
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA salt-concentrated electrolyte for aqueous ammonium-ion hybrid batteries†
Jianming
Meng
a,
Yu
Song
 *ab,
Jing
Wang
*d,
Peng
Hei
a,
Chang
Liu
a,
Mengxue
Li
a,
Yulai
Lin
a and
Xiao-Xia
Liu
*abc
*ab,
Jing
Wang
*d,
Peng
Hei
a,
Chang
Liu
a,
Mengxue
Li
a,
Yulai
Lin
a and
Xiao-Xia
Liu
*abc
aDepartment of Chemistry, Northeastern University, 3-11, Wenhua Road, Heping district, Shenyang, 110819, China. E-mail: songyu@mail.neu.edu.cn; xxliu@mail.neu.edu.cn
bNational Frontiers Science Center for Industrial Intelligence and Systems Optimization, Northeastern University, 3-11, Wenhua Road, Heping district, Shenyang, 110819, China
cKey Laboratory of Data Analytics and Optimization for Smart Industry (Northeastern University), Ministry of Education, 3-11, Wenhua Road, Heping district, Shenyang, 110819, China
dState Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao, 066004, China. E-mail: jwang6027@ysu.edu.cn
First published on 29th November 2023
Abstract
The development of aqueous ammonium-ion batteries (AAIBs) is currently attracting great attention because of the interesting electrochemical features induced by the charge carrier NH4+. One possible way to improve the performance of AAIBs is increasing the salt concentration in the electrolyte. Yet, few studies focus on the complex electrode–electrolyte interface behaviors in highly concentrated electrolytes, which affect the electrochemical performance of AAIBs significantly. Herein, we aim to understand the impact of CH3COONH4 electrolyte concentration on the NH4+ storage performance of a bimetallic hydroxide material. Experimental and theoretical simulation results indicate that the acetate anion will participate in the construction of the solvated NH4+ in a highly concentrated electrolyte, facilitating the adsorption of the solvated NH4+ cluster on the electrode surface. Besides, a new partial de-solvation model is also proposed, demonstrating an energy favorable de-solvation process. Finally, an ammonium-ion hybrid battery is designed, which provides a high average discharge voltage of 1.7 V and good energy density of 368 W h kg(cathode)−1, outperforming most of the state-of-the-art aqueous batteries. This work provides new understanding about the electrode's interfacial chemistry in different concentrated CH3COONH4 electrolytes, establishes a correlation between the electrolyte concentration and the electrode's performances, and demonstrates the superiority of the hybrid ammonium-ion battery design.
Introduction
Aqueous ammonium-ion batteries (AAIBs) using non-metallic ammonium ions (NH4+) as charge carriers are receiving increasing attention, due to the fast diffusion kinetics of NH4+ and the interesting H-bonding chemistry between NH4+ and host materials.1–4 Current research in AAIBs mainly focuses on fabricating high-performance electrode materials, such as metal oxides,5–9 polyanionic compounds,10 conducting polymers,11,12 Prussian blue analogues,13–16etc. Yet, the systematic study of electrolytes which are compatible with these NH4+ hosting materials is still lagging, limiting the development of competitive AAIBs.A recent breakthrough in electrolyte engineering is achieved by simply increasing the salt concentration in aqueous solution, offering distinct merits over dilute electrolytes.17 In a highly concentrated aqueous electrolyte, the content of free H2O molecules decreases, leading to less corrosion (dissolution) of electrode materials and a wider electrochemical stability window (ESW) of the electrolyte.18 For instance, active material's dissolution was significantly suppressed in an AAIB using the concentrated electrolyte [5.8 m (NH4)2SO4], exhibiting 72% capacity retention and good coulombic efficiency over 1000 cycles. The ESW was also broadened from 1.34 to 2.39 V upon increasing the electrolyte concentration from 1 to 5.8 m.18 Recently, Niu et al.19 reported a highly concentrated NH4CF3SO3 electrolyte (24 m) for high performance AAIBs. H2O molecules in the electrolyte were confined by NH4+ and CF3SO3− ions in the solvation sheath structure, weakening the water activity, and thus resulting in a large ESW and good cycling stability.
It is known that battery performance is highly associated with the solvation sheath structure of cations.20 In AAIBs, the high population of anions in a concentrated electrolyte will force them into the solvation sheath of NH4+, leading to the formation of a new NH4+-anion-H2O solvation sheath structure.19 The unique solvated NH4+ clusters would adsorb and get de-solvated at the electrolyte–electrode interface, making it the center of charge transfer reactions.21 Different from other metal cations, NH4+ could interact with H2O molecules, anions and electrode materials via H bonding, constituting unusual energy barriers for NH4+ de-solvation and intercalation, resulting in distinct physical and electrochemical properties for ammonium-ion batteries.22 Though previous reports have studied the solvation sheath structures of NH4+ in highly concentrated electrolytes [i.e., (NH4)2SO4 and NH4CF3SO3],18,19 the complicated electrolyte–electrode interface processes, including the solvated NH4+ adsorption and de-solvation, have never been studied, to the best of our knowledge.
In this work, starting from the concentration of electrolyte, we discussed how various concentrations of ammonium acetate (CH3COONH4) electrolytes led to different electrochemical performances of an electrochemically activated Co–Ni double hydroxide material (A-CoNi DH). Electrochemical and spectroscopic studies combined with theoretical simulations indicated that the enhanced NH4+ storage performance in 15 m CH3COONH4 electrolyte was mainly due to the facilitated adsorption/de-solvation processes of the unique solvated NH4+ on the electrode surface. Compared to the diluted electrolyte (1 m CH3COONH4), the solvated NH4+ exhibited stronger adsorption on the electrode surface in the concentrated electrolyte. In addition, a partial de-solvation model was also proposed to demonstrate an energy favorable de-solvation process in the highly concentrated CH3COONH4 electrolyte. To illustrate the practical application of the highly concentrated electrolyte, an ammonium-ion hybrid battery [Zn|15 m CH3COONH4 + 2 m Zn(CH3COO)2|A-CoNi DH] was assembled and delivered a high energy density of 368 W h kg(cathode)−1, which is better than those of the assembled Zn-ion battery [Zn|2 m Zn(CH3COO)2|A-CoNi DH] and other reported ammonium-based batteries, indicating the superiority of the highly concentrated CH3COONH4 electrolyte and the hybrid ammonium-ion battery design.
Results and discussion
Electrochemistry of A-CoNi DH in CH3COONH4 electrolytes with different concentrations
As a study prototype, an electrochemically activated Co–Ni double hydroxide (A-CoNi DH) was deposited on a 3D exfoliated graphite substrate (EG, Fig. S1†) using an electrochemical method reported before.21 The electrochemical activation strategy could significantly enhance the electrochemical performance of the double hydroxide material (Fig. S2 and S3†).23,24 Ultrathin nanosheets were uniformly grown on the exfoliated graphene/graphite sheets, forming a hierarchically porous structure (Fig. S4†). The XRD pattern of the A-CoNi DH electrode indicates the amorphous structure of the deposited material (Fig. S5a†). The chemical composition of A-CoNi DH was further characterized via energy dispersive X-ray spectroscopy (EDX, Fig. S5b†), Fourier transform infrared spectroscopy (FTIR, Fig. S5c†) and X-ray photoelectron spectroscopy (XPS, Fig. S5d–f†). The results suggest that the possible composition of A-CoNi DH is Co0.78Ni0.22O0.6(OH)1.27(NO3)0.05 (for details please see ESI, Fig. S5†).To investigate the effect of salt concentration on aqueous NH4+ storage, we first tested the electrochemical performance of A-CoNi DH in a three-electrode cell using ammonium acetate electrolytes with different concentrations [1 to 20 mol kg−1 (m)], and a saturated calomel electrode (SCE) as the reference electrode. Interestingly, we observe that the concentration of the electrolyte significantly affects the NH4+ storage performance of the A-CoNi DH electrode (Fig. 1a). The electrode tested in the concentrated 15 m electrolyte exhibits the optimized performance, delivering a high capacity of 220 mA h g−1 at a current density of 0.85 A g−1, which is much higher than that of 172 mA h g−1 obtained in 1 m electrolyte (Fig. 1b and S6†). Further increasing the electrolyte concentration to 20 m would lead to capacity decay at a high discharge rate, which could be due to the high viscosity and poor ion conduction in the highly concentrated solution.25
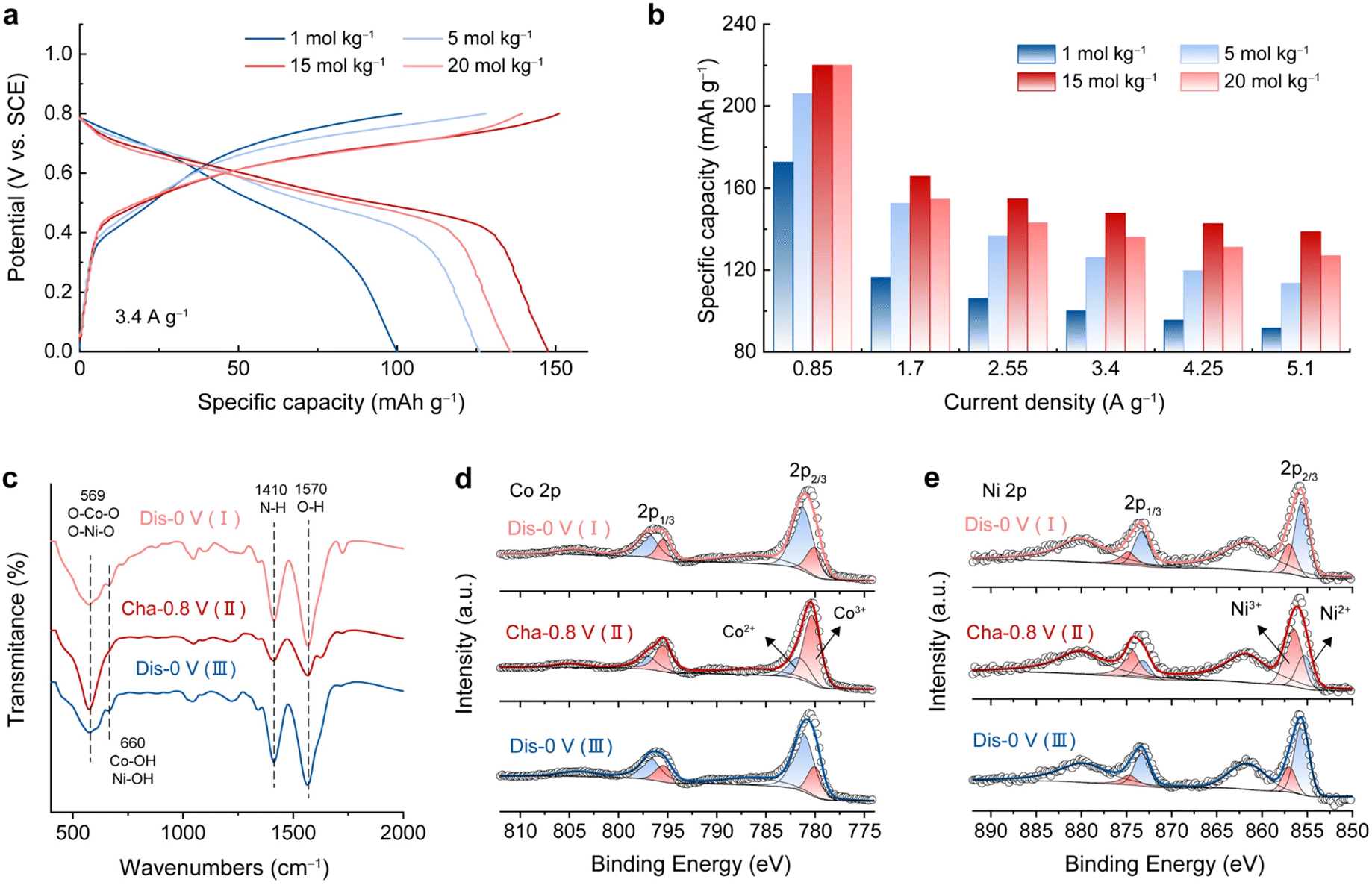 | ||
| Fig. 1 (a) Galvanostatic charge/discharge curves of the A-CoNi DH electrode in different concentrations of ammonium acetate electrolyte at the current density of 3.4 A g−1. (b) Rate performance of the A-CoNi DH electrode in different electrolytes. (c) FTIR, (d) XPS Co 2p, and (e) XPS Ni 2p spectra of the A-CoNi DH electrode at different states of charge. | ||
The charge storage mechanism of A-CoNi DH in NH4Ac electrolyte was studied using FTIR and XPS. Upon charging from 0 to 0.8 V vs. SCE (Fig. S7†), the intensity of the N–H vibration at 1410 cm−1 decreased, and recovered during the subsequent discharging process from 0.8–0 V vs. SCE (Fig. 1c). This result indicates that NH4+ participated in the charge storage of A-CoNi DH. XPS Co 2p (Fig. 1d) and Ni 2p (Fig. 1e) spectra indicate that both Co and Ni atoms are active sites for charge storage. XPS O 1s spectra of the samples can be fitted into three O containing components, i.e., Co/Ni–O (529.8 eV), Co/Ni–O–H (531.4 eV), and H–O–H (533 eV), as shown in Fig. S8.† Upon discharging from 0.8 to 0 V vs. SCE, the content of Co/Ni–O (529.8 eV) slightly decreased, suggesting that a small number of Co/Ni–O species were converted to Co/Ni–O–H due to the inevitable H+ insertion in aqueous electrolyte. These results suggest that A-CoNi DH experienced a NH4+/H+ co-insertion mechanism, and NH4+ is the main charge carrier in this work, which could be due to the highly concentrated NH4+ as well as the neutral pH value of the electrolyte. These results are also in accordance with our previous studies.26,27
Solvation structure of the concentrated ammonium acetate electrolytes
The above results suggest that the concentration of the electrolyte significantly affects the NH4+ storage performance of the A-CoNi DH electrode (Fig. 1a). Therefore, we studied the structural features of the electrolytes with different concentrations. Fig. 2a indicates that the CH3COONH4 electrolytes from 1 to 20 m all exhibit good ionic conductivity (higher than 60 mS cm−1), which is higher than those of most of the concentrated aqueous electrolytes, such as 10 m NH4CF3SO3 (18 mS cm−1),19 30 m ZnCl2 (3 mS cm−1),28 and 5 M Zn(ClO4)2 (46 mS cm−1).29 The high ionic conductivity of the electrolytes ensures fast ion transfer during charge/discharge processes.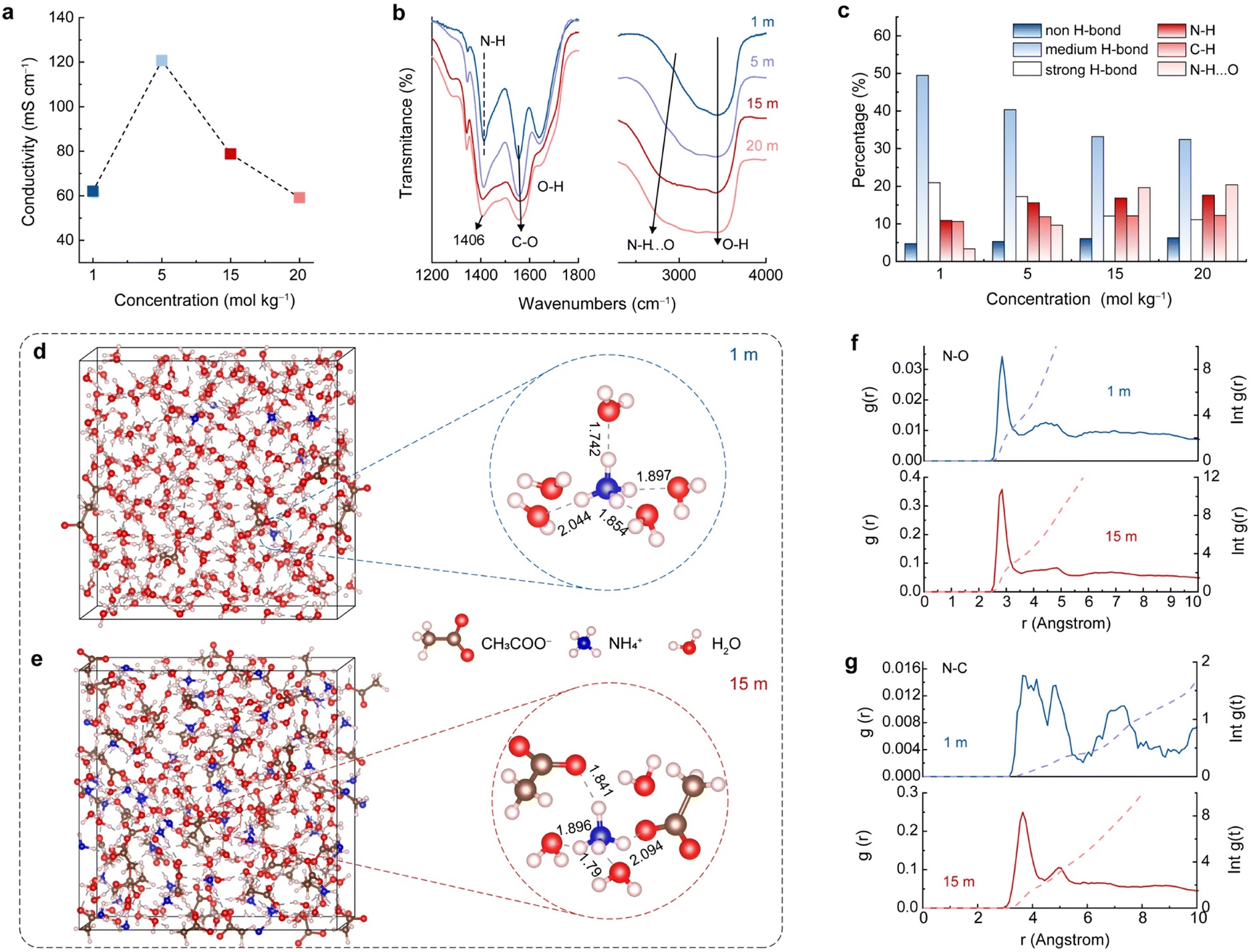 | ||
| Fig. 2 (a) Electrical conductivity and (b) FTIR spectra of CH3COONH4 electrolytes with different concentrations. (c) The corresponding percentage of the chemical bonds summarized in Fig. S9.† (d) and (e) MD simulation. (f) and (g) RDF plots of N–O and N–C, and the corresponding average coordination number of 1 and 15 m CH3COONH4 electrolytes. | ||
In addition to ionic conductivity, the solvation structure of NH4+ in these electrolytes plays an important role in the promising electrochemical NH4+ storage performances, which is further studied via Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and theoretical simulation. The specific infrared active bending modes of NH4+ and CH3COO− were observed at 1414 cm−1 and 1552 cm−1 for 1 m CH3COONH4 electrolyte, respectively (Fig. 2b).26,30 Interestingly, as the electrolyte concentration increased from 1 to 20 m, the N–H vibration shifts to lower wavenumbers (from 1414 to 1407 cm−1), while the C–O mode moves to higher wavenumbers (from 1552 to 1562 cm−1). These results indicate a strong interaction between NH4+ and CH3COO− in the electrolytes, and this interaction is strengthened by increasing the concentration of CH3COONH4.31 In order to obtain more band-related information of the overlapped O–H, N–H, and C–H stretching bands at high-wavenumber positions (Fig. 2b), we classified these peaks in Fig. S9† and the results are summarized in Fig. 2c. For O-H components, the higher wavenumber peak at 3620 cm-1 can be attributed to the non-hydrogen bond. Two intermediate vibrations at 3515 cm−1 and 3350 cm−1 are assigned to the weak hydrogen bond, and the lowest wavenumber at 3195 cm−1 is labeled as the strong hydrogen bond (Fig. S9†).32,33 With the increase of CH3COONH4 concentration, the proportion of strong and weak hydrogen bonds decreased, and non-hydrogen bonds increased, indicating that the H bond network of the aqueous solution was broken, due to the high proportion of solute in the highly concentrated electrolytes.19 Besides, the peaks for H bonded NH4+ (N–H⋯O) shifted to lower wavenumbers when the concentration of CH3COONH4 increased (Fig. 2b right column and Fig. S9†). In addition to the H bonds between N–H and H2O, NH4+ can also interact with CH3COO− upon increasing the CH3COONH4 concentration, resulting in the N–H⋯O peak shift and intensity enhancement. This strong interaction of NH4+ with H2O and CH3COO− in the electrolytes plays a crucial role in constructing the solvation structure. In the concentrated electrolytes, CH3COO− participates in the construction of the NH4+ solvation sheath and expels water molecules. Moreover, as indicated by Raman spectroscopy (Fig. S10†), when the CH3COONH4 concentration increased, the characteristic peaks for N–H and C–O shifted, and the water activity reduced, in agreement with the FTIR results.34,35
Ab initio molecular dynamics (AIMD) simulation was performed to further study the NH4+ solvation structures in different electrolytes. As study prototypes, we selected 1 and 15 m CH3COONH4 aqueous solutions to conduct the calculation. The snapshots in Fig. 2d and e show the simulated NH4+ solvation structures. In 1 m CH3COONH4 electrolyte, NH4+ coordinated with five H2O molecules, and four H2O molecules bonded with NH4+via H bonding, forming a long-lived tetrahedral cage. The fifth H2O molecule was mobile and flexible exchanging with one of the four H2O in the tetrahedral cage.36 The average bond length between the H-bonded water molecules and the central NH4+ in the first solvation sheath is 1.88 Å. The radial distribution function (RDF) result suggests that the number of ligands varied in the range of four to six for N–O interaction (NH4+-H2O) in 1 m CH3COONH4 solution (Fig. 2f, blue lines). The preferred coordination number was five, which was further confirmed by the density functional theory (DFT) results (Fig. S11†). The larger N–C distance and the low coordination number as shown in Fig. 2g (blue lines) indicate that CH3COO− did not participate in the NH4+ solvation sheath in 1 m CH3COONH4 solution. By contrast, CH3COO− anions tend to supersede partial water molecules and participate in constructing the solvation sheath of the central NH4+ in 15 m CH3COONH4 electrolyte (Fig. 2e and S12†). RDFs results in Fig. 2f and g (red lines) suggest that one NH4+ is coordinated with three H2O molecules and two CH3COO− in the solvation sheath. The simulation snapshot of 15 m CH3COONH4 shows that the average distance between NH4+ and the H-bonded species is 1.91 Å, which is larger than that in 1 m electrolyte. The increased distance leads to smaller bonding energy and weaker interatomic bonding interaction, facilitating the subsequent de-solvation process during discharging.37 The above experimental and theoretical results indicate that a higher electrolyte concentration will promote the CH3COO− involving in the first solvation sheath of NH4+, forming different solvation sheath structures compared with diluted electrolyte. The unique solvated NH4+ clusters would adsorb and get de-solvated at the electrolyte–electrode interface, leading to distinct electrochemical features.
Solvation structure–electrochemical performance relationship
Electrochemical impedance spectra (EIS) of the A-CoNi DH electrode were collected in different concentrated NH4Ac electrolytes (Fig. 3a and S13†). The equivalent series resistances (Rs, Z′-intercept) of the plots are almost the same, indicating that the solution resistances of the NH4Ac electrolytes have little effect on the electrochemical performance. Yet, the charge transfer resistances (Rct, semicircle diameter) decrease with the increase of electrolyte concentration from 1 to 15 m, and slightly increase when the concentration is higher than 15 m (Fig. S13†). In addition, Fig. 3b and c show that the A-CoNi DH electrode exhibits a much lower redox polarization potential in 15 m CH3COONH4 electrolyte, suggesting fast reaction kinetics in the highly concentrated electrolyte. These results explain why the A-CoNi DH electrode shows optimized electrochemical performance in 15 m electrolyte. | ||
| Fig. 3 (a) Nyquist plots of the A-CoNi DH electrode in different concentrated CH3COONH4 electrolytes. (b) CV curves of the A-CoNi DH electrodes in different electrolytes at the scan rate of 0.5 mV s−1. (c)The redox polarization potentials of A-CoNi DH's CV curves (b) in different electrolytes. The simulated electrolyte–electrode interface processes upon discharging in (d) 1 m, and (e) 15 m CH3COONH4 electrolytes, respectively. | ||
In general, the electrode's discharging process usually involves ion diffusion in bulk electrolytes, interfacial behaviors (including ion adsorption and de-solvation) on the electrolyte–electrode interface, and subsequent ionic solid-state migration in the electrode matrix. In this work, all the CH3COONH4 electrolytes exhibit good ionic conductivity, and the electrolyte concentration will not affect the NH4+ solid-state migration in the A-CoNi DH bulk. Therefore, the distinct interfacial processes in different concentrated electrolytes are believed to be the primary factor that affects the electrochemical performances.
To further study the complicated interfacial behavior and disclose the solvation structure-electrochemical performance relationship in aqueous NH4+ storage, we present a specific interfacial model that describes the adsorption and de-solvation behaviors of solvated cations (NH4+) on the electrode surface. Different from the conventional isolated ion adsorption model,21 this specific interfacial model could better describe the complicated interaction between cation–anion–solvent and electrode materials. For the diluted electrolyte (1 m CH3COONH4, as shown in Fig. 3d), upon discharging, solvated NH4+ (optimized from the solvation structure of NH4+ in Fig. 2) would attach on the electrode surface, and the adsorption energy is calculated to be −1.54 eV. The differential charge density distribution of the specific adsorption model suggests strong interaction between NH4+–H2O and the electrode material, which demands extra energy as large as 3.21 eV for solvent H2O molecules de-solvating before NH4+ insertion.
By contrast, in the concentrated 15 m electrolyte, the solvated NH4+ cluster exhibits a much lower adsorption energy of −3.05 eV compared with that in the diluted electrolyte (Fig. 3e). This can be ascribed to the strong coordination effect of acetate ions that participated in the solvation sheath of NH4+ in the concentrated electrolyte, leading to compact interaction between the solvated NH4+ and the electrode surface. As a result, a high de-solvation energy of 3.86 eV is required in a complete de-solvation process (Fig. 3e, Path I), which is unfavorable in thermodynamics. However, it is noticed that the solvation structure of NH4+ in 15 m electrolyte is loose due to the large volume of CH3COO−, which inspires us to propose a partial de-solvation process. DFT calculations were performed to further study this process. The partial de-solvation model (Fig. 3e, Path II) reveals that the solvated NH4+ loses one CH3COO− and one H2O molecule away from the electrode surface and the interaction between NH4+ and the H2O/CH3COO− attached on the electrode surface is broken, demanding only 1.07 eV in energy, which is less than that in the complete de-solvation pathway I. As shown in the model, the strong H bonding between the left H2O/CH3COO− and the hydroxide electrode remains (gray dashed lines). These surface adsorbed H2O and CH3COO− will be stable (in the form of O⋯H–O–metal) even after NH4+ insertion. As shown in Fig. S14,† after discharging, the electrode's XPS C 1s signal at higher binding energy (288∼291 eV for –COO−) was significantly enhanced compared with that of the pristine electrode. Another advantage of the partial de-solvation model is that the NH4+ is closer to the electrode surface due to the surface re-arrangement, which is beneficial for the insertion into the electrode material. Therefore, in the highly concentrated electrolyte, acetate anions would participate in the solvation structure of NH4+, and therefore improve the adsorption thermodynamics of the solvated NH4+ on the electrode surface. The proposed partial de-solvation model also provides an energy favorable NH4+ de-solvation process with low energy cost. All these features lead to the decreased charge transfer resistance and electrochemical polarization, and therefore make the highly concentrated electrolyte attractive in aqueous NH4+ storage.
High-performance NH4+-ion hybrid battery
To test the practical application of the highly concentrated CH3COONH4 electrolyte, an ammonium ion hybrid battery was assembled using A-CoNi DH as the cathode and a piece of zinc foil as the anode (Fig. 4a). To make sure that there are enough Zn2+ ions in the electrolyte for Zn deposition, a small amount of anhydrous Zn(CH3COO)2 was added into 15 m CH3COONH4 electrolyte to form the optimized hybrid electrolyte, i.e., 15 m CH3COONH4 + 2 m Zn(CH3COO)2 (Fig. S15†). SEM, XRD, XPS and EDX results suggest that the Zn2+ in the hybrid electrolyte did not participate the charge storage of the A-CoNi DH cathode (Fig. S16–S19†). Theoretical simulation was performed to further study the NH4+ solvation structures as well as the cation diffusion barrier (Tables S1 and S2†) in the hybrid electrolyte (Fig. 4b). The snapshot of the solvated NH4+ indicates that the small amount of Zn2+ additives in the hybrid electrolyte did not change the solvation structure of NH4+. In addition, the calculated adsorption energy of NH4+ (−0.08 eV) on the electrode surface is lower than that of Zn2+ (+0.65 eV), suggesting the favorable thermodynamic adsorption of NH4+ on A-CoNi DH. These results explain why Zn2+ did not participate in the charge storage of A-CoNi DH in the hybrid electrolyte. The possible chemical reactions occurring in the hybrid device are summarized as below: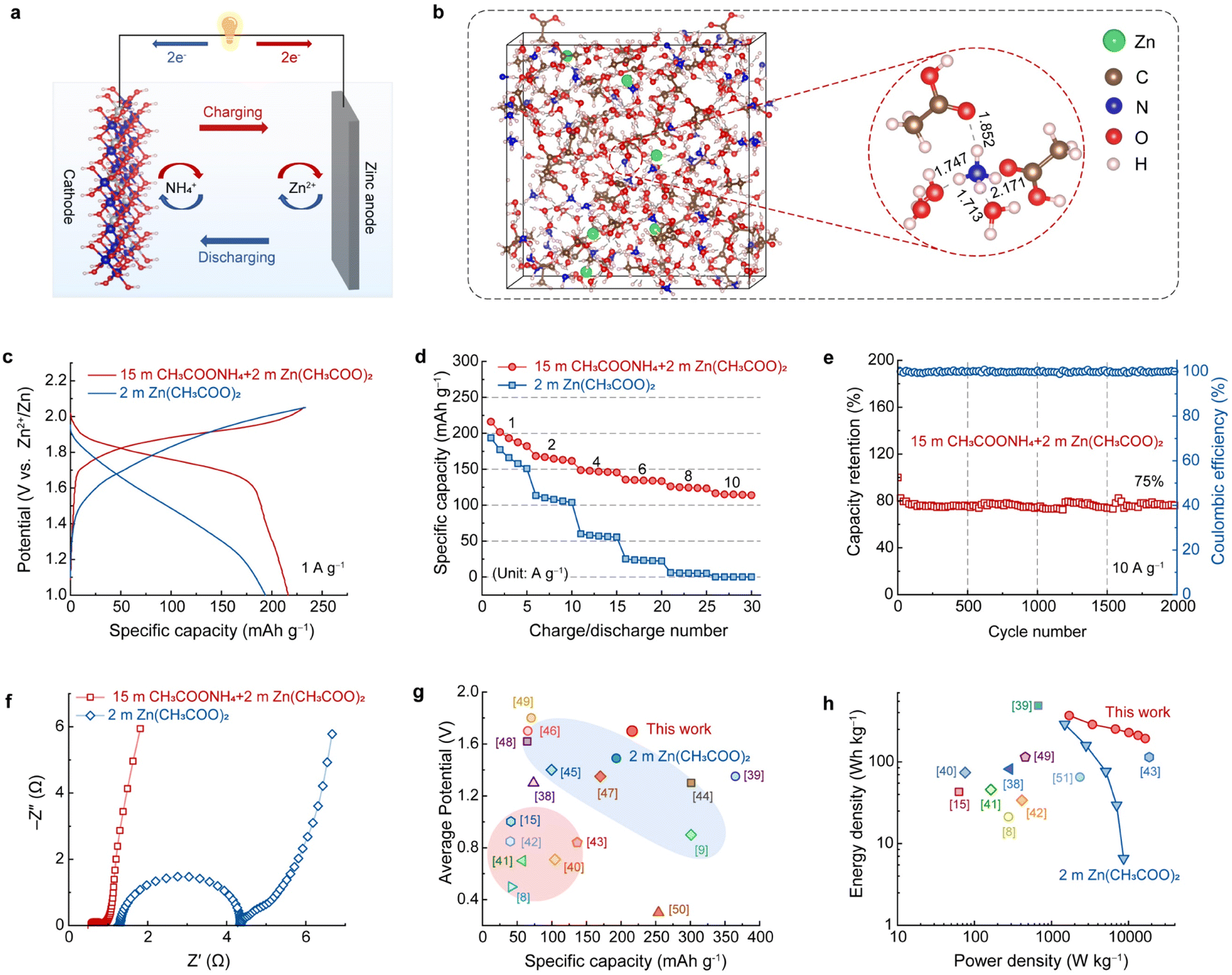 | ||
| Fig. 4 (a) Schematic illustration of a hybrid ammonium ion battery. (b) MD simulation of 15 m CH3COONH4 + 2 m Zn(CH3COO)2 electrolyte. (c) Galvanostatic charge/discharge curves, and (d) rate performance of the hybrid NH4+-ion battery and the assembled Zn2+-ion battery. (e) Nyquist plots of the A-CoNi DH electrode in 15 m CH3COONH4 + 2 m Zn(CH3COO)2 and 2 m Zn(CH3COO)2 electrolytes. (f) Cycling stability of the hybrid device at the current density of 10 A g−1. (g) The average potential vs. discharge capacity, and (h) Ragone plots of the hybrid battery and recently reported aqueous batteries for comparison. | ||
Cathode:
| Co0.78Ni0.22O0.6(OH)1.27(NO3)0.05 + NH4+ + e− ↔ (NH4)Co0.78Ni0.22O0.6(OH)1.27(NO3)0.05 |
Anode:
| 1/2Zn ↔ 1/2Zn2+ + e− |
Overall:
| Co0.78Ni0.22O0.6(OH)1.27(NO3)0.05 + NH4+ + 1/2Zn ↔ (NH4)Co0.78Ni0.22O0.6(OH)1.27(NO3)0.05 + 1/2Zn2+ |
To demonstrate the superiority of this hybrid electrolyte design, we also tested the electrochemical performance of a Zn-ion battery for comparison, using the 2 m Zn(CH3COO)2 electrolyte, A-CoNi DH cathode and Zn anode, respectively. The hybrid NH4+ ion battery exhibited a high capacity of 216 mA h g−1 at current density of 1 A g−1 and 116 mA h g−1 at 10 A g−1, which are higher than those (193.7 mA h g−1@1 A g−1 and 0.2 mA h g−1@10 A g−1) of the Zn-ion battery using 2 m Zn(CH3COO)2 electrolyte (Fig. 4c, d and S20†). Importantly, the charge/discharge curves and the corresponding differential capacity profile of the hybrid device display a high discharge plateau around 1.76 V (Fig. 4c and S21†), because of the high NH4+ insertion potential vs. Zn2+/Zn. This value is much higher than that of the assembled Zn-ion battery in this work. The electrochemical impedance spectra (EIS) of the A-CoNi DH electrode in the hybrid electrolyte and 2 m Zn(CH3COO)2 electrolyte were collected and are shown in Fig. 4e. The concentrated hybrid electrolyte exhibits a smaller equivalent series resistance (Rs, the intercept of the x-axis) than 2 m Zn(CH3COO)2, indicating the higher electrical conductivity of the hybrid electrolyte. In addition, the electrode in the hybrid electrolyte also shows a much smaller charge transfer resistance (Rct) of 0.3 Ω, which can well explain the superior rate performance of the hybrid device. This hybrid battery can deliver a good capacity retention of 75% after 2000 charge–discharge cycles at the current density of 10 A g−1 (Fig. 4f). The capacity degradation could be due to the slight dissolution of the active materials in the electrolyte during the charge/discharge processes (Fig. S22†). The average discharge voltage vs. the discharge capacity of the hybrid NH4+-ion battery with other aqueous batteries, including aqueous NH4+-ion and Zn2+-ion batteries, is shown in Fig. 4g and Table S3.† The hybrid battery exhibited a high average voltage of about 1.7 V and a relatively good gravimetric capacity of 216 mA h g−1, which are higher than or comparable with those of other aqueous NH4+ and Zn2+-ion batteries.8,9,15,38–50 Therefore, the hybrid device exhibits a high energy density of 368 W h kg(cathode)−1 at a power density of 1703 and 192 W h kg(cathode)−1 at an excellent power density of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 531 W kg−1, outperforming most of the reported aqueous batteries (Fig. 4h and Table S4†).8,15,38–43,49,51
531 W kg−1, outperforming most of the reported aqueous batteries (Fig. 4h and Table S4†).8,15,38–43,49,51
Conclusions
In summary, we fabricated an electrochemically activated CoNi hydroxide material and tested the electrochemical performances of the electrode in different concentrated ammonium acetate (CH3COONH4, from 1 to 20 m) electrolytes. The A-CoNi DH electrode exhibited the optimized electrochemical performance in 15 m CH3COONH4 electrolyte. The theoretical calculation, and spectroscopic and electrochemical studies established a direct correlation between the NH4+ solvation-sheath structure and the electrode's performances in different concentrated electrolytes. In 15 m electrolyte, acetate will replace water molecules and participate in the NH4+ solvation sheath, leading to a stronger specific adsorption between the solvated NH4+ and the A-CoNi DH electrode. In addition, a partial de-solvation model was proposed, in which the interaction between NH4+ and the surrounding solvated molecules, i.e., acetate and water, was broken. The electrode surface adsorbed acetate and water molecules do not necessarily de-solvated into the electrolytes, but bound to the hydroxide on the electrode surface, leading to a low energy cost process. To illustrate the practical application of the highly concentrated electrolyte, an ammonium-ion hybrid battery [Zn|15 m CH3COONH4 + 2 m Zn(CH3COO)2|A-CoNi DH] was assembled, which provided a high energy density of 368 W h kg(cathode)−1, a high working voltage of 1.7 V and good rate performance, outperforming most of the reported aqueous batteries. This work provides new understanding about the electrode–electrolyte interface processes in differently concentrated NH4+ based electrolytes, opening a viable route to the development of promising NH4+-ion batteries, which may also be extended to other aqueous batteries.Author contributions
J. M., Y. S., and J. W. conceived the project and designed this work. The experiment was performed by J. M. under the guidance of Y. S. and X.-X. L. with the help of P. H., C. L., M. L. and Y. L. carried out the SEM and FTIR tests. J. W. carried out the DFT calculations. J. M. and Y. S. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. S. acknowledges the financial support from the Fundamental Research Funds for the Central Universities (Grant No. N2205003) and Natural Science Foundation of Liaoning Province (no. 2023-MS-080). J. W. is thankful for the financial support from the Hebei Science Foundation (no. B2021203016 and E2021203005), Hebei provincial Department of Human Resources and Social Security (C20210503) and Hebei Education department (BJ2021042). The authors thank Y. Dong from the Analytical and Testing Center of Northeastern University for data acquisition.References
- S. Dong, W. Shin, H. Jiang, X. Wu, Z. Li, J. Holoubek, W. F. Stickle, B. Key, C. Liu, J. Lu, P. A. Greaney, X. Zhang and X. Ji, Ultra-fast NH4+ storage: strong H bonding between NH4+ and bi-layered V2O5, Chem, 2019, 5, 1537–1551 CAS.
- T. H. Lu, C. Zeng, H. Zhang, X. Shi, Y. Yu and X. Lu, Valence engineering enhancing NH4+ storage capacity of manganese oxides, Small, 2023, 19, 2206727 CrossRef CAS.
- L. Han, J. Luo, R. Zhang, W. Gong, L. Chen, F. Liu, Y. Ling, Y. Dong, Z. Yong, Y. Zhang, L. Wei, X. Zhang, Q. Zhang and Q. Li, Arrayed heterostructures of MoS2 nanosheets anchored TiN nanowires as efficient pseudocapacitive anodes for fiber-shaped ammonium-ion asymmetric supercapacitors, ACS Nano, 2022, 16, 14951–14962 CrossRef CAS.
- R. Zheng, Y. Li, H. Yu, X. Zhang, D. Yang, L. Yan, Y. Li, J. Shu and B. L. Su, Ammonium ion batteries: material, electrochemistry and strategy, Angew. Chem., Int. Ed., 2023, 62, e202301629 CrossRef CAS.
- Q. Chen, M. Song, X. Zhang, J. Zhang, G. Hou and Y. Tang, Ammonium ion pre-intercalation stabilized tunnel h-WO3 for fast NH4+ storage, J. Mater. Chem. A, 2022, 10, 15614–15622 RSC.
- S. F. Kuchena and Y. Wang, V2O5 intercalated with polyaniline for improved kinetics in aqueous ammonium-ion batteries, Electrochim. Acta, 2022, 425, 140751 CrossRef CAS.
- Y. Wu, S. Dong, N. Lv, Z. Xu, R. Ren, G. Zhu, B. Huang, Y. Zhang and X. Dong, Unlocking the high capacity ammonium-ion storage in defective vanadium dioxide, Small, 2022, 18, 2204888 CrossRef CAS.
- G. Liang, Y. Wang, Z. Huang, F. Mo, X. Li, Q. Yang, D. Wang, H. Li, S. Chen and C. Zhi, Initiating hexagonal MoO3 for superb-stable and fast NH4+ storage based on hydrogen bond chemistry, Adv. Mater., 2020, 32, 1907802 CrossRef CAS.
- D. Yu, Z. Wei, X. Zhang, Y. Zeng, C. Wang, G. Chen, Z. X. Shen and F. Du, Boosting Zn2+ and NH4+ storage in aqueous media via in situ electrochemical induced VS2/VOx heterostructures, Adv. Funct. Mater., 2020, 31, 2008743 CrossRef.
- F. Ye, R. Pang, C. Lu, Q. Liu, Y. Wu, R. Ma and L. Hu, Reversible ammonium ion intercalation/de-intercalation with crystal water promotion effect in layered VOPO4·2H2O, Angew. Chem., Int. Ed., 2023, 62, e202303480 CrossRef CAS.
- S. Zhang, K. Zhu, Y. Gao and D. Cao, A long cycle stability and high rate performance organic anode for rechargeable aqueous ammonium-ion battery, ACS Energy Lett., 2023, 8, 889–897 CrossRef CAS.
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Organic ammonium ion battery: A new strategy for a nonmetallic ion energy storage system, Angew. Chem., Int. Ed., 2022, 61, e202204351 CrossRef CAS PubMed.
- S. Li, M. Xia, C. Xiao, X. Zhang, H. Yu, L. Zhang and J. Shu, Common ion effect enhanced prussian blue analogues for aqueous ammonium ion storage, Dalton Trans., 2021, 50, 6520–6527 RSC.
- H. Yu, L. Fan, H. Yan, C. Deng, L. Yan, J. Shu and Z. B. Wang, Optimizing NH4+ storage capability of nickel ferrocyanide by regulating coordination anion in aqueous electrolytes, ChemElectroChem, 2022, 9, e202200492 CrossRef CAS.
- X. Wu, Y. Qi, J. J. Hong, Z. Li, A. S. Hernandez and X. Ji, Rocking-chair ammonium-ion battery: A highly reversible aqueous energy storage system, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- X. Zhang, M. Xia, T. Liu, N. Peng, H. Yu, R. Zheng, L. Zhang, M. Shui and J. Shu, Copper hexacyanoferrate as ultra-high rate host for aqueous ammonium ion storage, Chem. Eng. J., 2021, 421, 127767 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Highly reversible zinc metal anode for aqueous batteries, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- J. Han, M. Zarrabeitia, A. Mariani, M. Kuenzel, A. Mullaliu, A. Varzi and S. Passerini, Concentrated electrolytes enabling stable aqueous ammonium-ion batteries, Adv. Mater., 2022, 34, 2201877 CrossRef CAS PubMed.
- L. Du, S. Bi, M. Yang, Z. Tie, M. Zhang and Z. Niu, Coupling dual metal active sites and low-solvation architecture toward high-performance aqueous ammonium-ion batteries, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2214545119 CrossRef CAS.
- Z. Tian, J. Yin, T. Guo, Z. Zhao, Y. Zhu, Y. Wang, J. Yin, Y. Zou, Y. Lei, J. Ming, O. Bakr, O. F. Mohammed and H. N. Alshareef, A sustainable NH4+ ion battery by electrolyte engineering, Angew. Chem., Int. Ed., 2022, 61, e202213757 CrossRef CAS.
- M. Huang, Q. He, J. Wang, X. Liu, F. Xiong, Y. Liu, R. Guo, Y. Zhao, J. Yang and L. Mai, NH4+ deprotonation at interfaces induced reversible H3O+/NH4+ co-insertion/extraction, Angew. Chem., Int. Ed., 2023, 62, e202218922 CrossRef CAS PubMed.
- Z. Tian, V. S. Kale, Y. Wang, S. Kandambeth, J. Czaban-Jóźwiak, O. Shekhah, M. Eddaoudi and H. N. Alshareef, High-capacity NH4+ charge storage in covalent organic frameworks, J. Am. Chem. Soc., 2021, 143, 19178–19186 CrossRef CAS PubMed.
- J. Meng, Y. Song, Z. Qin, Z. Wang, X. Mu, J. Wang and X. X. Liu, Cobalt-nickel double hydroxide toward mild aqueous zinc-ion batteries, Adv. Funct. Mater., 2022, 32, 2204026 CrossRef CAS.
- C. Liu, M. Li, J. Meng, P. Hei, J. Wang, Y. Song and X. X. Liu, Electrochemical storage of ammonium versus metal ions in bimetallic hydroxide for sustainable aqueous batteries, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202310437.
- S. Gao, B. Li, H. Tan, F. Xia, O. Dahunsi, W. Xu, Y. Liu, R. Wang and Y. Cheng, High-energy and stable subfreezing aqueous Zn-MnO2 batteries with selective and pseudocapacitive zn-ion insertion in MnO2, Adv. Mater., 2022, 34, 2201510 CrossRef CAS.
- D. Yang, Y. Song, M. Y. Zhang, Z. Qin, J. Liu and X. X. Liu, Solid-liquid interfacial coordination chemistry enables high-capacity ammonium storage in amorphous manganese phosphate, Angew. Chem., Int. Ed., 2022, 61, e202207711 CrossRef CAS.
- Q. Pan, H. Peng, Y. Song, J. Meng, C. Liu and X. X. Liu, Electrochemically activated nickel-cobalt double hydroxide for aqueous ammonium-zinc hybrid battery, Nano Res., 2023, 16, 2495–2501 CrossRef CAS.
- L. Li, S. Liu, W. Liu, D. Ba, W. Liu, Q. Gui, Y. Chen, Z. Hu, Y. Li and J. Liu, Electrolyte concentration regulation boosting zinc storage stability of high-capacity K0.486V2O5 cathode for bendable quasi-solid-state zinc ion batteries, Nano-Micro Lett., 2021, 13 CAS.
- G. Yang, J. Huang, X. Wan, B. Liu, Y. Zhu, J. Wang, O. Fontaine, S. Luo, P. Hiralal, Y. Guo and H. Zhou, An aqueous zinc-ion battery working at −50°C enabled by low-concentration perchlorate-based chaotropic salt electrolyte, EcoMat, 2022, 4, e12165 CrossRef CAS.
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Efficient visible-light-driven CO2 reduction mediated by defect-engineered BiOBr atomic layers, Angew. Chem., Int. Ed., 2018, 130, 8855–8859 CrossRef.
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Tuning the electrolyte solvation structure to suppress cathode dissolution, water reactivity, and zn dendrite growth in zinc-ion batteries, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS.
- Q. Li, C. Yang, J. Zhang, X. Ji, J. Xu, X. He, L. Chen, S. Hou, J. Uddin, D. Addison, D. Sun, C. Wang and F. Wang, Controlling intermolecular interaction and interphase chemistry enabled sustainable water-tolerance LiMn2O4‖Li4Ti5O12 batteries, Angew. Chem., Int. Ed., 2022, 61, e202214126 CrossRef CAS PubMed.
- Y. Chen, Y.-H. Zhang and L.-J. Zhao, ATR-FTIR spectroscopic studies on aqueous LiClO4, NaClO4, and Mg(ClO4)2 solutions, Phys. Chem. Chem. Phys., 2004, 6, 537–542 RSC.
- H. Bhatt, C. Murli, A. K. Mishra, A. K. Verma, N. Garg, M. N. Deo, R. Chitra and S. M. Sharma, Hydrogen bond symmetrization in glycinium oxalate under pressure, J. Phys. Chem. B, 2016, 120, 851–859 CrossRef CAS.
- J. L. Dong, X. H. Li, L. J. Zhao, H. S. Xiao, F. Wang, X. Guo and Y. H. Zhang, Raman observation of the interactions between NH4+, SO42−, and H2O in supersaturated (NH4)2SO4 droplets, J. Phys. Chem. B, 2007, 111, 12170–12176 CrossRef CAS PubMed.
- F. Brugé, M. Bernasconi and M. Parrinello, Ab initio simulation of rotational dynamics of solvated ammonium ion in water, J. Am. Chem. Soc., 1999, 121, 10883–10888 CrossRef.
- M. Li, Y. Zhang, J. Hu, X. Wang, J. Zhu, C. Niu, C. Han and L. Mai, Universal multifunctional hydrogen bond network construction strategy for enhanced aqueous Zn2+/proton hybrid batteries, Nano Energy, 2022, 100, 107539 CrossRef CAS.
- C. Li, D. Zhang, F. Ma, T. Ma, J. Wang, Y. Chen, Y. Zhu, L. Fu, Y. Wu and W. Huang, A high-rate and long-life aqueous rechargeable ammonium zinc hybrid battery, ChemSusChem, 2019, 12, 3732–3736 CrossRef CAS.
- S. Wang, Z. Yuan, X. Zhang, S. Bi, Z. Zhou, J. Tian, Q. Zhang and Z. Niu, Non-metal ion co-insertion chemistry in aqueous Zn/MnO2 batteries, Angew. Chem., Int. Ed., 2021, 60, 7056–7060 CrossRef CAS PubMed.
- X. Mu, Y. Song, Z. Qin, J. Meng, Z. Wang and X. X. Liu, Core-shell structural vanadium oxide/polypyrrole anode for aqueous ammonium-ion batteries, Chem. Eng. J., 2023, 453, 139575 CrossRef CAS.
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, MnAl layered double hydroxides: a robust host for aqueous ammonium-ion storage with stable plateau and high capacity, Adv. Energy Mater., 2022, 13, 2202908 CrossRef.
- Y. Z. Zhang, J. Liang, Z. Huang, Q. Wang, G. Zhu, S. Dong, H. Liang and X. Dong, Ionically conductive tunnels in h-WO3 enable high-rate NH4+ storage, Adv. Sci., 2022, 9, 2105158 CrossRef CAS PubMed.
- G. Zhou, X. An, C. Zhou, Y. Wu, Y.-E. Miao and T. Liu, Highly porous electroactive polyimide-based nanofibrous composite anode for all-organic aqueous ammonium dual-ion batteries, Compos. Commun., 2020, 22, 100519 CrossRef.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Graphene scroll-coated α-MnO2 nanowires as high-performance cathode materials for aqueous zn-ion battery, Small, 2018, 14, 1703850 CrossRef.
- Y. Zeng, X. F. Lu, S. L. Zhang, D. Luan, S. Li and X. W. Lou, Construction of Co-Mn prussian blue analog hollow spheres for efficient aqueous zn-ion batteries, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Towards high-voltage aqueous metal-ion batteries beyond 1.5 V: the zinc/zinc hexacyanoferrate system, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- H. Y. Shi, Y. Song, Z. Qin, C. Li, D. Guo, X. X. Liu and X. Sun, Inhibiting VOPO4·xH2O decomposition and dissolution in rechargeable aqueous zinc batteries to promote voltage and capacity stabilities, Angew. Chem., Int. Ed., 2019, 58, 16057–16061 CrossRef CAS.
- W. Li, K. Wang, S. Cheng and K. Jiang, A long-life aqueous zn-ion battery based on Na3V2(PO4)2F3 cathode, Energy Storage Mater., 2018, 15, 14–21 CrossRef.
- C. Li, J. Wu, F. Ma, Y. Chen, L. Fu, Y. Zhu, Y. Zhang, P. Wang, Y. Wu and W. Huang, High-rate and high-voltage aqueous rechargeable zinc ammonium hybrid battery from selective cation intercalation cathode, ACS Appl. Energy Mater., 2019, 2, 6984–6989 CrossRef CAS.
- C. Huang, Y. Liu, J. Li, Z. Miao, X. Cai, Z. Wu, H. Yu, L. Yan, L. Zhang and J. Shu, Organic interlayer engineering of TiS2 for enhanced aqueous Zn ions storage, J. Mater. Sci. Technol., 2023, 140, 135–141 CrossRef CAS.
- X. Wen, J. Lou, K. Xiang, W. Zhou, C. Zhang and H. Chen, High-performance monoclinic WO3 nanospheres with the novel NH4+ diffusion behaviors for aqueous ammonium-ion batteries, Chem. Eng. J., 2023, 458, 141381 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05318k |
| This journal is © The Royal Society of Chemistry 2024 |