
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new era of LMCT: leveraging ligand-to-metal charge transfer excited states for photochemical reactions
Ann Marie
May
 and
Jillian L.
Dempsey
*
and
Jillian L.
Dempsey
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA. E-mail: dempseyj@email.unc.edu
First published on 17th April 2024
Abstract
Ligand-to-metal charge transfer (LMCT) excited states are capable of undergoing a wide array of photochemical reactions, yet receive minimal attention compared to other charge transfer excited states. This work provides general criteria for designing transition metal complexes that exhibit low energy LMCT excited states and routes to drive photochemistry from these excited states. General design principles regarding metal identity, oxidation state, geometry, and ligand sets are summarized. Fundamental photoreactions from these states including visible light-induced homolysis, excited state electron transfer, and other photoinduced chemical transformations are discussed and key design principles for enabling these photochemical reactions are further highlighted. Guided by these fundamentals, this review outlines critical considerations for the future design and application of coordination complexes with LMCT excited states.
 Ann Marie May | Ann Marie May obtained her B.S. in chemistry from Virginia Tech in 2019, where she was introduced to physical inorganic chemistry while working under Prof. Amanda Morris. She has continued pursuing these interests throughout her PhD studies at the University of North Carolina at Chapel Hill under the direction of Prof. Jillian Dempsey. Her current research interests focus on exploring the electronic structure, photophysics, and photochemistry of transition metal complexes with low-energy ligand-to-metal charge transfer excited states. |
 Jillian L. Dempsey | Jillian L. Dempsey obtained her S.B. in chemistry at the Massachusetts Institute of Technology, completing research with Daniel Nocera, and her PhD in chemistry at the California Institute of Technology, where she studied with Harry Gray and Jay Winkler. After completing postdoctoral research at the University of Washington with Daniel Gamelin, she began her independent career at the University of North Carolina at Chapel Hill where she now holds the position of Professor. Her research program is driven by deep curiosities about charge transfer reactions and a commitment to advancing the fundamental science that underpins renewable energy technologies. |
1 Introduction
Charge transfer excited states drive most photochemical reactions. Upon irradiation with light, these highly absorptive transitions redistribute electron density across a molecule, generating a chemical potential in the excited state capable of initiating reactivity including electron transfer, proton transfer, bond homolysis, and more.1–11 Leveraging charge transfer excited states provides a viable pathway to induce new reactivity and harness solar energy to drive chemical reactions. Charge transfer excited states are categorized into specific classes, including metal-to-ligand charge transfer (MLCT), ligand-to-metal charge transfer (LMCT), intraligand charge transfer (ILCT) and ligand-to-ligand charge transfer (LLCT).12–16 For MLCT, ILCT, and LLCT excited states, donor orbitals populate ligand-based acceptor orbitals. However, in LMCT excited states, metal-based acceptor orbitals are populated and leave behind oxidized ligand-based orbitals, imparting unique properties to the excited state.Transition metal complexes with LMCT excited states have historically been underexplored compared to other charge transfer excited states, namely due to their lack of photoluminescence, short excited state lifetimes, and/or propensity for degradation in the excited state. Due to these challenges, photoactive molecules with LMCT excited states were historically disregarded and photochemists turned to complexes with MLCT excited states, like tris(2,2′-bipyridine) ruthenium(II), [Ru(bpy)3]2+, for their attractive long-lived excited states, reversible excited state electron transfer, and (photo)chemical stability.13,17
Recently, a new era for LMCT excited states has begun. A handful of transition metal complexes are now known to display chemical and photochemical stability, exhibit nanosecond lifetimes, and undergo photochemical reactions.2,16,18–23 Despite these promising results, substantial work is still required for LMCT excited states to become robust and versatile participants in photochemical reactions. Inspired by this resurgence and overall promise of LMCT excited states, this article aims to highlight transition metal complexes with LMCT excited states and understand the photochemistry accessible from these states. This article will also highlight synthetic strategies to design complexes with low energy LMCT transitions suitable for photochemical reactions. Furthermore, strategies to enhance transition metal complex stability and extend excited state lifetimes will also be discussed, highlighting state-of-the-art literature examples. Specific classes of reactions promoted by LMCT excited states, including excited state electron transfer (ES-ET), visible light-induced homolysis (VLIH), excited state proton transfer (ES-PT), and photo-induced chemical rearrangements, will be presented.
2 Designing transition metal complexes with low-lying LMCT excited states for photochemical reactions
LMCT transitions occur when light promotes an electron from a ligand-based molecular orbital to a metal-based molecular orbital. These excitations are facilitated by specific electronic structures, where transition metal complexes exhibit electron-deficient metal centers and have strongly donating ligand scaffolds.2,24 Electron deficient metal centers provide vacant low energy metal-based orbitals ripe for population. In result, excitation from a ligand-based molecular orbital to an antibonding metal-based d orbital (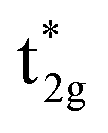 or
or  ) is accessible at low energies.2,24 Strong π donor ligands are known to promote weak to moderate ligand field splitting, where bonding orbitals are primarily ligand-based and antibonding orbitals are primarily metal-based (Fig. 1, for Oh symmetry).24 Furthermore, their anionic character supports highly oxidized metal centers. Strong σ donating ligands also give rise to coordination complexes with low-lying vacant metal-based orbitals when paired with electron deficient metal centers, albeit these metal-based orbitals exhibit non-bonding (n.b.) character. Therefore, to facilitate access to LMCT excited states, strongly π and σ donating ligands are commonly employed alongside electron deficient metal centers.
) is accessible at low energies.2,24 Strong π donor ligands are known to promote weak to moderate ligand field splitting, where bonding orbitals are primarily ligand-based and antibonding orbitals are primarily metal-based (Fig. 1, for Oh symmetry).24 Furthermore, their anionic character supports highly oxidized metal centers. Strong σ donating ligands also give rise to coordination complexes with low-lying vacant metal-based orbitals when paired with electron deficient metal centers, albeit these metal-based orbitals exhibit non-bonding (n.b.) character. Therefore, to facilitate access to LMCT excited states, strongly π and σ donating ligands are commonly employed alongside electron deficient metal centers.
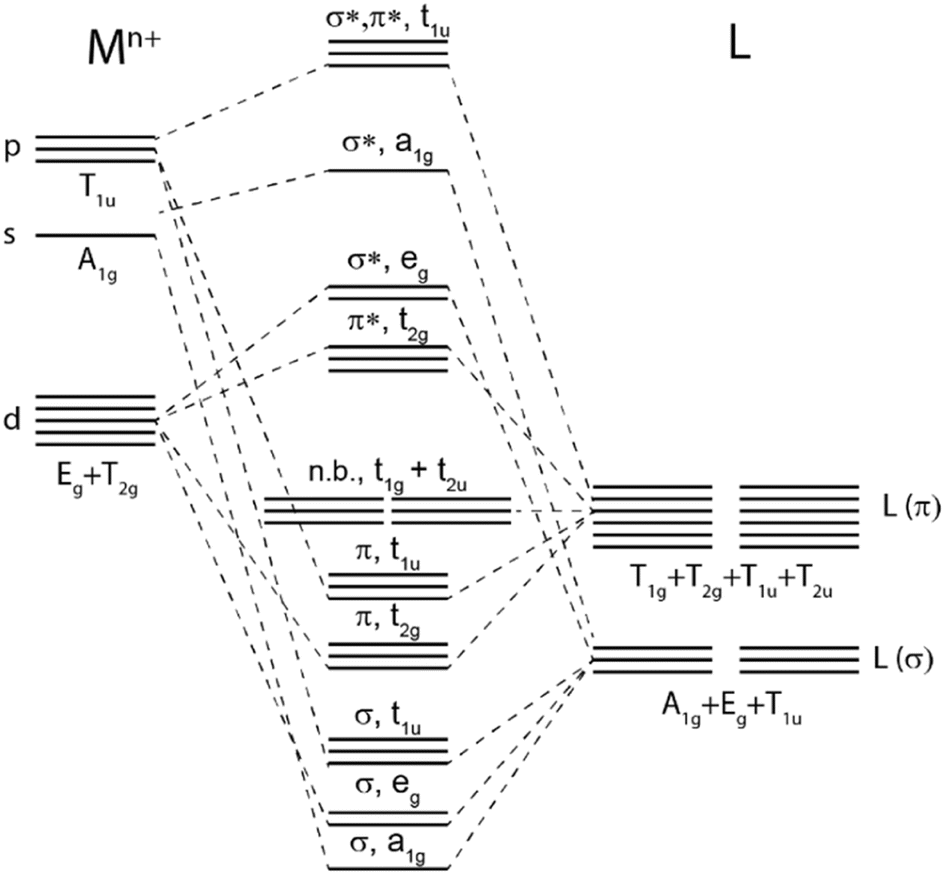 | ||
| Fig. 1 Molecular orbital diagram of an octahedral transition metal complex with π donating ligands. | ||
Classic examples of such LMCT-containing complexes include permanganate, [MnO4]−, and iridium(IV) hexabromide, [IrBr6]2−. For [MnO4]−, its strong π donating oxo ligands facilitate a low energy absorption within the visible region centered at 550 nm, assigned to promotion of an electron from a ligand-based t1 non-bonding orbital to the metal-based e* orbital.25–27 Similarly, π donating bromide ligands facilitate two sets of LMCT transitions in [IrBr6]2−, one in the visible (ranging from 500 to 900 nm) attributed to population of the 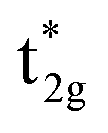 orbital and a second in the UV (spanning 250 nm to 450 nm) attributed to population of the
orbital and a second in the UV (spanning 250 nm to 450 nm) attributed to population of the 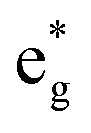 orbital.28–30
orbital.28–30
While a large number of coordination complexes exhibit LMCT transitions, very few undergo excited state reactivity from their LMCT excited state, and thus only a handful have been employed as photosensitizers or photocatalysts. When identifying coordination complexes with LMCT excited states for photochemical applications, there are several considerations. The first consideration is excited state character. The nature, geometry, and (de)localization of an excited state greatly dictates its photoreactivity. For LMCT excited states, this reactivity is dictated by a shift in electron density from the ligand scaffold to the metal, producing a formally reduced metal center and oxidized ligand(s). Careful consideration of a LMCT excited states' donor and acceptor orbitals can further tune excited state properties, such as excited state reduction potentials and ligand lability.
The second consideration for enabling photosensitization and photocatalysis is excited state energetics. The relative energy of the LMCT electronic transition, with respect to other optical transitions, is important. For most photochemical reactions, photoreactivity stems from the lowest energy excited state, necessitating the construction of complexes with low energy LMCT excited states. This criterion is rationalized by Kasha's rule which states that photoluminescence, and by extension photoreactions, occur from the lowest energy excited state of a given multiplicity – a result of fast internal conversion to the lowest energy excited state upon photoexcitation.31,32 While there are exceptions to this rule (some of which will be discussed herein), Kasha's rule is generally applicable to most photochemical reactions and highlights a common design criterion considered when designing photoactive molecules. To meet this criterion, many complexes that exhibit low energy LMCT excited states contain strong π donating ligands. The anionic nature of π donating ligands supports high valent metal centers, enabling low-lying, metal-based antibonding orbitals that can be filled upon photoexcitation. Simultaneously, strong π donors raise the energy of the t2g orbitals, resulting in small octahedral field (ΔOh) splittings, where both metal-based orbitals are antibonding. In result, this pairing provides ligand-based HOMOs and metal-based LUMOs (or SOMOs) poised for low energy LMCT transitions.
However, incorporating strong π donors and high valent metal centers does not always ensure low energy LMCT excited states. The weak field splittings of these complexes promote small ΔOh splitting, and can favor low energy d–d transitions for complexes with one or more d electrons. In addition, small ΔOh splitting may also enable high spin configurations for some complexes, similarly supporting low energy d–d transitions. To combat these low-lying d–d transitions, strong σ donors can be incorporated to stabilize the t2g orbitals and promote low spin electronic configurations. This stabilization results in larger ΔOh splitting relative to π donors and the ligand field (metal centered, d–d) excited states that commonly deactivate LMCT excited states increase in energy relative to the charge transfer transition.2,15 In addition to carefully tuning ligand identity and electronics, metal identity and oxidation state can similarly manipulate ΔOh splittings, where ΔOh splittings increase with increasing oxidation state number and principal quantum number, and change the energetics of metal-based acceptor orbitals in LMCT transitions. As a result, careful consideration of ligand and metal identities can enable the design of transition metal complexes with low energy LMCT excited states.
The third consideration for constructing effective photosensitizers and photocatalysts is excited state lifetime. For some reactions, ultrafast lifetimes are sufficient to enable photochemistry, particularly when the photoreaction is unimolecular in nature. However, lifetimes on the nanosecond timescale are typically required for bimolecular reactions with substrates in the millimolar concentration range, where excited states must persist on the timescale of diffusion. A variety of factors play into the lifetimes of these excited states, including excited state spin multiplicity and accessible nonradiative decay pathways. Metal complexes capable of intersystem crossing (e.g., from singlet to triplet excited states) are generally more likely to exhibit long-lived excited state lifetimes than complexes that cannot. Furthermore, complexes comprised of second and third row transition metals have larger ligand field splitting than first row metals. Large ligand field splittings can prevent deactivation of a charge transfer excited state through metal-centered excited states. Similarly, ligand choice also impacts ligand field splittings, as illustrated in Tanube–Sugano diagrams for specific electronic configurations.
The fourth design criterion for constructing photosensitizers and photocatalysts is (photo)stability. Most electronic transitions result in the population of antibonding orbitals upon photoexcitation. Therefore, in the case of many LMCT transitions, the metal–ligand bond lengths elongate and can result in ligand dissociation. For example, despite the short excited state lifetime of [MnO4]−, this molecule is capable of undergoing photoreactivity to unimolecularly produce O2 and [MnO2]− upon irradiation.25 This ultrafast photodecomposition rationalizes why it is not commonly employed as a photocatalyst or photosensitizer in photochemical reactions, as this irreversible reaction will inherently compete with more complex photoreactions of interest. Conversely, while the LMCT excitation of the d5 octahedral complex, [IrBr6]2−, also populates an antibonding t2g orbital, it does not showcase photochemical instability. Instead, [IrBr6]2− is unstable in the ground state, where it is known to undergo substitution with water molecules and is only stable in the presence of excess bromide ions.28 Given the challenges that arise from populating metal–ligand antibonding orbitals and transiently oxidizing ligands, it is important to employ ligands that are strongly coordinating and stable to oxidation to access transition metal complexes capable of photochemistry from low-lying LMCT excited states.
While the four criteria outlined above—excited state character, excited state energy, excited state lifetime, and photochemical stability—appear as straightforward design principles, designing transition metal complexes that possess low energy LMCT excited states capable of photochemical reactions is difficult. Targeting complexes with electron deficient metal centers and strongly π and/or σ donating ligands provides a sufficient starting point for designing complexes with LMCT excited states capable of photochemistry, yet these design principles are complex in practice. Molecular geometry, ligand donor ability, metal identity, and oxidation state all impact molecular orbital energetics, relative ordering of excited states, and even the metal vs. ligand character of excited states. In result, transition metal complexes that have low-lying LMCT excited states and undergo photochemical reactions exhibit diverse structures, as they have found design constraints that balance each of the outlined criteria.
Motivated by the complexity of photosensitizer and photocatalyst design, we have chosen to briefly highlight trends observed for complexes with low-lying LMCT excited states and general precedence for engendering LMCT excited states through coordination complex design. While only some of these complexes have been employed for photochemical applications, understanding ways to systematically design complexes with low-lying LMCT excited states is a key first step in achieving widespread photochemical reactivity. Below, we discuss general ligand properties, then review trends within families of coordination complexes with the same d-electron configurations.
A variety of ligands are well known to support LMCT excited states, including π donating halides, oxos, sulfides, and alkoxy ligands, as well as σ donating diphosphines, pyridinedipyrrolides, and N-heterocyclic carbenes.18,20,22,33–54 However, in addition to classically categorized σ and π donors, other ligands typically perceived as π acceptors have also been reported to support LMCT excited states. For example, complexes with cyanide and isocyanide ligands are known to have low-lying LMCT excited states.55–61 Furthermore, in a recent study polypyridyl nitrogen-based ligands, traditionally viewed as π accepting aromatic ligands, were found to engage in bonding as π donors when coordinated to first row transition metals.62 This observation, in conjunction with other examples, showcases the diversity of ligands that are known to support complexes with LMCT excited states. It also further underscores the opportunity to leverage TD-DFT to help identify and visualize the ligand-based orbitals that participate in LMCT transitions, and gain a deeper understanding of the ligand properties needed to support low energy LMCT excited states.22,47,55,63–65
The d electron configurations for complexes with LMCT excited states are also diverse. In particular, d0 complexes are well known for low-lying LMCT excited states, resulting from their vacant metal orbitals which can easily be populated by filled ligand orbitals. When d orbitals are filled, d electrons are capable of participating in their own excitation processes, such as d–d transitions, metal-to-ligand charge transfer, and other metal-based charge transfer processes (select d configurations shown in Fig. 2). For d0 complexes, these competing transitions (and the resultant excited states) are absent, rationalizing their abundance within the field of LMCT excited states. Quintessential examples of low-lying LMCT excited states include d0 complexes, such as vanadate, chromate, permanganate, and Group (IV) metallocenes.23,26,66–73
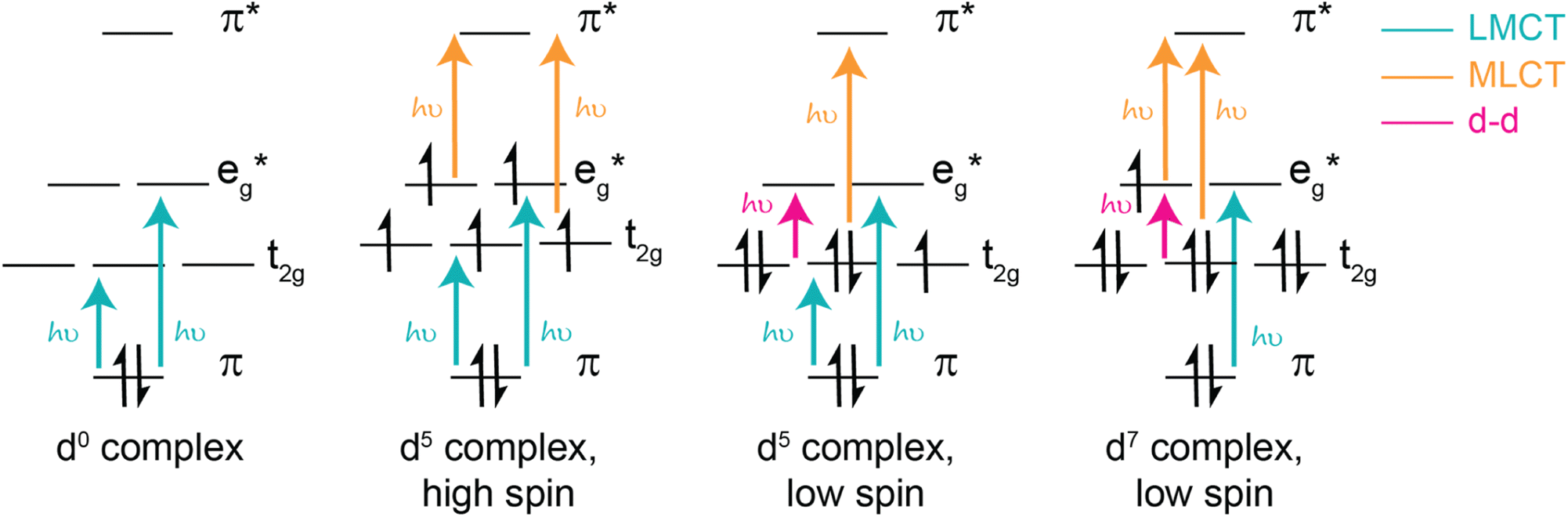 | ||
| Fig. 2 Examples of accessible excited states from d0, d5, and d7 complexes, which most commonly exhibit LMCT. In addition, it is important to note the bonding character of the metal-based t2g orbitals depends on the nature of the coordinating ligands. Strong π donors facilitate antibonding t2g orbitals, σ donors promote non-bonding t2g orbitals, and π acceptors facilitate bonding t2g orbitals. | ||
In addition to supporting low energy LMCT transitions, the d0 electronic configuration is also advantageous because the absence of d–d transitions eliminates a common pathway for excited state deactivation and simultaneously enables the opportunity for intersystem crossing of a singlet excited state to a triplet excited state — two qualities that are known to increase excited state lifetimes. However, despite these promising features, only a few examples of long-lived excited states from d0 LMCT excited states have been reported, including pentamethylcyclopentadienyltantalum(V)-based complexes, tantalum(V) imido complexes, scandocene derivatives, and zirconium(IV) bis(pyrroyl)pyridine-based complexes, highlighting an opportunity for further growth within the field.46,47,74–78 Regardless, several d0 complexes are known to promote photochemical reactions, several of which will be highlighted below.
Coordination complexes with other d electron configurations that have low energy LMCT excited states are also capable of promoting photochemical reactions. While d0 complexes are by far the most common transition metal complex to exhibit low-lying LMCT excited states, the second most common electronic configuration is likely d5, highlighted in Fig. 2. For many d5 complexes with low-lying LMCT excited states, strong σ donor ligands help deactivate ligand field states. In many cases, this strong σ donation results in low spin configurations, such as for iron(III) N-heterocyclic carbene complexes.2,49 However, it is important to note that high spin configurations are also possible for d5 complexes and can similarly facilitate photochemical reactions, as showcased by the visible light-induced homolysis of ferrioxalate. In addition to incorporating strong σ donors to destabilize ligand field states, another common design strategy is to access low spin configurations by employing second or third row transition metals, which exhibit larger ligand field splitting compared to first row counterparts. Recently, this low spin d5 electronic configuration has been termed a “sweet spot” for designing complexes with LMCT excited states and has shown great promise in enabling photochemical reactions, several of which are highlighted herein.2 Notably, however, low spin d5 complexes have excited states with doublet character. As such, intersystem crossing is not accessible and these complexes often have short excited state lifetimes.
In addition to d0 and d5 electronic configurations, LMCT excited states are also possible from other electronic configurations. Complexes with lower d electron configurations (i.e., d1-d4 electron configurations) are known to have LMCT transitions;18,79–83 however, most of these known complexes, have low-lying d–d transitions that deactivate low energy LMCT excited states and limit photochemistry from LMCT states. For example, [CrIII(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′-dipyridin-2-ylpyridine-2,6-diamine) and other complexes in the molecular ruby family have recently gained notoriety for their long-lived near-IR photoluminescence. However, it is important to note that this luminescence stems from intersystem crossing from metal-based or LMCT excited states to d–d excited states, generating long-lived, spin-forbidden near-IR emissions.53,80,81,84 Analogous emission has also recently been reported a Mn(IV) N-heterocyclic carbene complex.53 One notable exception to this trend is a homoleptic Mn(IV) complex with 2,6-diguanidylpyridine ligands which was recently reported to undergo excited state reductions from its low-energy 2LMCT and 4LMCT excited states.65 Other photochemical applications from low energy LMCT excited states of d1-d4 complexes are quite rare, and if present result from anti-Kasha behavior.
LMCT excited states for complexes with higher d electron configurations (i.e., d6-d9) are also reported, and a few have been utilized as photocatalysts. Many of these complexes are comprised of first row transition metals with strong π and σ donors and notable examples include cobalamin, cobaloxime, tris(bipyridine)cobalt(III), bis(2,6-diguanidylpyridine)cobalt(III) complexes, copper(II) phenanthroline-based complexes, and a variety of metal halide complexes.62,85–89 Several of these complexes, particularly metal halide complexes, have also gained traction as photoredox catalysts to drive a variety of organic transformations.1,86,89–93 Like d5 complexes, these electronic configurations enable a variety of d–d transitions and MLCT excited states, as illustrated in Fig. 2.1,19,62,94–98 Therefore, careful control of orbital energetics is required to preferentially enable low energy LMCT excited states.
Finally, it is worth noting that LMCT transitions are also accessible from lanthanide-based complexes, where LMCT transitions result in the population of low-lying f orbitals instead of d orbitals. While this topic will not be directly covered within this perspective article, several f-block metal complexes are known to facilitate low-energy LMCT excited states. Examples include cerium(IV) hexachloride, cerium(IV) carboxylates, cerium(IV) mixed-ligand guanidinate–amide complexes, ytterbocene complexes, europium(III) terpyridine-based complexes, among others.99–104 Recent reports have also showcased their role in dictating the luminescent properties of f-block metal complexes and their utility in driving photoredox reactions via VLIH, illustrating analogous applications to their d-block brethren.105–108
Collectively, these examples showcase the diversity of complexes known to support low energy LMCT excited states. d0 complexes are well-known to promote low energy LMCT excited states, whereas d1-d9 complexes rely on a careful balance of metal identity, oxidation state, molecular geometry, and ligand sets to support low-lying LMCT transitions. Accessing photochemistry from these excited states is similarly complex and requires careful consideration of each of these criteria to enable desired photoreactivity. As such, we have chosen to briefly review a selection of coordination complexes with LMCT excited states and discuss the photochemical reactions that they promote. This selection aims to demonstrate the breadth of photochemistry accessible by LMCT excited states and provide perspectives on opportunities to drive photochemistry from LMCT excited states within a broader scope.
3 Photochemical reactivity from LMCT excited states
Transition metal complexes with LMCT excited states are known to undergo several types of photoreactions, including visible light-induced homolysis (VLIH), excited state electron transfer (ES-ET), and photoinduced chemical transformations.3.1 Visible light-induced homolysis
One of the most common photoreactions that transition metal complexes with LMCT excited states undergo is visible light-induced homolysis (VLIH). In VLIH reactions, homolysis of a metal–ligand bond is promoted by irradiation, producing a formally reduced metal center and oxidized ligand radical. While the net observed chemistry is bond homolysis of a metal–ligand bond, the chemistry is best thought of as a two-step process. First, light promotes intramolecular electron transfer (ET) from a coordinating ligand to a metal center, producing a formally reduced metal center and oxidized ligand radical. Second, the ligand radical dissociates. VLIH reactions are capable of forming a variety of reactive radical species, including carboxylate, halogen, and azide radicals, among others, and commonly initiate follow-up chemical reactions in the ground state. Common applications of VLIH include actinometry and photoredox catalysis.1,21,92,97,98,109–111 Herein, we highlight two examples and discuss their mechanistic details.The photochemistry of ferrioxalate is promoted from the irradiation of a broad absorption centered at λmax = 260 nm, spanning toward the red (to ∼550 nm).115 This feature exhibits a molar extinction coefficient of ∼6000 M−1 cm−1 consistent with a charge transfer excited state.115 Further visualization of this excited state was provided by Ogi et al., who showed through TD-DFT calculations that two ligands donate electron density to an iron d orbital, albeit the visualized transition is one of many that make up this absorption band.116 Other natural transition orbitals are reported to have analogous LMCT character and the calculated UV-vis absorption spectrum constructed from these identified transitions is consistent with the experimental spectrum.116 The broad absorption is ideal for actinometry experiments and further showcases the widespread utility of this compound.
Photochemical actinometry experiments exploit the LMCT excited state of ferrioxalate to drive reactivity that forms ferrous oxalate, CO2, and oxalate, as summarized in Fig. 3. Most commonly, a bond homolysis mechanism is used to rationalize this observed photochemical reactivity, where light promotes electron transfer from the oxalate ligand to the Fe center, followed by dissociation of an unstable oxalate radical anion (C2O4˙−), see Fig. 3.115,117–119 Recent ultrafast time-resolved mid-IR spectroscopy experiments showed that the C–C bond of C2O4˙− rapidly cleaves to form CO2 and CO2˙−, the latter is capable of thermally reducing another equivalent of ferrioxalate.120 Complimentary time-resolved X-ray absorption and computational reports further support these claims.116 Similar reactivity has been noted across a series of iron(III) carboxalato complexes.121 This general photoreactivity showcases a prosperous route to form CO2˙− and other potent radical anions.117,121
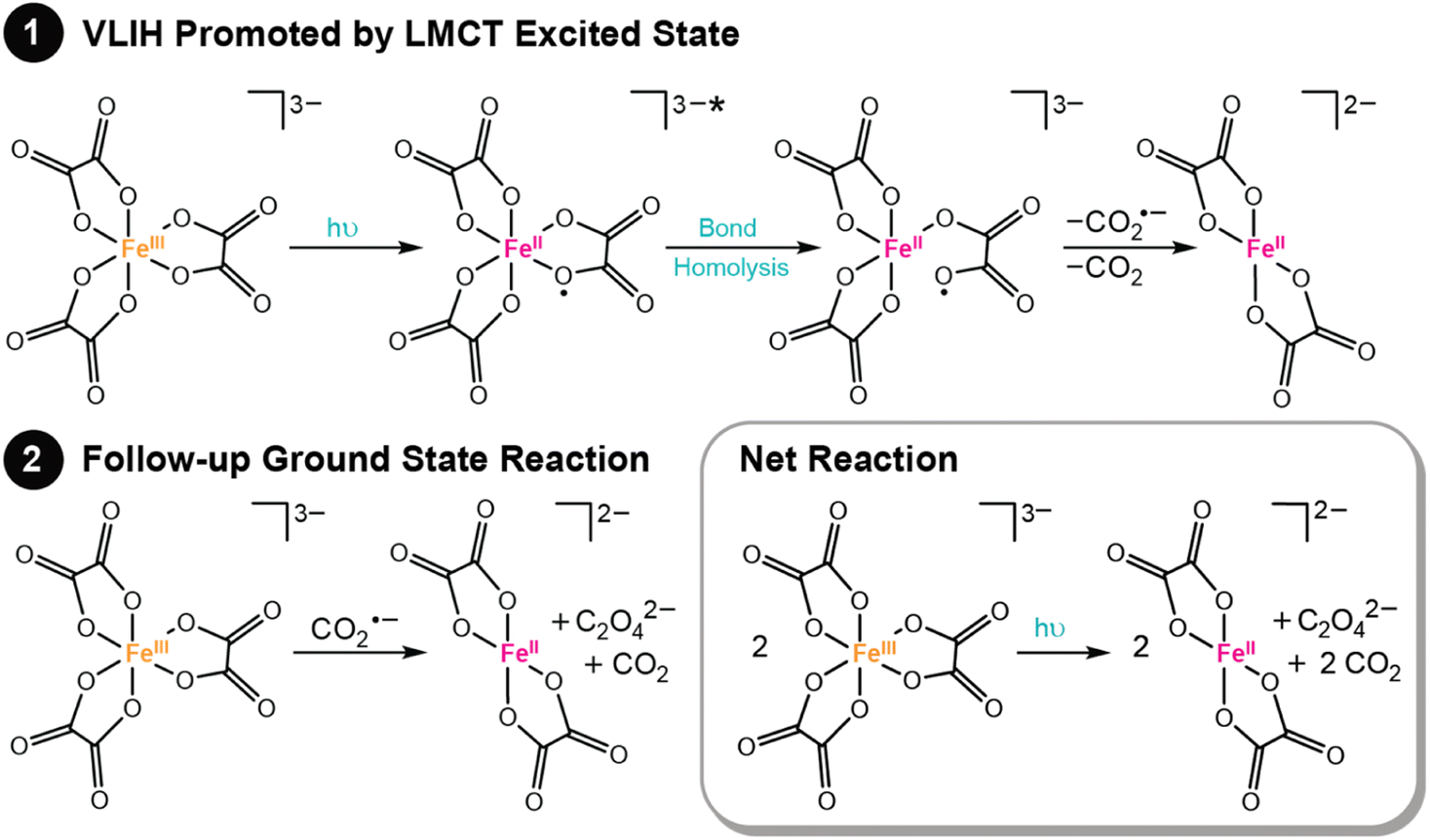 | ||
| Fig. 3 VLIH mechanism of ferrioxalate to form ferrous oxalate. Further decomposition of the C2O4˙− with a second equivalence of ferric oxalate yields an additional equivalence of ferrous oxalate and two equivalents of CO2. | ||
Nocera's original report of NiIIIX3(PP) photochemistry investigated the stabilizing effect of the 1,2-bis(diphenylphosphino)ethane ligand (dppe) on the released halogen radical (Fig. 4). Within <40 ns, transient absorption measurements of NiCl3(dppe) revealed a species with absorption maxima at 366 nm and 525 nm, assigned to an arene-to-chlorine atom charge-transfer adduct.19 Analogous signals have been observed across a variety of aryl-containing NiX3(PP) analogues and support that the photogenerated chlorine radical is stabilized on the aryl substituents for up to several microseconds depending on the diphosphine substituents.19,94 Furthermore, the aryl groups significantly increase the quantum yield for halogen release with aliphatic groups producing quantum yields of 20% and aryl-containing groups exhibiting quantum yields up to 96%.
 | ||
| Fig. 4 Irradiation of NiIIIX3(dppe), shown here, and other NiIIIX3PP complexes results in VLIH of the Ni–X bond, where the resulting halogen radical is stabilized by arene substituents on the diphosphine ligand. | ||
While many complexes have been reported to liberate halogen atoms following photoexcitation, including many metal complexes with LMCT excited states, these nickel(III) trihalide complexes incorporate an important design principle not described in other systems – halide radical stabilizing substituents in the secondary coordination sphere. This finding suggests that simple manipulation of ligand properties can impart stabilizing interactions with halogen radicals and improve photochemical outcomes. These interactions tune the quantum yields of halogen radical production and increase the lifetime of these halogen radical intermediates – parameters critical for dictating the efficiency of photoredox catalysis reactions.
3.2 Excited state electron transfer
One of the most fundamental photochemical reactions for LMCT excited states is excited state electron transfer (ES-ET). Excited states, regardless of their character, are inherently better reductants and oxidants than their ground state counterparts, as the chemical potential generated in the excited state promotes reduction and oxidation at milder potentials. Due to the higher chemical potential of the excited state, promoted electrons (holes) can more easily transfer to (from) quenching substrates. The potency of these excited state reduction potentials is dictated by two factors: the ground state reduction potential (E°′) and the energy stored in the excited state (ΔGES).122 This relationship is summarized in eqn (1) and (2), where E°′(M(n+1)/n) is the reduction potential corresponding to the oxidation of the photosensitizer and E°′(Mn/(n−1)) is the reduction potential for the reduction of the photosensitizer. For each case, these ground state reduction potentials are modified by ΔGES to yield the excited state reduction potentials relevant for photoreductive and photo-oxidative processes (eqn (1) and (2), respectively, where ΔGES is in units of V).122| E°′(M(n+1)/n*) = E°′(M(n+1)/n) − ΔGES | (1) |
| E°′(Mn*/(n−1)) = E°′(Mn*/(n−1)) + ΔGES | (2) |
While thermodynamics dictate the reducing and oxidizing power of excited states, kinetic factors also drive the accessibility of ES-ET reactions. In many cases, the lifetime of the excited state dictates whether or not ES-ET can occur by dynamic quenching before the excited state relaxes via radiative or non-radiative pathways. If an excited state is too short-lived, the photosensitizer is unable to accept (donate) an electron from (to) a substrate. Furthermore, the stability of the oxidized (reduced) photosensitizer is also essential to drive photocatalytic systems with photosensitizing compounds. If ES-ET results in a chemically unstable photosensitizer product, degradation reactions will inhibit desired reactivity, reducing quantum yields and preventing photosensitizer recyclability.5 While ES-ET is theoretically possible from all LMCT excited states, some LMCT excited states exhibit sub-nanosecond lifetimes at room temperature (including those that cannot undergo intersystem crossing, such as low spin d5 species with doublet ground states) and/or promote metal complex degradation via pathways like VLIH (see above), hindering their widespread use as photosensitizers. While extended reaction times and higher photon flux can enable photosensitization in some cases, the utility of these short-lived excited states is still limited compared to other state-of-the-art photosensitizers, like [Ru(bpy)3]2+. Despite these limitations, a handful of LMCT-based photosensitizers are known, including rhenium(II) tris(diphosphine), zirconium(IV) bis(pyrrolyl)pyridine, iron(III) N-heterocyclic carbene complexes (Fig. 5).45,47,123
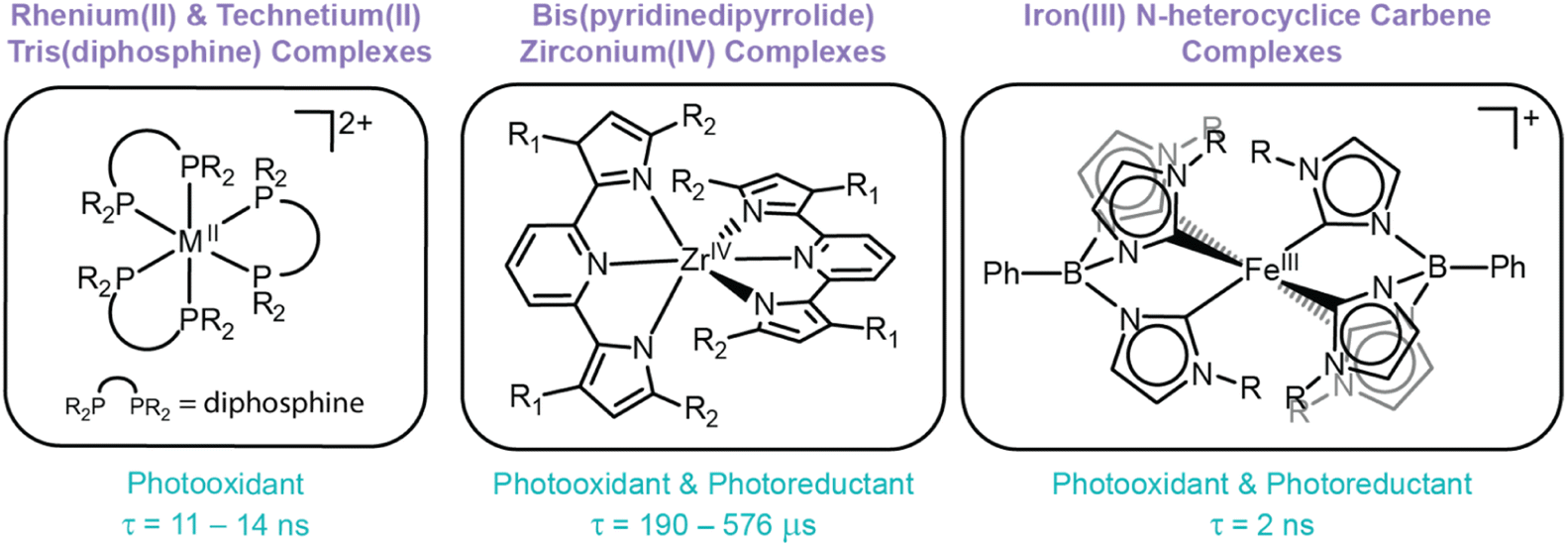 | ||
| Fig. 5 Structures of three classes of compounds known to facilitate ES-ET reactions (where M = Re or Tc). A summary of their role as photooxidants and/or photoreductants are included, alongside their respective excited state lifetimes. | ||
Thorough studies of rhenium(II) and technetium(II) diphosphine complexes have illustrated their predilection for undergoing excited state reductions. However, their propensity to undergo degradation upon (photo)oxidation exposes their primary weakness: ligand loss. Unfortunately, this characteristic is not isolated to rhenium(II) and technetium(II) tris(diphosphine) complexes, but instead is a recurring theme within the field of LMCT photochemistry. In this specific case, ligand flexibility permits solvent-assisted photodissociation of a diphosphine ligand and prevents reversible excited state oxidations to drive photoreduction reactions.20,41,45,124 Other examples of photoinstability are also well-reported, either via VLIH reactions or dissociation pathways, typically from monodentate ligands.1,2,55,92 This predisposition emphasizes the pervasive need to identify and/or design ligand scaffolds that facilitate oxidative stability and prevent dissociative pathways to enable photosensitizing applications.
These complexes have been employed as photosensitizers across a variety of photocatalytic reactions, including dehalogenation, trifluoromethylation of electron-rich arenes, and atom transfer radical addition reactions. In these reactions, Zr(PDP)2 undergoes ES-ET to (from) a substrate and follow-up reactivity of the activated substrate in the ground state yields targeted products.22,47,77,125 Addition of mild reductants (or oxidants) enables regeneration of Zr(PDP)2 and promotes catalytic turnover. Interestingly, the PDP ligands not only serve to direct electronics and tune the potency of excited state reduction potentials, but also to stabilize the complex itself. Early studies showed the Zr(MePDPPh)2 derivative (MePDPPh = 2,6-bis(5-methyl-3-phenyl-1H-pyrrol-2-yl)pyridine) easily hydrolyzes at its basic pyrrolide nitrogens.77 Furthermore, while this complex exhibits reversible ground state reductions, the oxidation of Zr(MePDPPh)2 is irreversible (reminiscent of rhenium(II) tris(diphosphine) complexes).22 As such, excited state reduction of Zr(MePDPPh)2 to [Zr(MePDPPh)2]− was accessible with a E°′([Zr]0*/−) = −0.07 V vs. Fc+/0, but excited state oxidation to form [Zr(MePDPPh)2]+ was associated with decomposition and prevented its use as a photoreductant.22 Interestingly, slight modification of this ligand, where methyl groups on the pyrrole rings were replaced with mesityl groups (denoted Zr(MesPDPPh)2), resulted in a complex that was resistant to hydrolysis and exhibited greater thermal and oxidative stabilities. Unlike its predecessors, Zr(MesPDPPh)2 exhibits two reversible redox features and, as such, both oxidative and reductive quenching pathways for this class of compounds were reported for the first time (E°′([Zr]+/0*) = −1.41 V and E°′([Zr]0*/–) = −0.31 V vs. Fc+/0, respectively).77
Zr(PDP)2 complexes are robust and powerful photoredox reagents that promote oxidation and reduction of substrates from their excited states. The absence of d electrons prevents deleterious deactivation pathways from ligand field states to support long-lived LMCT excited states. First generation complexes experienced oxidative instability, but synthetic tuning enabled a second generation of robust Zr(PDP)2 complexes capable of supporting stable excited state charge transfer products. This example showcases the importance of ligand design in enabling photostable LMCT excited states capable of undergoing ES-ET and serves as a direct testament for targeting robust oxidizable ligands to support LMCT excited states.
Due to the nanosecond timescales required to access bimolecular reactions at modest substrate concentrations and reaction length, only [FeIII(phtmeimb)2]+ complexes have exhibited ES-ET reactivity. In fact, [FeIII(phtmeimb)2]+ was the first known iron complex with a charge-transfer excited state to undergo excited state quenching.49,130 This complex is both potent as a photo-oxidant and photoreductant (E°′([Fe]IV/III*) = −1.9 V and E°′([Fe]III*/II) = 1.0 V vs. Fc+/0). Since the seminal report, several studies have further investigated this class of compounds for ES-ET reactions, including photocatalytic dehalogenation reactions.49,123,131 Follow-up synthetic studies by Wärnmark and coworkers have attempted to further tune the excited state reduction potentials and identify structure-(photo)functional relationships through modulation of the 4-position of the phenyl group within the scorpionate ligand with electron-donating and electron-withdrawing groups. However, the excited state reduction potentials and lifetimes were extremely similar, suggesting that the modification at this position was not impactful in modulating the excited state.130 While this observation did not provide synthetic strategies for improving its photophysical properties, it does showcase that the 4-position of the scorpionate's phenyl substituent can be exploited as an attachment point to construct molecular or hybrid assemblies, a strategy commonly invoked for photosensitizing architectures.
The photostability of substituted [Fe(phtmeim)2]+ complexes have also been investigated within a visible-light-mediated dehalogenation catalysis reaction of 4-bromobenzyl-2-chloro-2-phenylacetate. [Fe(phtmeim)2]+ was employed as a reductive quencher to facilitate the dehalogenation reaction of 4-bromobenzyl-2-chloro-2-phenylacetate and compared to a prototypically employed photosensitizer, [Ru(bpy)3]2+. Significant ligand loss was observed for [Ru(bpy)3]2+, whereas [Fe(phtmeim)2]+ showed no noticeable degradation.130 Collectively, these studies highlight a transition metal complex that exhibits a lifetime on the nanosecond timescale, a tailorable ligand scaffold, and photochemical stability – three characteristics that have been historically difficult to simultaneously achieve for complexes that exhibit LMCT excited states. Therefore, future studies within the field of LMCT should greatly consider the design features implemented within this scaffold to inspire next-generation photosensitizing and photocatalytic systems.
3.3 Photochemical transformations
VLIH and ES-ET reactions from LMCT excited states enable routes to generate reactive species, such as electron rich radicals or oxidized/reduced analytes. However, a less explored strategy to exploit LMCT excited states is to promote chemical transformations at the light absorber itself. Enabling these reactions is essential to access photocatalytic transformations, where the metal center can bind substrates, chemically transform them into value-added products, and release these products to enable catalytic turnover. Two such reactions, excited state protonation and photoinduced reductive elimination, are highlighted herein. While simple in nature, these proton transfer and reductive elimination steps are central to a variety of catalytic reactions and when coupled to light absorption could revolutionize our approaches to chemical synthesis.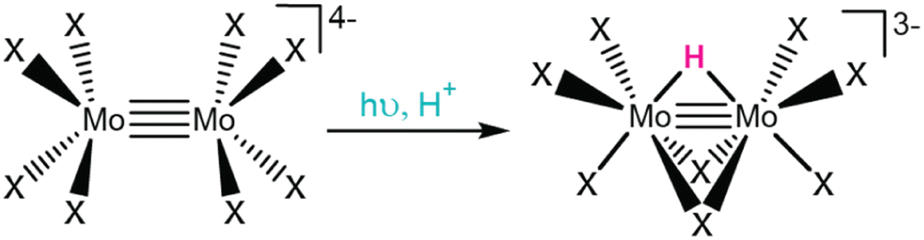 | ||
| Fig. 6 Excited state protonation of [Mo2X8]4− yields [Mo2(μ-H)(μ-X)2(X)6]3−. | ||
While excited state protonation and associated ground state reactivity was observed for [Mo2X8]4−, it is important to note the conditions of this photolysis experiment. Under dark conditions, ground state protonation for [Mo2X8]4− is known, where reaction kinetics to form [Mo2(μ-H)(μ-X)2(X)6]3− are sluggish at low mineral acid concentrations and fast at high concentrations. Therefore, photolytic conditions employed low acid concentrations to minimize competitive protonation in the ground state, due to the irreversibility of these photoreactions. By irradiating aqueous solutions of [Mo2X8]4− with UV light, the formation of [Mo2(μ-H)(μ-X)2(X)6]3− occurred much faster than in the dark, suggesting a new light-driven mechanism was operable.18 These observations are consistent with the formation of an excited state that is more basic than its ground state analogue and the more facile generation of [Mo2(μ-H)(μ-X)2(X)6]3− with UV irradiation.
Gray and coworkers attributed this change in basicity to the nature of the irradiated state.18 They assign an UV absorbance feature at 270 nm (320 nm) for [Mo2Cl8]4− ([Mo2Br8]4−) as a LMCT excited state, where halide (π) ligand orbitals populate a Mo–Mo δ* orbital.18,134 This electronic transition significantly shifts the electron density toward the Mo–Mo centers and increases their relative basicity in the excited state.134 Surprisingly, photochemistry from this charge transfer excited state disobeys Kasha's rule, as photochemical [Mo2(μ-H)(μ-X)2(X)6]3− formation was only observed upon irradiation of the high energy LMCT features, not at optical features observed at lower energies. Control photolytic studies showed no photochemical [Mo2(μ-H)(μ-X)2(X)6]3− formation from lower energy δ → δ* transitions (centered at 510 nm and 518 nm for [Mo2Cl8]4− and [Mo2Br8]4−, respectively).18 This uncommon photochemical occurrence was rationalized by slow internal conversion rates to metal-centered orbitals. While several follow-up studies have further investigated these complexes (e.g., ground state protonation and electronic structure via TD-DFT),135,136 many questions still remain, including the geometry of the orbitals involved in the excited state protonation and exact mechanism of protonation to form the metal hydride (i.e., protonation to a bridging or terminal position). This example illustrates the unique reactivity of octahalodimolybdates in driving excited state protonation by LMCT and further showcases the promise of these excited states to enable fundamental photochemical reactivity, like transition metal hydride formation, and pursuit of more complex photocatalytic schemes.
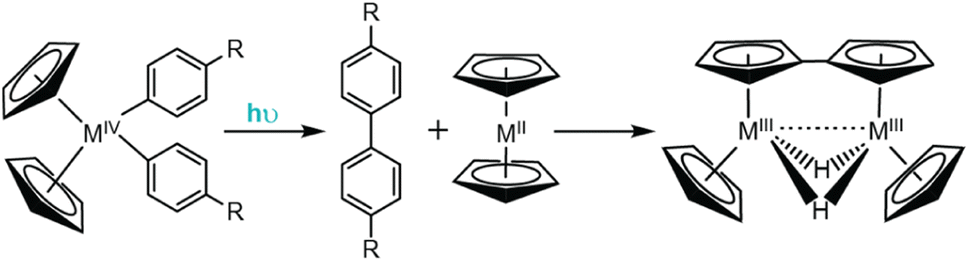 | ||
| Fig. 7 Irradiation of Cp2MR2 complexes (R = Ar) results in photo-induced reductive elimination to form biphenyl derivatives. | ||
Since these original reports, the photophysics and photochemistry of Cp2TiR2 complexes (where R = arylethynyl ligands) have been extensively studied.140–142 Wagenknecht and coworkers employed TD-DFT studies to computationally rationalize the observed photoreactivity and discovered the lowest energy transition for these complexes is a LMCT transition dominated by donation from the arylalkynyl ligands to the Ti dz2−y2 orbital.140 Photo-induced population of the Ti dz2−y2 antibonding orbital weakens the bond between the Ti metal and arylalkynyl π orbitals and facilitates the experimentally observed reductive elimination of the arylalkynyl ligands. Interestingly, for this class of Cp2TiR2 compounds, the major product is not a diyne (the anticipated reductive coupling product), but instead an enyne. Control studies revealed the resulting titanocene by-product is non-innocent following the photo-induced reductive elimination. Accounts from Brintzinger and Bercaw show that titanocene is not stable and readily undergoes dimerization to yield a strongly reducing, bridging dihydride fulvalene complex (Fig. 8).144 Upon formation of the diyne, it is proposed to undergo rapid reduction to the enyne by the fulvalene complex either through a stepwise or concerted pathway, and has since been shown to occur across several synthetic derivatives.138,140,141
 | ||
| Fig. 8 Irradiation of arylalkynyl titanocene complexes results in photo-induced reductive elimination to form an enyne product. | ||
Further derivatization of these Cp2TiR2 complexes supports that the LMCT excited state is responsible for driving the reductive photochemistry observed. When ferrocene moieties are appended to the alkynyl ligands of Ti, the lowest energy excited state exhibits metal-to-metal charge transfer (MMCT) character (instead of LMCT character), as the donor orbitals are primarily localized on the ferrocene moiety. As a result, this complex does not undergo photoinduced reductive elimination of its arylalkynyl ligands.141 These examples support the assignment that photoinduced reductive elimination from Cp2TiR2 and Cp2ZrR2 is promoted by LMCT.
While many accounts report the photoreductive elimination of Cp2MR2 complexes as decomposition pathways, Chirik and coworkers describe the utility in harnessing this reactivity to access a reactive intermediate capable of activating N2.68 UV irradiation of [(η5-C5Me4H)2Zr(Tol)2] (Tol = 4-Me-C6H4, depicted in Fig. 9A) under N2 atmosphere resulted in quantitative conversion to 4,4′-dimethylbiphenyl and [{(η5-C5Me4H)2Zr}2(μ2,η2,η2-N2)], a complex known to undergo hydrogenation and CO-induced N2 cleavage.68,145 Carbon monoxide and bis(trimethylsilyl)acetylene can similarly be trapped by this reactive synthon to form [(η5-C5Me4H)2Zr(CO)2] or [(η5-C5Me4H)2Zr(η2-Me3SiCCSiMe3)], respectively. Detailed mechanistic studies support this photoreaction follows a photoinduced unimolecular, two-electron reductive elimination to produce 4,4′-dimethylbiphenyl and [(η5-C5Me4H)2Zr]. The resulting 4,4′-dimethylbiphenyl photoproduct then likely associates to [(η5-C5Me4H)2Zr], forming a stabilizing Zr–arene interaction, and enabling facile displacement by strong π donors. In the absence of π donors, like N2, these Zr–arene complexes react further with the biphenyl photoproducts or aromatic hydrocarbon solvents via oxidative addition of the C–H bond to stabilize the undercoordinated metal center.68
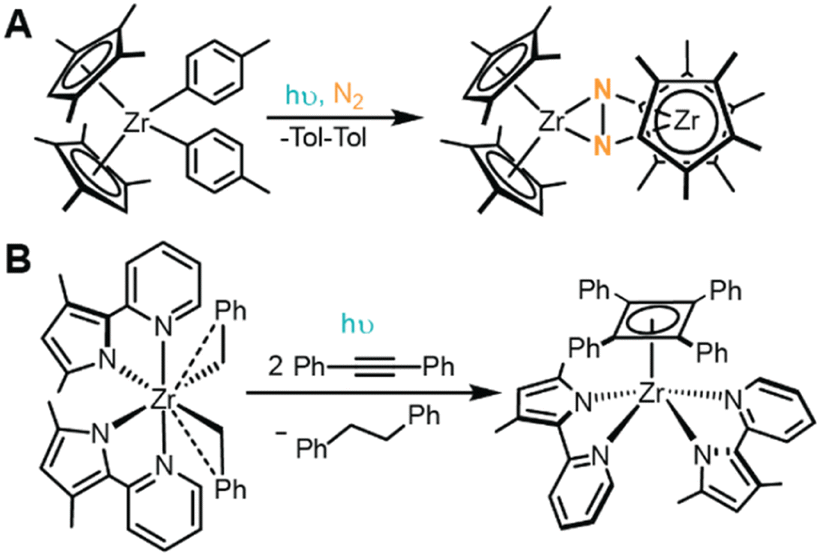 | ||
| Fig. 9 Summary of reactions for (A) [(η5-C5Me4H)2Zr(Tol)2] and (B) (MePMPMe)2ZrBn2. | ||
The Milsmann group recently reported a similar photoinduced reductive elimination from (MePMPMe)2ZrBn2 (MePMPMe = 3,5-dimethyl-2-(2-pyridyl)pyrrol-ide and Bn = benzyl), illustrated in Fig. 9B.146 Upon irradiation to populate a LMCT excited state, this complex releases an equivalent of 4,4′-biphenyl as the only byproduct, akin to the reactivity observed in Cp2MR2 complexes described above. Interestingly, in the presence of diphenylacetylene, the resulting (MePMPMe)2Zr species was shown to form a rare η4-cyclobutadienyl zirconium complex, (MePMPMe)2Zr(η4-C4Ph4).146 Such reactivity is reminiscent of Chirik's work,68 as it showcases the stabilizing effect of a π donating ligand which binds to the Zr center following photoinduced reductive elimination. However, unlike the prototypical Cp2MR2 case, mechanistic studies within this work suggest that (MePMPMe)2ZrBn2 does not undergo concerted reductive elimination but instead follows a stepwise pathway, where it is proposed that Zr–C bond homolysis occurs to liberate a benzyl radical and accompanying ZrIII intermediate.146 Subsequent ground state reactivity yields the observed 4,4′-biphenyl products, although the mechanistic details are still under investigation. This work showcases the intricacies of photoinduced reductive elimination steps. In each of these highlighted examples, similar reactions occur, yet their mechanistic details and accessible follow-up reactivity are different. This known disparity emphasizes the fundamental need to continue exploring the operative mechanisms for photochemical transformations promoted by LMCT.
4 Conclusions and outlook
While LMCT excited states once received a forlorn reputation of instability and degradation, their redemption is underway. Over the past several decades, a range of reactivity has been seen for complexes with LMCT excited states, including photoinduced bond homolysis, excited state electron transfer, excited state proton transfer, and photoinduced reductive eliminations. This diverse reactivity offers a wide variety of opportunities to leverage the energy stored in excited states for complex, energy-intensive transformations and enable new photochemical and photocatalytic reactions with broad applications. Some examples have already achieved widespread notoriety, such as ferrioxalate with its widespread use as a chemical actinometer and halogen radical-generating complexes with their roles in photoredox catalysis. However, a variety of other photoreactions are known and the applications for these reactions have not been realized.Challenges in the widespread adoption of LMCT-based photocatalysts and photosensitizers primarily stem from the absence of design principles for complexes with LMCT excited states. The structures of these LMCT-containing metal complexes are typically disparate and difficult to rationalize, making it difficult to design new photosensitizers and photocatalysts for niche applications. Some ligands are well known to support LMCT excited states, such as oxos and halides, whereas many other ligand scaffolds can facilitate a variety of charge transfer processes depending on the metal center identity, metal oxidation, and coordination geometry. As such, it is important to consider what factors are important in designing complexes to facilitate LMCT, but simultaneously consider the desired applications of these complexes for designing systems from the top-down.
First, the metal center identity, oxidation state, and coordination geometry are key to enabling LMCT excited states. Complexes must exhibit electron deficient metal centers that can easily be reduced upon photoexcitation. As a result, complexes with LMCT excited states are often d0 and d5, as their low energy t2g orbitals are fully or partially unfilled. Other examples with higher d electron configurations, such as d7 NiIII and d9 CuII, are typically supported by strongly σ donating ligands which lower e2g orbitals sufficiently to enable LMCT. However, in addition to preferentially tailoring these excited states to promote photochemistry, additional factors must also be considered. For example, d electron configurations are extremely important in dictating accessible photoproducts and photocatalytic intermediates generated from follow-up ground state reactivity. Photosensitizers must exhibit stability in their singly reduced and oxidized forms to enable turnover in photoredox catalytic reactions. Furthermore, if more complex photochemical transformations, like ES-PT and photoinduced reductive elimination reactions, are integrated into photocatalytic architectures, additional oxidation states must be accessed, as both processes result in oxidation states that decrease/increase by two, respectively. In addition to d electron configuration, the metal identity and oxidation state also impact ground state reduction potentials, and in turn excited state reduction potentials, enabling yet another synthetic variable to modulate photochemical reactivity. Absorption profiles and excitation energies of LMCT excited states may similarly change, allowing access to a breadth of photochemical applications.
The geometry of the orbitals that facilitate LMCT must also be considered. For photoinduced bond homolysis reactions, studies showed that charge transfer to a dz2 orbital liberates an apical ligand due to perturbation of a σ-interaction in the excited state. However, this observation is extremely dependent on the geometry of the complex, its d-orbital splitting, orientation of the coordinating ligands, and energy of the incident photon. For applications targeting photoinduced bond homolysis, complexes that facilitate population of a dz2 orbital would be more attractive and rationalizes the commonality of d7 and d8 complexes in facilitating photoinduced bond homolysis of apical ligands (as dz2 orbitals are common SOMOs/LUMOs for these complexes). The metal complex geometry must also be considered for photocatalytic applications, as substrate binding to a metal is likely required. Therefore, complexes must either (1) exhibit an open coordination site in the ground state or (2) generate an open coordination site upon photoexcitation.
Finding a balance between each of these design criteria is critical for enabling more complex photochemical and photocatalytic reactions. In many cases outlined herein, the transition metal complexes with known photochemistry from LMCT excited states were studied via TD-DFT to rationalize the orbitals involved in the electronic transitions. While many of these studies retroactively determine the nature of the excited state, the converse approach is also feasible. TD-DFT calculations can provide essential predictive power in understanding a complex's electronic structure, specifically the ordering of excited states and orbital character, without synthesis or experiment. These calculations are especially useful when targeting unexplored and/or complex photochemical reactions, where TD-DFT calculations can expedite the design of complexes that will exhibit low-lying LMCT transitions and exhibit electronic structures that promote photochemical reactivity of interest. As such, TD-DFT calculations should be added to every chemist's toolbox prior to designing novel complexes with LMCT excited states and targeting specific photochemistry from those states.
In addition to tailoring the metal center identity, ligand scaffolds are also critical for dictating the reactivity of LMCT excited states. Complexes with LMCT excited states must exhibit oxidizable ligands, as they transfer electron density from ligands to the metal center upon irradiation. However, this irradiation may result in bond homolysis or reductive elimination. For some applications, this photoreactivity is advantageous. For other applications, like ES-ET reactions, this reactivity is deleterious. In these cases, stable chelating ligands that donate electron density in the excited state should be targeted. However, it is important to note that not all chelating ligands are stable, as highlighted by the chemical actinometer ferrioxalate which undergoes bond homolysis and further decomposes, due to the instability of the carboxylate radical anion. Similarly, diphosphines are known to dissociate from rhenium(II) tris(diphosphine) complexes. Therefore, chelating ligand scaffolds that are donating, oxidizable, and stable are required to enable routes to photostabilize metal complexes.
Finally, the excited state lifetime and reaction quantum yields must be considered. Many coordination complexes with LMCT excited states are known to have short-lived excited states, often due to the lack of accessible intersystem crossing to long-lived excited states and/or fast deactivation by metal-centered states, as in the case of d5 complexes with doublet ground and excited states. Short excited state lifetimes can limit bimolecular reactivity, such as ES-ET and ES-PT reactions, where excited molecules must diffuse to their co-reactants. Conversely, intermolecular processes, such as photoinduced bond homolysis and reductive elimination reactions, are not limited by diffusion. Instead, these reactions typically occur on the femtosecond to picosecond timescales, rationalizing their proclivity within the reactivity arena of LMCT excited states. However, it is important to note that while these reactions occur on a fast timescale, the lifetime of these excited states is still important. Many of these reactions are proposed to occur on the vibrational timescale, such that if the excited state is longer-lived, a higher probability of a successful photoreaction is anticipated.
Overall, the design principles presented above must harmonize to enable the orchestration of complex photochemical reactions from LMCT excited states. Metal centers and their coordinating ligands must delicately balance one another to foster molecular geometries and electronic structures that enable low-lying LMCT excited states. From these excited states, a wealth of photochemical reactions are possible. Common photochemical examples include VLIH, ES-ET, ES-PT, and photoinduced reductive eliminations, but many other chemical transformations could also be envisioned. Driven by these prospects and the guiding synthetic design principles outlined herein, complexes with low energy LMCT excited states are poised to overcome their proposed shortcomings and enable a new era of LMCT-driven photochemistry.
Author contributions
A. M. M. drafted the original manuscript, prepared figures, and revised the manuscript. J. L. D. formally reviewed and edited the manuscript. Both authors reviewed and revised the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1954868). A.M.M. acknowledges support from the Department of Defense through the National Defense Science & Engineering Graduate (NDSEG) Fellowship Program.References
- F. Juliá, Ligand-to-Metal Charge Transfer (LMCT) Photochemistry at 3d-Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis, ChemCatChem, 2022, 14(19), e202200916, DOI:10.1002/cctc.202200916.
- P. Chábera, L. Lindh, N. W. Rosemann, O. Prakash, J. Uhlig, A. Yartsev, K. Wärnmark, V. Sundström and P. Persson, Photofunctionality of Iron(III) N-Heterocyclic Carbenes and Related d5 Transition Metal Complexes, Coord. Chem. Rev., 2021, 426, 213517, DOI:10.1016/j.ccr.2020.213517.
- C. Li, X. Y. Kong, Z. H. Tan, C. T. Yang and H. S. Soo, Emergence of Ligand-to-Metal Charge Transfer in Homogeneous Photocatalysis and Photosensitization, Chem. Phys. Rev., 2022, 3(2), 021303, DOI:10.1063/5.0086718.
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140(42), 13522–13533, DOI:10.1021/jacs.8b08822.
- D. M. Arias-Rotondo and J. K. McCusker, The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design, Chem. Soc. Rev., 2016, 45(21), 5803–5820, 10.1039/C6CS00526H.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, MLCT Excited States of Cuprous Bis-Phenanthroline Coordination Compounds, Coord. Chem. Rev., 2000, 208(1), 243–266, DOI:10.1016/S0010-8545(00)00309-X.
- A. Vogler and H. Kunkely, Photoreactivity of Metal-to-Ligand Charge Transfer Excited States, Coord. Chem. Rev., 1998, 177(1), 81–96, DOI:10.1016/S0010-8545(98)00131-3.
- A. Vlček, Mechanistic Roles of Metal-to-Ligand Charge-Transfer Excited States in Organometallic Photochemistry, Coord. Chem. Rev., 1998, 177(1), 219–256, DOI:10.1016/S0010-8545(98)00187-8.
- A. Vogler and H. Kunkely, Excited State Properties of Transition Metal Phosphine Complexes, Coord. Chem. Rev., 2002, 230(1), 243–251, DOI:10.1016/S0010-8545(01)00438-6.
- O. S. Wenger, Proton-Coupled Electron Transfer Originating from Excited States of Luminescent Transition-Metal Complexes, Chem. –Euro. J., 2011, 17(42), 11692–11702, DOI:10.1002/chem.201102011.
- C. Hicks, G. Ye, C. Levi, M. Gonzales, I. Rutenburg, J. Fan, R. Helmy, A. Kassis and H. D. Gafney, Excited-State Acid–Base Chemistry of Coordination Complexes, Coord. Chem. Rev., 2001, 211(1), 207–222, DOI:10.1016/S0010-8545(00)00279-4.
- O. S. Wenger, A Bright Future for Photosensitizers, Nat. Chem., 2020, 12(4), 323–324, DOI:10.1038/s41557-020-0448-x.
- N. Sinha and O. S. Wenger, Photoactive Metal-to-Ligand Charge Transfer Excited States in 3d6 Complexes with Cr0, MnI, FeII, and CoIII, J. Am. Chem. Soc., 2023, 145(9), 4903–4920, DOI:10.1021/jacs.2c13432.
- C. Wegeberg and O. S. Wenger, Luminescent First-Row Transition Metal Complexes, JACS Au, 2021, 1(11), 1860–1876, DOI:10.1021/jacsau.1c00353.
- J. K. McCusker, Electronic Structure in the Transition Metal Block and Its Implications for Light Harvesting, Science, 2019, 363(6426), 484–488, DOI:10.1126/science.aav9104.
- O. S. Wenger, Is Iron the New Ruthenium?, Chem. –Euro. J., 2019, 25(24), 6043–6052, DOI:10.1002/chem.201806148.
- D. W. Thompson, A. Ito and T. J. Meyer, [Ru(bpy)3]2+* and other remarkable metal-to-ligand charge transfer (MLCT) excited states, Pure Appl. Chem., 2013, 85(7), 1257–1305, DOI:10.1351/PAC-CON-13-03-04.
- W. C. Trogler, D. K. Erwin, G. L. Geoffroy and H. B. Gray, Production of Hydrogen by Ultraviolet Irradiation of Binuclear Molybdenum(II) Complexes in Acidic Aqueous Solutions. Observation of Molybdenum Hydride Intermediates in Octahalodimolybdate(II) Photoreactions, J. Am. Chem. Soc., 1978, 100(4), 1160–1163, DOI:10.1021/ja00472a020.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, Trap-Free Halogen Photoelimination from Mononuclear Ni(III) Complexes, J. Am. Chem. Soc., 2015, 137(20), 6472–6475, DOI:10.1021/jacs.5b03192.
- Y. F. Lee and J. R. Kirchhoff, Absorption and Luminescence Spectroelectrochemical Characterization of a Highly Luminescent Rhenium(II) Complex, J. Am. Chem. Soc., 1994, 116(8), 3599–3600, DOI:10.1021/ja00087a056.
- A. J. Allmand and W. W. Webb, CXCVII.—The Photolysis of Potassium Ferrioxalate Solutions. Part I. Experimental, J. Chem. Soc., 1929, 1518–1531, 10.1039/JR9290001518.
- Y. Zhang, J. L. Petersen and C. Milsmann, A Luminescent Zirconium(IV) Complex as a Molecular Photosensitizer for Visible Light Photoredox Catalysis, J. Am. Chem. Soc., 2016, 138(40), 13115–13118, DOI:10.1021/jacs.6b05934.
- G. Erker, The Reaction of Intermediate Zirconocene-Aryne Complexes with C—H Bonds in the Thermolysis of Diarylzirconocenes, J. Organomet. Chem., 1977, 134(2), 189–202, DOI:10.1016/S0022-328X(00)81419-9.
- R. Carter, Molecular Symmetry and Group Theory, John Wiley & Sons, Ltd, Danvers, MA, 1998 Search PubMed.
- O. S. Haggag, P. Malakar, P. Pokhilko, J. F. Stanton, A. I. Krylov and S. Ruhman, The Elusive Dynamics of Aqueous Permanganate Photochemistry, Phys. Chem. Chem. Phys., 2020, 22(18), 10043–10055, 10.1039/C9CP07028A.
- A. Viste and H. B. Gray, The Electronic Structure of Permanganate Ion, Inorg. Chem., 1964, 3(8), 1113–1123, DOI:10.1021/ic50018a011.
- S. L. Holt and C. J. Ballhausen, Low Temperature Absorption Spectra of KMnO4 in KClO4, Theor. Chim. Acta, 1967, 7(4), 313–320, DOI:10.1007/BF00537508.
- E. M. Glebov, I. P. Pozdnyakov, V. F. Plyusnin and I. Khmelinskii, Primary Reactions in the Photochemistry of Hexahalide Complexes of Platinum Group Metals: A Minireview, J. Photochem. Photobiol., C, 2015, 24, 1–15, DOI:10.1016/j.jphotochemrev.2015.05.003.
- C. K. Jørgensen, Electron Transfer Spectra of Hexahalide Complexes, Mol. Phys., 1959, 2(3), 309–332, DOI:10.1080/00268975900100291.
- D. S. Budkina, F. T. Gemeda, S. M. Matveev and A. N. Tarnovsky, Ultrafast Dynamics in LMCT and Intraconfigurational Excited States in Hexahaloiridates(IV), Models for Heavy Transition Metal Complexes and Building Blocks of Quantum Correlated Materials, Phys. Chem. Chem. Phys., 2020, 22(30), 17351–17364, 10.1039/D0CP00438C.
- M. Kasha, Characterization of Electronic Transitions in Complex Molecules, Discuss. Faraday Soc., 1950, 9, 14–19, 10.1039/DF9500900014.
- S. E. Braslavsky, Glossary of Terms Used in Photochemistry, 3rd Edition (IUPAC Recommendations 2006), Pure Appl. Chem., 2007, 79(3), 293–465, DOI:10.1351/pac200779030293.
- A. B. P. Lever, Charge Transfer Spectra of Transition Metal Complexes, J. Chem. Educ., 1974, 51(9), 612, DOI:10.1021/ed051p612.
- S. M. Matveev, D. S. Budkina, I. L. Zheldakov, M. R. Phelan, C. M. Hicks and A. N. Tarnovsky, Femtosecond Dynamics of Metal-Centered and Ligand-to-Metal Charge-Transfer (t2g-Based) Electronic Excited States in Various Solvents: A Comprehensive Study of IrBr62-, J. Chem. Phys., 2019, 150(5), 054302, DOI:10.1063/1.5079754.
- H. Kunkely and A. Vogler, Photoredox Reactivity of Iron(III) Phenolates in Aqueous Solution Induced by Ligand-to-Metal Charge Transfer Excitation, Inorg. Chem. Commun., 2003, 6(10), 1335–1337, DOI:10.1016/j.inoche.2003.07.004.
- E. H. Choi, D.-S. Ahn, S. Park, C. Kim, C. W. Ahn, S. Kim, M. Choi, C. Yang, T. W. Kim, H. Ki, J. Choi, M. N. Pedersen, M. Wulff, J. Kim and H. Ihee, Structural Dynamics of Bismuth Triiodide in Solution Triggered by Photoinduced Ligand-to-Metal Charge Transfer, J. Phys. Chem. Lett., 2019, 10(6), 1279–1285, DOI:10.1021/acs.jpclett.9b00365.
- I. A. Tonks, A. C. Durrell, H. B. Gray and J. E. Bercaw, Groups 5 and 6 Terminal Hydrazido(2−) Complexes: Nβ Substituent Effects on Ligand-to-Metal Charge-Transfer Energies and Oxidation States, J. Am. Chem. Soc., 2012, 134(17), 7301–7304, DOI:10.1021/ja302275j.
- H. Kunkely and A. Vogler, Photolysis of CH3Re(O2)2O Induced by Ligand-to-Metal Charge Transfer and by Peroxide Intraligand Excitation, Inorg. Chem. Commun., 2005, 8(5), 467–470, DOI:10.1016/j.inoche.2005.02.014.
- K. S. Lim, D. Y. Lee, G. M. Valencia, D. A. Bull and Y.-W. Won, Direct Incorporation of Functional Peptides into M-DNA through Ligand-to-Metal Charge Transfer, ACS Macro Lett., 2017, 6(2), 98–102, DOI:10.1021/acsmacrolett.6b00865.
- H. Kunkely and A. Vogler, Photochemistry of Tris(Diethyldithiocarbamato)Iron(III). Reduction to a Stable Iron(II) Complex Induced by Ligand-to-Metal Charge Transfer Excitation, Inorg. Chem. Commun., 2002, 5(9), 730–732, DOI:10.1016/S1387-7003(02)00555-5.
- J. R. Kirchhoff, M. R. Allen, B. V. Cheesman, K. Okamoto, W. R. Heineman and E. Deutsch, Electrochemistry and Spectroelectrochemistry of [Re(1,2-Bis(Dimethylphosphino)Ethane)3]+, Inorg. Chim. Acta, 1997, 262(2), 195–202, DOI:10.1016/S0020-1693(97)05523-0.
- A. S. Del Negro, C. J. Seliskar, W. R. Heineman, S. E. Hightower, S. A. Bryan and B. P. Sullivan, Highly Oxidizing Excited States of Re and Tc Complexes, J. Am. Chem. Soc., 2006, 128(51), 16494–16495, DOI:10.1021/ja067114g.
- S. J. Messersmith, K. Kirschbaum and J. R. Kirchhoff, Luminescent Low-Valent Rhenium Complexes with 1,2-Bis(Dialkylphosphino)Ethane Ligands. Synthesis and X-Ray Crystallographic, Electrochemical, and Spectroscopic Characterization, Inorg. Chem., 2010, 49(8), 3857–3865, DOI:10.1021/ic902549b.
- S. Chatterjee, A. S. Del Negro, F. N. Smith, Z. Wang, S. E. Hightower, B. P. Sullivan, W. R. Heineman, C. J. Seliskar and S. A. Bryan, Photophysics and Luminescence Spectroelectrochemistry of [Tc(Dmpe)3]+/2+ (Dmpe = 1,2-Bis(Dimethylphosphino)Ethane), J. Phys. Chem. A, 2013, 117(48), 12749–12758, DOI:10.1021/jp406365c.
- J. J. Adams, N. Arulsamy, B. P. Sullivan, D. M. Roddick, A. Neuberger and R. H. Schmehl, Homoleptic Tris-Diphosphine Re(I) and Re(II) Complexes and Re(II) Photophysics and Photochemistry, Inorg. Chem., 2015, 54(23), 11136–11149, DOI:10.1021/acs.inorgchem.5b01395.
- Y. Zhang, N. G. Akhmedov, J. L. Petersen and C. Milsmann, Photoluminescence of Seven-Coordinate Zirconium and Hafnium Complexes with 2,2′-Pyridylpyrrolide Ligands, Chem. –Euro. J., 2019, 25(12), 3042–3052, DOI:10.1002/chem.201804671.
- Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, A Zirconium Photosensitizer with a Long-Lived Excited State: Mechanistic Insight into Photoinduced Single-Electron Transfer, J. Am. Chem. Soc., 2018, 140(18), 5934–5947, DOI:10.1021/jacs.8b00742.
- C. Romain, S. Choua, J.-P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Redox and Luminescent Properties of Robust and Air-Stable N-Heterocyclic Carbene Group 4 Metal Complexes, Inorg. Chem., 2014, 53(14), 7371–7376, DOI:10.1021/ic500718y.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Luminescence and Reactivity of a Charge-Transfer Excited Iron Complex with Nanosecond Lifetime, Science, 2019, 363(6424), 249–253, DOI:10.1126/science.aau7160.
- P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. E. Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, A Low-Spin Fe(III) Complex with 100-ps Ligand-to-Metal Charge Transfer Photoluminescence, Nature, 2017, 543(7647), 695–699, DOI:10.1038/nature21430.
- S. Kaufhold and K. Wärnmark, Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes, Catalysts, 2020, 10(1), 132, DOI:10.3390/catal10010132.
- L. Lindh, P. Chábera, N. W. Rosemann, J. Uhlig, K. Wärnmark, A. Yartsev, V. Sundström and P. Persson, Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis, Catalysts, 2020, 10(3), 315, DOI:10.3390/catal10030315.
- J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Near-Infrared 2Eg → 4A2g and Visible LMCT Luminescence from a Molecular Bis-(Tris(Carbene)Borate) Manganese(IV) Complex, Can. J. Chem., 2017, 95(5), 547–552, DOI:10.1139/cjc-2016-0607.
- F. Ericson, A. Honarfar, O. Prakash, H. Tatsuno, L. A. Fredin, K. Handrup, P. Chabera, O. Gordivska, K. S. Kjær, Y. Liu, J. Schnadt, K. Wärnmark, V. Sundström, P. Persson and J. Uhlig, Electronic Structure and Excited State Properties of Iron Carbene Photosensitizers – A Combined X-Ray Absorption and Quantum Chemical Investigation, Chem. Phys. Lett., 2017, 683, 559–566, DOI:10.1016/j.cplett.2017.03.085.
- T. M. Rodriguez, M. Deegbey, C.-H. Chen, E. Jakubikova and J. L. Dempsey, Isocyanide Ligands Promote Ligand-to-Metal Charge Transfer Excited States in a Rhenium(II) Complex, Inorg. Chem., 2023, 62(17), 6576–6585, DOI:10.1021/acs.inorgchem.2c03193.
- A. Vogler and H. Kunkely, Luminescence from Hexacyanoruthenate(III), Inorg. Chim. Acta, 1981, 53, L215–L216, DOI:10.1016/S0020-1693(00)84799-4.
- J. J. Alexander and H. B. Gray, Electronic Structures of Hexacyanometalate Complexes, J. Am. Chem. Soc., 1968, 90(16), 4260–4271, DOI:10.1021/ja01018a013.
- A. W. Hahn, B. E. Van Kuiken, V. G. Chilkuri, N. Levin, E. Bill, T. Weyhermüller, A. Nicolaou, J. Miyawaki, Y. Harada and S. DeBeer, Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X-Ray Scattering (RIXS), Inorg. Chem., 2018, 57(15), 9515–9530, DOI:10.1021/acs.inorgchem.8b01550.
- W. Zhang, M. Ji, Z. Sun and K. J. Gaffney, Dynamics of Solvent-Mediated Electron Localization in Electronically Excited Hexacyanoferrate(III), J. Am. Chem. Soc., 2012, 134(5), 2581–2588, DOI:10.1021/ja207306t.
- Y. Avila, P. Acevedo-Peña, L. Reguera and E. Reguera, Recent Progress in Transition Metal Hexacyanometallates: From Structure to Properties and Functionality, Coord. Chem. Rev., 2022, 453, 214274, DOI:10.1016/j.ccr.2021.214274.
- K. R. Mann, M. Cimolino, G. L. Geoffroy, G. S. Hammond, A. A. Orio, G. Albertin and H. B. Gray, Electronic Structures and Spectra of Hexakisphenylisocyanide Complexes of Cr(0), Mo(0), W(0), Mn(I), and Mn(II), Inorg. Chim. Acta, 1976, 16, 97–101, DOI:10.1016/S0020-1693(00)91697-9.
- J. T. Yarranton and J. K. McCusker, Ligand-Field Spectroscopy of Co(III) Complexes and the Development of a Spectrochemical Series for Low-Spin d6 Charge-Transfer Chromophores, J. Am. Chem. Soc., 2022, 144(27), 12488–12500, DOI:10.1021/jacs.2c04945.
- T. M. Rodriguez, M. Deegbey, E. Jakubikova and J. L. Dempsey, The Ligand-to-Metal Charge Transfer Excited State of [Re(dmpe)3]2+, Photosynth. Res., 2022, 151(2), 155–161, DOI:10.1007/s11120-021-00859-7.
- A. M. May, M. Deegbey, K. Edme, K. J. Lee, R. N. Perutz, E. Jakubikova and J. L. Dempsey, Electronic Structure and Photophysics of Low Spin d5 Metallocenes, Inorg. Chem., 2024, 63(4), 1858–1866, DOI:10.1021/acs.inorgchem.3c03451.
- N. R. East, R. Naumann, C. Förster, C. Ramanan, G. Diezemann and K. Heinze, Oxidative Two-State Photoreactivity of a Manganese(IV) Complex Using near-Infrared Light, Nat. Chem., 2024, 1–8, DOI:10.1038/s41557-024-01446-8.
- G. V. Loukova, W. Huhn, V. P. Vasiliev and V. A. Smirnov, Ligand-to-Metal Charge Transfer Excited States with Unprecedented Luminescence Yield in Fluid Solution, J. Phys. Chem. A, 2007, 111(20), 4117–4121, DOI:10.1021/jp0721797.
- H. Alt and M. D. Rausch, Photochemical Reactions of Dimethyl Derivatives of Titanocene, Zirconocene, and Hafnocene, J. Am. Chem. Soc., 1974, 96(18), 5936–5937, DOI:10.1021/ja00825a041.
- G. W. Margulieux, S. P. Semproni and P. J. Chirik, Photochemically Induced Reductive Elimination as a Route to a Zirconocene Complex with a Strongly Activated N2 Ligand, Angew. Chem., Int. Ed., 2014, 53(35), 9189–9192, DOI:10.1002/anie.201402401.
- Z.-T. Tsai and C. H. Brubaker, Photolysis of Titanocene Dichloride, J. Organomet. Chem., 1979, 166(2), 199–210, DOI:10.1016/S0022-328X(00)91634-6.
- J. W. Kenney, D. R. Boone, D. R. Striplin, Y. H. Chen and K. B. Hamar, Electronic Luminescence Spectra of Charge Transfer States of Titanium(IV) Metallocenes, Organometallics, 1993, 12(9), 3671–3676, DOI:10.1021/om00033a045.
- R. W. Harrigan, G. S. Hammond and H. B. Gray, Photochemistry of Titanocene(IV) Derivatives, J. Organomet. Chem., 1974, 81(1), 79–85, DOI:10.1016/S0022-328X(00)87889-4.
- W. Tumas, D. R. Wheeler and R. H. Grubbs, Photochemistry of Titanacyclobutanes. Evidence for a Metal-Centered 1,4-Biradical Intermediate, J. Am. Chem. Soc., 1987, 109(20), 6182–6184, DOI:10.1021/ja00254a048.
- E. L. Patrick, C. J. Ray, G. D. Meyer, T. P. Ortiz, J. A. Marshall, J. A. Brozik, M. A. Summers and J. W. Kenney, Non-Localized Ligand-to-Metal Charge Transfer Excited States in (Cp)2Ti(IV)(NCS)2, J. Am. Chem. Soc., 2003, 125(18), 5461–5470, DOI:10.1021/ja028769u.
- B. W. Pfennig, M. E. Thompson and A. B. Bocarsly, A New Class of Room Temperature Luminescent Organometallic Complexes: Luminescence and Photophysical Properties of Permethylscandocene Chloride in Fluid Solution, J. Am. Chem. Soc., 1989, 111(24), 8947–8948, DOI:10.1021/ja00206a044.
- B. W. Pfennig, M. E. Thompson and A. B. Bocarsly, Luminescent d0 Scandocene Complexes: Photophysical Studies and Electronic Structure Calculations on Cp*2ScX (X = Cl, I, Me), Organometallics, 1993, 12(3), 649–655, DOI:10.1021/om00027a014.
- S. Paulson, B. P. Sullivan and J. V. Caspar, Luminescent Ligand-to-Metal Charge-Transfer Excited States Based on Pentamethylcyclopentadienyl Complexes of Tantalum, J. Am. Chem. Soc., 1992, 114(17), 6905–6906, DOI:10.1021/ja00043a040.
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano and C. Milsmann, Delayed Fluorescence from a Zirconium(IV) Photosensitizer with Ligand-to-Metal Charge-Transfer Excited States, Nat. Chem., 2020, 12(4), 345–352, DOI:10.1038/s41557-020-0430-7.
- K. S. Heinselman and M. D. Hopkins, Luminescence Properties of d0 Metal-Imido Compounds, J. Am. Chem. Soc., 1995, 117(49), 12340–12341, DOI:10.1021/ja00154a039.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. A. Büldt, N. H. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, A Vanadium(III) Complex with Blue and NIR-II Spin-Flip Luminescence in Solution, J. Am. Chem. Soc., 2020, 142(17), 7947–7955, DOI:10.1021/jacs.0c02122.
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Báez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, Strongly Red-Emissive Molecular Ruby [Cr(bpmp)2]3+ Surpasses [Ru(bpy)3]2+, J. Am. Chem. Soc., 2021, 143(30), 11843–11855, DOI:10.1021/jacs.1c05971.
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Chiral Molecular Ruby [Cr(dqp)2]3+ with Long-Lived Circularly Polarized Luminescence, J. Am. Chem. Soc., 2019, 141(33), 13244–13252, DOI:10.1021/jacs.9b06524.
- W. R. Kitzmann, J. Moll and K. Heinze, Spin-Flip Luminescence, Photochem. Photobiol. Sci., 2022, 21(7), 1309–1331, DOI:10.1007/s43630-022-00186-3.
- B. E. Bursten, F. A. Cotton, P. E. Fanwick and G. G. Stanley, A Molecular Orbital Calculation of the [Re2Cl8]2- Ion by the Relativistic SCF-Xα-SW Method. Redetermination and Reassignment of the Electronic Absorption Spectrum, J. Am. Chem. Soc., 1983, 105(10), 3082–3087, DOI:10.1021/ja00348a022.
- W. R. Kitzmann and K. Heinze, Charge-Transfer and Spin-Flip States: Thriving as Complements, Angew. Chem., Int. Ed., 2023, 62(15), e202213207, DOI:10.1002/anie.202213207.
- J. J. Shiang, A. G. Cole, R. J. Sension, K. Hang, Y. Weng, J. S. Trommel, L. G. Marzilli and T. Lian, Ultrafast Excited-State Dynamics in Vitamin B12 and Related Cob(III)Alamins, J. Am. Chem. Soc., 2006, 128(3), 801–808, DOI:10.1021/ja054374+.
- A. K. Pal, C. Li, G. S. Hanan and E. Zysman-Colman, Blue-Emissive Cobalt(III) Complexes and Their Use in the Photocatalytic Trifluoromethylation of Polycyclic Aromatic Hydrocarbons, Angew. Chem., Int. Ed., 2018, 57(27), 8027–8031, DOI:10.1002/anie.201802532.
- G. N. Schrauzer, L.-P. Lee and J. W. Sibert, Alkylcobalamins and Alkylcobaloximes. Electronic Structure, Spectra, and Mechanism of Photodealkylation, J. Am. Chem. Soc., 1970, 92(10), 2997–3005, DOI:10.1021/ja00713a012.
- S. Engl and O. Reiser, Making Copper Photocatalysis Even More Robust and Economic: Photoredox Catalysis with [CuII(dmp)2Cl]Cl, Eur. J. Org Chem., 2020, 2020(10), 1523–1533, DOI:10.1002/ejoc.201900839.
- A. Reichle and O. Reiser, Light-Induced Homolysis of Copper(II)-Complexes – a Perspective for Photocatalysis, Chem. Sci., 2023, 14(17), 4449–4462, 10.1039/D3SC00388D.
- B. D. Ravetz, J. Y. Wang, K. E. Ruhl and T. Rovis, Photoinduced Ligand-to-Metal Charge Transfer Enables Photocatalyst-Independent Light-Gated Activation of Co(II), ACS Catal., 2019, 9(1), 200–204, DOI:10.1021/acscatal.8b04326.
- P. Xu, P. López-Rojas and T. Ritter, Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination, J. Am. Chem. Soc., 2021, 143(14), 5349–5354, DOI:10.1021/jacs.1c02490.
- Y. Abderrazak, A. Bhattacharyya and O. Reiser, Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis, Angew. Chem., Int. Ed., 2021, 60(39), 21100–21115, DOI:10.1002/anie.202100270.
- A. Hossain, A. Vidyasagar, C. Eichinger, C. Lankes, J. Phan, J. Rehbein and O. Reiser, Visible-Light-Accelerated Copper(II)-Catalyzed Regio- and Chemoselective Oxo-Azidation of Vinyl Arenes, Angew. Chem., Int. Ed., 2018, 57(27), 8288–8292, DOI:10.1002/anie.201801678.
- S. J. Hwang, B. L. Anderson, D. C. Powers, A. G. Maher, R. G. Hadt and D. G. Nocera, Halogen Photoelimination from Monomeric Nickel(III) Complexes Enabled by the Secondary Coordination Sphere, Organometallics, 2015, 34(19), 4766–4774, DOI:10.1021/acs.organomet.5b00568.
- M. I. Gonzalez, D. Gygi, Y. Qin, Q. Zhu, E. J. Johnson, Y.-S. Chen and D. G. Nocera, Taming the Chlorine Radical: Enforcing Steric Control over Chlorine-Radical-Mediated C–H Activation, J. Am. Chem. Soc., 2022, 144(3), 1464–1472, DOI:10.1021/jacs.1c13333.
- G. Balakrishnan, A. V. Soldatova, P. J. Reid and T. G. Spiro, Ultrafast Charge Transfer in Nickel Phthalocyanine Probed by Femtosecond Raman-Induced Kerr Effect Spectroscopy, J. Am. Chem. Soc., 2014, 136(24), 8746–8754, DOI:10.1021/ja503541v.
- K. V. Prather and E. Y. Tsui, Photoinduced Ligand-to-Metal Charge Transfer of Cobaltocene: Radical Release and Catalytic Cyclotrimerization, Inorg. Chem., 2023, 62(5), 2128–2134, DOI:10.1021/acs.inorgchem.2c03779.
- S. M. Treacy and T. Rovis, Copper Catalyzed C(sp3)–H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer, J. Am. Chem. Soc., 2021, 143(7), 2729–2735, DOI:10.1021/jacs.1c00687.
- L. L. Costanzo, S. Pistarà and G. Condorelli, Photoreduction of Cerium(IV) Hexachloride in Acetonitrile, J. Photochem., 1983, 21(1), 45–51, DOI:10.1016/0047-2670(83)80006-9.
- Y. Qiao, H. Yin, L. M. Moreau, R. Feng, R. F. Higgins, B. J. Manor, P. H. Carroll, C. H. Booth, J. Autschbach and E. J. Schelter, Cerium(IV) Complexes with Guanidinate Ligands: Intense Colors and Anomalous Electronic Structures, Chem. Sci., 2021, 12(10), 3558–3567, 10.1039/D0SC05193D.
- R. E. Da Re, C. J. Kuehl, M. G. Brown, R. C. Rocha, E. D. Bauer, K. D. John, D. E. Morris, A. P. Shreve and J. L. Sarrao, Electrochemical and Spectroscopic Characterization of the Novel Charge-Transfer Ground State in Diimine Complexes of Ytterbocene, Inorg. Chem., 2003, 42(18), 5551–5559, DOI:10.1021/ic030069i.
- S. Petoud, J.-C. G. Bünzli, T. Glanzman, C. Piguet, Q. Xiang and R. P. Thummel, Influence of Charge-Transfer States on the Eu(III) Luminescence in Mononuclear Triple Helical Complexes with Tridentate Aromatic Ligands, J. Lumin., 1999, 82(1), 69–79, DOI:10.1016/S0022-2313(99)00015-0.
- H.-R. Mürner, E. Chassat, R. P. Thummel and J.-C. G. Bünzli, Strong Enhancement of the Lanthanide-Centred Luminescence in Complexes with 4-Alkylated 2,2′;6′,2′′-Terpyridines, J. Chem. Soc., Dalton Trans., 2000, 16, 2809–2816, 10.1039/B003577G.
- S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, Cerium(IV) Carboxylate Photocatalyst for Catalytic Radical Formation from Carboxylic Acids: Decarboxylative Oxygenation of Aliphatic Carboxylic Acids and Lactonization of Aromatic Carboxylic Acids, J. Am. Chem. Soc., 2020, 142(12), 5668–5675, DOI:10.1021/jacs.9b12918.
- E. Rousset, M. Piccardo, R. W. Gable, M. Massi, L. Sorace, A. Soncini and C. Boskovic, Elucidation of LMCT Excited States for Lanthanoid Complexes: A Theoretical and Solid-State Experimental Framework, Inorg. Chem., 2022, 61(35), 14004–14018, DOI:10.1021/acs.inorgchem.2c01985.
- Y. Qiao, Q. Yang and E. J. Schelter, Photoinduced Miyaura Borylation by a Rare-Earth-Metal Photoreductant: The Hexachlorocerate(III) Anion, Angew. Chem., Int. Ed., 2018, 57(34), 10999–11003, DOI:10.1002/anie.201804022.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Photocatalytic C–H Activation and the Subtle Role of Chlorine Radical Complexation in Reactivity, Science, 2021, 372(6544), 847–852, DOI:10.1126/science.abd8408.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, Dehydroxymethylation of Alcohols Enabled by Cerium Photocatalysis, J. Am. Chem. Soc., 2019, 141(26), 10556–10564, DOI:10.1021/jacs.9b05932.
- K.-J. Bian, S.-C. Kao, D. Nemoto, X.-W. Chen and J. G. West, Photochemical Diazidation of Alkenes Enabled by Ligand-to-Metal Charge Transfer and Radical Ligand Transfer, Nat. Commun., 2022, 13(1), 7881, DOI:10.1038/s41467-022-35560-3.
- M. Zhang, J. Zhang, Q. Li and Y. Shi, Iron-Mediated Ligand-to-Metal Charge Transfer Enables 1,2-Diazidation of Alkenes, Nat. Commun., 2022, 13(1), 7880, DOI:10.1038/s41467-022-35344-9.
- Y.-C. Lu and J. G. West, Chemoselective Decarboxylative Protonation Enabled by Cooperative Earth-Abundant Element Catalysis, Angew. Chem., Int. Ed., 2023, 62(3), e202213055, DOI:10.1002/anie.202213055.
- C. G. Hatchard and C. A. Parker, A New Sensitive Chemical Actinometer - II. Potassium Ferrioxalate as a Standard Chemical Actinometer, Proc. R. Soc. London, Ser. A, 1956, 235(1203), 518–536, DOI:10.1098/rspa.1956.0102.
- C. A. Parker, A New Sensitive Chemical Actinometer. I. Some Trials with Potassium Ferrioxalate, Proc. R. Soc. London, Ser. A, 1953, 220(1140), 104–116, DOI:10.1098/rspa.1953.0175.
- W. D. Bowman and J. N. Demas, Ferrioxalate Actinometry. A Warning on Its Correct Use, J. Phys. Chem., 1976, 80(21), 2434–2435, DOI:10.1021/j100562a025.
- I. P. Pozdnyakov, O. V. Kel, V. F. Plyusnin, V. P. Grivin and N. M. Bazhin, New Insight into Photochemistry of Ferrioxalate, J. Phys. Chem. A, 2008, 112(36), 8316–8322, DOI:10.1021/jp8040583.
- Y. Ogi, Y. Obara, T. Katayama, Y.-I. Suzuki, S. Y. Liu, N. C.-M. Bartlett, N. Kurahashi, S. Karashima, T. Togashi, Y. Inubushi, K. Ogawa, S. Owada, M. Rubešová, M. Yabashi, K. Misawa, P. Slavíček and T. Suzuki, Ultraviolet Photochemical Reaction of [Fe(III)(C2O4)3]3− in Aqueous Solutions Studied by Femtosecond Time-Resolved X-Ray Absorption Spectroscopy Using an X-Ray Free Electron Laser, Struct. Dyn., 2015, 2(3), 034901, DOI:10.1063/1.4918803.
- X. Li, M. Hou, Y. Fu, L. Wang, Y. Wang, D. Lin, Q. Li, D. Hu and Z. Wang, A Chronological Review of Photochemical Reactions of Ferrioxalate at the Molecular Level: New Insights into an Old Story, Chin. Chem. Lett., 2023, 34(5), 107752, DOI:10.1016/j.cclet.2022.107752.
- C. A. Parker, Induced Autoxidation of Oxalate in Relation to the Photolysis of Potassium Ferrioxalate, Trans. Faraday Soc., 1954, 50, 1213–1221, 10.1039/TF9545001213.
- J. Chen and W. R. Browne, Photochemistry of Iron Complexes, Coord. Chem. Rev., 2018, 374, 15–35, DOI:10.1016/j.ccr.2018.06.008.
- D. M. Mangiante, R. D. Schaller, P. Zarzycki, D. J. F. Banfield and B. Gilbert, Mechanism of Ferric Oxalate Photolysis, ACS Earth Space Chem., 2017, 1(5), 270–276, DOI:10.1021/acsearthspacechem.7b00026.
- C. Weller, S. Horn and H. Herrmann, Photolysis of Fe(III) Carboxylato Complexes: Fe(II) Quantum Yields and Reaction Mechanisms, J. Photochem. Photobiol., A, 2013, 268, 24–36, DOI:10.1016/j.jphotochem.2013.06.022.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, Estimation of Excited-State Redox Potentials by Electron-Transfer Quenching. Application of Electron-Transfer Theory to Excited-State Redox Processes, J. Am. Chem. Soc., 1979, 101(17), 4815–4824, DOI:10.1021/ja00511a007.
- A. Aydogan, R. E. Bangle, A. Cadranel, M. D. Turlington, D. T. Conroy, E. Cauët, M. L. Singleton, G. J. Meyer, R. N. Sampaio, B. Elias and L. Troian-Gautier, Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light, J. Am. Chem. Soc., 2021, 143(38), 15661–15673, DOI:10.1021/jacs.1c06081.
- Y. Deng, C. J. Seliskar and W. R. Heineman, Electrochemical Behavior of [ReI(DMPE)3]+, Where DMPE = 1,2-Bis(dimethylphosphino)ethane, at Perfluorosulfonated Ionomer-Modified Electrodes, Anal. Chem., 1997, 69(19), 4045–4050, DOI:10.1021/ac961295v.
- Y. Zhang, D. C. Leary, A. M. Belldina, J. L. Petersen and C. Milsmann, Effects of Ligand Substitution on the Optical and Electrochemical Properties of (Pyridinedipyrrolide)Zirconium Photosensitizers, Inorg. Chem., 2020, 59(20), 14716–14730, DOI:10.1021/acs.inorgchem.0c02343.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, Photochemistry of [Ru(bpy)3]2+, J. Am. Chem. Soc., 1982, 104(18), 4803–4810, DOI:10.1021/ja00382a012.
- J. A. Bandy, F. G. N. Cloke, G. Copper, J. P. Day, R. B. Girling, R. G. Graham, J. C. Green, R. Grinter and R. N. Perutz, Decamethylrhenocene, (η5-C5Me5)2Re, J. Am. Chem. Soc., 1988, 110(15), 5039–5050, DOI:10.1021/ja00223a023.
- S. Gazi, W. K. H. Ng, R. Ganguly, A. M. P. Moeljadi, H. Hirao and H. S. Soo, Selective Photocatalytic C–C Bond Cleavage under Ambient Conditions with Earth Abundant Vanadium Complexes, Chem. Sci., 2015, 6(12), 7130–7142, 10.1039/C5SC02923F.
- M. Barker, T. J. Whittemore, H. C. London, J. M. Sledesky, E. A. Harris, T. M. Smith Pellizzeri, C. D. McMillen and P. S. Wagenknecht, Design Strategies for Luminescent Titanocenes: Improving the Photoluminescence and Photostability of Arylethynyltitanocenes, Inorg. Chem., 2023, 62(43), 17870–17882, DOI:10.1021/acs.inorgchem.3c02712.
- O. Prakash, L. Lindh, N. Kaul, N. W. Rosemann, I. B. Losada, C. Johnson, P. Chábera, A. Ilic, J. Schwarz, A. K. Gupta, J. Uhlig, T. Ericsson, L. Häggström, P. Huang, J. Bendix, D. Strand, A. Yartsev, R. Lomoth, P. Persson and K. Wärnmark, Photophysical Integrity of the Iron(III) Scorpionate Framework in Iron(III)–NHC Complexes with Long-Lived 2LMCT Excited States, Inorg. Chem., 2022, 61(44), 17515–17526, DOI:10.1021/acs.inorgchem.2c02410.
- N. W. Rosemann, P. Chábera, O. Prakash, S. Kaufhold, K. Wärnmark, A. Yartsev and P. Persson, Tracing the Full Bimolecular Photocycle of Iron(III)–Carbene Light Harvesters in Electron-Donating Solvents, J. Am. Chem. Soc., 2020, 142(19), 8565–8569, DOI:10.1021/jacs.0c00755.
- A. Bino and M. Ardon, Unusual Reactions of a Bridged Hydride Ligand in Aqueous Solution, J. Am. Chem. Soc., 1977, 99(19), 6446–6447, DOI:10.1021/ja00461a049.
- F. A. Cotton and B. J. Kalbacher, Oxidative Addition of Hydrohalic Acids to Dimolybdenum(II) Species. Reformulation of Mo2X83- as Mo2X8H3-, Inorg. Chem., 1976, 15(3), 522–524, DOI:10.1021/ic50157a007.
- J. G. Norman Jr and H. J. Kolari, Electronic Structure of Octachlorodimolybdate(II), J. Am. Chem. Soc., 1975, 97(1), 33–37, DOI:10.1021/ja00834a008.
- E. D. Simandiras, N. Psaroudakis and K. Mertis, Which Component of the Quadruple Bond Breaks First upon Protonation of the Octachlorodimetallate Anions [MM′Cl8]4−, M,M′=Mo, W? A Theoretical Study of Reactivity, Mechanism and Bonding, Polyhedron, 2013, 54, 173–179, DOI:10.1016/j.poly.2013.02.019.
- P. Vilarrubias, Electronic Structure and Spectroscopy of Homoleptic Compounds of Dimolybdenum Using TDDFT, Can. J. Chem., 2016, 94(1), 20–27, DOI:10.1139/cjc-2015-0210.
- K. I. Gell, T. V. Harris and J. Schwartz, Synthesis and Characterization of Dimeric Zirconium(III) Complexes. Structure of “Zirconocene.”, Inorg. Chem., 1981, 20(2), 481–488, DOI:10.1021/ic50216a033.
- H. C. London, D. Y. Pritchett, J. A. Pienkos, C. D. McMillen, T. J. Whittemore, C. J. Bready, A. R. Myers, N. C. Vieira, S. Harold, G. C. Shields and P. S. Wagenknecht, Photochemistry and Photophysics of Charge-Transfer Excited States in Emissive d10/d0 Heterobimetallic Titanocene Tweezer Complexes, Inorg. Chem., 2022, 61(28), 10986–10998, DOI:10.1021/acs.inorgchem.2c01746.
- A. Roloff, K. Meier and M. Riediker, Synthetic and Metal Organic Photochemistry in Industry, Pure Appl. Chem., 1986, 58(9), 1267–1272, DOI:10.1351/pac198658091267.
- H. C. London, T. J. Whittemore, A. G. Gale, C. D. McMillen, D. Y. Pritchett, A. R. Myers, H. D. Thomas, G. C. Shields and P. S. Wagenknecht, Ligand-to-Metal Charge-Transfer Photophysics and Photochemistry of Emissive d0 Titanocenes: A Spectroscopic and Computational Investigation, Inorg. Chem., 2021, 60(18), 14399–14409, DOI:10.1021/acs.inorgchem.1c02182.
- M. D. Turlington, J. A. Pienkos, E. S. Carlton, K. N. Wroblewski, A. R. Myers, C. O. Trindle, Z. Altun, J. J. Rack and P. S. Wagenknecht, Complexes with Tunable Intramolecular Ferrocene to TiIV Electronic Transitions: Models for Solid State FeII to TiIV Charge Transfer, Inorg. Chem., 2016, 55(5), 2200–2211, DOI:10.1021/acs.inorgchem.5b02587.
- D. B. Pourreau and G. L. Geoffroy, Photochemistry of Alkyl, Alkylidene, and Alkylidyne Complexes of the Transition Metals, in Advances in Organometallic Chemistry, ed. F. G. A. Stone, R. West, Academic Press, 1985, vol. 24, pp. 249–352, DOI:10.1016/S0065-3055(08)60417-7.
- D. J. Sikora and M. D. Rausch, Reactions of Cp2M(CO)2 and Cp2M(CO)(PPh3) (M = Zr, Hf) with Acetylenes: Formation of Some Metallacyclopentadiene Complexes of Zirconocene and Hafnocene, J. Organomet. Chem., 1984, 276(1), 21–37, DOI:10.1016/0022-328X(84)80601-4.
- H. Brintzinger and J. E. Bercaw, Nature of So-Called Titanocene, (C10H10Ti)2, J. Am. Chem. Soc., 1970, 92(21), 6182–6185, DOI:10.1021/ja00724a013.
- J. A. Pool, W. H. Bernskoetter and P. J. Chirik, On the Origin of Dinitrogen Hydrogenation Promoted by [(η5-C5Me4H)2Zr]2(μ2,η2,η2-N2), J. Am. Chem. Soc., 2004, 126(44), 14326–14327, DOI:10.1021/ja045566s.
- P. M. N. Đỗ, N. G. Akhmedov, J. L. Petersen, B. S. Dolinar and C. Milsmann, Photochemical Synthesis of a Zirconium Cyclobutadienyl Complex, Chem. Commun., 2020, 56(40), 5397–5400, 10.1039/D0CC01104E.
| This journal is © The Royal Society of Chemistry 2024 |