DOI:
10.1039/D3SC05034C
(Edge Article)
Chem. Sci., 2024,
15, 906-913
Symmetry breaking charge transfer leading to charge separation in a far-red absorbing bisstyryl-BODIPY dimer†
Received
23rd September 2023
, Accepted 14th December 2023
First published on 15th December 2023
Abstract
Symmetry breaking charge transfer is one of the important photo-events occurring in photosynthetic reaction centers that is responsible for initiating electron transfer leading to a long-lived charge-separated state and has been successfully employed in light-to-electricity converting optoelectronic devices. In the present study, we report a newly synthesized, far-red absorbing and emitting BODIPY-dimer to undergo symmetry-breaking charge transfer leading to charge-separated states of appreciable lifetimes in polar solvents. Compared to its monomer analog, both steady-state and time-resolved fluorescence originating from the S1 state of the dimer revealed quenching which increased with an increase in solvent polarity. The electrostatic potential map from DFT and the time-dependent DFT calculations suggested the existence of a quadrupolar type charge transfer state in polar solvents, and the singlet excited state to be involved in the charge separation process. The electrochemically determined redox gap being smaller than the energy of the S1 state supported the thermodynamic feasibility of the envisioned symmetry-breaking charge transfer and separation. The spectrum of the charge-separated state arrived from spectroelectrochemical studies, revealing diagnostic peaks helpful for transient spectral interpretation. Finally, ultrafast transient pump-probe spectroscopy provided conclusive evidence of diabatic charge separation in polar solvents by far-red pulsed laser light irradiation. The measured lifetime of the final charge-separated states was found to be 165 ps in dichlorobenzene, 140 ps in benzonitrile, and 43 ps in dimethyl sulfoxide, revealing their significance in light energy harvesting, especially from the less-explored far-red region.
Introduction
Symmetry breaking charge transfer and charge separation (SBCT and SBCS) are processes in which a pair of identical chromophores (dimer) absorb a photon and use its energy to transfer an electron from one chromophore to the other, breaking the symmetry of the chromophore in the dimer.1–20 This excited state phenomenon is rationalized to occur in the photosynthetic reaction center enabling the efficient formation of charge-separated states that are ultimately utilized in the synthesis of adenine triphosphate (ATP).21 The concept of SBCT and SBCS has also been successfully exploited for developing photocatalytic and photovoltaic systems operating with minimal energy loss.22,23 Also, it has been successfully employed in light-to-electricity converting optoelectronic devices.24,25 In symmetric molecular dimers, upon photoexcitation, the weakly coupled dimers form an excited state that dynamically localizes on one of the chromophore units of the dimer, rapidly producing a localized excited state (LE). Fluctuations in the local solvent environment surrounding the dimer create an asymmetry in the two units and are mainly responsible for driving charge transfer (SBCT) and solvent-stabilized charge separation (SBCS) with the radical cation and radical ion species. This mechanism of producing radical pairs is different from the asymmetric donor–acceptor (D–A) systems where the electron (or hole) transfer is energetically dictated to be a vectorial process producing D˙+–A˙− species through differences in redox potentials. Experimentally, the emission properties of the charge transfer state produced in the dimer are used to distinguish SBCT and SBCS. SBCT is often an emissive state, while SBCS is known to produce charge-separated states decaying nonradiatively. In SBCS the electron and holes are fully decoupled and behave independently while in SBCT electron–hole pairs are weakly coupled, often referred to as charge transfer excitons.1–20
From the above discussion, it is clear that there is a need for synthetic model systems to explore SBCT and SBCS to advance our ability to create symmetric systems that separate charges efficiently. However, photoinduced SBCS in synthetic dimers composed of the same chromophores is rare, mainly due to the energy gaps of the charge-separated states are not negative enough to drive SBCS.26–40 Among the well-known chromophores, SBCS is observed in 4,4-difluoro-4-bora-3a,4adiaza-s-indacene34,35 (also known as BODIPY, and its BF2-smaragdyrin analog36 (only SBCT), subphthalocyanine,37 and perylene diimide dimers.37–40 In the present study, instead of using ‘simple’ BODIPYs that absorb and fluoresce in the 510 nm range,41,42 we target far-red absorbing and emitting π-extended BODIPY dimers to check whether far-red excitation of the dimer would produce SBCS. The structure of the newly synthesized dimer along with the monomer used as a control is shown in Fig. 1. In the molecular design, two phenyl-styryl entities on each BODIPY are appended to shift the absorption and emission to the 650 nm range.43–46 As summarized below, efficient SBCS is indeed observed in the dimer in polar solvents with appreciable lifetimes of the charge-separated species.
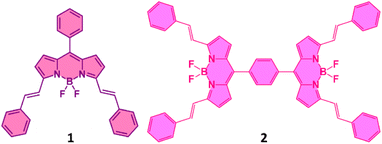 |
| | Fig. 1 Structure of the bis-styrylBODIPY monomer, 1 and dimer, 2 synthesized to probe SBCS. | |
Results and discussion
Synthesis
The synthesis of dimer 2 is shown in Scheme 1. Synthesis of 1 followed a similar procedure. That is, the synthesis of meso-phenyl BODIPY, 1a and its conversion to 1 (see ESI† for synthetic details). The structural integrity was confirmed by 1H and 13C NMR, and MALDI-TOF-mass analysis (see Fig. S1–S10 in ESI†).
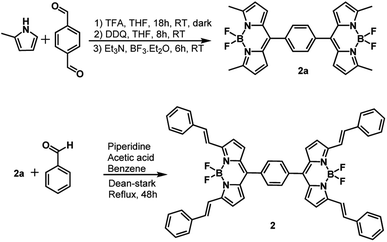 |
| | Scheme 1 Synthesis of far-IR absorbing and emitting bisstyrylBODIPY dimer, 2. | |
Spectroscopic studies
Fig. 2 shows the absorption spectrum of 1 and 2, normalized to the main far-red peak maxima, in solvents of varying polarity. Five solvents, viz., toluene (dielectric constant, ε = 2.38), o-dichlorobenzene (DCB, ε = 9.93), benzonitrile (PhCN, ε = 26.0) dimethyl formamide (DMF, ε = 36.7), and dimethyl sulfoxide (DMSO, ε = 46.7) were employed. The monomeric compound 1 was characterized by three main peaks located in the 360, 586, and 638 nm region while the dimer 2 was also characterized by three, slightly red-shifted, peaks in the 365, 592, and 640 nm region. The main far-red peak revealed a shift of 10 nm in both cases among the investigated solvents. Interestingly, compound 2 had an additional shoulder-type band around 378 nm in all solvents, likely due to an excitonic process occurring in the dimer. Importantly, a red-shifted absorption spectrum due to the presence of two styryl entities on the BODIPY π-system as compared to pristine BODIPY having no styryl entities (peak maxima was around 500 nm) in both monomer and dimer was noted. Additionally, no new peaks corresponding to charge transfer in the ground state including in polar solvents were observed. This suggests that if excited state charge separation indeed occurs in 2 that would be diabatic.
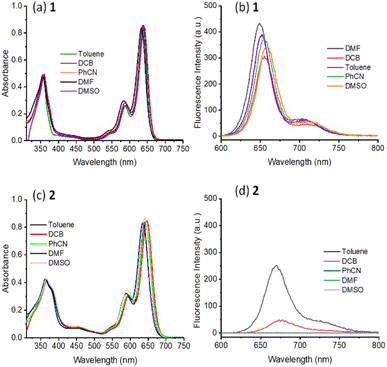 |
| | Fig. 2 Normalized absorption (a and c) and fluorescence (b and d) of 1 (a and b) and 2 (c and d) in the indicated solvents. The compounds were excited corresponding to their far-red peak maxima listed in Table 1. | |
Fig. 2b and d show the fluorescence spectrum of 1 and 2 in the indicated solvents. In the case of 1, a main emission band in the 648–656 nm range with a shoulder-type band in the 712 nm range was observed. This band was found to be red-shifted in 2 and appeared in the range of 670–676 nm, however, with substantial quenching. Going from non-polar to polar solvent, the quenching in the case of 1 was about 30% while such quenching in 2 reached 100% in polar solvents. In the least polar toluene used here, the emission intensity of 2 was about 40% less compared to the emission of equimolar concentration of 1. These results indicate the occurrence of excited state events in 2. Such was the case for the measured singlet lifetimes from the time-correlated single photon counting (TCSPC) method. Here, nanoLED operating at 495 nm was used to excite the samples, and the emission was collected at the emission band maxima. The singlet lifetimes of 1 and 2 in toluene were about 6.1 ns which decreased slightly with increasing the solvent polarity in the case of 1 while such decrease was substantial in the case of 2 (see Fig. S11† for decay curves). In fact, we could measure the lifetimes of 2 only in toluene and DCB, and in other polar solvents, the decay was beyond the detection limit of our TCSPC setup. Both steady-state and time-resolved emission results support the occurrence of excited state events in 2 in polar solvents. The singlet state energies, E0,0 were measured as the average energy of the 0,0 transitions of the absorption and fluorescence bands. An E0,0 value of ∼1.92 eV for 1 and ∼1.88 eV for 2 were possible to arrive. Further, the radiative, kr, and nonradiative, knr rate constants were also measured for the monomer and dimer in the investigated solvents using the standard procedure,48 and such results are given in Table 1. Faster knr for the dimer with an increase in solvent polarity was witnessed.
Table 1 Absorption and fluorescence peak maxima, singlet excited state energy, singlet lifetime, radiative and nonradiative rate constants of the investigated compounds in various solvents
| Compound |
Solvent |
λ
abs (nm) |
λ
em (nm) |
E
00 (ev) |
τ
av (ns) |
Φ
f
|
k
r (107 s−1) |
k
nr (107 s−1) |
|
Φ
f was calculated with reference to pristine BODIPY.47
|
|
1
|
Toluene |
638 |
655 |
1.92 |
6.12 |
0.46 |
7.52 |
8.82 |
| DCB |
640 |
660 |
1.91 |
5.68 |
0.39 |
6.87 |
10.74 |
| PhCN |
640 |
659 |
1.91 |
5.52 |
0.46 |
8.33 |
9.78 |
| DMF |
633 |
653 |
1.93 |
5.29 |
0.52 |
9.83 |
9.07 |
| DMSO |
639 |
660 |
1.91 |
5.44 |
0.43 |
7.90 |
10.48 |
|
2
|
Toluene |
642 |
670 |
1.89 |
6.18 |
0.38 |
6.15 |
10.03 |
| DCB |
646 |
675 |
1.88 |
3.79 |
0.09 |
2.37 |
24.01 |
| PhCN |
643 |
— |
— |
— |
— |
— |
— |
| DMF |
635 |
— |
— |
— |
— |
— |
— |
| DMSO |
640 |
— |
— |
— |
— |
— |
— |
Electrochemical and spectroelectrochemical studies
In the area of excited state electron and energy transfer, results gathered from electrochemical and spectroelectrochemical studies provide information that is a necessity for arriving at the energetics and subsequent product identification and evaluating kinetic information of photo-events. With this in mind, first, electrochemical studies on both the monomer and dimer in DCB were performed and their differential pulse voltammograms (DPVs) are shown in Fig. 3a. Both 1 and 2 revealed reversible one-electron reduction and quasi-reversible one-electron oxidation (as determined from cyclic voltammetry, see Fig. S12†). The first oxidation and first reduction of 1 are located at −0.66 and 1.12 V vs. Ag/AgCl while these processes for 2 appeared at −0.60 and 1.08 V vs. Ag/AgCl, respectively. This resulted in an electrochemical redox gap of 1.78 V for 1 and 1.68 V for 2. A smaller redox gap for 2 compared to 1 and the redox gap of 2 being smaller by 200 mV compared to the calculated E0,0 from optical studies (1.88 eV in DCB) are borne out from these studies.
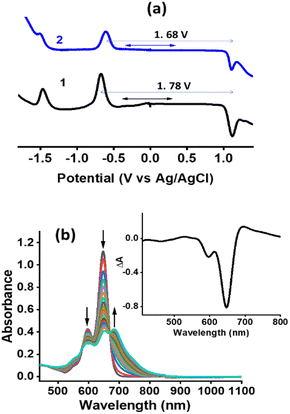 |
| | Fig. 3 (a) DPVs of the investigated compounds in DCB containing 0.1 M TBAClO4. (b) Spectral changes observed during the first oxidation of 2 in DCB containing 0.2 M TBAClO4. Fig. 3b inset shows the spectrum deduced for the charge-separated state by averaging the spectrum of the radical cation and radical anion minus that of the neutral compound for 2. | |
Spectral changes during the first oxidation of 2 in an optically transparent thin-layer cell are shown in Fig. 3b. During the process, diminished peak intensities of neutral 2 are accompanied by new peaks at 599, 652, and 686 nm. A clear isosbestic point at 575 nm suggesting the existence of only one equilibrium was also clear. During the process of the first reduction, the diminished intensity of the neutral compound did not result in new bands perhaps due to low molar absorptivity of such transitions. In order to generate the spectrum of the electron transfer product, the spectrum of the radical cation and radical anion were digitally added and subtracted with that of the neutral compound. Such a spectrum is shown as a figure inset of Fig. 3b. In the event of charge separation in 2, a spectrum closely resembling this one is expected.
Computational studies
In an effort to understand the geometry and electronic structure of 2 in relation to the anticipated SBCT and SBCS events, DFT calculations at the B3LYP/6-311+G (2df, pd) level49,50 were performed. The structures were fully optimized on the Born-Oppenheimer potential energy surface both in vacuo and DMSO solvent media (SCRF = CPCM). As shown in Fig. 4a, the bridging phenyl ring was perpendicular to the two bisstrylBODIPY entities without causing any steric hindrance. The center-to-center distance was 17.1 Å while the edge-to-edge distance was 6.0 Å between the two chromophores. The presence of DMSO did not make noticeable changes in the geometry.
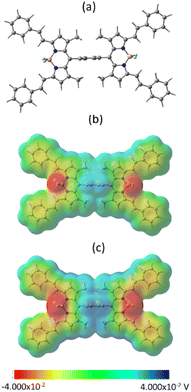 |
| | Fig. 4 (a) Optimized geometry in vacuo, (b and c) molecular electrostatic potential map in vacuo and DMSO, respectively. | |
Fig. 4b and c show the molecular electrostatic potential map (MEP) of 2in vacuo and DMSO. The positive and negative potentials were symmetrically distributed over the two bisstyryl entities irrespective of the media. This was also the case with frontier (HOMO-1, HOMO, LUMO, and LUMO+1, see Fig. S13†) orbitals. Initially, we assumed that in polar media, unequal electron distribution would result in favoring charge transfer (dipolar δ+–δ− state), however, to our surprise this was not the case. Interestingly, a close examination of the MEP map shows different degrees of potential, that is, higher positive potentials on the BF2 segments and higher negative potentials on the bridging phenyl entity in polar DMSO. This suggests the existence of a quadrupolar (δ+–δ−–δ+) state in polar media which could facilitate charge transfer upon photoexcitation.
To confirm this prediction, time dependent-DFT (TD-DFT) studies were performed for the dimer in DMSO to test whether an SBCS state forms when relaxing the dyad excited state. Evidence of CS was clear from the 4th (HOMO-1 → LUMO+1, at 605.15 nm, f = 2.0809), and 5th (HOMO-2 → LUMO, at 419.25 nm, f = 0.3011) excited states of the dimer. Evidence of CS for monomer 1 was rather slim. The natural transition orbitals (NTOs) were also produced as this provides an intuitive picture of the orbitals involved in electron–hole excitation. Here, separate unitary transformations were performed on the occupied and virtual set of orbitals in order to get a localized picture of the transition density matrix. The oscillator strength, f, is used as a measure of the probability of a given excitation. Table S1 in ESI† lists the key findings. Clear evidence of CS from HOMO → LUMO with contributions from other orbitals was witnessed.
Using the electrochemical and computational data, the thermodynamic driving force for charge separation and recombination as a function of solvent polarity was also calculated§51 and is given in Table 2. Using this data and the calculated excitation energy, a Jablonski-type energy diagram was constructed, as shown in Fig. 5. From this, SBCT in DCB, PhCN, and DMSO but not in toluene was clear from this study.
Table 2 The thermodynamic driving force for charge separation, ΔGCS and recombination, ΔGCR as a function of solvent polarity for 2
| Solvent |
ΔGsol |
ΔGCS |
ΔGCR |
| Toluene |
0.432 |
0.12 |
−2.20 |
| DCB |
−0.083 |
−0.39 |
−1.69 |
| PhCN |
−0.183 |
−0.49 |
−1.59 |
| DMSO |
−0.210 |
−0.52 |
−1.56 |
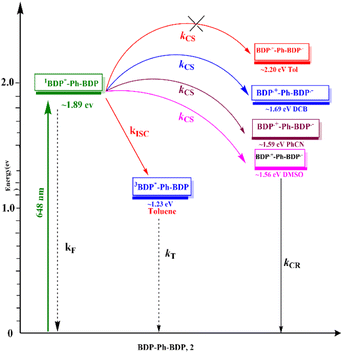 |
| | Fig. 5 Jablonski-type energy diagram revealing SBCS in 2 as a function of solvent polarity. (BDP = bisstrylBODIPY). | |
An attempt was also made to estimate the electronic coupling between the two BODIPY entities through the phenylene spacer. Kochi et al. reported an electronic coupling, H of 0.40 × 103 cm−1 in a phenothiazine dimer held by a phenylene linker (almost orthogonal geometry).52 Using the procedure given in the paper, H was estimated and found to range between 2.8–3.4 × 104 cm−1 for 2 depending on the solvent (see Table S2 in ESI†). Although more studies are warranted to confirm this, the higher values of H could be attributed to a smaller tilt angle (40° between the BODIPY entities) and higher extinction coefficient values for BODIPY compared with phenothiazine.
Pump-probe studies to probe SBCS
The systematic studies performed so far clearly suggested the possibility of the occurrence of SBCS in the investigated dimer. Support for this conclusion was primarily derived from a smaller redox gap compared to E0,0 values, and computational studies revealing a quadrupolar state in polar solvent. Femtosecond transient absorption (fs-TA) studies on both 1 and 2 in nonpolar toluene and polar solvents were performed to witness such photo events. Fs-TA studies on 1 serve as a control where no SBCT or SBCS is anticipated in both nonpolar and polar solvents. Fig. 6a shows fs-TA at the indicated delay times of monomer 1 in toluene. Upon excitation at 648 nm laser light, the instantaneously formed within the first couple of picoseconds singlet excited state revealed excited state absorption bands at 476, 524, 555, 603, 707, and 780 nm. Negative peaks at 590 and 642 nm were also observed. By comparing the peak positions of the absorption and fluorescence spectra, the 590 nm peak was attributed to ground state bleaching (GSB) while the 642 nm peak to both GSB and stimulated emission (SE). Initial decay and recovery of the positive and negative peaks were accompanied by a new peak at 465 nm and have been attributed to the relaxed S1 state from the initial hot S1 state. The recovery of the SE peak was slow and lasted over 3 ns, in agreement with earlier discussed fluorescence lifetimes. The data was subjected to GloTarAn analysis53,54 where a simple model representing S0 → S1(hot) → S1 was satisfactory. The species-associated spectra (SAS) for these states are shown in Fig. 6b. Lifetimes of 2.6 ps for S1* (hot state), and > 3 ns for the S1 state were possible to arrive. Similar spectral trends were found in the other two utilized solvents (see Fig. S14†).
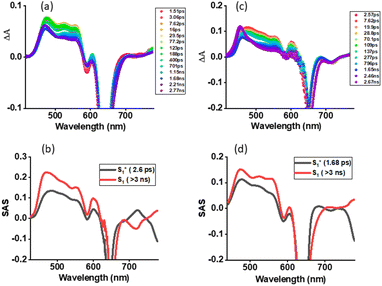 |
| | Fig. 6 Femtosecond transient absorption spectra at the indicated delay times of (a) 1 and (c) 2 in toluene (λex = 648 nm). Species associated spectrum from Glotaran analysis is shown beneath for each spectrum. | |
Fs-TA spectra at the indicated delay times for the dimer 2 in toluene are shown in Fig. 6c. The earlier discussed steady-state and time-resolved emission revealed no quenching in toluene suggesting a lack of SBCT or SBCS events. This was confirmed by the fs-TA studies. The hot S1 state of 2, 12*(hot) formed within the first couple of picoseconds, had ESA, GSB, and SE peaks resembling close to those of 11*. This was also the case for the S1 state developed in the latter stage. GloTarAn analysis of the data revealed lifetimes of 1.68 ps for S1* (hot state), and > 3 ns for the S1 by fitting the data to an S0 → S1(hot) → S1 model (see Fig. 6d for SASs). A close resemblance of SAS for different states of 1 and 2 is noteworthy here.
Next, fs-TA studies of 1 and 2 were performed in the most polar solvent utilized here, DMSO. Fig. 7a shows spectral changes observed for 1 with different delay times. The spectral changes tracked those observed in toluene for this compound. That is the initial formation of the S1(hot) state that relaxes to the S1 state. Glotaran analysis fitting the data to an S1(hot) → S1 model resulted in lifetimes of 2.64 ps for S1* (hot state), and > 3 ns for the S1 state (see Fig. 7b). Interestingly, the spectral changes observed for 2 in DMSO were different from those observed in toluene for this compound and also for 1. As shown in Fig. 7c, the S1 state revealed ESA peaks at 475, 521, 557, 606, and 692 nm. In addition, GSB at 590 and a GSB/SE peak at 640 nm were observed (see spectrum at 1.44 ps). In the next 5–6 ps, this spectrum transformed into a new spectrum with ESA peaks at 472, 560, and 692 nm, closely resembling the earlier spectrum deduced for the charge-separated species from spectroelectrochemical studies (shown in Fig. 3b inset) thus confirming excited state charge separation in polar DMSO. With additional delay times, peaks corresponding to the charge-separated state started decaying, and by about 200 ps, no peaks were visible, especially in the 400, 500, and 710 nm range corresponding to the triplet state.55 These results indicate the charge-separated state to nonradiatively relax to the ground state instead of populating the triplet state. The data was subjected to Glotaran analysis for the S1 → CS → S0 model. Lifetimes for the S1 state of 3 ps and the CS state of 43 ps were obtained from such analysis (see Fig. 7d).
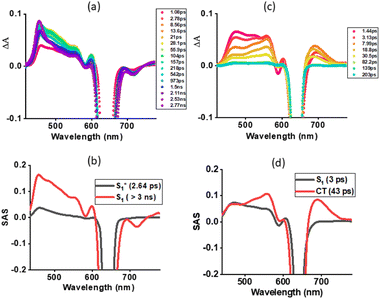 |
| | Fig. 7 Femtosecond transient absorption spectra at the indicated delay times of (a) 1 and (c) 2 in DMSO (λex = 648 nm). Species associated spectrum from Glotaran analysis is shown beneath for each spectrum. | |
The ability of 2 undergoing SBCS in other polar solvents was also investigated. As shown in Fig. S15,† in both DCB and PhCN spectral data supporting charge separation was witnessed. From Glotaran analysis, a lifetime of the charge-separated states of 140 ps in benzonitrile and 165 ps in DCB were obtained.
Conclusions
In summary, the occurrence of SBCS in a bisstyrylBODIPY dimer, 2 in polar solvents by far-red light laser illumination has been successfully demonstrated. The newly synthesized dimer for this purpose revealed strong absorption and emission in the far-red region, however, no charge transfer peak in the ground state was observed. Both steady-state and time-resolved emissions revealed quenching in polar solvents supporting the possibility of SBCS in the dimer, 2. Such an observation was also supported by the measured redox gap which was found to be smaller than the singlet state energy. Interestingly, the computational studies suggested the existence of a quadrupolar ground state in the dimer, and the TD-DFT studies supported charge separation from the S1 state in polar media. Femtosecond transient absorption studies provided ultimate evidence of SBCS in the dimer wherein the spectrum corresponding to the charge-separated state largely matched the spectrum deduced from spectroelectrochemical studies. The lifetime of the final charge-separated state ranged between 40 to 200 ps depending upon the polarity of the solvent. The present study brings out the significance of a far-red absorbing BODIPY dimer in generating charge-separated states of appreciable lifetimes, useful as model compounds for artificial photosynthesis and building optoelectronic devices. Further studies along this line are in progress in our laboratory.
Data availability
All the data supporting this article have been included in ESI.†
Author contributions
Methodology: synthesis, AY and SK; photo- and electrochemistry, RRK; computational studies, PAK, funding acquisition, FD; FD conceived the project and supervised the work. All authors have approved the published version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was financially supported by the National Science Foundation (Grant No. 2000988 to FD). The computational work was completed at the Holland Computing Center of the University of Nebraska, which receives support from the Nebraska Research Initiative.
Notes and references
- L. Estergreen, A. R. Mencke, D. E. Cotton, N. V. Korovina, J. Michl, S. T. Roberts, M. E. Thompson and S. E. Bradforth, Acc. Chem. Res., 2022, 55, 1561–1572 CrossRef CAS PubMed.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- E. Vauthey, ChemPhysChem, 2012, 13, 2001–2011 CrossRef CAS PubMed.
- H. Detert, E. Sugiono and G. Kruse, J. Phys. Org. Chem., 2002, 15, 638–641 CrossRef CAS.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Chem. –Eur. J., 2006, 12, 2358–2370 CrossRef CAS PubMed.
- A. Rebane, M. Drobizhev, N. S. Makarov, G. Wicks, P. Wnuk, Y. Stepanenko, J. E. Haley, D. M. Krein, J. L. Fore, A. R. Burke, J. E. Slagle, D. G. McLean and T. M. Cooper, J. Phys. Chem. A, 2014, 118, 3749–3759 CrossRef CAS PubMed.
- B. Carlotti, E. Benassi, C. G. Fortuna, V. Barone, A. Spalletti and F. Elisei, ChemPhysChem, 2016, 17, 136–146 CrossRef CAS.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- F. Terenziani, O. V. Przhonska, S. Webster, L. A. Padilha, Y. L. Slominsky, I. G. Davydenko, A. O. Gerasov, Y. P. Kovtun, M. P. Shandura, A. D. Kachkovski, D. Hagan, E. W. Stryland and A. Painelli, J. Phys. Chem. Lett., 2010, 1, 1800–1804 CrossRef CAS.
- S. Das, W. G. Thornbury, A. N. Bartynski, M. E. Thompson and S. E. Bradforth, J. Phys. Chem. Lett., 2018, 9, 3264–3270 CrossRef CAS PubMed.
- B. Dereka, D. Svechkarev, A. Rosspeintner, A. Aster, M. Lunzer, R. Liska, A. M. Mohs and E. Vauthey, Nat. Commun., 2020, 11, 1925 CrossRef CAS PubMed.
- V. Markovic, D. Villamaina, I. Barabanov, L. M. Lawson Daku and E. Vauthey, Angew. Chem., Int. Ed., 2011, 50, 7596–7598 CrossRef CAS PubMed.
- H. Yao, T. Okada and N. Mataga, J. Phys. Chem., 1989, 93, 7388–7394 CrossRef CAS.
- N. Mataga, H. Yao, T. Okada and W. Rettig, J. Phys. Chem., 1989, 93, 3383–3386 CrossRef CAS.
- J. J. Piet, W. Schuddeboom, B. R. Wegewijs, F. C. Grozema and J. M. Warman, J. Am. Chem. Soc., 2001, 123, 5337–5347 CrossRef CAS PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- C. E. Ramirez, S. Chen, N. E. Powers-Riggs, I. Schlesinger, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2020, 142, 18243–18250 CrossRef CAS PubMed.
- T. Zaima, W. Ota, N. Haruta, M. Uejima, H. Ohkita and T. Sato, J. Phys. Chem. Lett., 2023, 14, 9706–9712 CrossRef CAS PubMed.
- K. Wang, G. Shao, S. Peng, X. You, X. Chen, J. Xu, H. Huang, H. Wang, D. Wu and J. Xia, J. Phys. Chem. B, 2022, 126, 3758–3767 CrossRef CAS PubMed.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- A. N. Bartynski, M. Gruber, S. Das, S. Rangan, S. Mollinger, C. Trinh, S. E. Bradforth, K. Vandewal, A. Salleo, R. A. Bartynski, W. Bruetting and M. E. Thompson, J. Am. Chem. Soc., 2015, 137, 5397–5405 CrossRef CAS PubMed.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329–10339 CrossRef CAS PubMed.
- J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, M. R. D. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, Appl. Energy Mater., 2018, 1, 1083–1095 CrossRef CAS.
- E. Collet, M. Lemée-Cailleau, M. Buron-Le Cointe, H. Cailleau, M. Wulff, T. Luty, S. Koshihara, M. Meyer, L. Toupet, P. Rabiller and S. Techert, Science, 2003, 300, 612–615 CrossRef CAS PubMed.
- J. M. Giaimo, A. V. Gusev and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 8530–8531 CrossRef CAS PubMed.
- R. W. Zijlstra, F. C. Grozema, M. Swart, B. L. Feringa and P. T. van Duijnen, J. Phys. Chem. A, 2001, 105, 3583–3590 CrossRef CAS.
- M. W. Holman, P. Yan, D. M. Adams, S. Westenhoff and C. Silva, J. Phys. Chem. A, 2005, 109, 8548–8552 CrossRef CAS PubMed.
- B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 12268–12269 CrossRef CAS PubMed.
- S. Bhosale, A. L. Sisson, P. Talukda, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- A. L. Sisson, N. Sakai, N. Banerji, A. Furstenberg, E. Vauthey and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 3727–3729 CrossRef CAS.
- Y. Wu, R. M. Young, M. Frasconi, S. T. Schneebeli, P. Spenst, D. M. Gardner, K. E. Brown, F. Wurthner, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2015, 137, 13236–13239 CrossRef CAS PubMed.
- Z. Szakács and E. Vauthey, J. Phys. Chem. Lett., 2021, 12, 4067–4071 CrossRef.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284–286 RSC.
- J. S. Sandoval, Q. Gong, L. Jiao and D. W. McCamant, J. Phys. Chem. A, 2023, 127, 7156–7167 CrossRef CAS PubMed.
- L. Wen, X. Cao, S. Lee, L. Xu, Y. Rao, S. Kang, D. Kim, A. Osuka and J. Song, Commun. Chem., 2023, 6, 25 CrossRef CAS PubMed.
- P. Roy, G. Bressan, J. Gretton, A. N. Cammidge and S. R. Meech, Angew. Chem., Int. Ed., 2021, 60, 10568–10572 CrossRef CAS PubMed.
- N. E. Powers-Riggs, X. Zuo, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2019, 141, 17512–17516 CrossRef CAS PubMed.
- E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2021, 143, 13769–13781 CrossRef CAS PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 11, 4891–4932 CrossRef.
- M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30–47 CrossRef CAS PubMed.
- S. Shao, M. B. Thomas, K. H. Park, Z. Mahaffey, D. Kim and F. D'Souza, Chem. Commun., 2018, 54, 54–57 RSC.
- N. Zarrabi, C. O. Obondi, G. N. Lim, S. Seetharaman, B. G. Boe, F. D'Souza and P. K. Poddutoori, Nanoscale, 2018, 10, 20723–20739 RSC.
- C. V. Ileperuma, J. Garcés-Garcés, S. Shao, F. Fernández-Lázaro, Á. Sastre-Santos, P. A Karr and F. D'Souza, Chem. – Eur. J., 2023, 29, e202301686 CrossRef CAS PubMed.
- S. Kazemi, Y. Jang, A. Liyanage, P. A. Karr and F. D'Souza, Angew. Chem., Int. Ed., 2022, 61, e202212474 CrossRef CAS PubMed.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC.
- C. Cooper, R. Paul, A. Alsaleh, S. Washburn, W. Rackers, S. Kumar, V. N. Nesterov, F. D'Souza, S. A. Vinogradov and H. Wang, Chem. – Eur. J., 2023, 29, e202302013 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16 (Revision A.03), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
-
R. Dennington, T. A. Keith, and J. M. Millam, GaussView (Version 6.0.16), Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 7, 259–276 CrossRef.
- D. Sun, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 1388–1401 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
-
Glotaran, http://glotaran.org/ Search PubMed.
- A. Fatima, J. Rabah, E. Allard, H. Fensterbank, K. Wright, G. Burdzinski, G. Clavier, M. Sliwa, T. Pino, R. Meallet-Renault, K. Streenkeste and M.-H. Ha-Thi, Photochem. Photobiol. Sci., 2022, 21, 1573–1584 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and experimental details, additional CV and fs-TA spectral data. See DOI: https://doi.org/10.1039/d3sc05034c |
| ‡ Equal contribution. |
§ The free energy change for charge separation (ΔGCS) from the singlet excited state of the modified BODIPY within the donor-acceptor system was calculated using spectroscopic, computational and electrochemical data following eqn (1)–(3).50| | | ΔGCR = Eox − Ered + ΔGS | (1) |
where E00 and ΔGCS correspond to the energy of excited singlet state and electrostatic energy, respectively. The Eox and Ered represent the oxidation and reduction potentials of 2. The term ΔGs refers to the static Coulombic energy, calculated by using the “dielectric continuum model” according to eqn (3):| | 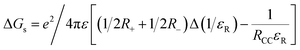 | (3) |
The symbols ε0, and εR represent the vacuum permittivity and dielectric constant of the solvent used for photochemical and electrochemical studies, respectively. RCC is the center-to-center distance between pull-pull entities from computational data. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a