
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic ammonia synthesis on HY-zeolite-supported angstrom-size molybdenum cluster†
Satoshi
Kamiguchi
 *ab,
Kiyotaka
Asakura
c,
Tamaki
Shibayama
d,
Tomoko
Yokaichiya
e,
Tatsushi
Ikeda
e,
Akira
Nakayama
*e,
Ken-ichi
Shimizu
c and
Zhaomin
Hou
ab
*ab,
Kiyotaka
Asakura
c,
Tamaki
Shibayama
d,
Tomoko
Yokaichiya
e,
Tatsushi
Ikeda
e,
Akira
Nakayama
*e,
Ken-ichi
Shimizu
c and
Zhaomin
Hou
ab
aAdvanced Catalysis Research Group, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama, 351-0198, Japan. E-mail: kamigu@riken.jp
bOrganometallic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako, Saitama, 351-0198, Japan
cInstitute for Catalysis, Hokkaido University, Sapporo, 001-0021, Japan
dCenter for Advanced Research of Energy Conversion Materials, Hokkaido University, Sapporo, 060-8628, Japan
eDepartment of Chemical System Engineering, Graduate School of Engineering, The University of Tokyo, Tokyo, 113-8656, Japan. E-mail: nakayama@chemsys.t.u-tokyo.ac.jp
First published on 22nd January 2024
Abstract
The development of new catalysts with high N2 activation ability is an effective approach for low-temperature ammonia synthesis. Herein, we report a novel angstrom-size molybdenum metal cluster catalyst for efficient ammonia synthesis. This catalyst is prepared by the impregnation of a molybdenum halide cluster complex with an octahedral Mo6 metal core on HY zeolite, followed by the removal of all the halide ligands by activation with hydrogen. In this activation, the size of the Mo6 cluster (ca. 7 Å) is almost retained. The resulting angstrom-size cluster shows catalytic activity for ammonia synthesis from N2 and H2, and the reaction proceeds continuously even at 200 °C under 5.0 MPa. DFT calculations suggest that N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage is promoted by the cooperation of the multiple molybdenum sites.
N bond cleavage is promoted by the cooperation of the multiple molybdenum sites.
Introduction
Ammonia production from atmospherically abundant dinitrogen (N2) is an essential chemical process for human beings, because ammonia is a feedstock for globally used fertilizer and a wide variety of nitrogen-containing chemicals.1 Recently, ammonia has also been expected to be used in hydrogen (H2) storage owing to its high hydrogen content ratio (17.6 wt%)2 and as a fuel that does not emit carbon dioxide during combustion.3 The worldwide production of ammonia is increasing year by year and reached 0.18 Gton in 2022.4 Industrially, ammonia is produced from N2 and H2via the Haber–Bosch process using iron (Fe)-based catalysts under high pressure (10–30 MPa) and high temperature (400–500 °C) conditions.5 The reaction conditions of industrial ruthenium (Ru)-based catalysts developed later are still harsh (<10 MPa and 325–450 °C).6 Making these reaction conditions milder (e.g. <5 MPa and <200 °C) is in high demand to reduce the high energy consumption.7 In ammonia synthesis on these industrial catalysts, the cleavage of the chemically inert NN triple bond has the highest energy barrier,8 and acceleration of this triple bond cleavage facilitates efficient ammonia synthesis at lower temperatures.2c,9 Recently, it has been reported that trinuclear titanium (Ti) and chromium (Cr) hydride cluster complexes achieve NN bond cleavage and subsequent hydrogenation of N at ambient temperature and pressure, based on cooperation by multiple metal sites.10 Biologically, nitrogenase enzymes in certain microbial organisms produce ammonia from N2 under ambient conditions,11 and the cooperation by multiple metal sites is considered to be responsible for the reaction.12 These results suggest the potential of clusters for efficient catalytic ammonia synthesis.
Previously, Kamiguchi, one of the authors of this paper, reported that transition-metal cluster compounds with chloride or bromide ligands had catalyzed various reactions since 2002, although there had been no reports on these clusters as catalysts for more than 140 years.13 A molecular molybdenum (Mo) chloride cluster with an octahedral metal framework, (H3O)2[(Mo6Cl8)Cl6]·6H2O (1), exhibits catalytic activity by partial elimination of halide ligands.14 In this activation, however, only some of the metallic sites of the Mo6 cluster can participate in the catalytic reactions. When all the halide ligands are removed by H2-activation in the anticipation of the participation of more metallic sites, the Mo6 cluster aggregates to form bulk molybdenum metal.15 Thus, we expect that the H2-activation of 1 dispersed on a porous material would form an isolated ultra-small molybdenum metal cluster without aggregation of the Mo6 cluster core, leading to the cooperation by more molybdenum sites for the NN bond cleavage and further efficient ammonia synthesis. Moreover, in comparison with Fe and Ru, Mo is more active in N2 activation, as deduced from the calculated N2 dissociative adsorption energy on the metal surface.16 Several molecular Mo complexes afford ammonia from N2 using proton sources and reducing agents at ambient temperature and pressure in a homogeneous system.17 From these results, it is expected that the ultra-small Mo cluster will show high catalytic activity for Haber–Bosch-like ammonia synthesis from N2 and H2. Herein, we report the preparation of an angstrom-size Mo metal cluster from HY-zeolite-supported 1. The resulting cluster produced ammonia with high stability, and the reaction proceeded continuously even at 200 °C under 5.0 MPa. The cooperation of the multiple Mo sites promotes the NN bond cleavage and efficient ammonia synthesis.
Results and discussion
Activation of cluster
As previously reported, when unsupported 1 is heated progressively in flowing hydrogen, 1 is converted to bulk molybdenum metal up to 600 °C, with complete removal of the chloride ligands as hydrogen chloride.15 When HY-supported 1 was analyzed by H2-temperature-programmed reduction (H2-TPR), a large reduction peak was observed at 450–600 °C (Fig. S1†), suggesting that 1 on HY also releases chloride ligands as hydrogen chloride up to 600 °C. Thus, supported 1 was heated in flowing hydrogen at 600 °C for 3 h at atmospheric pressure before ammonia synthesis. Elemental analyses showed a decrease in Cl-content from 1.9 to 0 wt% without the loss of Mo-content after the H2-treatment (Table S1†), indicating the complete removal of the chloride ligands of 1 on HY by H2-activation. HZSM5- and MCM41-supported 1 also showed complete removal of the chloride ligands, as confirmed by elemental analyses (Table S1†).The change in the local structures of HY-supported 1 by impregnation and H2-activation was investigated using the X-ray absorption fine structure (XAFS) technique. The results are summarized in Fig. 1 and Table 1. Cluster 1 impregnated on HY exhibited very similar XAFS (Fig. 1A(b)), X-ray absorption near edge structure (XANES) (Fig. 1A(b), inset), and Fourier transforms of k3-weighted extended XAFS (FT-EXAFS) spectra (Fig. 1B(b)) to those of 1 before impregnation (Fig. 1A(a) and B(a)). The fitted parameters of 1 after impregnation were almost the same as those before impregnation (Table 1). Thus, impregnation did not change the molecular structure of 1 or the size of the Mo6 cluster (ca. 7 Å). However, the XAFS (Fig. 1A(c)), XANES (Fig. 1A(c), inset), and FT-EXAFS spectra (Fig. 1B(c)) changed after H2-activation. The FT-EXAFS spectrum had two small peaks at 1.5 and 2.5 Å. As shown by curve fitting analysis (Table 1), while the former peak was attributed to Mo–O (oxygen of a silanol of HY), the latter was assigned to the nearest Mo–Mo with a bond length of 2.84 Å. No significant peaks attributed to the next nearest Mo–Mo shell were observed over the longer range (Fig. 1B(c)), indicating that the Mo6 cluster on HY did not aggregate to form a larger molybdenum particle with long Mo–Mo distances. The coordination number (CN) of the nearest Mo–Mo was 3.6 (Table 1), and this CN value was close to that of 1 before and after impregnation (4.0 and 4.2, respectively), indicating that the average nuclearity of the Mo cluster was almost retained after H2-activation. The XAFS (Fig. 1A(e)), XANES (Fig. 1A(e), inset), and FT-EXAFS spectra (Fig. 1B(e)) of Mo foil were quite different from those of 1/HY after H2-activation, which also demonstrates that the impregnation of 1 on HY prevented the aggregation of 1 after activation.
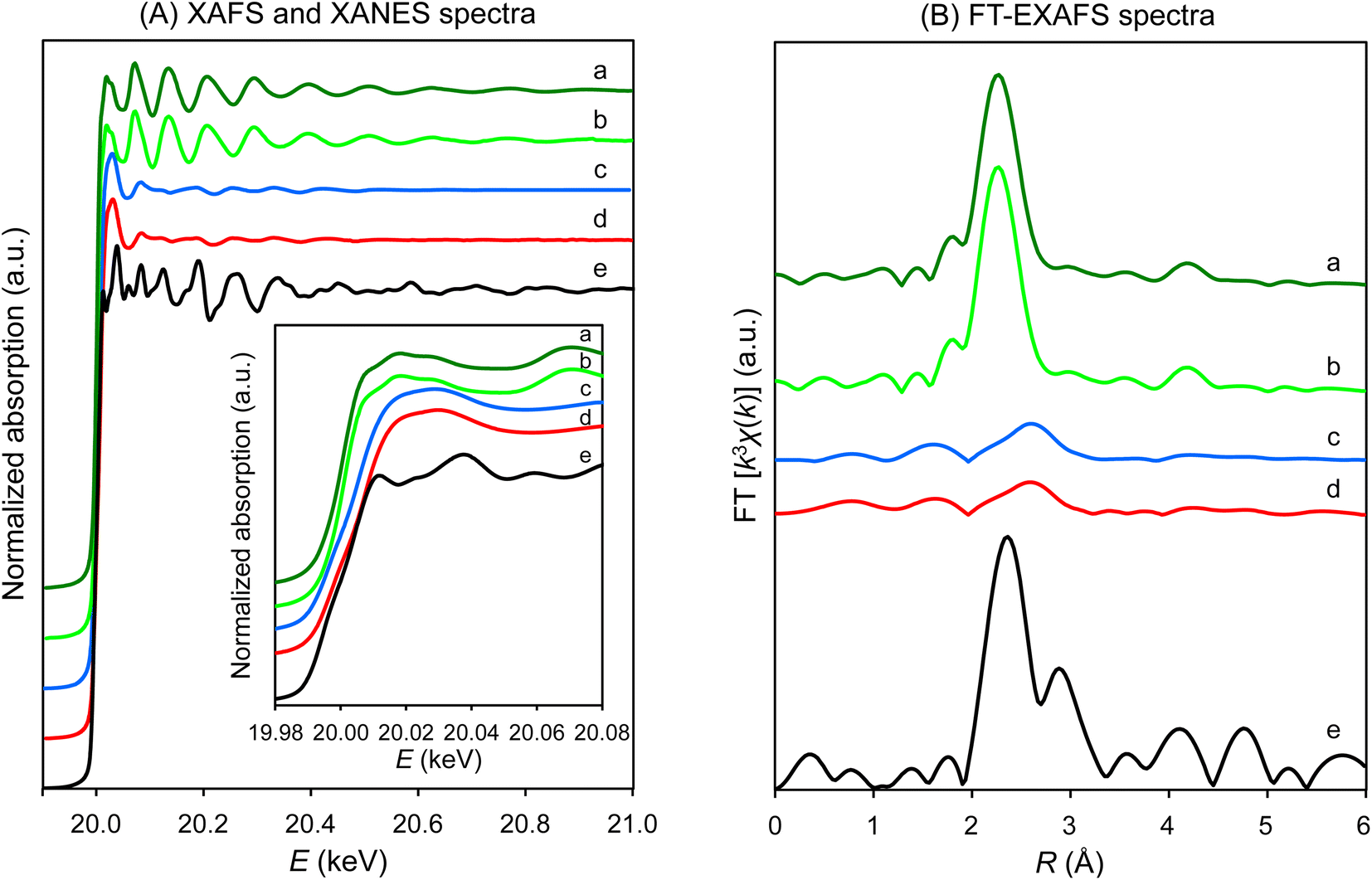 | ||
| Fig. 1 (A) XAFS spectra of (H3O)2[(Mo6Cl8)Cl6]·6H2O (1). The inset shows the XANES region. (B) FT-EXAFS spectra of 1: (a) as prepared, (b) 1/HY after impregnation, (c) 1/HY after H2-activation, and (d) 1/HY after NH3-synthesis. Spectrum of Mo foil from the Spring-8 BENTEN database (https://doi.org/10.48505/nims.2249) (e) is also shown. | ||
| Sample | Conditions | Shell | CNa | R (Å) | σ /10−2 (Å) | R f (%) |
|---|---|---|---|---|---|---|
| a Numbers in parentheses are errors estimated using the Hamilton ratio test with a significance level of 0.317.38 b The good fit of the observed and calculated data was also demonstrated by the EXAFS-fitting curves shown in Fig. S13(a)–(d). c Sample diluted with boron nitride was analyzed. | ||||||
| (H3O)2[(Mo6Cl8)Cl6]·6H2O (1) | As preparedc | Mo–Mo | 4.0 (fixed) | 2.65 (0.01) | 2.2 (0.7) | 0.14 |
| Mo–Cl | 5.0 (fixed) | 2.54 (0.01) | 1.3 (1.3) | |||
| (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HY | After impregnation | Mo–Mo | 4.2 (0.3) | 2.65 (0.01) | 2.2 (0.7) | 0.12 |
| Mo–Cl | 5.3 (0.2) | 2.54 (0.01) | 1.3 (1.3) | |||
| After H2-activation | Mo–Mo | 3.6 (0.8) | 2.84 (0.01) | 7.0 (0.8) | 3.2 | |
| Mo–O | 1.3 (0.5) | 2.04 (0.02) | 5.8 (3.2) | |||
| After NH3-synthesis | Mo–Mo | 3.6 (0.7) | 2.83 (0.01) | 7.7 (0.6) | 2.4 | |
| Mo–O | 1.0 (0.3) | 2.04 (0.02) | 3.5 (3.3) | |||
The morphologies of the HY-supported 1 after impregnation and H2-activation were observed using high-magnification Cs-corrected scanning transmission electron microscopy (Cs-STEM). The results are shown in Fig. S2, S3, and S11(a), (b).† In the STEM images after impregnation (Fig. S2†), each of the white particles corresponds to intact 1, as the XAFS confirmed the retention of the structure of 1 (see above). The average particle size of 9.7 Å (Fig. S11a†) was smaller than the molecular size of 1 including the chloride ligands (ca. 11 Å), which is attributed to the lower intensity of Cl than that of Mo, since the clarity of the STEM image is proportional to the square of the atomic weight (36.5 for Cl vs. 96.0 for Mo).18 Although we tried to obtain a clear atomic image of the Mo clusters, it was impossible because of the fluctuations of the clusters under high energy electron irradiations. The size of the zeolite micropore (7.5 Å) was smaller than that of 1, and the particles on the edge of the sample projected into the black area (Fig. S2b†). These results show that 1 was not embedded in the micropore after impregnation. After H2-activation, the STEM average particle size decreased from 9.7 to 7.9 Å (Fig. S11b†), which was close to the size of the Mo6 cluster (ca. 7 Å), and this decrease is attributed to the removal of chloride ligands by activation. The STEM image shows that, except for some large particles overlapping a stripe, the particles were observed between pore stripes or on the edge of a stripe (Fig. S3a†). The pore volume measurements also confirmed the embedding of Mo particles in the micropores after activation (Table S2† and description therein). These results indicate that the encapsulation of the metal cluster in the zeolite pore after activation prevented the aggregation of the cluster and retained its average particle size by the interaction of the cluster with silanols in the pore.19 In spite of some reports on supported molybdenum nitride and carbide clusters,20,21 there have been no reports on supported molybdenum metal clusters.
We also analyzed the HZSM5- and MCM41-supported clusters. The XAFS and STEM of 1/HZSM5 after H2-activation showed the formation of body-centered cubic (bcc) structured large Mo particles (Fig. S12Bc†) with an average particle size of 37 Å (Fig. S11e†). All the particle sizes (>10 Å) were larger than the zeolite pore size (ca. 5.5 Å). These results indicate that the Mo6 metal cluster formed by activation was not encapsulated in the small pore but was aggregated to form large bcc-Mo metal particles outside the pore. Conversely, the MCM41-supported cluster after activation exhibited a smaller CN of Mo–Mo (3.0) in the XAFS (Table S3†) and a smaller STEM average particle size (5.5 Å) (Fig. S11h†) than the HY-supported cluster. All the particles were smaller (<20 Å) (Fig. S11h†) than the MCM41 pore size (ca. 24 Å) and were observed on the edge of a stripe (Fig. S9†). These results suggest that the Mo6 metal cluster was converted to a smaller cluster inside the large mesopore. It is reported that H2-activation of a silica-supported dinuclear Mo complex and its ligand elimination causes coordination of silanol-oxygen atoms to the Mo atoms and cleavage of the Mo–Mo bond.22 The reduction in nuclearity of the Mo6 metal cluster in the MCM41 mesopore can be explained in the same way. In contrast, the micropore of HY just fits the Mo6 metal cluster, and therefore, the cluster is embedded without decomposition even when silanol-oxygen atoms coordinate to the Mo atoms. Thus, the size of the Mo metal cluster after H2-activation depends on the pore size of the support, and HY with a pore size of ca. 7.5 Å is suitable for the retention of the size of the Mo6 cluster of 1.23
Catalytic performance
The H2-activated clusters on the three supports were applied to ammonia synthesis. After the preparation of the H2-activated clusters, they were subsequently subjected to the reaction of a mixture of N2 and H2 with a flow ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 at 400 °C and 1.0 MPa (absolute pressure) without exposure to air. Ammonia was continuously formed for 8 h after the start of the reaction for the three supports (Fig. 2). On all these three supports, the clusters after ammonia synthesis showed very similar elemental analysis data (Table S1†), XAFS results (Fig. 1, Table 1, Fig. S12, and Table S3†), and STEM data (Fig. S4, S7, S10, S11c, S11f, and S11i†) to those after H2-activation. Thus, the structures of the cluster catalysts were stable during the ammonia synthesis.
:3 at 400 °C and 1.0 MPa (absolute pressure) without exposure to air. Ammonia was continuously formed for 8 h after the start of the reaction for the three supports (Fig. 2). On all these three supports, the clusters after ammonia synthesis showed very similar elemental analysis data (Table S1†), XAFS results (Fig. 1, Table 1, Fig. S12, and Table S3†), and STEM data (Fig. S4, S7, S10, S11c, S11f, and S11i†) to those after H2-activation. Thus, the structures of the cluster catalysts were stable during the ammonia synthesis.
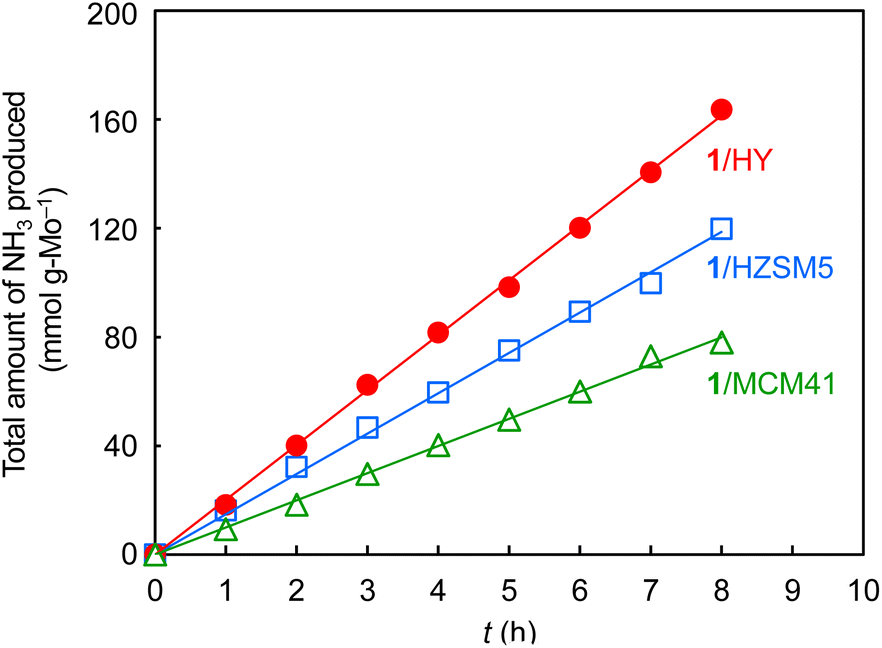 | ||
| Fig. 2 Catalytic performance of ammonia synthesis at 400 °C and 1.0 MPa (absolute pressure) using (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HY, (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HZSM5, and (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/MCM41 as precursors. Catalyst amount, 0.2 g; N2/H2 (1/3-mixture) flow rate, 60 mL min−1. | ||
Table 2 lists ammonia synthesis rates of various supported Mo catalysts at 400 °C. In comparison with the ammonia synthesis rates of H2-activated 1/MCM41 (10.2 mmol gMo−1 h−1, entry 1) and 1/HZSM5 (14.7 mmol gMo−1 h−1, entry 2), that of activated 1/HY (20.5 mmol gMo−1 h−1, entry 3) was higher at an absolute pressure of 1.0 MPa. This indicates that the Mo cluster with an averaged structure of Mo6 was most effective for ammonia synthesis. As Table S4† shows, the cluster on HY after H2-activation adsorbed more ammonia (0.58 cm3 gcat−1) than those on HZSM5 or MCM41 (0.39 and 0.42 cm3 gcat−1, respectively), and hence had the largest number of catalytically-active Mo atoms, which could be a cause of the higher ammonia synthesis rate. The smaller number of active sites of the larger-sized Mo cluster on HZSM5 is attributable to the embedding of some Mo atoms inside the large cluster, while that of the smaller-sized cluster on MCM41 is ascribed to the larger ratio of silica-coordinated Mo atoms, as confirmed by the higher CN of Mo–O (2.0) than that for the cluster on HY (1.3) in the XAFS analysis (Tables 1 and S3†).24 It is reported in the case of carbon-supported Ru catalysts that a suitable subnanometer-sized metal cluster shows higher activity than larger- and smaller-sized metal particles.25Table 2 also shows that, when the pressure was increased to 2.0 MPa, the rate of the activated 1/HY increased by about twofold (37.1 mmol gMo−1 h−1, entry 4). This rate was significantly higher than those of previously-reported supported Mo catalysts at the same reaction pressure even when different weight hourly space velocity (WHSV) values are considered: a silica-supported single-metal catalyst prepared from Mo(CBut)(Np)3 (6.8 mmol gMo−1 h−1, entry 5)26 and HZSM5-supported MoNx (4.3 mmol gMo−1 h−1, entry 6) as well as MoCx (4.5 mmol gMo−1 h−1, entry 7) prepared by nitridation and carbonization of MoO3, respectively.21 The lower rate of supported MoNx can be attributed to the weaker N2 dissociation ability of the nitrided Mo surface than that of the metallic Mo surface.27 Furthermore, our supported Mo metal clusters prepared from 1 have advantages in terms of the stability of the precursor in air and capability of ammonia synthesis after simple H2-activation using the same reaction tube, in contrast to the use of highly air-sensitive Mo(CBut)(Np)3 as a precursor or the need to transfer the sample from a quartz tube for air-calcination and succeeding nitridation or carbonization to a metal tube for ammonia synthesis under pressurized conditions.
| Entry | Catalyst (precursor) | Mo ratio (wt%) | NH3 yielda (mmol gMo−1 h−1) | Reaction pressure | WHSV (mL gcat−1 h−1) | Ref. |
|---|---|---|---|---|---|---|
| a The experiments were performed at least three times, and the values in parentheses are standard deviations. b Absolute pressure. c SiO2/Al2O3 molar ratio = 70 (Si/Al molar ratio = 35). | ||||||
| 1 | (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/MCM41 | 2.36 | 10.2 (0.7) | 1.0b MPa | 18000 |
This work |
| 2 | (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HZSM5 | 2.36 | 14.7 (1.1) | 1.0b MPa | 18000 |
This work |
| 3 | (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HY | 2.36 | 20.5 (0.7) | 1.0b MPa | 18000 |
This work |
| 4 | (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HY | 2.36 | 37.1 (1.2) | 2.0b MPa | 18000 |
This work |
| 5 | Mo(CBut)(Np)3/SiO2 |
2.0 | 6.8 | 2.0 MPa | 12000 |
26 |
| 6 | MoNx/HZSM5c | 2.17 | 4.3 | 2.0 MPa | 9000 | 21 |
| 7 | MoCx/HZSM5c | 2.17 | 4.5 | 2.0 MPa | 9000 | 21 |
The turnover frequency (TOF) of activated 1/HY (0.006, Table S4†) was 30% of that of a commercially-used Ru catalyst, Ba–Ru/C (0.02), based on the number of active metal sites.28 As Table S5† shows, whereas the rate of the activated 1/HY per metal weight (20.5 mmol gMo−1 h−1, entry 3) was higher than or comparable to that of an Fe catalyst (<16 mmol gFe−1 h−1, entry 8) and about a quarter of that of an Ru catalyst (91 mmol gRu−1 h,1 entry 9). The rate per catalyst weight (0.483 mmol gcat−1 h−1, entry 3) was much lower than those of the Fe (14 mmol gcat−1 h−1, entry 8) and Ru catalysts (8.2 mmol gcat−1 h−1, entry 9), under similar reaction conditions. The rate of 1/HY per catalyst weight allows a lot of room for increase by improvements such as the addition of promoters for industrialization.
The catalytic behavior of activated 1/HY with the highest synthesis rate among the supported Mo catalysts (Table 2, entry 3) was further investigated. As shown in Fig. S14,† the synthesis rate (20.5 mmol gMo−1 h−1) remained constant for 258 h at 1.0 MPa, indicating that the activated supported cluster is highly durable for long-term ammonia synthesis. As shown in the Arrhenius plots (Fig. S15†), the apparent activation energy of activated 1/HY (89 kJ mol−1) was lower than those of activated 1/HZSM5 (92 kJ mol−1) and 1/MCM41 (110 kJ mol−1), indicating that activated 1/HY is most effective for ammonia synthesis at lower temperatures. At a higher reaction pressure (5.0 MPa), 1/HY afforded ammonia catalytically even at 200 °C with a turnover number of more than 4 per Mo-atom (Fig. 3). Various catalysts with ammonia synthesis activity at 200 °C have been reported, with TOF values at around 1.0 MPa ranging in the orders of 10−3 to 10−4 s−1.29,30 In comparison with these values, the TOF of 1/HY at 5.0 MPa (2.4 × 10−5 s−1) (Fig. 3) was lower by one or two orders of magnitude. However, the ammonia synthesis rate of 1/HY was stable for at least as long as 520 h, while the stability of the rate at 200 °C (for up to 26–480 h) has been reported for only a few catalysts.7d,31 In the case of 1/HY, no pretreatment with a mixture of N2 and H2 at a higher temperature was necessary before the stable formation of ammonia at 200 °C, which is indicative of the high and sustainable N2 dissociation ability of the supported Mo metal cluster at low temperatures.
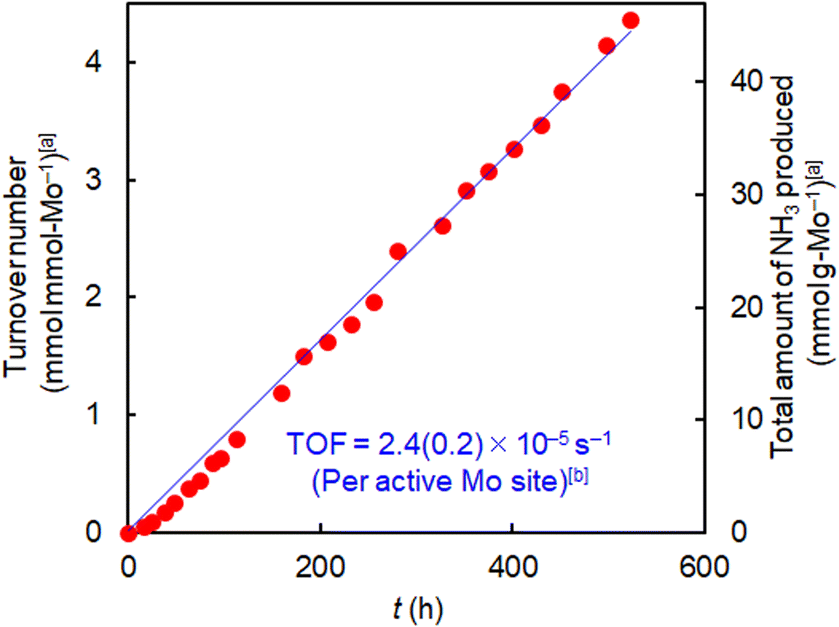 | ||
| Fig. 3 Ammonia synthesis using (H3O)2[(Mo6Cl8)Cl6]·6H2O (1)/HY at 200 °C and 5.0 MPa (absolute pressure). Catalyst amount, 0.2 g; N2/H2 (1/3-mixture) flow rate, 60 mL min−1. [a] Per Mo included in the catalysts. [b] Per NH3-adsorbing Mo (see Table S4†). | ||
Kinetic studies
To investigate the reaction mechanism for ammonia synthesis over the supported Mo metal clusters, kinetic and density functional theory (DFT) studies were carried out. In the kinetic studies, reaction orders with respect to N2, H2, and NH3 for the clusters were measured (Fig. 4). The Mo metal cluster on HY as well as HZSM5 and MCM41 showed N2 and H2 reaction orders of around 0.5–0.6 and 1.0, respectively. This suggests that the reaction of dissociated N with H2 forming N–H bonds is the rate-determining step (RDS), while the N2 dissociation step is no longer the RDS.32 In contrast, the RDS of the supported Mo catalysts reported previously (listed in entries 5 and 6 in Table 2) is ascribed to N2 dissociation,21,26 as for most conventional catalysts.33 Low N2 reaction orders of around 0.5 have been reported for catalysts with electrides,28,31a,34 oxyhydrides,35 a nitride-hydride,31c and alkali- or alkaline earth-hydrides,29a,36 and the favorable N2 dissociation has been attributed to strong electron donation by these catalyst components to the NN bond. Our supported Mo metal clusters achieved low N2 reaction orders without using such strongly electron-donating components.
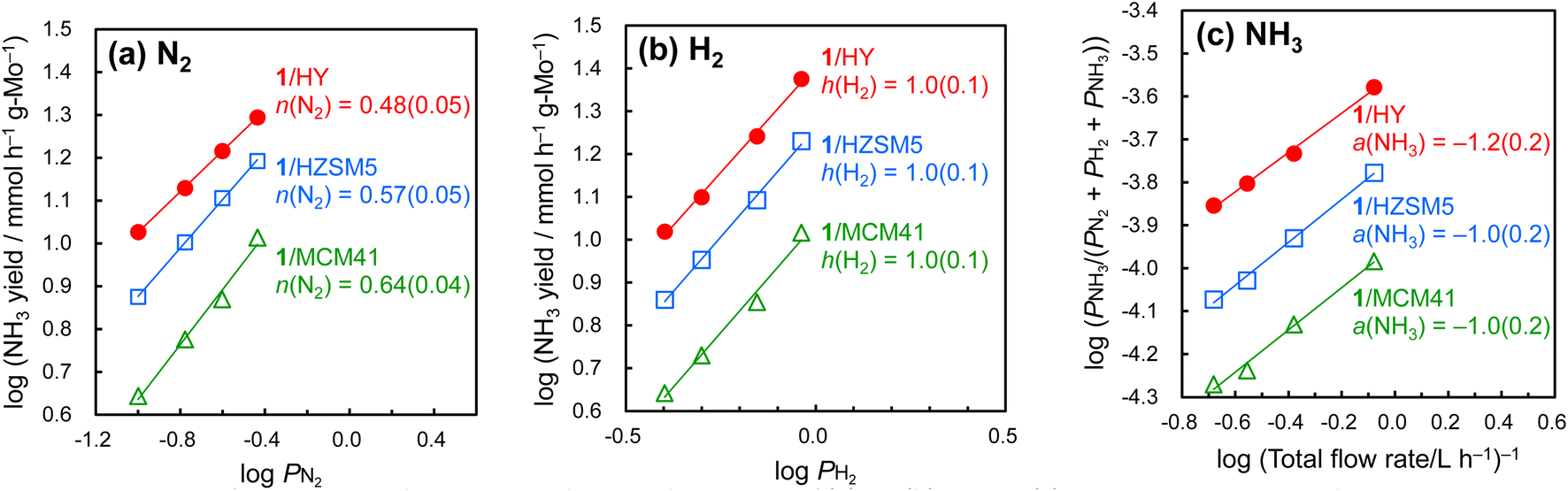 | ||
| Fig. 4 Dependence of ammonia synthesis rate on the partial pressures of (a) N2, (b) H2, and (c) NH3 over H2-activated clusters on various supports at 400 °C and 1 MPa. | ||
The RDS of ammonia synthesis over the HY-supported cluster was investigated by comparing the experimental reaction rates with calculated ones. When the synthesis reaction is assumed to obey the Langmuir–Hinshelwood (dissociative) mechanism, in which cleavage of the N–N bond takes place before the formation of N–H bonds,33a the eight elementary reaction steps are established (eqn (S6)–(S13)†). Among these eight steps, the dissociation of N2 or the formation of NH, NH2, or NH3 (eqn (S9)–(S12)†) can be attributed to the RDS because of its high activation energy, and the four corresponding calculated reaction rates can be deduced (eqn (S14)–(S17)†).29c,34b These equations were fitted to a set of experimental rates obtained under various reaction gas ratios, using a least-squares method. Fig. S17† shows the best-fit of the calculated rates to the experimental ones. When the dissociation of N2 was assumed to be the RDS, the fitting was poor with a negative value of determination coefficient. In contrast, when the formation of NH, NH2, and NH3 was assumed to be the RDS, larger determination coefficient values (0.71–0.99) were obtained. These observations suggest that the RDS of ammonia synthesis over the HY-supported cluster is the formation step of NH, NH2, or NH3 rather than the dissociation step of N2. This conclusion is also supported by the DFT results shown below.
DFT calculations
The reaction mechanism for the HY-supported cluster was studied in more detail using DFT calculations. As suggested by the XAFS and STEM analysis, after H2-activation and ammonia synthesis, the molybdenum cluster sizes were almost the same as that of hexanuclear precursor 1. Thus, we assumed that the cluster during the ammonia synthesis also has a hexanuclear structure. We examined various local structures of HY zeolite to accommodate the Mo6 metal cluster. When the cluster was located on the two adjacent four-membered oxygen rings with three Mo–oxygen interactions, the most stable structure was obtained (see Fig. S18 and Table S6† for details). Then, this model was used for further investigation of the reaction intermediates (Fig. S19†) and mechanism.We determined several reaction pathways for ammonia synthesis through dissociative and associative mechanisms37 (Fig. S20† and description therein), followed by microkinetic analysis using the potential energies of intermediates and transition states of these pathways (see Fig. S21† and description therein). Fig. 5 shows the dominant reaction pathway determined by microkinetic analysis. According to this, an N2 molecule is first adsorbed on an Mo atom in a terminal end-on mode (A1), followed by a configurational change of the Mo-bonded N into the μ2-bridging adsorption mode (A2). Then, the N–N bond cleavage by participation of three Mo atoms takes place to afford two μ2-bridging N atoms (A3), followed by a configurational change of one N atom from μ2- to μ3-bridging (B1). Here, we use A and B notations to distinguish the pathway depending on the coordination mode of the N atom (μ2- or μ3-bridging) that is not involved in the first hydrogenation step (see Fig. S20† for details). The first H2 molecule is subsequently introduced by dissociative adsorption (B2), followed by the transfer of one H to the μ2-bridging N to form a μ2-bridging NH (B3). After the migration of the remaining H atom (B3 → B3′), the second H transfers to NH to yield NH2 (B4). After a minor configurational change of NH2 from μ2-bridging to the terminal site (B4 → B5), the second H2 molecule is introduced by dissociative adsorption (B6). Then, the third H transfer to the NH2 group affords a terminal NH3 (B7), followed by release of the first NH3 molecule (B8). After that, configurational change of the μ3-bridging N to a μ2-bridging mode takes place (A9). Further, after the migration of the H atom (A9 → A9′), the N atom accepts the fourth H atom to form a μ2-bridging NH (A10). After the third introduction of the H2 molecule (A11), the fifth H transfer gives a μ2-bridging NH2 (A12). Then, after the configurational change of the μ2-bridging NH2 to terminal NH2 (A12 → A13), the sixth H transfer occurs to afford a terminal NH3 (A14). Finally, the second NH3 molecule is released. In comparison with the energy barrier of N2 dissociation (119 kJ mol−1 for TSA2A3), the barriers of the first and second NH formation (115 kJ mol−1 for TSB2B3 and 119 kJ mol−1 for TSA9′A10, respectively) are comparable, and those for the first and second NH2 formation (156 kJ mol−1 for TSB3′B4 and 163 kJ mol−1 for TSA11A12, respectively) and the first and second NH3 formation (177 kJ mol−1 for TSB6B7 and 173 kJ mol−1 for TSA13A14, respectively) are higher. Thus, the RDS is not N–N bond cleavage but N–H bond formation, which is consistent with the experimental results. These results demonstrate that the angstrom-size Mo cluster prefers the dissociative pathway, in which the N–N bond cleavage promoted by the Mo6 multinuclear structure is not the RDS.
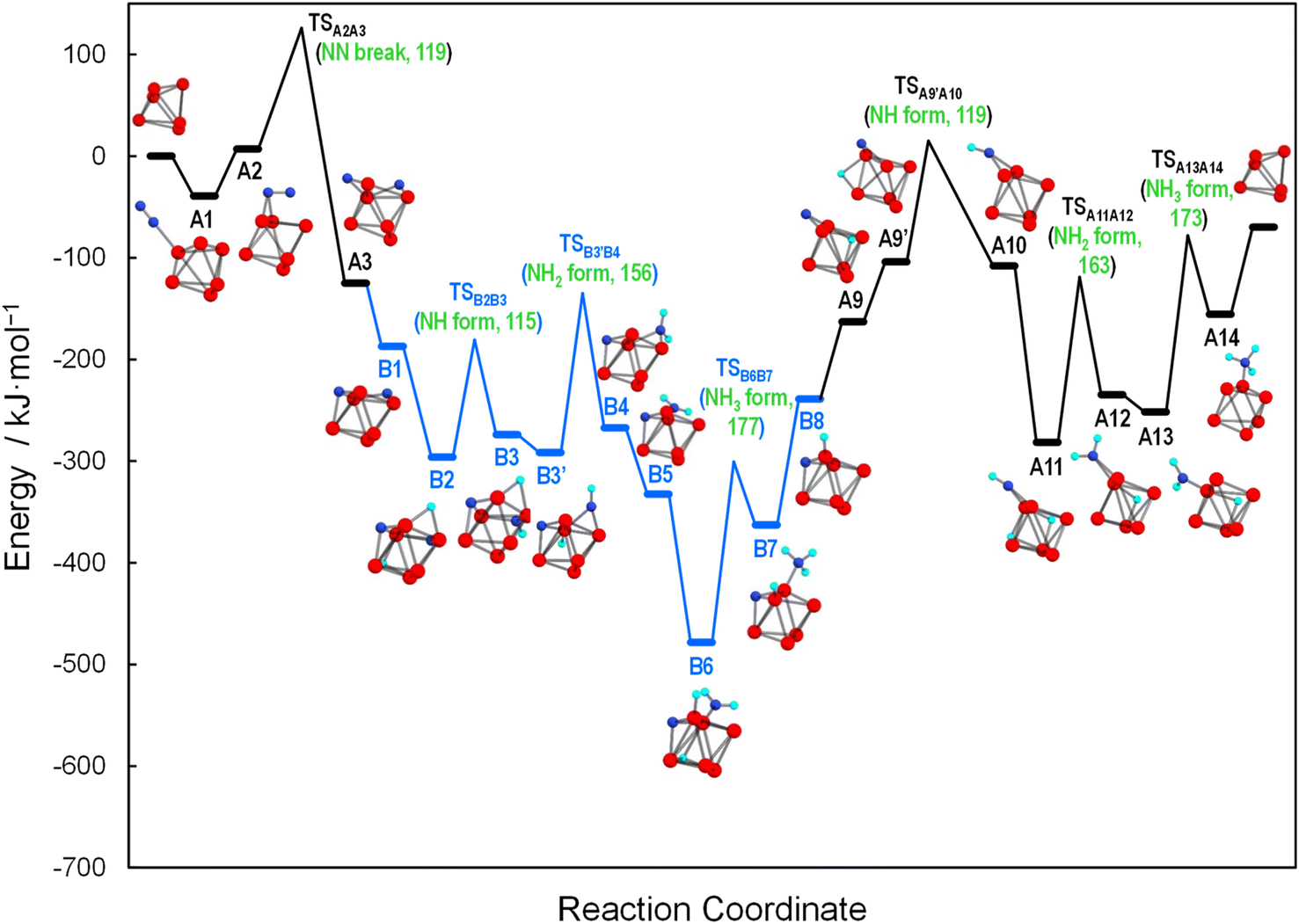 | ||
| Fig. 5 Potential energy profiles and structural change along the dominant reaction pathway determined by microkinetic analysis. The HY zeolite and gaseous N2, H2, and NH3 are omitted for clarity. | ||
Conclusions
In summary, supported angstrom-scale Mo metal clusters were prepared by the impregnation of a hexanuclear molecular halide cluster on various porous supports and subsequent activation with H2 and were characterized by XAFS and STEM analysis. When HY zeolite was used as the support, the Mo cluster size of precursor 1 was retained even after activation. The resulting angstrom-size metal cluster catalyzed ammonia synthesis from N2 and H2. The catalytic activity was highly durable even at 200 °C. The NN bond was effectively cleaved by the cooperation of multiple Mo sites, and the RDS shifted from N2 dissociation to N–H formation, as confirmed by kinetic and computational studies. This work has expanded the scope of the application of a halide cluster for catalysis and developed a novel ultra-small Mo metal cluster catalyst for efficient ammonia synthesis, based on the multinuclearity of a metal cluster of suitable size.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Satoshi Kamiguchi: conceptualization (lead), data curation (lead), formal analysis (lead), funding acquisition (lead), investigation (lead), methodology (lead), project administration (lead), supervision (lead), validation (lead), and writing – original draft (lead). Kiyotaka Asakura: formal analysis (supporting), investigation (supporting), resources (lead), validation (supporting), and writing – original draft (supporting) on XAFS analysis. Tamaki Shibayama: formal analysis (supporting), investigation (supporting), resources (lead), validation (supporting), and writing – original draft (supporting) on STEM measurements. Tomoko Yokaichiya: formal analysis (supporting), investigation (lead), and writing – original draft (supporting) on DFT calculations. Tatsushi Ikeda: formal analysis (lead), investigation (supporting), and writing – original draft (supporting) on DFT calculations. Akira Nakayama: data curation (lead), funding acquisition (lead), project administration (lead), supervision (lead), validation (lead), and writing – original draft (lead) on DFT calculations. Ken-ichi Shimizu: formal analysis (supporting), resources (lead), investigation (supporting), validation (supporting), and writing – original draft (supporting) on catalyst analysis. Zhaomin Hou: conceptualization (supporting), project administration (supporting), supervision (supporting), validation (supporting), and writing – original draft (supporting).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by KAKENHI grant 19K05515 from the JSPS. This work was also supported by the Joint Usage/Research Center for Catalysis, Hokkaido University (Proposal No. 16B1016, 18B1012, 19B1023, and 20B1038). The calculations were partly performed on supercomputers at RCCS (Okazaki), RIIT (Kyushu Univ.), ACCMS (Kyoto Univ.), and the Center for Computational Materials Science, Institute for Materials Research (Tohoku University, Proposal No. 202012-SCKXX-0002). The XAFS experiments were conducted at the BLS51 and BL11S2 of Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation, Aichi, Japan (Proposal No. 201602001, 201905002, and 202002012). The STEM measurements were supported by the “Nanotechnology Platform Program” at Hokkaido University (A-19-HK-0055 and A-20-HK-0013). The authors thank the technical division of the Institute for Catalysis (Hokkaido University) for manufacturing glass cells for the XAFS measurements. The authors also thank the Materials Characterization Support Unit, RIKEN CEMS for the elemental analysis, and the MC Evolve Technologies Corporation for measurements of pore volume and BET surface area.Notes and references
- (a) J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a century of ammonia synthesis changed the world, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS; (b) J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, Wiley, Hoboken, 2014 Search PubMed.
- (a) A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, Ammonia for hydrogen storage: challenges and opportunities, J. Mater. Chem., 2008, 18, 2304–2310 RSC; (b) F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Ammonia as a possible element in an energy infrastructure: catalysts for ammonia decomposition, Energy Environ. Sci., 2012, 5, 6278–6289 RSC; (c) J. Guo and P. Chen, Interplay of Alkali, Transition Metals, Nitrogen, and Hydrogen in Ammonia Synthesis and Decomposition Reactions, Acc. Chem. Res., 2021, 54, 2434–2444 CrossRef CAS PubMed.
- D. Erdemir and I. Dincer, A perspective on the use of ammonia as a clean fuel: challenges and solutions, Int. J. Energy Res., 2021, 45, 4827–4834 CrossRef.
- L. E. Apodaca, Mineral Commodity Summaries, U.S. Department of the Interior, U.S. Geological Survey, 2022, pp. 126–127, available at https://pubs.usgs.gov/periodicals/mcs2022/mcs2022.pdf Search PubMed.
- (a) A. Mittasch, Early Studies of Multicomponent Catalysts, Adv. Catal., 1950, 2, 81–104 Search PubMed; (b) C. W. Hooper, Ammonia synthesis: commercial practice, in Catalytic Ammonia Synthesis, Fundamentals and Practice, ed. J. R. Jennings, Springer, US, 1991, pp. 253–283 Search PubMed.
- C. W. Hooper, Ammonia synthesis: commercial practice, in Catalytic Ammonia Synthesis, Fundamentals and Practice, ed. J. R. Jennings, Springer, US, 1991, pp. 303–348 Search PubMed.
- (a) Y. V. Larichev, D. A. Shlyapin, P. G. Tsyrul'nikov and V. I. Bukhtiyarov, Comparative Study of Rubidium and Cesium as Promoters in Carbon-supported Ruthenium Catalysts for Ammonia Synthesis, Catal. Lett., 2008, 120, 204–209 CrossRef CAS; (b) C. Fernández, C. Sassoye, D. P. Debecker, C. Sanchez and P. Ruiz, Effect of the size and distribution of supported Ru nano particles on their activity in ammonia synthesis under mild reaction conditions, Appl. Catal., A, 2014, 474, 194–202 CrossRef; (c) C. Fernández, C. Pezzotta, E. M. Gaigneaux, N. Bion, D. Duprez and P. Ruiz, Disclosing the synergistic mechanism in the catalytic activity of different-sized Ru nanoparticles for ammonia synthesis at mild reaction conditions, Catal. Today, 2015, 251, 88–95 CrossRef; (d) M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Solid solution for catalytic ammonia synthesis from nitrogen and hydrogen gases at 50 °C, Nat. Commun., 2020, 11, 2001 CrossRef CAS PubMed; (e) K. H. R. Rouwenhorst, A. G. J. Van der Ham and L. Lefferts, Beyond Haber-Bosch: the renaissance of the Claude process, Int. J. Hydrogen Energy, 2021, 46, 21566–21579 CrossRef CAS.
- C. C. Ahn and O. L. Krivanek, EELS Atlas: A Reference Collection of Electron Energy Loss Spectra Covering All Stable Elements, Gatan, Inc., 1983 Search PubMed.
- (a) K. Aika, Role of alkali promoter in ammonia synthesis over ruthenium catalysts—Effect on reaction mechanism, Catal. Today, 2017, 286, 14–20 CrossRef CAS; (b) J. Humphreys, R. Lan and S. Tao, Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS; (c) H. Hosono and M. Kitano, Advances in Materials and Applications of Inorganic Electrides, Chem. Rev., 2021, 121, 3121–3185 CrossRef CAS PubMed.
- (a) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Dinitrogen Cleavage and Hydrogenation by a Trinuclear Titanium Polyhydride Complex, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed; (b) T. Shima, G. Luo, S. Hu and Z. Hou, Experimental and Computational Studies of Dinitrogen Activation and Hydrogenation at a Tetranuclear Titanium Imide/Hydride Framework, J. Am. Chem. Soc., 2019, 141, 2713–2720 CrossRef CAS; (c) T. Shima, J. Yang, G. Luo, Y. Luo and Z. Hou, Dinitrogen Activation and Hydrogenation by C5Me4SiMe3-Ligated Di- and Trinuclear Chromium Hydride Complexes, J. Am. Chem. Soc., 2020, 142, 9007–9016 CrossRef CAS.
- J. B. Howard and D. C. Rees, Structural Basis of Biological Nitrogen Fixation, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- (a) I. Dance, Elucidating the Coordination Chemistry and Mechanism of Biological Nitrogen Fixation, Chem.–Asian J., 2007, 2, 936–946 CrossRef CAS PubMed; (b) J. Kästner and P. E. Blöchl, Ammonia Production at the FeMo Cofactor of Nitrogenase: Results from Density Functional Theory, J. Am. Chem. Soc., 2007, 129, 2998–3006 CrossRef PubMed.
- (a) S. Kamiguchi, S. Nagashima and T. Chihara, Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes, Metals, 2014, 4, 84–107 CrossRef; (b) S. Nagashima, S. Kamiguchi and T. Chihara, Catalytic Reactions over Halide Cluster Complexes of Group 5–7 Metals, Metals, 2014, 4, 235–313 CrossRef.
- S. Kamiguchi, T. Mori, M. Watanabe, A. Suzuki, M. Kodomari, M. Nomura, Y. Iwasawa and T. Chihara, Retention of the octahedral metal framework of Nb and Mo halide clusters in catalytic decomposition of phenyl acetate to phenol and ketene, J. Mol. Catal. A, 2006, 253, 176–186 CrossRef CAS.
- S. Nagashima, S. Kamiguchi, S. Ohguchi and T. Chihara, Gas-Phase Alkylation of Pyridine and Phenol with Alcohols Over Halide Clusters of Group 5–7 Transition Metals as Solid Acid Catalysts, J. Cluster Sci., 2011, 22, 647–660 CrossRef CAS.
- (a) A. Logadottir, T. H. Rod, J. K. Nørskov, B. Hammer, S. Dahl and C. J. H. Jacobsen, The Brønsted–Evans–Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts, J. Catal., 2001, 197, 229–231 CrossRef CAS; (b) C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center, Science, 2003, 301, 76–78 CrossRef CAS PubMed; (b) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed; (c) Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- S. J. Pennycook, Z-contrast stem for materials science, Ultramicroscopy, 1989, 30, 58–69 CrossRef.
- D. Hou, L. Grajciar, P. Nachtigall and C. J. Heard, Origin of the Unusual Stability of Zeolite-Encapsulated Sub-Nanometer Platinum, ACS Catal., 2020, 10, 11057–11068 CrossRef CAS.
- W. Ding, S. Li, G. D. Meitzner and E. Iglesia, Methane Conversion to Aromatics on Mo/H-ZSM5: Structure of Molybdenum Species in Working Catalysts, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS.
- N. Liu, L. Nie, N. Xue, H. Dong, L. Peng, X. Guo and W. Ding, Catalytic Ammonia Synthesis over Mo Nitride/ZSM-5, ChemCatChem, 2010, 2, 167–174 CrossRef CAS.
- Y. Iwasawa and M. Yamaguchi, New SiO2-Attached Mo-Pair Catalysts–preparations, Surface Structures, and Chemical Nature, J. Catal., 1983, 82, 373–381 CrossRef CAS.
- C. Aydin, J. Lu, M. Shirai, N. D. Browning and B. C. Gates, Ir6 Clusters Compartmentalized in the Supercages of Zeolite NaY: Direct Imaging of a Catalyst with Aberration-Corrected Scanning Transmission Electron Microscopy, ACS Catal., 2011, 1, 1613–1620 CrossRef CAS.
- (a) A. Beniya, S. Higashi, N. Ohba, R. Jinnouchi, H. Hirata and Y. Watanabe, CO oxidation activity of non-reducible oxide-supported mass-selected few-atom Pt single-clusters, Nat. Commun., 2020, 11, 1888 CrossRef CAS PubMed; (b) A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity, Nat. Chem., 2013, 11, 775 CrossRef.
- (a) Y. Zhou, Q. Sai, Z. Tan, C. Wang, X. Wang, B. Lin, J. Ni, J. Lin and L. Jiang, Highly efficient subnanometer Ru-based catalyst for ammonia synthesis via an associative mechanism, Chin. J. Chem. Eng., 2022, 43, 177–184 CrossRef CAS; (b) L. Lin, Y.-F. Jiang, T. Zhang, H. Cai, Y. Zhou, B. Lin, X. Lin, Y. Zheng, L. Zheng, X. Wang, C.-Q. Xu, C. Au, L. Jiang and J. Li, Size sensitivity of supported Ru catalysts for ammonia synthesis: from nanoparticles to subnanometric clusters and atomic clusters, Chem, 2022, 8, 749–768 CrossRef.
- L. M. Azofra, N. Morlanes, A. Poater, M. K. Samantaray, B. Vidjayacoumar, K. Albahily, L. Cavallo and J.-M. Basset, Single-Site Molybdenum on Solid Support Materials for Catalytic Hydrogenation of N2-into-NH3, Angew. Chem., Int. Ed., 2018, 57, 15812–15816 CrossRef CAS PubMed.
- J. Zhao, C. Cui, H. Wang, J. Han, X. Zhu and Q. Ge, Anisotropic N-Modification of the Mo4 Ensemble for Efficient Ammonia Synthesis on Molybdenum Nitrides, J. Phys. Chem. C, 2020, 124, 616–624 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- (a) W. Gao, P. Wang, J. Guo, F. Chang, T. He, Q. Wang, G. Wu and P. Chen, Barium Hydride-Mediated Nitrogen Transfer and Hydrogenation for Ammonia Synthesis: A Case Study of Cobalt, ACS Catal., 2017, 7, 3654–3661 CrossRef CAS; (b) P. Wang, H. Xie, J. Guo, Z. Zhao, X. Kong, W. Gao, F. Chang, T. He, G. Wu, M. Chen, L. Jiang and P. Chen, The Formation of Surface Lithium–Iron Ternary Hydride and its Function on Catalytic Ammonia Synthesis at Low Temperatures, Angew. Chem., Int. Ed., 2017, 56, 8716–8720 CrossRef CAS PubMed; (c) M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Self-organized Ruthenium–Barium Core–Shell Nanoparticles on a Mesoporous Calcium Amide Matrix for Efficient Low-Temperature Ammonia Synthesis, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS PubMed; (d) K. Sato, S. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, Barium Oxide Encapsulating Cobalt Nanoparticles Supported on Magnesium Oxide: Active Non-Noble Metal Catalysts for Ammonia Synthesis under Mild Reaction Conditions, ACS Catal., 2021, 11, 13050–13061 CrossRef CAS; (e) S. Miyahara, K. Sato, K. Tsujimaru, Y. Wada, Y. Ogura, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu and K. Nagaoka, Co Nanoparticle Catalysts Encapsulated by BaO−La2O3 Nanofractions for Efficient Ammonia Synthesis Under Mild Reaction Conditions, ACS Omega, 2022, 7, 24452–24460 CrossRef CAS PubMed; (f) Z. Li, Y. Lu, J. Li, M. Xu, Y. Qi, S.-W. Park, M. Kitano, H. Hosono, J.-S. Chen and T.-N. Ye, Multiple reaction pathway on alkaline earth imide supported catalysts for efficient ammonia synthesis, Nat. Commun., 2023, 14, 6373 CrossRef CAS PubMed; (g) K. Era, K. Sato, S. Miyahara, T. Naito, K. De Silva, S. Akrami, H. Yamada, T. Toriyama, T. Yamamoto, Y. Murakami, K. Aika, K. Inazu and K. Nagaoka, Catalytic Behavior of K-doped Fe/MgO Catalysts for Ammonia Synthesis Under Mild Reaction Conditions, ChemSusChem, 2023, 16, e202300942 CrossRef CAS PubMed.
- When the TOF at a different temperature (TOFT) was reported, the TOF at 200 °C (TOF200) was calculated using the ammonia synthesis rates at these temperatures (rT and r200) as follows: TOF200 = TOFT × (r200/rT).
- (a) Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama, M. Hara and H. Hosono, Efficient and Stable Ammonia Synthesis by Self-Organized Flat Ru Nanoparticles on Calcium Amide, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS; (b) W. Gao, S. Feng, H. Yan, Q. Wang, H. Xie, L. Jiang, W. Zhang, Y. Guan, H. Wu, H. Cao, J. Guo and P. Chen, In situ formed Co from a Co–Mg–O solid solution synergizing with LiH for efficient ammonia synthesis, Chem. Commun., 2021, 57, 8576–8579 RSC; (c) Y. Guan, W. Zhang, Q. Wang, C. Weidenthaler, A. Wu, W. Gao, Q. Pei, H. Yan, J. Cui, H. Wu, S. Feng, R. Wang, H. Cao, X. Ju, L. Liu, T. He, J. Guo and P. Chen, Barium chromium nitride-hydride for ammonia Synthesis, Chem Catal., 2021, 1, 1042–1054 CrossRef CAS; (d) M. Hattori, N. Okuyama, H. Kurosawa and M. Hara, Low-Temperature Ammonia Synthesis on Iron Catalyst with an Electron Donor, J. Am. Chem. Soc., 2023, 145, 7888–7897 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Electride support boosts nitrogen dissociation over ruthenium catalyst and shifts the bottleneck in ammonia synthesis, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- (a) K. Aika, M. Kumasaki, T. Oma, O. Kato, H. Matsuda, N. Watanabe, K. Yamazaki, A. Ozaki and T. Onishi, Support and Promoter Effect of Ruthenium Catalyst. III. Kinetics of Ammonia Synthesis over Various Ru Catalysts, Appl. Catal., A, 1986, 28, 57–68 CrossRef CAS; (b) F. Rosowski, A. Hornung, O. Hinrichsen, D. Herein, M. Muhler and G. Ertl, Appl. Catal., A, 1997, 151, 443–460 CrossRef CAS; (c) S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen and I. Chorkendorff, Ruthenium catalysts for ammonia synthesis at high pressures: preparation, characterization, and power-law kinetics, J. Catal., 2003, 214, 327–335 CrossRef CAS; (d) S. E. Siporin and R. J. Davis, Use of kinetic models to explore the role of base promoters on Ru/MgO ammonia synthesis catalysts, J. Catal., 2004, 225, 359–368 CrossRef CAS.
- (a) M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Essential role of hydride ion in ruthenium-based ammonia synthesis catalysts, Chem. Sci., 2016, 7, 4036–4043 RSC; (b) Y. Gong, J. Wu, M. Kitano, J. Wang, T.-N. Ye, J. Li, Y. Kobayashi, K. Kishida, H. Abe, Y. Niwa, H. Yang, T. Tada and H. Hosono, Ternary intermetallic LaCoSi as a catalyst for N2 Activation, Nat. Catal., 2018, 1, 178–185 CrossRef CAS; (c) H. Mizoguchi, M. Okunaka, M. Kitano, S. Matsuishi, T. Yokoyama and H. Hosono, Hydride-Based Electride Material, LnH2 (Ln = La, Ce, or Y), Inorg. Chem., 2016, 55, 8833–8838 CrossRef CAS PubMed.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Layer, K. Mitsuhara, K. Yamanaka, Y. Tamenori, C. Tassel, T. Yamamoto, T. Tanaka and H. Kageyama, Metal-Dependent Support Effects of Oxyhydride-Supported Ru, Fe, Co Catalysts for Ammonia Synthesis, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- P. K. Wang, F. Chang, W. B. Gao, J. P. Guo, G. T. Wu, T. He and P. Chen, Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- C. D. Zeinalipour-Yazdi, J. S. Hargreaves and C. R. A. Catlow, Low-T Mechanisms of Ammonia Synthesis on Co3Mo3N, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef CAS.
- W. C. Hamilton, Significance Tests on the Crystallographic R Factor, Acta Crystallogr., 1965, 18, 502–510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05447k |
| This journal is © The Royal Society of Chemistry 2024 |