
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cleavage of ketoximes to ketones using photoexcited nitroarenes†
Lucas T.
Göttemann‡
,
Stefan
Wiesler‡
and
Richmond
Sarpong
 *
*
Department of Chemistry, Latimer Hall, University of California, Berkeley, California 94720, USA. E-mail: rsarpong@berkeley.edu
First published on 24th November 2023
Abstract
The methoxime group has emerged as a versatile directing group for a variety of C–H functionalizations. Despite its importance as a powerful functional handle, conversion of methoximes to the parent ketone, which is often desired, usually requires harsh and functional group intolerant reaction conditions. Therefore, the application of methoximes and their subsequent conversion to the corresponding ketone in a late-stage context can be problematic. Here, we present an alternative set of conditions to achieve mild and functional group tolerant conversion of methoximes to the parent ketones using photoexcited nitroarenes. The utility of this methodology is showcased in its application in the total synthesis of cephanolide D. Furthermore, mechanistic insight into this transformation obtained using isotope labeling studies as well as the analysis of reaction byproducts is provided.
Introduction
The total synthesis of natural products continues to be an exercise that highlights unaddressed gaps in synthetic methodology.1 In particular, the high density of functional groups in complex natural products necessitates the development of functional group tolerant methods that can effect the selective reaction of a targeted reactive group among many others.2 Recently, as a part of a total synthesis study of natural products in the cephalotane family,3,4 we required a method that would convert a methoxime to the corresponding ketone. Over the last decade, methoximes have emerged as superior directing groups for C–H functionalization.5–10 leading to value-added products where a variety of groups are installed. Despite their important role in directing C–H functionalizations, methoximes, unlike O-acetyl oximes, are notoriously difficult to convert to the parent ketone, which is often the desired functional group. The conditions for the removal of methoximes are generally harsh and, in some instances, can result in low yields (Scheme 1).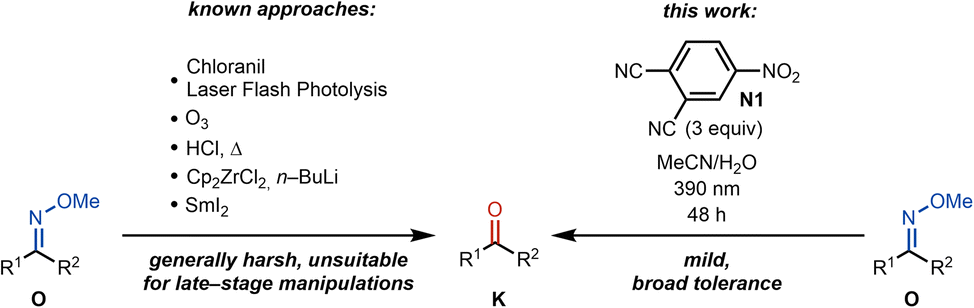 | ||
| Scheme 1 Known conditions for the cleavage of methoximes to ketones and our approach. | ||
Therefore, the established conditions that are required for the conversion of methoximes to ketones are incompatible with a wide range of functional groups, making their use in a late-stage context challenging.11–15
In our synthesis of cephanolide D (Scheme 2), a methoxime (1) that was prepared from a precursor ketone, had served as a directing group to effect carbomethoxylation at C15 giving rise to 2 through a palladated intermediate.16 However, myriad attempts to convert the methoxime moiety in 2 back to the ketone group, including using strongly acidic17 or reductive18 conditions, resulted in, among many other undesired outcomes, elimination of the tertiary hydroxy group in 2 and subsequent decarboxylation of the bridging lactone. The only successful approach that was identified for removing the methoxime group in 2 was to effect ozonolysis cleavage of the oxime19,20 followed by a reductive workup. Even in that case, the reaction proceeded capriciously and did not lead to full conversion. Typically, reaction yields in the 35% range were realized along with a significant amount of recovered starting material (64%).
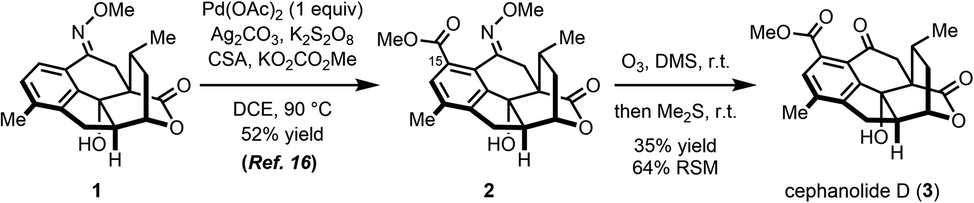 | ||
| Scheme 2 Ozonolytic cleavage of the cephanolide D methoxime. | ||
The yield of the desired product dropped precipitously upon prolonged exposure to ozone, likely due to competing oxidation at other susceptible sites on the molecule including the arene21 and benzylic positions.22
While effective, ozonolysis can be impractical, especially on a small scale where it is difficult to determine the precise stoichiometry of ozone.23 Furthermore, the inhalation and explosivity hazards of ozone and ozonides,24 respectively, can make their use a significant safety risk. As a result of these considerations, we sought to identify milder conditions that could be employed as an alternative for the cleavage of oximes — ideally, in a practical, safe, stoichiometrically precise and functional group tolerant fashion for applications in complex molecule synthesis.
Recently, concurrent reports from the groups of Leonori25 and Parasram26 described the use of photoexcited nitroarenes to cleave alkenes (Scheme 3). Notably, a range of nitroarenes were surveyed in these studies, whereby in some cases, the nature of the nitroarene significantly affected the yield of the oxidative cleavage product. Several functional groups were tolerated under these oxidative cleavage conditions including oxidation-susceptible groups such as a sulfide (see 4).27 These initial reports set the stage for additional developments such as the dihydroxylation of alkenes,28 hydroxylation of aliphatic substrates,29 and the oxidation of aldehydes and aldimines to carboxylic acids and amides, respectively.30 Furthermore, Li recently reported a photoinduced cascade process between nitroarenes and amines to form isoxazolidine derivatives.31
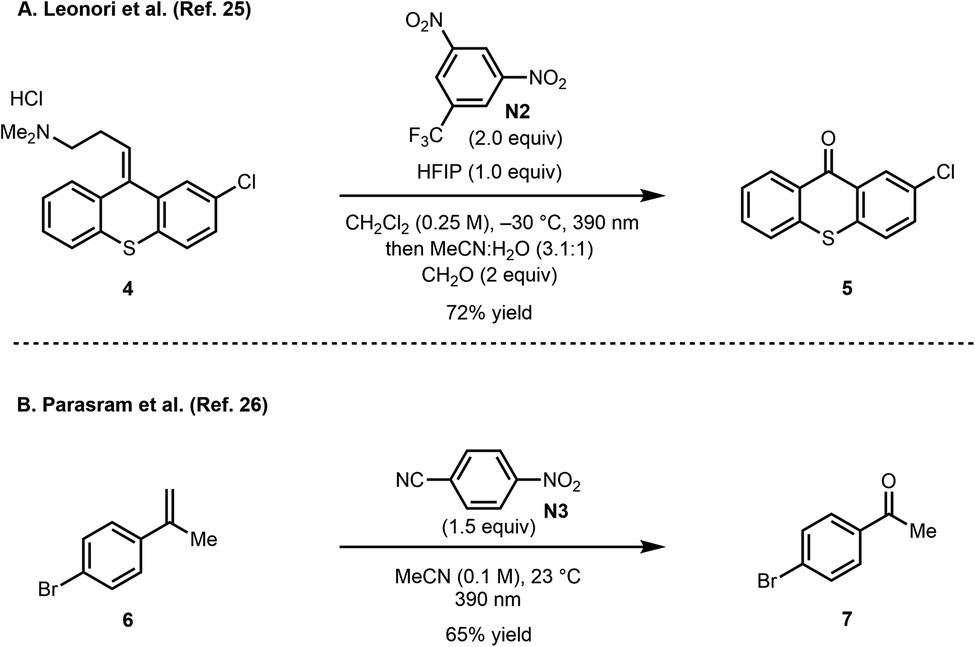 | ||
| Scheme 3 Pioneering work by Leonori and Parasram. | ||
Results and discussion
On the basis of the alkene oxidative cleavage reports from Leonori and Parasram, we envisioned a cleavage of oximes using photoexcited nitroarenes that might proceed through an analogous mechanism. However, we anticipated that this transformation was likely to be challenging due to the significantly different properties of the oxime double bond compared to a C–C double bond. We envisioned that for cyclohexanone-derived ketoxime 8 (Scheme 4), reaction with a photoexcited nitroarene32 might result in the formation of an exciplex (see 9), which upon cycloaddition would yield photoadduct 10. Several scenarios could then be envisioned for the fate of photoadduct 10 (Scheme 4). In the fragmentation A pathway, the anticipated ketone product (i.e., cyclohexanone, 11) could form directly along with byproduct 12, shown as its diradical and ylide resonance forms, which is expected to be a transient species. Alternatively, in fragmentation B, aza-carbonyl ylide 13 and alkyl nitrite 14 would be the anticipated products. Again, aza-carbonyl ylide 13 would be expected to be short-lived. In the fragmentation C pathway, direct formation of the ketone would be accompanied by alkyl nitrite 14 and aryl nitrene 15 as byproducts. Here, nitrene 15 would be expected to be fleeting. We reasoned that because a reactive intermediate would be formed in each of the fragmentation scenarios from photoadduct 10, a trapping agent should be introduced to quench the reactive byproducts. We rationalized that water would be ideal for these purposes. In the fragmentation A pathway, 12 would be expected to hydrolyze to alkyl nitrite 14 and an aryl hydroxylamine. The formation of an aryl hydroxylamine byproduct would also be expected from the hydrolysis of aza-carbonyl ylide 13 (fragmentation B) and upon reaction of nitrene 15 with water through an OH-insertion.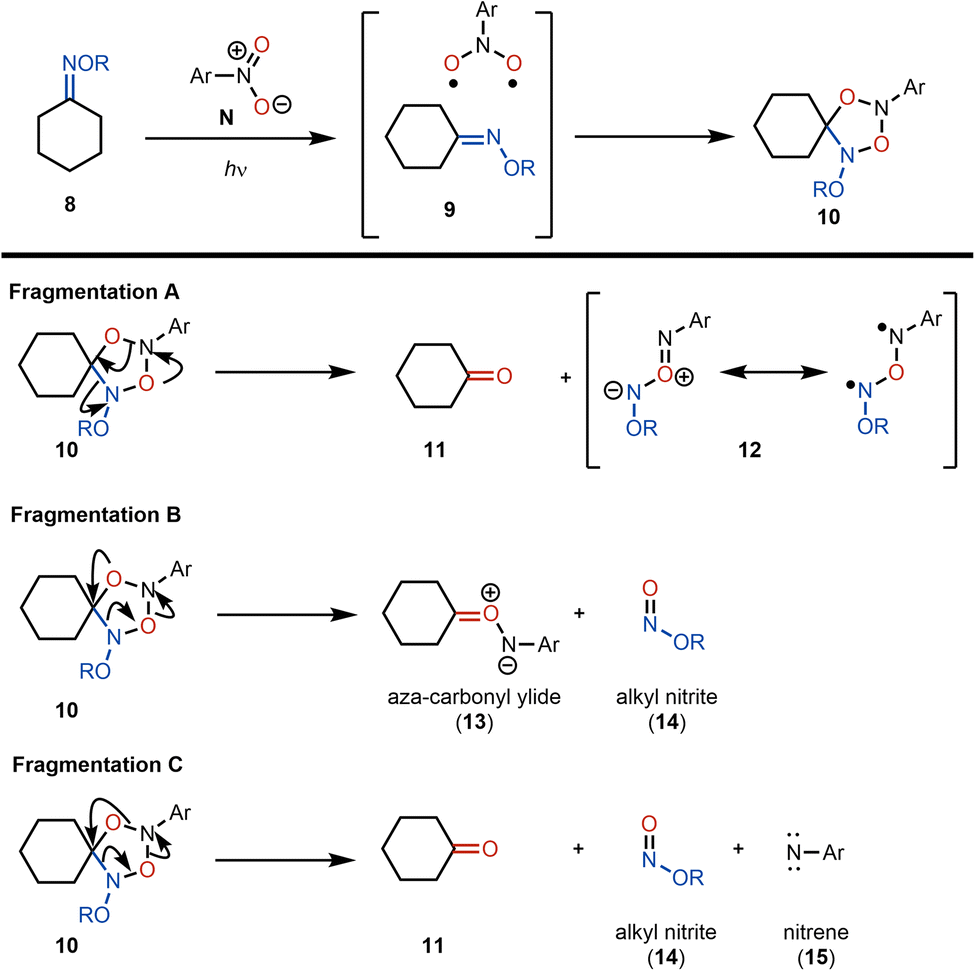 | ||
| Scheme 4 Possible fragmentation pathways for 10 in the oxidative cleavage. | ||
On the basis of this rationalization, we embarked on a survey of conditions to effect the photoexcited nitroarene-mediated oxidative cleavage of oximes using tetralone methoxime 16 (which we envisioned to be a suitable model substrate for our cephanolide D synthesis). In addition to varying reaction conditions (i.e., irradiation wavelength and duration, temperature, and solvent), we also tested several nitroarenes in our optimization studies (see the ESI† for details). Of note, in the cases where incomplete cleavage of the starting oxime was observed, a side-product observed in the mixture was the oxime diastereomer (Z-isomer), which presumably arises from photoisomerization under the reaction conditions.33 With an initial set of effective conditions in hand (see conditions in entry 1, Table 1), we began to examine the scope of this reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Wavelength | Nitroarene | Solvent | Timeb | Yield 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18:19 :18:19 |
| a A reaction time of 48 h is consistent with values reported for photochemical reactions with nitroarenes.25,26 b The reactions were carried out in a photoreactor.28 A reaction time of 48 h is consistent with values reported for photochemical reactions with nitroarenes.25,26 c The reaction was carried out using Kessil lamps (distance to LED = 3 cm). | |||||
| 1 | 420 nm | N3 (1.5 equiv) | MeCN/H2O 4:1 |
48 h | 6%/70%/20% |
| 2 | 405 nm | N3 (1.5 equiv) | MeCN/H2O 4:1 |
48 h | 7%/65%/23% |
| 3 | 390 nm | N3 (1.5 equiv) | MeCN/H2O 4:1 |
48 h | 10%/58%/29% |
| 4 | 390 nm | N2 (1.5 equiv) | MeCN/H2O 4:1 |
48 h | 14%/33%/47% |
| 5 | 390 nm | N1 (1.5 equiv) | MeCN/H2O 4:1 |
48 h | 7%/21%/65% |
| 6 | 390 nm | N1 (3.0 equiv) | MeCN/H2O 4:1 |
48 h | <5%/17%/74% |
| 7c | 390 nm | N1 (3.0 equiv) |
MeCN/H2O 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
48 h | –/trace/95% |
| 8 | 390 nm | — | MeCN/H2O 4:1 |
48 h | 90%/7%/– |
|
|||||
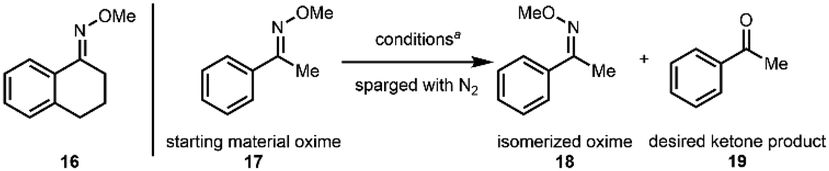
Unfortunately, these conditions turned out not to be generalizable and we observed low yields for additional substrates that were tested such as acetophenone-derived oxime 17 (20% yield, entry 1), necessitating a second round of optimization for less reactive compounds. We chose acetophenone methoxime (17) as the substrate for this fine-tuning to achieve more broadly applicable conditions (see the ESI† for the full screening table). We retained the solvent mixture (4:1 MeCN:H2O), which was shown to be significantly superior to other solvent mixtures used in our initial screening. We started by testing lower wavelength irradiation. LEDs centered at 390 nm showed an immediate improvement and resulted in a significant reduction of side reactions (entries 1–3). We then screened several nitroarenes and observed improved yields using nitroarenes N2 and N1, which possess more electrophilic character in their excited states as compared to N3 (entries 3–5).25 Increasing the nitroarene equivalents and using a setup with Kessil lamps instead of a photoreactor34 (see pictures in the ESI†) led to significantly higher yields (entries 5–7). Overall, the optimized conditions that we identified from these two surveys using tetralone methoxime (16) and acetophenone methoxime (17) are: using nitroarene N1 (3 equiv) in a 4:1 mixture of acetonitrile and water with irradiation using LEDs centered at 390 nm at 40 °C (the measured temperature of our setup using a household fan for cooling) for 48 h (Table 1, entry 7). Finally, as shown in entry 8, in the absence of the nitroarene, the starting oxime is recovered in 90% yield along with 7% of the isomerized oxime.
We then investigated the scope of the reaction to test the functional group tolerance of our method and its efficacy. Importantly, we selected substrates featuring groups that are often encountered in more complex settings (e.g., in total synthesis), where existing methods for converting methoximes to ketones have not been as effective. As shown in Fig. 1, a range of benzylic ketoximes are effectively cleaved under the conditions we have identified to afford the corresponding ketones.35 Of note, sterically encumbered methoximes can be cleaved (see 34) and oxidatively sensitive groups such as a thiomethyl group (45), which is oxidized to the sulfone under ozonolysis conditions (see the ESI†), are tolerated to some extent, emphasizing the complementarity of the current method to existing methods. Furthermore, tetralone, acetophenone, benzosuberone, and chromanone derived oximes all participate faithfully in this oxidative cleavage reaction. In addition, free oximes (see 39), acetylated oximes (see 40) and hydrazones (41) all undergo oxidative cleavage to the corresponding ketones under the conditions that we have identified. In general, we have found that substrates that proceed in low conversion, but high mass balance, can be further converted to the product over an extended reaction time, but the reaction rate decreases noticeably. In cases where poor mass balance was observed, longer wavelength irradiation (using LEDs centered at 420 nm) was beneficial to the reaction outcome. This is likely due reactivity of intermediates at the shorter wavelengths that were previously employed, which led to undefined side reactions. Mainly, the yields improved by irradiation at longer wavelengths for substrates with benzylic C–H groups that are known to undergo benzylic oxidation reactions. Albeit proceeding with lower mass balance upon irradiation at 420 nm over a prolonged period, we did not observe significant benzylic C–H hydroxylation products under these conditions.
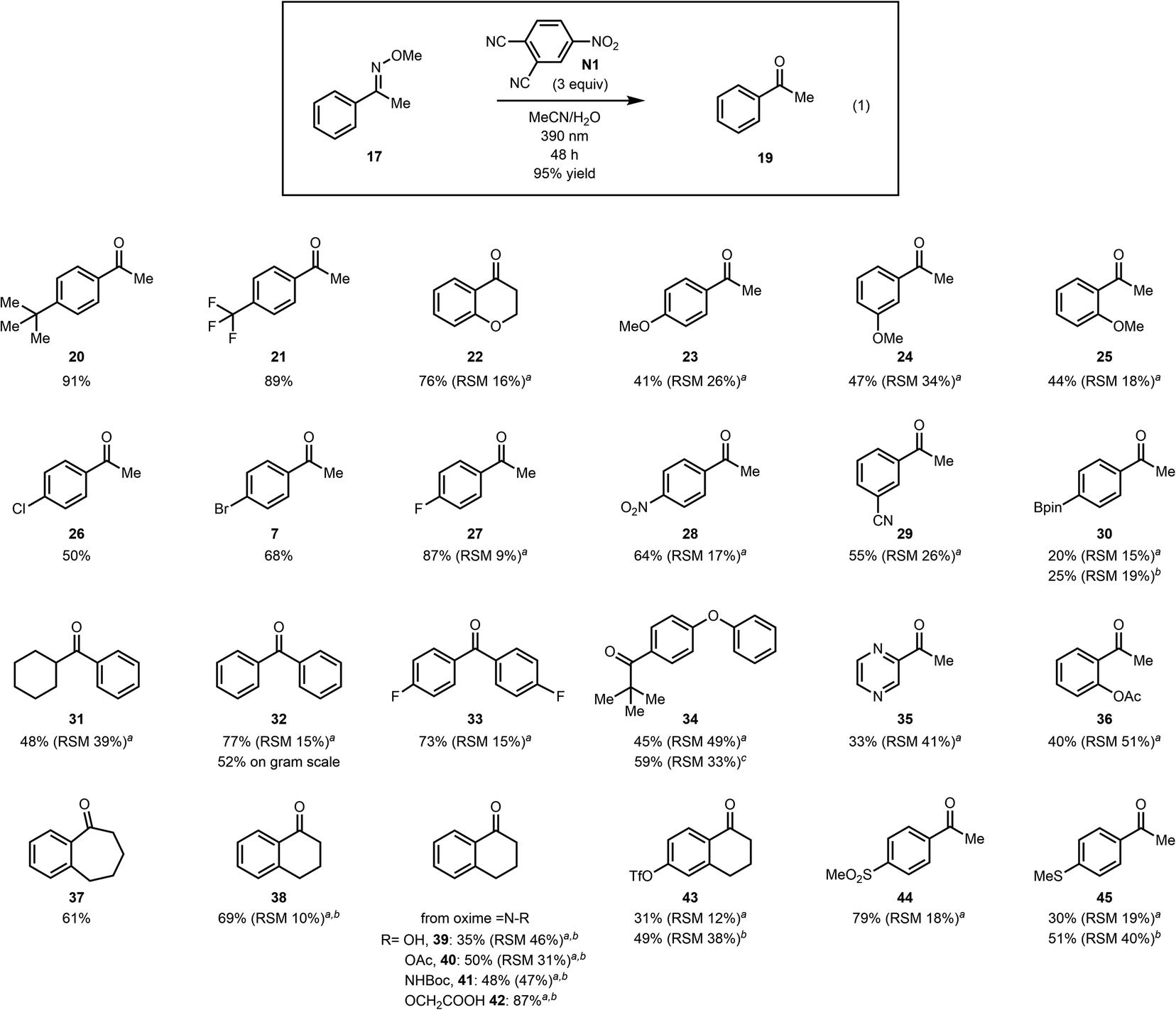 | ||
| Fig. 1 Scope of benzylic methoxime cleavage. aRSM = recovered starting material. The RSM is a combined yield of oxime diastereomers. bIrradiation was conducted at 420 nm using nitroarene N3 for 48 h. cThe reaction was irradiated for 96 h. | ||
The photoexcited nitroarene oxidative cleavage of a series of aliphatic methoximes was also investigated (Fig. 2). The reaction was found to work well for several aliphatic ketoximes including cyclic and sterically encumbered as well as acyclic substrates (see 46–49).
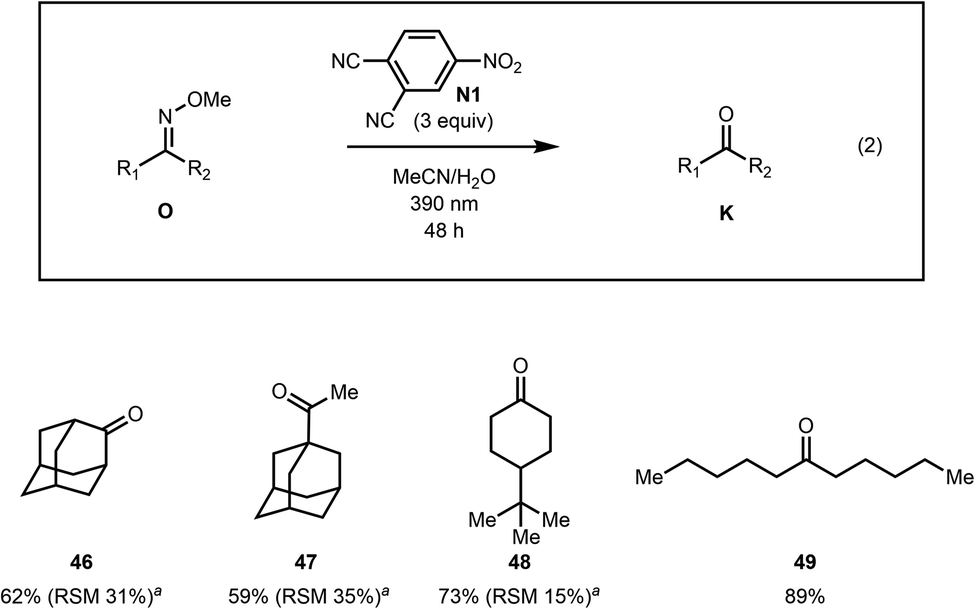 | ||
| Fig. 2 Methoxime cleavage of aliphatic compounds. aRSM = recovered starting material. The RSM is a combined yield of oxime diastereomers. | ||
However, the reaction does not tolerate alkenes, which are cleaved under the reaction conditions. For example, α,β-unsaturated oxime 50 leads to benzaldehyde (51) as the main detected product (Scheme 5A). Similarly, we found that acyloin derived methoxime 52 undergoes C–C bond cleavage under the reaction conditions to give acetophenone (Scheme 5B).
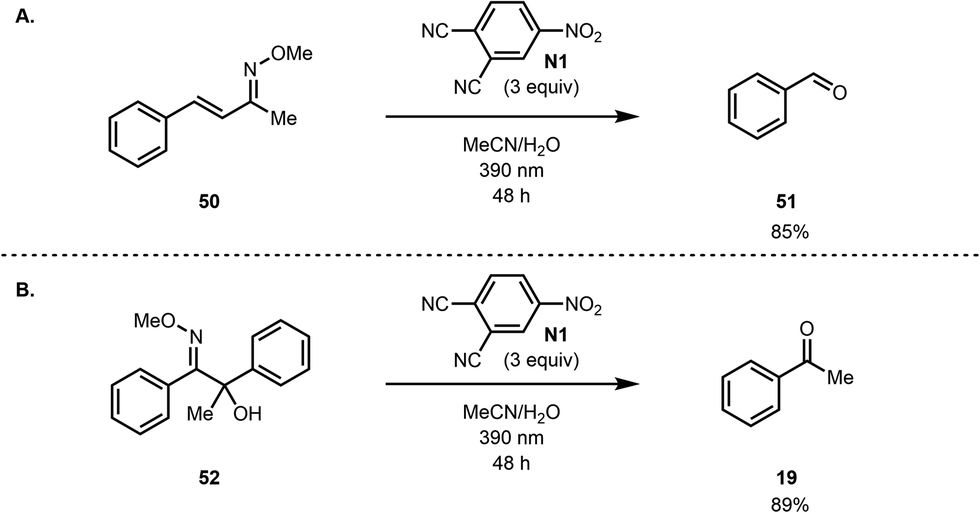 | ||
| Scheme 5 Limitations and side reactions. | ||
Of note, ozonolysis of 52 also results in the formation of acetophenone (19). Furthermore, we observed that substrates with free alcohol groups as well as amines were not tolerated under the reaction conditions, leading to a very low conversion of the starting material (see the ESI† for examples).
The oxime cleavage reaction has also been found to work well in several complex substrates (Fig. 3). For example, naproxen derived methylketone 53 is formed from the corresponding methoxime using our standard photoexcited nitroarene oxidative cleavage conditions in 21% yield (isolated along with 51% of the methoxime starting material). Cholestanone (54) is obtained in 31% yield (along with 52% of the methoxime starting material) upon subjection to the cleavage conditions. Similar to substrate 34 the yield for methylketone 53 could be improved upon irradiation for 96 h (see Fig. 3A), however cholestanone derived methoxime 54 suffered from solubility issues. On the other hand, the cleavage of the corresponding methoxime generated from kobusone (55) gives 55 in 67% yield (along with 22% of the methoxime starting material). Finally, in the context of our synthesis of cephanolide D, we subjected methoxime 5636 to the newly developed oxidative cleavage conditions (Fig. 3B). To our delight, our conditions were tolerated by the ester, lactone and even the TMS group, giving rise to ketone 57 — showcasing the use of photoexcited nitroarenes as an effective alternative for methoxime cleavage in compounds bearing various reactive groups.37 Cephanolide D (3) was then obtained from 57 in 93% yield upon treatment with TBAF.
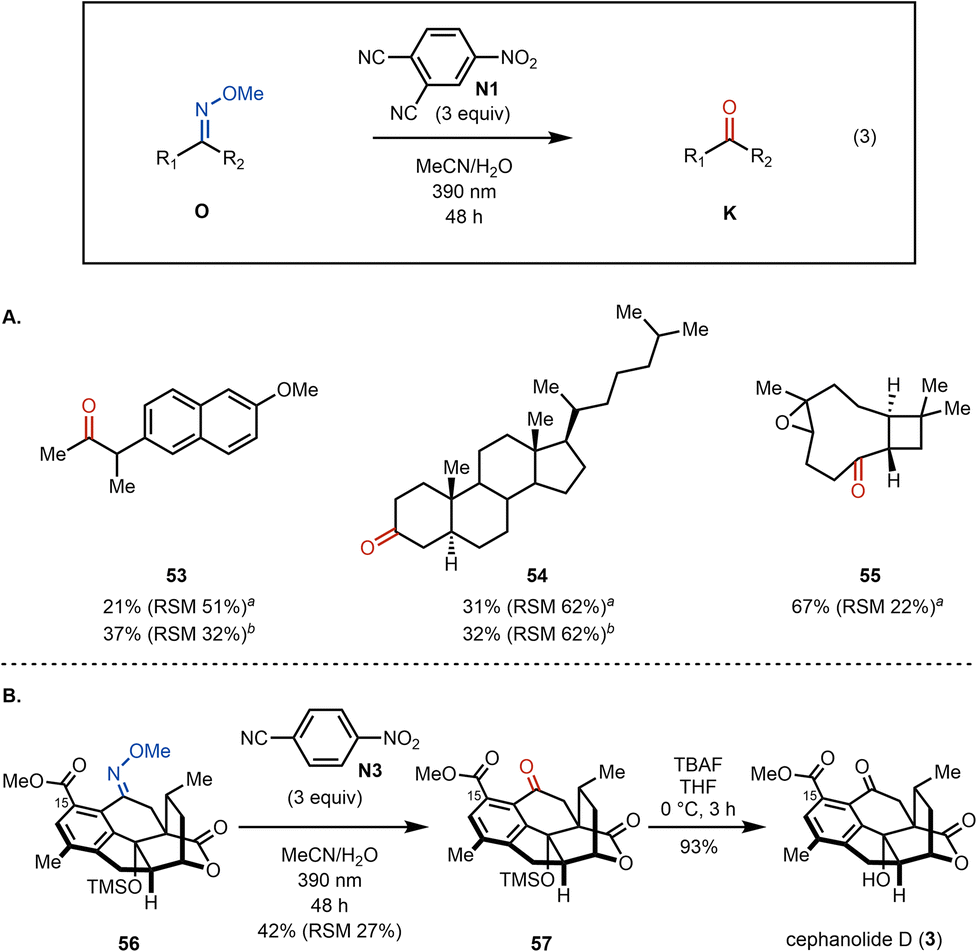 | ||
| Fig. 3 (A) Application of methoxime cleavage in bioactive and complex substrates. (B) Synthesis of cephanolide D (3). aRSM = recovered starting material. The RSM is a combined yield of oxime diastereomers. bThe reaction was irradiated for 96 h. | ||
In order to test our hypothesis of a photoexcited nitroarene mediated oxidative cleavage of methoximes to the corresponding ketones, a comprehensive photophysical study was carried out. As a part of this investigation, the UV-vis data, including the absorption maxima of all the methoxime starting materials, ketone products, and nitroarenes at various concentrations were obtained experimentally. Our observations indicated that at a concentration of 50 μM, the methoximes, ketones, and nitroarenes do not absorb light centered at 390 nm. However, at higher concentrations, which reflect our reaction conditions (0.2 M), the nitroarenes strongly absorb whereas ketones can partially absorb at 390 nm (Fig. 4B). These absorption profiles strongly support the hypothesis that the reaction is mediated by photoexcited nitroarenes. Furthermore, our observations also explain the relatively long reaction time of 48 h since the ketone, formed over the course of the reaction (i.e., continuously increasing concentration) partially absorbs light and reduces the efficiency of the absorption of the nitroarene and therefore reaction. This rationale is further supported by an increasing redshift over the course of the 48 h reaction duration, which suggests that an inner filter effect slows down the reaction.
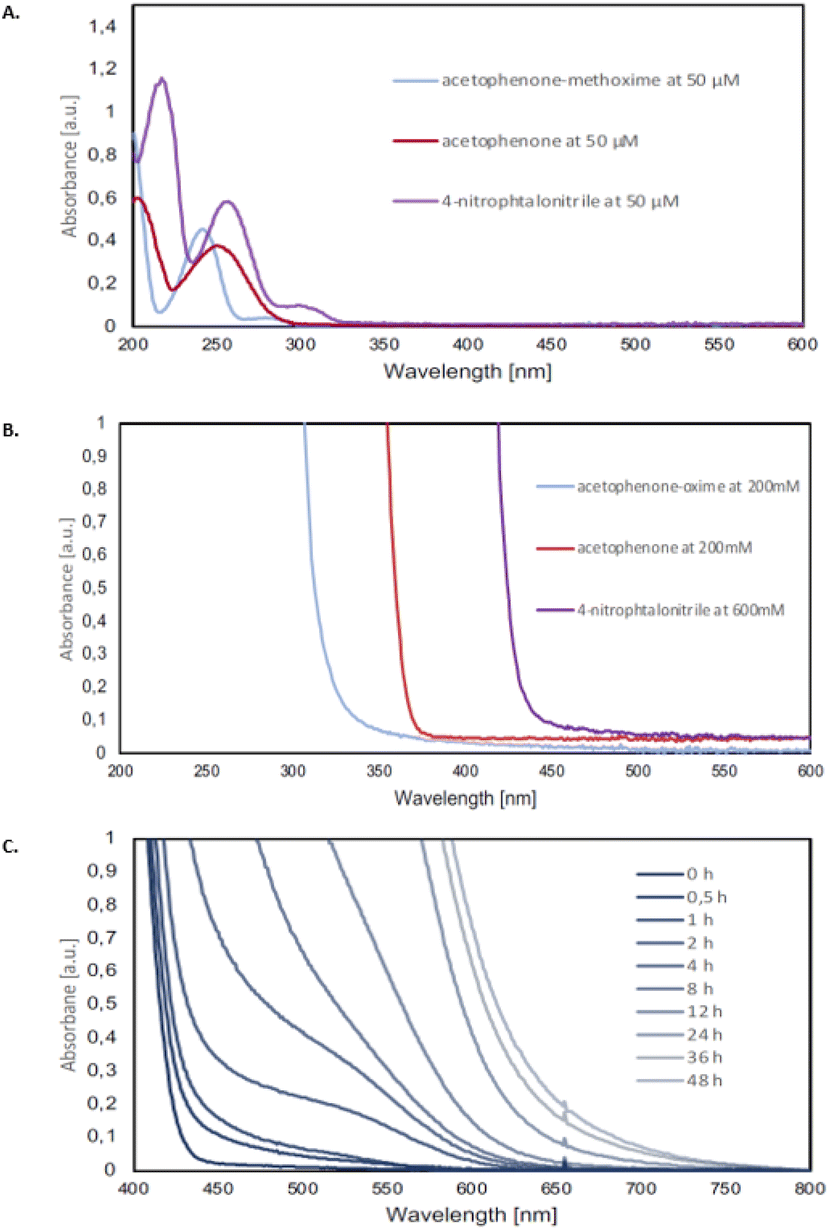 | ||
| Fig. 4 (A). Absorption maximum for 17, 19 and N1 at 50 μM. (B) UV-vis absorption spectra for 17 and 19 at 200 mM and N1 at 600 mM. C. UV-visible spectra for the reaction monitoring experiment indicating redshift using 17 and N1 under standard reaction conditions and taking aliquots at the indicated time. | ||
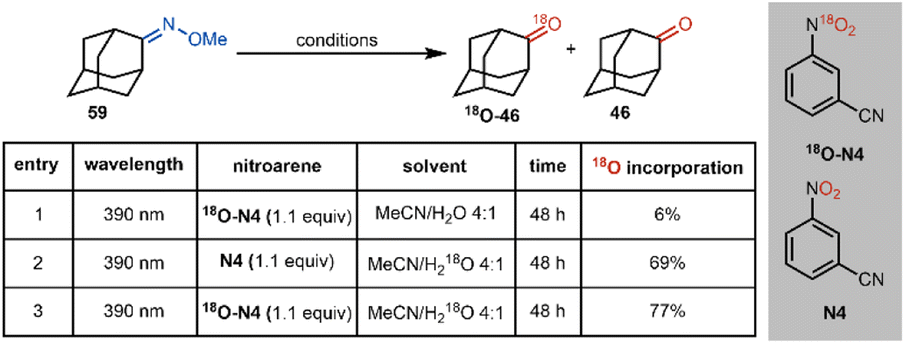
To gain further insight into the proposed mechanism for this oxime cleavage method, we have carried out a series of experiments using 18O-labeled nitroarene 18O-N4 and H218O (see Table 2). 18O-Labeled nitroarene 18O-N4 was prepared by nitration of benzonitrile using 18O-labeled nitric acid and sulfuric acid analogous to the protocol of Wibaut (Scheme 6).38
|
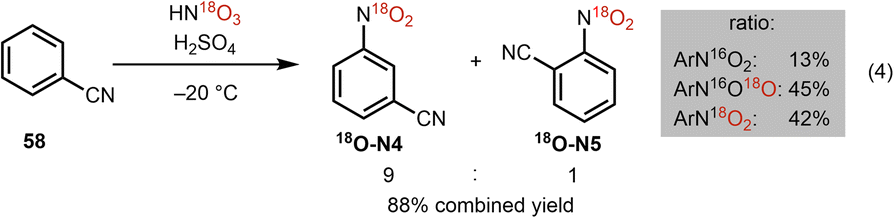 | ||
| Scheme 6 Preparation of 18O labelled nitroarene 18O-N4. | ||
In our mechanistic studies, we have found that subjecting adamantanone methoxime (59) to our standard conditions using the 18O-labeled 18O-N4 in a mixture of acetonitrile and 16O-labeled water leads to a 6% 18O incorporation in the ketone product (entry 1). The use of unlabeled (i.e., 16O) nitroarene N4 and 18O-labeled water leads to a 69% incorporation of 18O in the ketone product (entry 2), suggesting a fast O-atom exchange in the ketone group of adamantanone under the reaction conditions. The use of 18O-labeled nitroarene 18O-N4 and 18O-labeled water leads to the ketone product in a slightly more isotopically enriched form (77% incorporation of 18O; entry 3) than in the case where only 18O-labeled water is used (i.e., entry 2). While strict conclusions as to the source of 18O label in the observed products cannot be drawn (i.e., the oxygen atom in the ketone product might arise from the nitroarene or through fast exchange with water), the slight enrichment in 18O for the product under the conditions where only 18O-N4 is used (entry 1), and also the higher enrichment for the case where 18O-N4 and H218O are used (compare entry 3 to entry 2) may support the nitroarene, in addition to the water co-solvent, as sources of the oxygen atom in the product. These preliminary labeling studies support the proposed mechanism (Scheme 4) wherein a cycloaddition between the photoexcited nitroarene and oxime is proposed (see 9).
To gain further mechanistic insight, we sought to investigate the fate of the oxime residue. Two chemical probes (60 and 61) bearing a bulky, non-volatile, alkyl substituent (in place of the methyl group) were synthesized and subjected to our reaction conditions (Scheme 7). Assuming the formation of cycloadduct 62, fragmentation B pathway (vide supra) would give rise to aza-carbonyl ylide 63 and transient adamantyl nitrite 64. Subsequently, aza-carbonyl ylide 63 could be trapped by water yielding the desired ketone (19 or 32) as well as hydroxylamine 67, which can then be envisioned to undergo NO mediated reduction to aniline 67.39,40 We assumed that the NO arises from the photochemical decomposition of alkyl nitrite 64, resulting in the formation of 1-adamantanol (65) and NO.41 Both 65 and 67 were detected in the crude reaction mixture (using GC-MS and NMR; see the ESI† for more details), providing additional support for the fragmentation B pathway. Due to the reactive nature of the intermediates described, other concomitant decomposition pathways of 62 are also plausible (e.g., via the fragmentation A pathway), which would explain a partial incorporation of 18O from the nitroarene as observed in the labeling experiments.
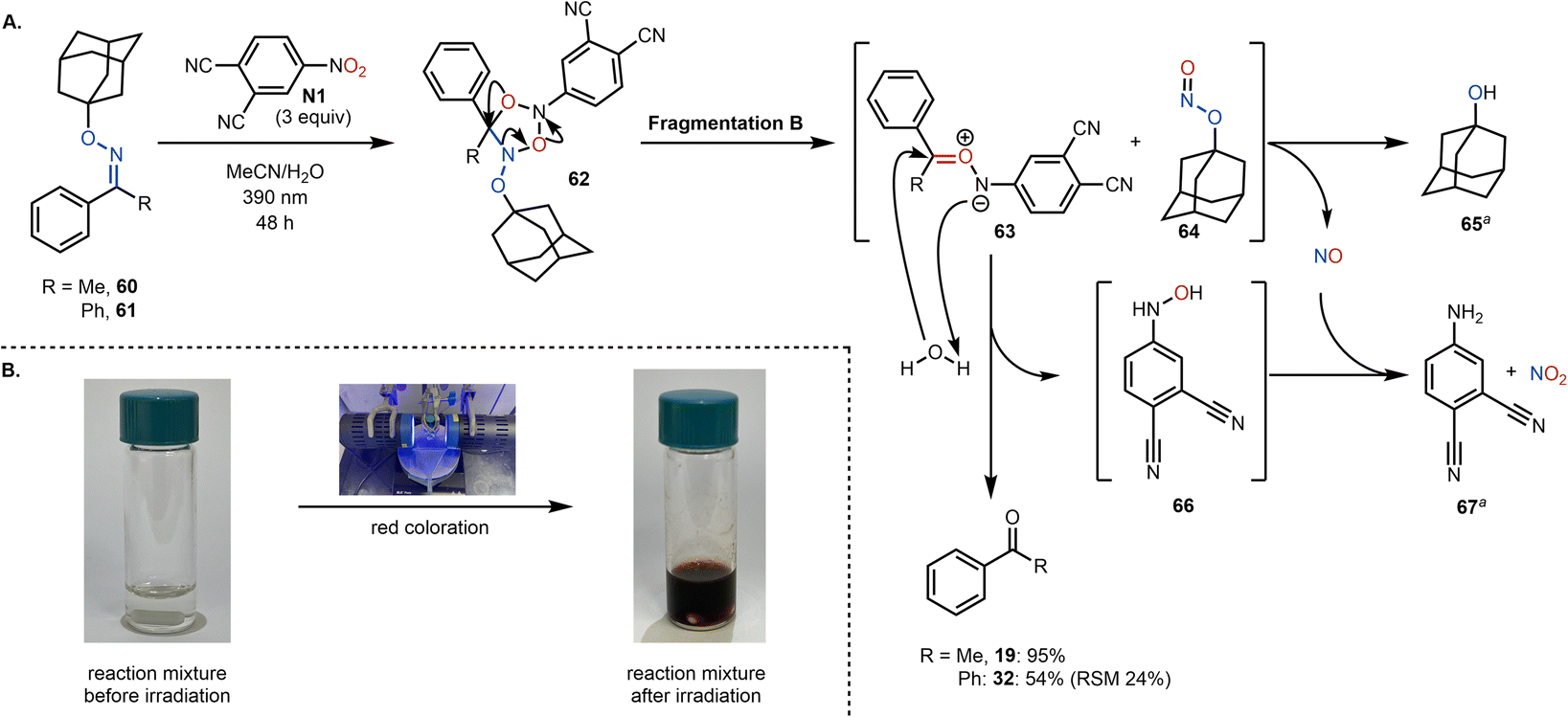 | ||
| Scheme 7 (A). Proposed mechanism of the oxidative cleavage. (B). Color change after 48 h reaction time. aByproducts were both detected by GC-MS and 1H NMR. | ||
Conclusions
In summary, building on recent studies that describe the use of photoexcited nitroarenes to effect oxidative cleavage of alkenes, we report here the removal of oxime groups to afford the corresponding ketones under a related set of conditions. The substrate scope of the transformation is relatively broad but is not tolerant of alkenes, which are cleaved under the reaction conditions (consistent with the precedent of Leonori and Parasram). The success of the current studies enabled the application of this new protocol to a preparation of cephanolide D from a precursor methoxime. While the mechanism of the reaction is yet to be fully elucidated, isotopic labelling studies and the detection of several by-products support both the water cosolvent and nitroarene as possible sources of the O-atom in the ketone product that is obtained. Overall, this protocol offers an alternative to the strongly acidic or reductive conditions as well as ozonolysis cleavage conditions that are usually employed in the conversion of ketoxime groups to ketones.Author contributions
The project was conceived by S. W. The experimental work was performed by L. G. and S.W. The initial draft was written by R. S. with input from all authors. L. G., S. W. and R. S. were involved in editing and finalizing the manuscript. R. S. supervised the project and secured funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the US National Science Foundation (CHE-1700982). S.W. thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, WI 5665/1-1) for a postdoctoral scholarship. The authors are grateful to Dr Hasan Celik and UC Berkeley's NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. The authors thank Magan Powell (UC Berkeley) for assistance with GC-MS.Notes and references
- K. C. Nicolaou, S. Rigol and R. Yu, CCS Chem., 2019, 3–37 CAS.
- R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530–541 CrossRef CAS.
- M. Haider, G. Sennari, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 2710–2715 CrossRef CAS PubMed.
- G. Sennari, K. E. Gardner, S. Wiesler, M. Haider, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 19173–19185 CrossRef CAS PubMed.
- Y.-F. Liang, X. Wang, Y. Yuan, Y. Liang, X. Li and N. Jiao, ACS Catal., 2015, 5, 6148–6152 CrossRef CAS.
- C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC.
- Y. Park, S. Jee, J. G. Kim and S. Chang, Org. Process Res. Dev., 2015, 19, 1024–1029 CrossRef CAS.
- Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956–1963 CrossRef CAS.
- B. S. Pilgrim, A. E. Gatland, C. H. A. Esteves, C. T. McTernan, G. R. Jones, M. R. Tatton, P. A. Procopiou and T. J. Donohoe, Org. Biomol. Chem., 2016, 14, 1065–1090 RSC.
- S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58–61 CrossRef CAS PubMed.
- H. J. P. De Lijser and C.-K. Tsai, J. Org. Chem., 2004, 69, 3057–3067 CrossRef CAS.
- R. E. Erickson, P. J. Andrulis, J. C. Collins, M. L. Lungle and G. D. Mercer, J. Org. Chem., 1969, 34, 2961–2966 CrossRef CAS.
- E. Tan, M. Zanini and A. M. Echavarren, Angew. Chem., Int. Ed., 2020, 59, 10470–10473 CrossRef CAS.
- M. Kitamura, Y. Shintaku, D. Kudo and T. Okauchi, Tetrahedron Lett., 2010, 51, 4890–4893 CrossRef CAS.
- F. Huang and S. Zhang, Org. Lett., 2019, 21, 7430–7434 CrossRef CAS.
- Z.-Y. Li and G.-W. Wang, Org. Lett., 2015, 17, 4866–4869 CrossRef CAS PubMed.
- R. J. Sharpe and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 4968–4971 CrossRef CAS.
- M. M. Majireck, J. A. Witek and S. M. Weinreb, Tetrahedron Lett., 2010, 51, 3555–3557 CrossRef CAS.
- (a) Y. Ito, H. Yokoya, Y. Umehara and T. Matsuura, Bull. Chem. Soc. Jpn., 1980, 53, 2407–2408 CrossRef CAS; (b) D. J. Weitz and M. D. Bednarski, J. Org. Chem., 1989, 54, 4957–4959 CrossRef CAS.
- K. Griesbaum, B. Övez, T. S. Huh and Y. Dong, Liebigs Ann., 1995, 1995, 1571–1574 CrossRef.
- E. A. Skrotzki, J. K. Vandavasi and S. G. Newman, J. Org. Chem., 2021, 86, 14169–14176 CrossRef CAS PubMed.
- W. Gao, L. Du, W. Jiao and Y. Liu, Chin. J. Chem. Eng., 2020, 28, 808–814 CrossRef CAS.
- A. L. Gomez, T. L. Lewis, S. A. Wilkinson and S. A. Nizkorodov, Environ. Sci. Technol., 2008, 42, 3582–3587 CrossRef CAS PubMed.
- A. Lazen, Emergency and Continuous Exposure Limits for Selected Airborne Contaminants, Defense Technical Information Center, Fort Belvoir, VA, 1984, vol. 1 Search PubMed.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86 CrossRef CAS.
- D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442 CrossRef CAS PubMed.
- A. Maggiolo and E. A. Blair, Ozone Chemistry and Technology, American Chemical Society, N.W. Washington 6, D.C., 1959 Search PubMed.
- C. Hampton, M. Simonetti and D. Leonori, Angew. Chem., Int. Ed., 2023, 62, e202214508 CrossRef CAS.
- J. M. Paolillo, A. D. Duke, E. S. Gogarnoiu, D. E. Wise and M. Parasram, J. Am. Chem. Soc., 2023, 145, 2794–2799 CrossRef CAS PubMed.
- J. K. Mitchell, W. A. Hussain, A. H. Bansode, R. M. O’Connor, D. E. Wise, M. H. Choe and M. Parasram, Org. Lett., 2023, 25, 6517–6521 CrossRef CAS.
- X.-Z. Shao, G.-Y. Xu, W. Fan, S. Zhang and M.-B. Li, Org. Chem. Front., 2023, 10, 624–631 RSC.
- R. Hurley and A. C. Testa, J. Am. Chem. Soc., 1966, 88, 4330–4332 CrossRef CAS.
- X. Zhang and T. Rovis, J. Am. Chem. Soc., 2021, 143, 21211–21217 CrossRef CAS PubMed.
- A. Penn PhD M2 was used. See ESI† for details.
- Generally, caution must be taken when isolating the product ketones since in some instances their volatility must be taken into consideration as this can result in diminished yields..
- As we mentioned before, free alcohols react poorly and are therefore a limitation of this method. For this reason we decided to use the TMS protected methoxime 62 instead of methoxime 10..
- Subsequently, we removed the TMS group in 63 using TBAF to yield cephanolide D (93% yield)..
- J. P. Wibaut and R. van Strik, Recueil, 1958, 77, 316–326 CrossRef CAS.
- E. Damiani, L. Greci and C. Rizzoli, J. Chem. Soc. Perkin Trans., 2001, 2, 1139–1144 RSC.
- H. Weber, A. Grzesiok, R. Sustmann and H.-G. Korth, Z. Naturforsch., 1994, 49b, 1041–1050 CrossRef.
- G. R. McMillan, J. Phys. Chem., 1963, 67, 931–932 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05414d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |