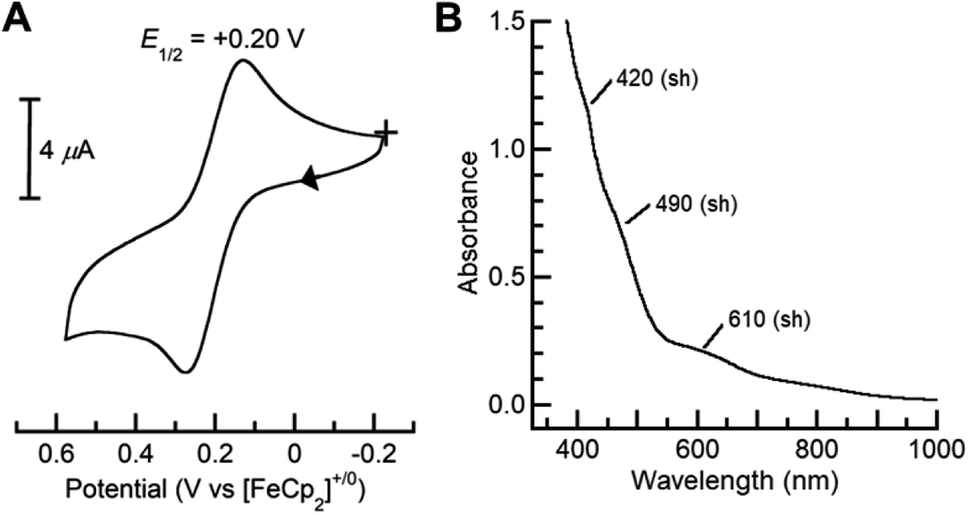 | ||
| Fig. 6 Cyclic voltammogram (A) of [FeIII(O)MnIIIpoat]+ collected at 100 mV s−1 in CH2Cl2, and electronic absorption spectrum of [FeIII(O)MnIVpoat]2+ (B) measured in a 0.20 mM CH2Cl2 solution at −60 °C. | ||
Variable-field 57Fe Mössbauer spectra and simulations of the oxidized species showed that the features associated with [FeIII(O)MnIIIpoat]+ are nearly absent (Fig. 7). The simulations were based on an S = 5/2 FeIII ion antiferromagnetically exchange-coupled to an S = 3/2 MnIV ion with J > 100 cm−1, producing a coupled system with an S = 1 state that is lowest in energy. The FeIII site has the parameters δ = 0.50 mm s−1, ΔEQ = −1.31 mm s−1, and an isotropic A-tensor of −20 T. These values are typical of a high-spin FeIII center and close to those of [FeIII(O)MnIIIpoat]+, indicating that the Mn center has been oxidized. The new species was therefore formulated as [(TMTACN)FeIII–(μ-O)–MnIVpoat]2+ ([FeIII(O)MnIVpoat]2+). Although this species has an S = 1 spin state, no signals were observed in parallel-mode EPR spectra. A minor species, assigned to [FeIII(O)MnIIIpoat]+, was also present in the Mössbauer spectra (15%) and is most evident in the 45 mT spectrum – it was included in all simulations. As observed in the spectra of [FeIII(O)MnIIIpoat]+, the Mössbauer spectrum of [FeIII(O)MnIVpoat]2+ at 45 mT was affected by the Mn nuclear spin (Fig. 7, dashed line). The effect is minor owing to the small spin expectation for the spin S = 1 state at low field (Fig. 7, inset).
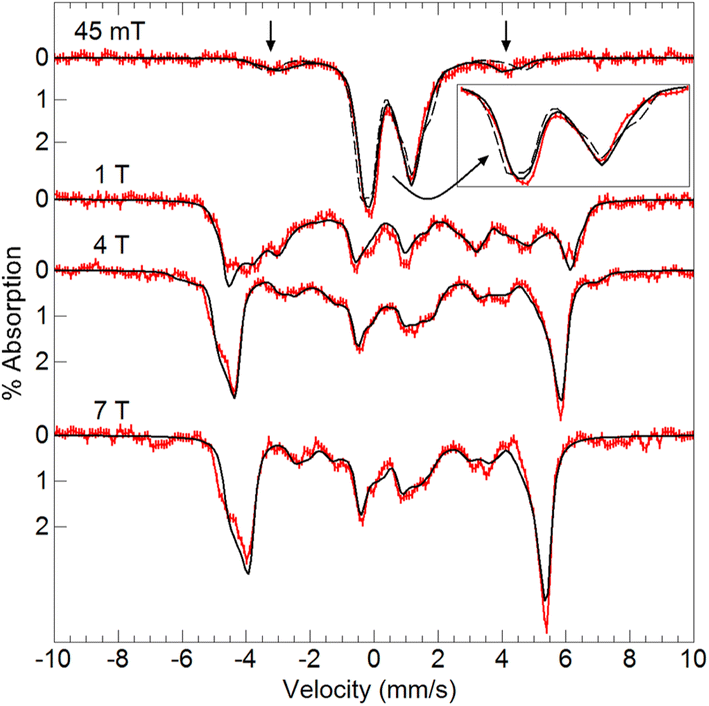 | ||
| Fig. 7 Mössbauer spectra (red traces) of [57FeIII(O)MnIVpoat]2+ in PrCN at 4.2 K and the magnetic fields listed. The simulations (black traces) are for an S = 5/2 FeIII ion antiferromagnetically exchange-coupled to S = 3/2 MnIV ion (see text for parameters). The vertical arrows mark spectral features from a minor amount of [FeIII(O)MnIIIpoat]+. The dashed line in the 45 mT spectrum is without the 55Mn hyperfine interaction (see inset). | ||
There are several examples of complexes with FeMn centers, and in each, the oxidation states of the metal centers are below 4+.23–29,31–34 Our knowledge of synthetic systems containing an FeIII–(μ-O)–MnIV core has been limited to a single previous report of one complex whose spectroscopic and magnetic characterizations are incomplete.31 In fact, the best-studied system prior to [FeIII(O)MnIVpoat]2+ is RNR Ic R2-X,6,7,12 the active intermediate that initiates long-range PCET from the R1 subunit for catalytic function. Comparison of the spectroscopic properties of these two FeIIIMnIV systems revealed several common features: similar electronic absorption spectra, Mössbauer parameters, and J-values (Table 3). As the PCET pathway between the R1/R2 subunits involves tyrosine residues, we tested the reactivity of [FeIII(O)MnIVpoat]2+ with various para-substituted phenols, such as 4-MeO-PhOH (BDFE = 79.9 kcal mol−1),52 4-tert-butyl-PhOH (83.5),53 and 4-F-PhOH (83.8),52 at −60 °C in CH2Cl2 (Scheme 1 and Fig. S5–S8†). For instance, upon treatment with an excess (≥5 equivalents) of 4-MeO-PhOH, the optical and EPR features of the FeIIIMnIV species were replaced by those of [FeIII(OH)MnIIIpoat]2+ in a near quantitative amount (>90% by EPR quantification; Fig. S5†); upon warming, the FeIII–(μ-OH)–MnIII core underwent intramolecular proton transfer to form [FeIII(OH)MnIIIpoat]2+, analogous to the rearrangement observed after the protonation of [FeIII(O)MnIIIpoat]+ (Scheme 1). The organic product was identified to be 5,5′-dimethoxy-[1,1′-biphenyl]-2,2′-diol (246.2 m/z; > 95% by GC-MS quantification; Fig. S6†), which is consistent with a PCET mechanism in which the para-substituted phenol reacts with [FeIII(O)MnIVpoat]2+ to first form the phenoxyl radical, which then undergoes a bimolecular homocoupling to produce the bisphenol.39,54 These results demonstrate for the first time that systems with an FeIII–(μ-O)–MnIV core are indeed competent to perform this type of PCET process.
| Complex | S T | λ (nm, εM) | 57Fe δ/ΔEQa | A x,y,z(57Fe)b | J |
|---|---|---|---|---|---|
| a mm s−1. b Tesla. c cm−1. d From Mössbauer spectroscopy using +JSFe·SMn. e Ref. 6, 7 and 12. | |||||
| [FeIII(O)MnIVpoat]2+ | 1 | 420 (sh) | 0.50/–1.31 | −20 | >100d |
| 490 (sh) | −20 | ||||
| 610 (sh) | −20 | ||||
| RNR Ic R2-Xe | 1 | 360 (sh) | 0.52/|1.32| | −23.0 | 200 |
| 408 (sh) | −22.2 | ||||
| 477 (1500) | −21.7 | ||||
Our reactivity studies additionally found that [FeIII(O)MnIVpoat]2+ did not react with compounds having relatively weak C–H bonds, such as 9,10-dihydroanthracene (72.9) and xanthene (70.2). This lack of reactivity was also found for the analogous [FeIII(O)FeIVpoat]2+ species, which also reacts with similar phenols to first produce [FeIII(OH)FeIIIpoat]2+.39 In both systems, the homolytic cleavage of the O–H bond of the phenolic substrate leads to the initial protonation of the oxido ligand, which is sterically hindered by the [poat]3− ligand and methyl groups of the TMTACN ligand. We proposed that the PCET process involved for homolytic ArO–H bond cleavage by [FeIII(O)FeIVpoat]2+ favors the protonation of a site near to the metal center even though it was hindered (that is, rather than protonating the ligand to form [FeIII(O)FeIIIHpoat]2+). A similar process appears to be operative for the FeIIIMnIV analog. However, [FeIII(O)MnIVpoat]2+ did not react with 2,6-tert-butyl-4-R-PhOH (R = –OMe (BDFE = 72.6 kcal mol−1), –tBu (75.5), and –H (77.0),52 substrates that are substantially more sterically hindered. Steric effects cannot be the only reason for this lack of reactivity because we have previously shown that the analogous [FeIII(O)FeIVpoat]2+ did react with these substrates.39 We do not completely understand the lack of reactivity for [FeIII(O)MnIVpoat]2+ and its apparent difference from that found for the FeIIIFeIV analog. With this said, the two bimetallic complexes have differing FeIIIMIV/FeIIIMIII redox potentials with [FeIII(O)FeIVpoat]2+ being the stronger oxidant that may contribute to its increased reactivity.39
Conclusions
In summary, the rigid [poat]3−/TMTACN ligand scaffold allowed us to construct discrete FeMn complexes and examine their individual or coupled proton and electron transfer steps. We described the reactivity, spectroscopic character, and electrochemical properties of a high-valent compound that contains an FeIII–(μ-O)–MnIV core. We have previously shown that mononuclear Mn–O(H) complexes in trigonal symmetry have lower MIV/III reduction potentials than their Fe counterparts,55–57 but, to our best knowledge, this work presents the first direct experimental comparison of the redox properties, and the resultant changes in the electronic structures, of related FeMn and di-Fe compounds at an oxidation state above 3+. Our studies found that the lower potentials of FeMn complexes compared to their di-Fe analog could be used to form a complex with an FeIII–(μ-O)–MnIV core. Within a biological context, a more thermodynamically accessible MnIV ion over an FeIV ion can be one important contributing factor leading to the selective binding of FeMn sites in Class Ic RNR and R2lox enzymes, especially when the key catalytic steps do not require an overly potent oxidant (such as di-FeIV) that may cause undesirable damage to the proteins. [FeIII(O)MnIVpoat]2+ shares similar electronic, magnetic, and spin-exchange features as RNR R2 Ic-X, suggesting the former to be the first well-characterized model for the biological FeIIIMnIV core.The Mössbauer findings on [FeIII(O)MnIVpoat]2+ and [FeIII(O)MnIIIpoat]+ revealed a mismatch between the data and simulations at low external magnetic fields that is caused by the hyperfine interaction of the 55Mn nuclear spin with the electronic system spin. This effect on the Mössbauer spectra of inorganic complexes is uncommon because it requires an electronic interaction between Fe and a second metal ion having a nuclear spin and large hyperfine values. The possible influence of the 55Mn nuclear spin has been suggested in the Mössbauer spectra of an MnIVFeIV intermediate in Chlamydia trachomatis ribonucleotide reductase.8 Our findings for [FeIII(O)MnIIIpoat]+ and [FeIII(O)MnIVpoat]2+ support this premise and demonstrate a new analysis to obtain quantitative agreement between Mössbauer data and simulations that incorporate the 55Mn A-tensor and rotation derived from EPR spectroscopy. To our knowledge, this is the first quantitative demonstration of the effect of the hyperfine interaction from a non-Fe nucleus on the Mössbauer spectra of inorganic complexes. Our work also highlights the importance of accessibility during PCET processes: the initial formation of the [FeIII(OH)MnIIIpoat]2+ species upon PCET with phenolic substrates, in which the proton transfers to the bridging oxido ligand and the electron reduces the MnIV center, appears to be necessary for reactivity to occur. Within a growing body of literature and improved understanding of FeMn enzymes and their oxidative chemistry, the [(TMTACN)Fem–(μ-O(H))–Mnn(H)poat]z+ system serves as a useful structural, spectroscopic, and functional model to complement biochemical investigations.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
The concept and experimental studies were performed by J. L. L. and A. S. B. The magnetic measurements and DFT calculations were performed by S. B., E. L. B., and M. P. H. The crystallographic measurements and analyses were performed by J. W. Z. All of the authors actively participated in manuscript preparation and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgement is made to the National Institutes of Health USA (GM050781 to A. S. B., and GM141948 to M. P. H.) for funding and the UCI Department of Chemistry for a fellowship to J. L. L.Notes and references
- C. E. Tinberg and S. J. Lippard, Dioxygen Activation in Soluble Methane Monooxygenase, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- J. D. Lipscomb and L. Que, MMO: P450 in wolf's clothing?, JBIC, J. Biol. Inorg. Chem., 1998, 3, 331–336 CrossRef CAS.
- M. O. Ross and A. C. Rosenzweig, A tale of two methane monooxygenases, JBIC, J. Biol. Inorg. Chem., 2017, 22, 307–319 CrossRef CAS PubMed.
- J. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer?, Chem. Rev., 2003, 103, 2167–2201 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que, Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- W. Jiang, D. Yun, L. Saleh, E. W. Barr, G. Xing, L. M. Hoffart, M.-A. A. Maslak, C. Krebs and J. M. Bollinger, A Manganese(IV)/Iron(III) Cofactor in Chlamydia trachomatis Ribonucleotide Reductase, Science, 2007, 316, 1188–1191 CrossRef CAS PubMed.
- W. Jiang, J. M. Bollinger and C. Krebs, The Active Form of Chlamydia trachomatis Ribonucleotide Reductase R2 Protein Contains a Heterodinuclear Mn(IV)/Fe(III) Cluster with S = 1 Ground State, J. Am. Chem. Soc., 2007, 129, 7504–7505 CrossRef CAS PubMed.
- W. Jiang, L. M. Hoffart, C. Krebs and J. M. Bollinger, A Manganese(IV)/Iron(IV) Intermediate in Assembly of the Manganese(IV)/Iron(III) Cofactor of Chlamydia trachomatis Ribonucleotide Reductase, Biochemistry, 2007, 46, 8709–8716 CrossRef CAS PubMed.
- J. M. Younker, C. M. Krest, W. Jiang, C. Krebs, J. M. Bollinger and M. T. Green, Structural Analysis of the Mn(IV)/Fe(III) Cofactor of Chlamydia trachomatis Ribonucleotide Reductase by Extended X-ray Absorption Fine Structure Spectroscopy and Density Functional Theory Calculations, J. Am. Chem. Soc., 2008, 130, 15022–15027 CrossRef CAS PubMed.
- J. M. Bollinger, W. Jiang, M. T. Green and C. Krebs, The manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase: structure, assembly, radical initiation, and evolution, Curr. Opin. Struct. Biol., 2008, 18, 650–657 CrossRef CAS PubMed.
- L. M. K. Dassama, C. Krebs, J. M. Bollinger, A. C. Rosenzweig and A. K. Boal, Structural Basis for Assembly of the MnIV/FeIII Cofactor in the Class Ic Ribonucleotide Reductase from Chlamydia trachomatis, Biochemistry, 2013, 52, 6424–6436 CrossRef CAS PubMed.
- Y. Kwak, W. Jiang, L. M. K. Dassama, K. Park, C. B. Bell, L. V. Liu, S. D. Wong, M. Saito, Y. Kobayashi, S. Kitao, M. Seto, Y. Yoda, E. E. Alp, J. Zhao, J. M. Bollinger, C. Krebs and E. I. Solomon, Geometric and Electronic Structure of the Mn(IV)Fe(III) Cofactor in Class Ic Ribonucleotide Reductase: Correlation to the Class Ia Binuclear Non-Heme Iron Enzyme, J. Am. Chem. Soc., 2013, 135, 17573–17584 CrossRef CAS PubMed.
- J. Livada, R. J. Martinie, L. M. K. Dassama, C. Krebs, J. M. Bollinger and A. Silakov, Direct Measurement of the Radical Translocation Distance in the Class I Ribonucleotide Reductase from Chlamydia trachomatis, J. Phys. Chem. B, 2015, 119, 13777–13784 CrossRef CAS PubMed.
- R. J. Martinie, E. J. Blaesi, C. Krebs, J. M. Bollinger, A. Silakov and C. J. Pollock, Evidence for a Di-μ-oxo Diamond Core in the Mn(IV)/Fe(IV) Activation Intermediate of Ribonucleotide Reductase from Chlamydia trachomatis, J. Am. Chem. Soc., 2017, 139, 1950–1957 CrossRef CAS PubMed.
- C. S. Andersson and M. Högbom, A Mycobacterium tuberculosis ligand-binding Mn/Fe protein reveals a new cofactor in a remodeled R2-protein scaffold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5633–5638 CrossRef CAS PubMed.
- J. J. Griese, K. Roos, N. Cox, H. S. Shafaat, R. M. M. Branca, J. Lehtiö, A. Gräslund, W. Lubitz, P. E. M. Siegbahn and M. Högbom, Direct observation of structurally encoded metal discrimination and ether bond formation in a heterodinuclear metalloprotein, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17189–17194 CrossRef CAS PubMed.
- E. C. Kisgeropoulos, J. J. Griese, Z. R. Smith, R. M. M. Branca, C. R. Schneider, M. Högbom and H. S. Shafaat, Key Structural Motifs Balance Metal Binding and Oxidative Reactivity in a Heterobimetallic Mn/Fe Protein, J. Am. Chem. Soc., 2020, 142, 5338–5354 CrossRef CAS PubMed.
- E. K. Miller, N. E. Trivelas, P. T. Maugeri, E. J. Blaesi and H. S. Shafaat, Time-Resolved Investigations of Heterobimetallic Cofactor Assembly in R2lox Reveal Distinct Mn/Fe Intermediates, Biochemistry, 2017, 56, 3369–3379 CrossRef CAS PubMed.
- E. C. Kisgeropoulos, Y. J. Gan, S. M. Greer, J. M. Hazel and H. S. Shafaat, Pulsed Multifrequency Electron Paramagnetic Resonance Spectroscopy Reveals Key Branch Points for One- vs. Two-Electron Reactivity in Mn/Fe Proteins, J. Am. Chem. Soc., 2022, 144, 11991–12006 CrossRef CAS PubMed.
- Biological Inorganic Chemistry: Structure and Reactivity, ed. I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, University Science Book, Sausalito, CA, 1st edn, 2006 Search PubMed.
- A. D. Anbar, Elements and Evolution, Science, 2008, 332, 1481–1483 CrossRef PubMed.
- H. Irving and R. J. P. Williams, The Stability of Transition-metal Complexes, J. Chem. Soc., 1953, 3192–3210 RSC.
- Y. Sano, A. C. Weitz, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Unsymmetrical Bimetallic Complexes with MII–(μ-OH)–MIII Cores (MIIMIII = FeIIFeIII, MnIIFeIII, MnIIMnIII): Structural, Magnetic, and Redox, Inorg. Chem., 2013, 52, 10229–10231 CrossRef CAS PubMed.
- Y. Sano, N. Lau, A. C. Weitz, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Models for Unsymmetrical Active Sites in Metalloproteins: Structural, Redox, and Magnetic Properties of Bimetallic Complexes with MII-(μ-OH)-FeIII Cores, Inorg. Chem., 2017, 56, 14118–14128 CrossRef CAS PubMed.
- T. R. Holman, Z. Wang, M. P. Hendrich and L. Que, Structural and Spectroscopic Properties of Antiferromagnetically Coupled FeIIIMnII and FeIIMnII Complexes, Inorg. Chem., 1995, 34, 134–139 CrossRef CAS.
- M. Carboni, M. Clémancey, F. Molton, J. Pécaut, C. Lebrun, L. Dubois, G. Blondin and J. M. Latour, Biologically Relevant Heterodinuclear Iron-Manganese Complexes, Inorg. Chem., 2012, 51, 10447–10460 CrossRef CAS PubMed.
- B. Das, H. Daver, A. Singh, R. Singh, M. Haukka, S. Demeshko, F. Meyer, G. Lisensky, M. Jarenmark, F. Himo and E. Nordlander, A Heterobimetallic FeIIIMnII Complex of an Unsymmetrical Dinucleating Ligand: A Structural and Functional Model Complex for the Active Site of Purple Acid Phosphatase of Sweet Potato, Eur. J. Inorg. Chem., 2014, 2204–2212 CrossRef CAS.
- A. L. Poptic, Y.-P. Chen, T. Chang, Y.-S. Chen, C. E. Moore and S. Zhang, Site-Differentiated MnIIFeII Complex Reproducing the Selective Assembly of Biological Heterobimetallic Mn/Fe Cofactors, J. Am. Chem. Soc., 2023, 145, 3491–3498 CrossRef CAS PubMed.
- S. J. Tereniak, R. K. Carlson, L. J. Clouston, V. G. Young, E. Bill, R. Maurice, Y.-S. Chen, H. J. Kim, L. Gagliardi and C. C. Lu, Role of the Metal in the Bonding and Properties of Bimetallic Complexes Involving Manganese, Iron, and Cobalt, J. Am. Chem. Soc., 2014, 136, 1842–1855 CrossRef CAS PubMed.
- U. Bossek, T. Weyhermüller, K. Wieghardt, J. Bonvoisin and J. J. Girerd, Synthesis, E.S.R. Spectrum and Magnetic Properties of a Heterobinuclear Complex Containing the {FeIII(μ-O)(μ-MeCO2)2MnIII}2+ Core, J. Chem. Soc., Chem. Commun., 1989, 633–636 RSC.
- R. Hotzelmann, K. Wieghardt, U. Flörke, H.-J. Haupt, D. C. Weatherburn, J. Bonvoisin, G. Blondin and J.-J. Girerd, Spin Exchange Coupling in Asymmetric Heterodinuclear Complexes Containing the μ-Oxo-bis(μ-acetato)dimetal Core, J. Am. Chem. Soc., 1992, 114, 1681–1696 CrossRef CAS.
- A. Zhou, P. M. Crossland, A. Draksharapu, A. J. Jasniewski, S. T. Kleespies and L. Que, Oxoiron(IV) complexes as synthons for the assembly of heterobimetallic centers such as the Fe/Mn active site of Class Ic ribonucleotide reductases, JBIC, J. Biol. Inorg. Chem., 2018, 23, 155–165 CrossRef CAS PubMed.
- P. M. Crossland, Y. Guo and L. Que, Spontaneous Formation of an Fe/Mn Diamond Core: Models for the Fe/Mn Sites in Class 1c Ribonucleotide Reductases, Inorg. Chem., 2021, 60, 8710–8721 CrossRef CAS PubMed.
- P. Karsten, A. Neves, A. J. Bortoluzzi, M. Lanznaster and V. Drago, Synthesis, structure, properties, and phosphatase-like activity of the first heterodinuclear FeIIIMnII complex with the unsymmetric ligand H2BPBPMP as a model for the PAP in sweet potato, Inorg. Chem., 2002, 41, 4624–4626 CrossRef CAS PubMed.
- S. Ross, T. Weyhermüller, E. Bill, E. Bothe, U. Flörke, K. Wieghardt and P. Chaudhuri, Asymmetric Heterodinuclear FeIIIMII (M = Zn, Cu, Ni, Fe, Mn), CoIIIFeII and FeIICoIII Species: Synthesis, Structure, Redox Behavior, and Magnetism, Eur. J. Inorg. Chem., 2004, 2004, 984–997 CrossRef.
- V. F. Oswald, J. L. Lee, S. Biswas, A. C. Weitz, K. Mittra, R. Fan, J. Li, J. Zhao, M. Y. Hu, E. E. Alp, E. L. Bominaar, Y. Guo, M. T. Green, M. P. Hendrich and A. S. Borovik, Effects of Noncovalent Interactions on High-Spin Fe(IV)–Oxido Complexes, J. Am. Chem. Soc., 2020, 142, 11804–11817 CrossRef CAS PubMed.
- J. L. Lee, V. F. Oswald, S. Biswas, E. A. Hill, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Stepwise assembly of heterobimetallic complexes: synthesis, structure, and physical properties, Dalton Trans., 2021, 50, 8111–8119 RSC.
- C. Sun, V. F. Oswald, E. A. Hill, J. W. Ziller and A. S. Borovik, Investigation of iron–ammine and amido complexes within a C3-symmetrical phosphinic amido tripodal ligand, Dalton Trans., 2021, 50, 11197–11205 RSC.
- J. L. Lee, S. Biswas, C. Sun, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Bioinspired Di-Fe Complexes: Correlating Structure and Proton Transfer over Four Oxidation States, J. Am. Chem. Soc., 2022, 144, 4559–4571 CrossRef CAS PubMed.
- C. Sun, J. L. Jaimes, A. H. Follmer, J. W. Ziller and A. S. Borovik, Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex, Molecules, 2023, 28, 4755 CrossRef CAS PubMed.
- J. A. Cotruvo and J. Stubbe, Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo, Annu. Rev. Biochem., 2011, 80, 733–767 CrossRef CAS PubMed.
- L. Vogt, D. J. Vinyard, S. Khan and G. W. Brudvig, Oxygen-evolving complex of Photosystem II: an analysis of second-shell residues and hydrogen-bonding networks, Curr. Opin. Chem. Biol., 2015, 25, 152–158 CrossRef CAS PubMed.
- S. Nagano and T. L. Poulos, Crystallographic Study on the Dioxygen Complex of Wild-type and Mutant Cytochrome P450cam: Implications for the Dioxygen Activation Mechanism, J. Biol. Chem., 2005, 280, 31659–31663 CrossRef CAS PubMed.
- I. Dance, The controlled relay of multiple protons required at the active site of nitrogenase, Dalton Trans., 2012, 41, 7647–7659 RSC.
- A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, Mechanism of Proton Ttransfer in [FeFe]-Hydrogenase from Clostridium pasteurianum, J. Biol. Chem., 2011, 286, 38341–38347 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, Moving Protons and Electrons in Biomimetic Systems, Biochemistry, 2015, 54, 1863–1878 CrossRef CAS PubMed.
- M. M. Powell, G. Rao, R. D. Britt and J. Rittle, Enzymatic Hydroxylation of Aliphatic C–H Bonds by a Mn/Fe Cofactor, J. Am. Chem. Soc., 2023, 145, 16526–16537 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, A. S. Borovik and M. P. Hendrich, Characterization of Monomeric MnII/III/IV-Hydroxo Complexes from X- and Q-Band Dual Mode Electron Paramagnetic Resonance (EPR) Spectroscopy, Inorg. Chem., 2013, 52, 12568–12575 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, High-spin Mn–oxo complexes and their relevance to the oxygen-evolving complex within photosystem II, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- V. F. Oswald, A. C. Weitz, S. Biswas, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Manganese−Hydroxido Complexes Supported by a Urea/Phosphinic Amide Tripodal Ligand, Inorg. Chem., 2018, 57, 13341–13350 CrossRef CAS PubMed.
- D. M. Kurtz, Oxo- and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit, Chem. Rev., 1990, 90, 585–606 CrossRef CAS.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications, Chem. Rev., 2021, 122, 1–49 CrossRef PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Taylor & Francis Group, Boca Raton, FL, 2007 Search PubMed.
- S. Steenken and P. Neta, in The Chemistry of Phenols, ed. Z. Rappoport, John Wiley & Sons, Ltd, New York, NY, 2003, pp. 1107–1152 Search PubMed.
- T. H. Parsell, R. K. Behan, M. T. Green, M. P. Hendrich and A. S. Borovik, Preparation and Properties of a Monomeric MnIV–Oxo Complex, J. Am. Chem. Soc., 2006, 128, 8728–8729 CrossRef CAS PubMed.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Formation, Structure, and EPR Detection of a High Spin FeIV—Oxo Species Derived from Either an FeIII—Oxo or FeIII—OH Complex, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS PubMed.
- T. Taguchi, K. L. Stone, R. Gupta, B. Kaiser-Lassalle, J. Yano, M. P. Hendrich and A. S. Borovik, Preparation and properties of an MnIV–hydroxide complex: proton and electron transfer at a mononuclear manganese site and its relationship to the oxygen evolving complex within photosystem II, Chem. Sci., 2014, 5, 3064–3071 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2287842–2287844. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04900k |
| ‡ [FeIII(O)MnIIIHpoat](OTf)2 can also be prepared via a different stepwise route from K[MnIIpoat] and [FeII(TMTACN)(OTf)2]; for details, see ESI, Scheme S1 and Fig. S9.† |
| This journal is © The Royal Society of Chemistry 2024 |