
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition from vehicle to Grotthuss proton transfer in a nanosized flask: cryogenic ion spectroscopy of protonated p-aminobenzoic acid solvated with D2O†
Keisuke
Hirata
 acd,
Kyota
Akasaka
ab,
Otto
Dopfer
*de,
Shun-ichi
Ishiuchi
*acd and
Masaaki
Fujii
*abd
acd,
Kyota
Akasaka
ab,
Otto
Dopfer
*de,
Shun-ichi
Ishiuchi
*acd and
Masaaki
Fujii
*abd
aLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan. E-mail: mfujii@res.titech.ac.jp; ishiuchi.s.aa@m.titech.ac.jp
bSchool of Life Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan
cDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
dInternational Research Frontiers Initiative, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, 226-8503, Japan
eInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstrasse 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 19th January 2024
Abstract
Proton transfer (PT) is one of the most ubiquitous reactions in chemistry and life science. The unique nature of PT has been rationalized not by the transport of a solvated proton (vehicle mechanism) but by the Grotthuss mechanism in which a proton is transported to the nearest proton acceptor along a hydrogen-bonded network. However, clear experimental evidence of the Grotthuss mechanism has not been reported yet. Herein we show by infrared spectroscopy that a vehicle-type PT occurs in the penta- and hexahydrated clusters of protonated p-aminobenzoic acid, while Grotthuss-type PT is observed in heptahydrated clusters, indicating a change in the PT mechanism depending on the degree of hydration. These findings emphasize the importance of the usually ignored vehicle mechanism as well as the degree of hydration. It highlights the possibility of controlling the PT mechanism by the number of water molecules in chemical and biological environments.
Introduction
Proton transfer (PT) is one of the most ubiquitous reactions prevailing in aqueous solutions,1,2 proton-conducting oxides/polymers,3,4 metal–organic frameworks (MOFs),5,6 and biological macromolecules.7–9 Much experimental and computational effort has been devoted to unravelling the nature of PT. An elemental reaction mechanism is the direct transport of a solvated proton, such as H3O+ in water, which is called the vehicle mechanism.10 However, this mechanism cannot rationalize several important properties often observed for PT. For example, it predicts almost identical diffusion coefficients (D) for the hydrated proton (or H3O+) and H2O in aqueous solution because of their similar molecular size. However, Dproton is around five times higher than DH2O at room temperature.2 The anomalously high Dproton has been ascribed to another mechanism, named Grotthuss mechanism,11 first proposed more than 200 years ago.12,13 It involves sequential proton shuttling to an adjacent proton acceptor site along a hydrogen-bonded (H-bonded) water network. Calculations14,15 using classical and quantum molecular dynamics together with chemical insight2,11 predict that the proton relay experiences a lower reaction barrier than the vehicle mechanism, suggesting that the high Dproton value shall indeed be ascribed to the Grotthuss mechanism. This mechanism is now considered to be the “golden” standard for PT in aqueous solutions and various other molecular systems mentioned above,1–9 although there is surprisingly little experimental validation at the molecular level.Experimental evidence for the Grotthuss mechanism has been obtained by the photo-induced PT dynamics of the rhodopsin and yellow proteins.7,16 However, it is limited only to photo-initiated PT processes in the excited electronic state. Significantly, it is not straightforward to transfer this conclusion to ground-state PT phenomena, like those in aqueous solutions, because of its stochastic character. Indeed, many studies on ground-state PT in the condensed phase suggest the Grotthuss mechanism simply because of the low barrier for ion conductivity without presenting direct evidence.17–19 One of the approaches to characterize the PT mechanism is the assignment of intermediates. In the theoretical framework of the Grotthuss mechanism in aqueous solution, a proton of an H3O+ ion hydrated by three water molecules (Eigen ion, H9O4+)1 is transferred to its nearest H2O to form another Eigen structure via the transient Zundel ion (H5O2+).20–22 Two-dimensional IR spectroscopy revealed that an asymmetric Zundel structure can be formed in aqueous solution.21 However, the presence of such intermediates is not always directly connected to the Grotthuss mechanism. The general difficulty in determining the PT mechanisms in various molecular systems prevents a deeper understanding of the mechanism–property correlations at the molecular level.
PT in hydrated clusters consisting of a finite number of water molecules23–28 and other solvent molecules29–31 provides a well-defined benchmark to explore the so far elusive PT mechanism. For example, p-aminobenzoic acid (PABA, Fig. 1) is a prototypical molecule for microhydration-induced PT.23,24,32–34 PABA has two protonation sites, i.e., the O- and N-protomers resulting from protonation of the carboxy and amino groups, respectively (Fig. 1). Their relative stability varies with the surrounding environment. The O-protomer is more stable in the gas phase while the N-protomer is more stable in a polar solvent.35,36 Studies using ion mobility mass spectrometry37 and infrared photodissociation (IRPD) spectroscopy23 claim that for the hydrated clusters of protonated PABA, PABAH+(H2O)n, generated by electrospray ionization (ESI), the N-protomer becomes detectable at n = 6, despite the spectral ambiguity in both the arrival time distributions and the IRPD spectra. In subsequent studies, we re-determined the threshold size (n) for the O → N protomer switching as n = 5 using cryogenic double ion trap IR spectroscopy.24 For the PT mechanism, none of previous studies (including ours) have provided any experimental evidence whether the vehicle or the Grotthuss mechanism takes place.23,24,37 The previous studies using ion mobility and IRPD claim the Grotthuss mechanism because their density functional theory (DFT) calculations show a stable structure for n = 6, in which the O- and N-protonation sites are bridged by a H-bonded water chain. However, no experimental evaluation for this structure was presented. We also applied DFT calculations for n = 5 and found that a structure locally hydrated around the protonated amino group is significantly more stable than the ones in which the O- and N-sites are bridged by an H-bonded water chain, and thus the only observed hydration motif. Based on this exclusively observed hydration motif, we suggested the vehicle rather than the Grotthuss mechanism, although again we had no direct experimental evidence regarding which PT mechanism is in operation for n ≥ 5.
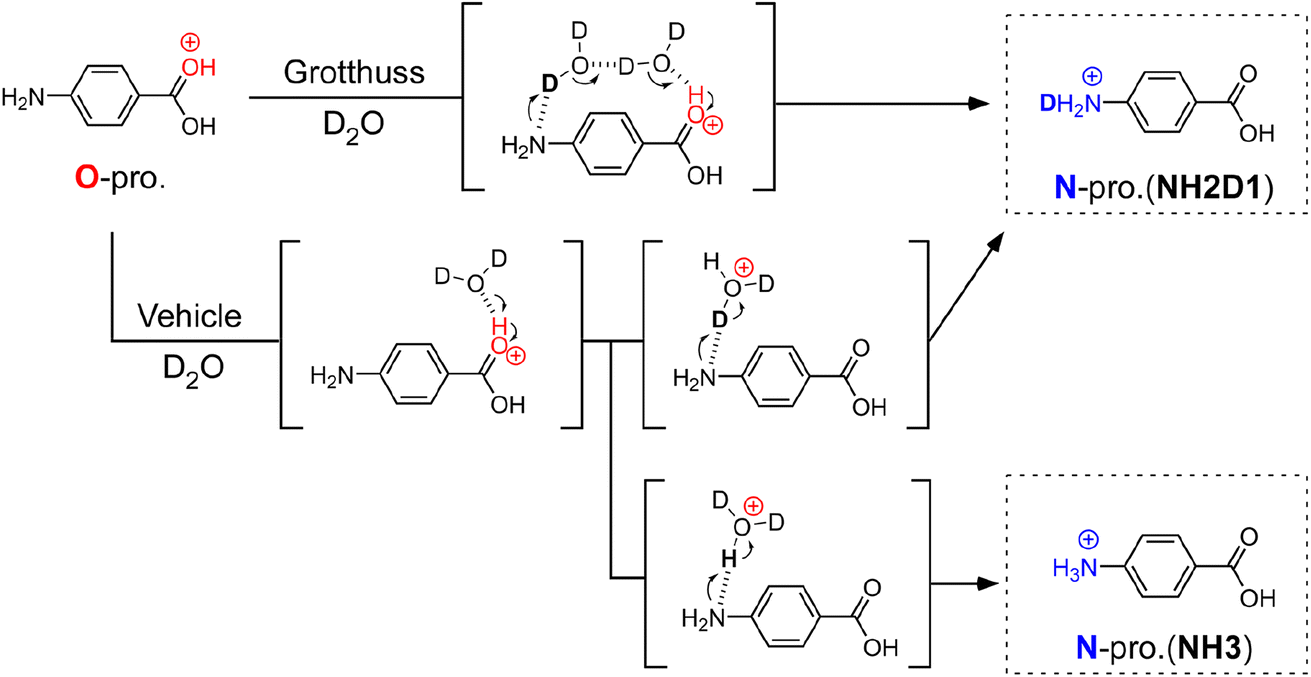 | ||
| Fig. 1 Schematic illustration of hydration-induced O → N proton transfer of PABAH+ by vehicle and Grotthuss mechanisms. The parentheses show a possible intermediate structure for the proton transfer. In the Grotthuss mechanism, proton transfer is mediated by a water bridge between CO and NH sites, resulting in a singly deuterated N-protomer denoted as NH2D1. On the other hand, vehicle-type proton transfer affords both NH2D1 and nondeuterated NH3. Note that the reaction intermediate in the scheme is simplified for clarity. For example, we do not exclude the possibility of bridged structures with more than two water molecules for the Grotthuss mechanism. | ||
To determine the PT mechanisms operating in hydrated PABAH+ at the molecular level, we combine herein cryogenic double ion trap IR spectroscopy38–42 with isotopic labelling. Deuterated hydrated clusters of PABAH+ can be produced by introducing D2O vapour into the reaction trap. One can readily expect that, starting from the O-protomer, the Grotthuss-type PT gives a singly deuterated N-protomer (NH2D1) in the deuterated hydrated clusters because the proton source is D2O (in Fig. 1). The vehicle-type PT yields both deuterated (NH2D1) and nondeuterated (NH3) N-protomers via the transfer of HD2O+ (Fig. 1). Clearly, the detection of NH3 provides direct evidence for the vehicle mechanism. However, the appearance of NH3 can hardly be probed by conventional IRPD spectroscopy because its vibrational signatures are largely red-shifted and broadened by H-bonding with water molecules, causing spectral congestion.23,43
This problem is overcome herein by applying the novel collision-assisted stripping (CAS) method, developed recently by our group.24,27,43 In CAS experiments, one measures IRPD spectra of the PABAH+ monomer core after removing all hydrated water molecules of the cluster by soft collisions in the ion trap, allowing for the clear spectroscopic assignment of deuterated isotopologues of PABAH+, as the IR spectra of the solvent-free ions are not congested. Herein, we perform CAS-IRPD of PABAH+(D2O)n to determine the PT mechanisms as a function of the degree of hydration (see Experimental in the ESI†). We obtain clear evidence of the vehicle mechanism for n = 5/6 and suggestions of the Grotthuss mechanism for n = 7, respectively, suggesting a switch in the PT mechanism between n = 5 and n = 7. The consistency with the IRPD spectra of the hydrated clusters is also discussed.
Results and discussion
Fig. 2 shows the CAS-IRPD spectra of PABAH+(D2O)n with n = 0 and 4–7. All bands observed for PABAH+(n = 0) are ascribed to vibrational modes of the O-protomer according to previous assignments: symmetric NH2 stretch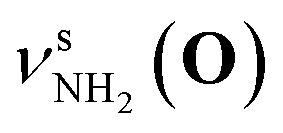 = 3435 cm−1; outward-facing OH stretch νZ-OH (O) = 3496 cm−1; antisymmetric NH2 stretch
= 3435 cm−1; outward-facing OH stretch νZ-OH (O) = 3496 cm−1; antisymmetric NH2 stretch 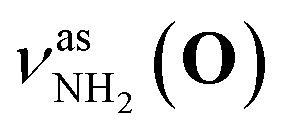 = 3535 cm−1; benzene-facing OH stretch νE-OH (O) = 3558 cm−1.24 The lack of any transitions in the 3200–3400 cm−1 range indicates the complete absence of the N-protomer of bare PABAH+ under the chosen experimental conditions.24 The ESI ion beam of bare PABAH+ with pure O-protomer population was injected into the reaction trap and exposed to deuterated water vapor and He buffer gas. Under such conditions, the hydration-induced O → N PT reaction can be directly probed by the generation of the N-protomer in the hydrated clusters.
= 3535 cm−1; benzene-facing OH stretch νE-OH (O) = 3558 cm−1.24 The lack of any transitions in the 3200–3400 cm−1 range indicates the complete absence of the N-protomer of bare PABAH+ under the chosen experimental conditions.24 The ESI ion beam of bare PABAH+ with pure O-protomer population was injected into the reaction trap and exposed to deuterated water vapor and He buffer gas. Under such conditions, the hydration-induced O → N PT reaction can be directly probed by the generation of the N-protomer in the hydrated clusters.
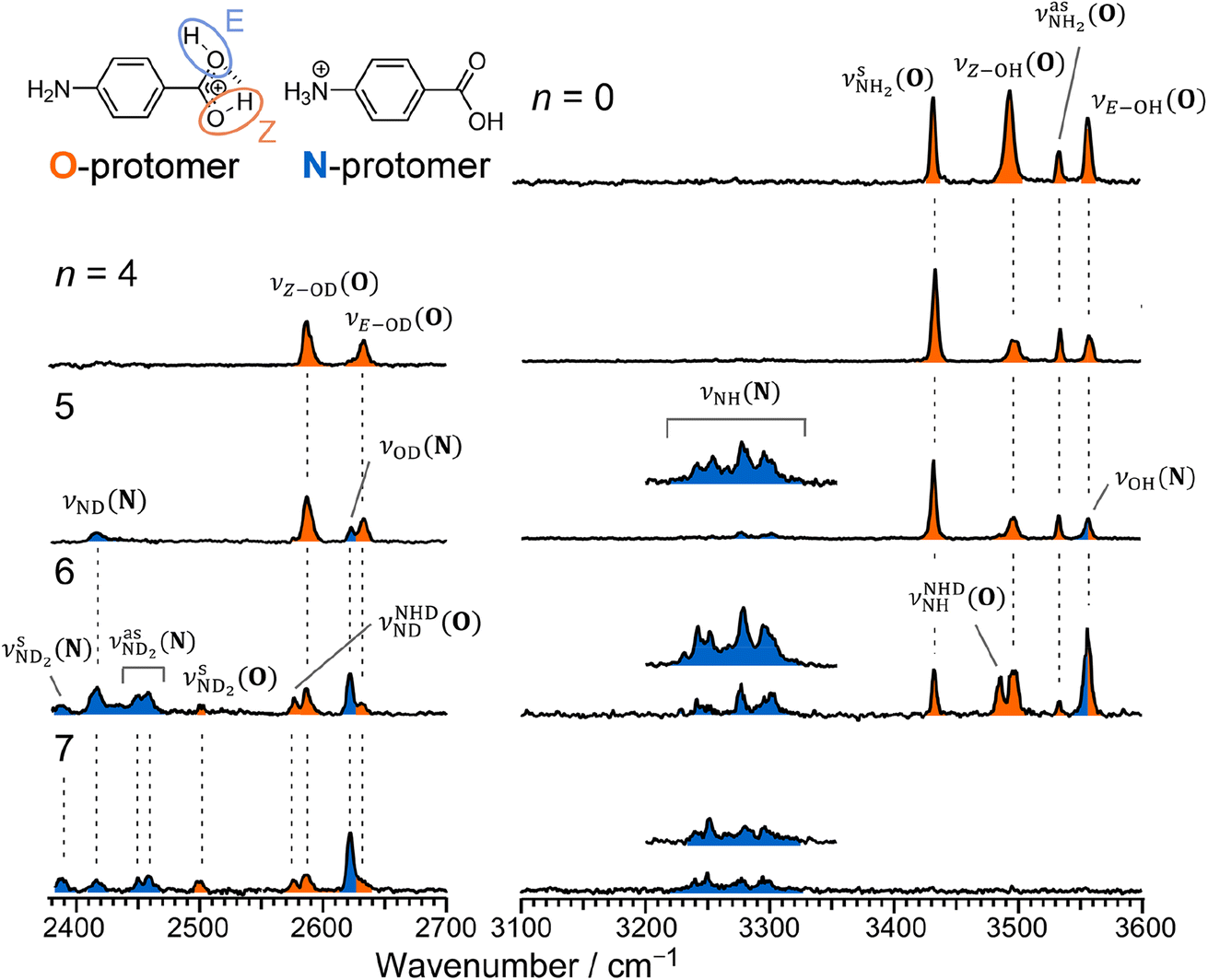 | ||
| Fig. 2 IRPD spectrum of PABAH+ and CAS-IRPD spectra of PABAH+(D2O)n (n = 4–7). The orange- and blue-colored bands are assigned to the vibrational modes of O- and N-protomers, respectively. The inset CAS-IRPD spectra in 3200–3360 cm−1 for n = 5–7 were re-measured with ∼two times higher IR-laser fluence. The indices N and O in the notation of vibrational modes indicate N- and O-protomers, respectively. | ||
The CAS-IRPD spectrum of PABAH+(D2O)5 shows new bands in the 3220–3320 cm−1 range, which are absent for n ≤ 4 (Fig. 2). The spectrum of these bands measured with a twice higher laser intensity is also shown as the inset. These bands are assigned to NH stretches of the N-protomer,24,44 indicating the appearance of the N-protomer core upon O → N PT. A further new band at 2416 cm−1 is attributed to an ND stretch of the N-protomer based on its frequency (see Fig. S1 in the ESI† for detailed band assignments), confirming the appearance of the N-protomer core at n = 5. The threshold size (n = 5)24 for O → N protomer switching does not change upon H2O → D2O substitution. Based on the given assignment, the other new band at 2620 cm−1 can be assigned to the OD stretching of the N-protomer core (νOD (N), Fig. S1†). The remaining two bands at 2585 and 2631 cm−1 are then attributed to νZ-OD (O) and νE-OD (O), respectively (Fig. S1†). Note that the OD stretches also appear for n = 4, indicating H/D exchange at the carboxylic group without protomer switching. One may wonder why H/D exchange happens at such low temperatures (T = 80 K), at which protonated glycine is not deuterated by clustering with D2O.45 This observation is rationalized here by the high acidity of the protonated carboxy group. Recently, the Johnson group reported IRPD spectra of hydrated deuterated isotopologues of the N-protomer (ND3+C6H4COOD(H2O)n) and found that the acidic COOD group is more susceptible to H/D exchange than the charged ND3+ group.46 The protonated C(OH)2+ group of the O-protomer may more readily replace H by D.
The CAS-IRPD spectra of PABAH+(D2O)6,7 also exhibit the ND/NH stretches of the N-protomer core in the 2380–2470/3220–3320 cm−1 ranges, but their band patterns are more complicated than that of n = 5 (Fig. 3a). To understand the PT mechanism, one must assign the vibrational bands of deuterated (NH2D1) and nondeuterated (NH3) N-protomers without ambiguity. For this purpose, we measured the IR spectra of nondeuterated (m/z 138), monodeuterated (m/z 139, NH3OD1 and NH2D1), and dideuterated (m/z 140, NH2D1OD1 and NH1D2) forms of the bare N-protomer in the NH stretch range, which are shown in Fig. 3b along with molecular structures. Details of the assignments are summarized in the ESI.† Briefly, three prominent bands in the spectrum of m/z 138 are assigned to NH3 stretching bands (coloured in green, 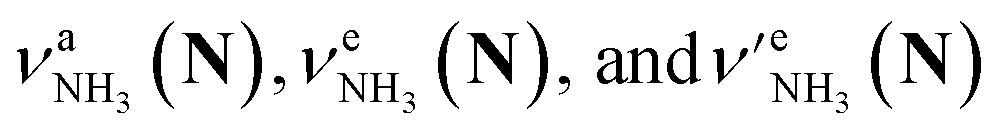 ) of the N-protomer of the fully hydrogenated species (NH3). The new bands indicated by blue colour in the spectrum of m/z 139 are NH2 stretching bands
) of the N-protomer of the fully hydrogenated species (NH3). The new bands indicated by blue colour in the spectrum of m/z 139 are NH2 stretching bands 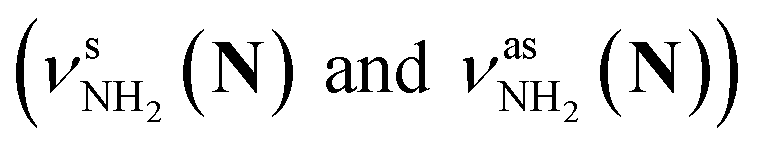 of the amino-deuterated N-protomer (NH2D1). The bands due to the NH3 stretching (green) are overlapped with transitions of the coexisting N-protomer deuterated at the carboxylate group (NH3OD1). The newly observed band (purple) in the spectrum of m/z 140 is an NH stretch mode of the dideuterated amino group of the N-protomer (NH1D2). Among these bands, the vibrational transitions of NH3 (
of the amino-deuterated N-protomer (NH2D1). The bands due to the NH3 stretching (green) are overlapped with transitions of the coexisting N-protomer deuterated at the carboxylate group (NH3OD1). The newly observed band (purple) in the spectrum of m/z 140 is an NH stretch mode of the dideuterated amino group of the N-protomer (NH1D2). Among these bands, the vibrational transitions of NH3 (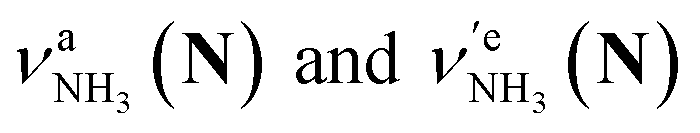 ) are the most important ones because they indicate the presence of the N-protomer (NH3) which can be generated only by the vehicle mechanism (Fig. 1).
) are the most important ones because they indicate the presence of the N-protomer (NH3) which can be generated only by the vehicle mechanism (Fig. 1).
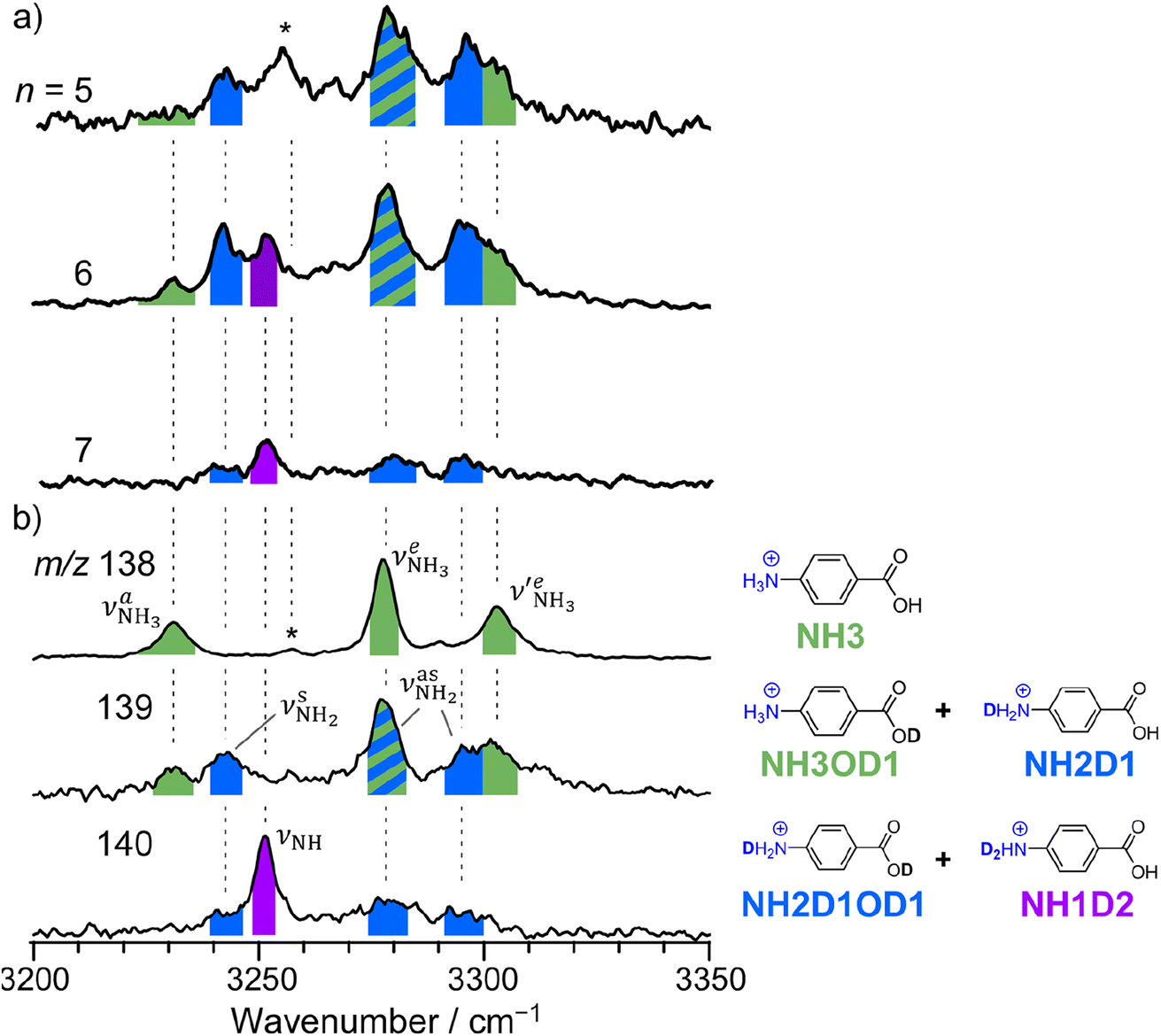 | ||
| Fig. 3 (a) CAS-IRPD spectra of PABAH+(D2O)n (n = 5–7) in the 3200–3350 cm−1 range (reprinted from the inset spectra in Fig. 2). (b) IRPD spectra of PABAH+ (m/z 138), singly deuterated protonated PABA (m/z 139), and doubly deuterated protonated PABA (m/z 140). These spectra are necessary to assign the CAS-IRPD spectra because the monomer core after the CAS event includes several deuterated isotopomers. The green-, blue-, and purple-colored bands are assigned to NH stretches of NH3, NH2D1, and NH1D2, respectively. The band marked by an asterisk is assigned to a combination band of the O-protomer (in-plane NH2 bend24,44 at ∼1660 cm−1 + aryl C–C stretch24,44 at ∼1600 cm−1). This type of combination band is also observed for an analog of PABA, ethyl p-aminobenzoate.43 | ||
Let us check the presence of 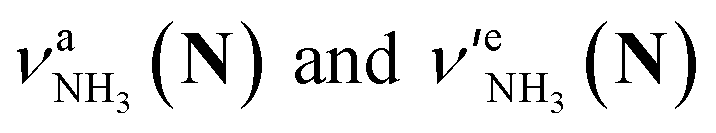 (green), the marker bands of the vehicle mechanism, in the CAS-IRPD spectra of PABAH+(D2O)n with n = 5–7. Fig. 3a reproduces expanded spectra in the 3200–3350 cm−1 range. The green marker bands obviously appear in the spectra of n = 5 and 6, but are clearly absent in the n = 7 spectrum. This result directly indicates that the vehicle mechanism operates in the n = 5 and 6 clusters but not for n = 7. One cannot directly conclude from the CAS-IRPD spectra whether the Grotthuss mechanism operates in n = 5 and 6. The observed bands in the n = 7 spectrum correspond to N-protomers of NH2D1 (blue) and NH1D2 (purple). The former, NH2D1, can be generated by both the vehicle and Grotthuss mechanisms. As the absence of the vehicle mechanism for n = 7 is clear, the blue bands can only arise from the Grotthuss PT. The latter, NH1D2, cannot be generated by a single O → N PT reaction via either the Grotthuss or the vehicle mechanism. The observed multiple deuteration thus is ascribed to H/D exchange by D2O or the interconversion between N- and O-protomers by O → N and N → O back reactions (Fig. S5 in the ESI†). Here, we cannot fully exclude the possibility that the absence of
(green), the marker bands of the vehicle mechanism, in the CAS-IRPD spectra of PABAH+(D2O)n with n = 5–7. Fig. 3a reproduces expanded spectra in the 3200–3350 cm−1 range. The green marker bands obviously appear in the spectra of n = 5 and 6, but are clearly absent in the n = 7 spectrum. This result directly indicates that the vehicle mechanism operates in the n = 5 and 6 clusters but not for n = 7. One cannot directly conclude from the CAS-IRPD spectra whether the Grotthuss mechanism operates in n = 5 and 6. The observed bands in the n = 7 spectrum correspond to N-protomers of NH2D1 (blue) and NH1D2 (purple). The former, NH2D1, can be generated by both the vehicle and Grotthuss mechanisms. As the absence of the vehicle mechanism for n = 7 is clear, the blue bands can only arise from the Grotthuss PT. The latter, NH1D2, cannot be generated by a single O → N PT reaction via either the Grotthuss or the vehicle mechanism. The observed multiple deuteration thus is ascribed to H/D exchange by D2O or the interconversion between N- and O-protomers by O → N and N → O back reactions (Fig. S5 in the ESI†). Here, we cannot fully exclude the possibility that the absence of 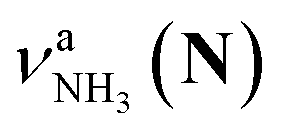 , the signature of the vehicle mechanism, may arise from the H/D exchange reaction because such an H/D exchange is observed for n ≥ 4 (see above). However, if this scenario were the case, H/D exchange also modulates the intensities of
, the signature of the vehicle mechanism, may arise from the H/D exchange reaction because such an H/D exchange is observed for n ≥ 4 (see above). However, if this scenario were the case, H/D exchange also modulates the intensities of 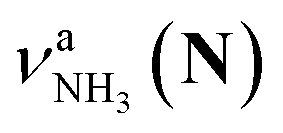 in the spectra of n = 5 and 6 where the band is clearly observed. This observation means that the relative intensity of
in the spectra of n = 5 and 6 where the band is clearly observed. This observation means that the relative intensity of 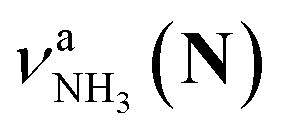 for n = 7 is rather weak in comparison to those for n = 5 and 6, and the vehicle PT mechanism is negligibly minor for n = 7.
for n = 7 is rather weak in comparison to those for n = 5 and 6, and the vehicle PT mechanism is negligibly minor for n = 7.
The size-dependent PT mechanisms proposed by the experiment are consistent with the molecular structures of microhydrated PABAH+. The bridged structures in which the NH3+ and COOH groups are connected by an H-bonded water chain are more abundant than the unbridged structures for the N-protomer at n = 7, whereas the bridged structures become minor for n = 6 and are not observed for n = 5 (Fig. S6 in the ESI†).24 This strongly suggests that the water-bridged intermediates for the Grotthuss-type PT are stabilized at n = 7 (and partly stabilized at n = 6). In this sense, the Grotthuss mechanism may be involved in part in the PT at n = 6. These observations show that the reaction barriers of the Grotthuss mechanism can be altered by the number of water molecules. The Grotthuss mechanism with a lowered reaction barrier would account for the minor contribution of the vehicle mechanism at n = 7. This finding gives suggestions for the poorly understood properties of PT in various systems. For example, it is known that proton conductivity in porous materials such as MOFs is sensitive to humidity.5,6 Some MOFs show a drastic decrease in the conductivity below a critical humidity,47,48 however, the reason remains unclear. The poor conductivity at low humidity has been hampering the application of MOFs to proton-conducting membranes in fuel cells. Our conclusion that the PT mechanisms would be changed from Grotthuss to vehicle with decreasing number of water molecules would rationalize such a drastic change in PT by humidity.
Conclusion
In summary, we spectroscopically determined the mechanisms for hydration-induced PT of PABAH+ by cryogenic double ion trap IR spectroscopy combined with deuterium labelling. The presence of NH3 for PABAH+(D2O)5,6 unambiguously indicates O → N PT via the vehicle mechanism, in sharp contrast to the computational assumption that the Grotthuss mechanism is dominant in this molecular system49,50 with inference from experimental results. The vehicle mechanism for solvent-mediated PT is poorly experimentally validated for water,51 including methanol29 and ammonia.30,31 We have spectroscopically validated the vehicle mechanism for water-containing molecular systems at the molecular level. This study experimentally demonstrates the significance of the vehicle mechanism in water as well, which has often been ignored in previous work. On the other hand, the absence of NH3 and the presence of NH2D1 for PABAH+(D2O)7 strongly suggest the Grotthuss mechanism, which is correlated with the stability of the water-bridged structures at this cluster size. The calculated bridged structure has three/four water molecules in the chain connecting the O and N sites. The remaining water molecules attach to this chain and the amino NH3+ group. This result highlights the importance of the second-shell water molecules in stabilizing the water bridge. Although the system we studied is not the same as solution in terms of the number of water molecules and temperature, the findings afford a clue to understanding the nature of PT in molecular systems consisting of a finite number of water molecules. In particular, PT in functional materials such as proton-conducting polymers4 and MOFs6,47,48 and biological macromolecules9 could be facilitated by residual water in their nanosized pores. The number of water molecules in the pores is pivotal to stabilize the water chain and control the PT mechanism.Data availability
Essential data are fully provided in the main text and ESI.† Further data in this study are available from the corresponding authors upon a reasonable request.Author contributions
K. H., O. D., S. I., and M. F. conceived and designed the project. K. A. and K. H. carried out the experiments and analyzed the data. All the authors contributed to writing the manuscript and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by KAKENHI (JP19K23624, JP20K20446, JP20H00372, JP21H04674, JP21K14585), Core-to-Core program (JPJSCCA20210004) from Japan Society for the Promotion of Science, research grant from World Research Hub Initiative (WRHI) of Tokyo Institute of Technology, Cooperative Research Program of the “Network Joint Research Center for Materials and Devices” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The computations were performed at the Research Center for Computational Science, Okazaki, Japan. M. F. is grateful for support from the Alexander von Humboldt foundation. O. D. acknowledges support from Deutsche Forschungsgemeinschaft (DFG, DO 729/10) and the World Research Hub Initiative (WRHI) of Tokyo Institute of Technology.References
- M. Eigen, Angew. Chem., Int. Ed., 1964, 3, 1–19 CrossRef.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- K.-D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- K.-D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- D.-W. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16–33 RSC.
- F. Garczarek and K. Gerwert, Nature, 2006, 439, 109–112 CrossRef CAS PubMed.
- J. R. Schnell and J. J. Chou, Nature, 2008, 451, 591–595 CrossRef CAS PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- K.-D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed., 1982, 21, 208–209 CrossRef.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- C. J. T. d. Grotthuss, Ann. Chim., 1806, LVIII, 54–74 Search PubMed.
- D. Marx, ChemPhysChem, 2006, 7, 1848–1870 CrossRef CAS PubMed.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- M. Tuckerman, K. Laasonen, M. Sprik and M. Parrinello, J. Chem. Phys., 1995, 103, 150–161 CrossRef CAS.
- H. Kuramochi, S. Takeuchi, K. Yonezawa, H. Kamikubo, M. Kataoka and T. Tahara, Nat. Chem., 2017, 9, 660–666 CrossRef CAS PubMed.
- I. Popov, Z. Zhu, A. R. Young-Gonzales, R. L. Sacci, E. Mamontov, C. Gainaru, S. J. Paddison and A. P. Sokolov, Commun. Chem., 2023, 6, 77 CrossRef CAS PubMed.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta and W. Huang, et al. , Nat. Energy, 2019, 4, 123–130 CrossRef CAS.
- Y. Yang, P. Zhang, L. Hao, P. Cheng, Y. Chen and Z. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21838–21845 CrossRef CAS PubMed.
- R. Janoschek, E. G. Weidemann, H. Pfeiffer and G. Zundel, J. Am. Chem. Soc., 1972, 94, 2387–2396 CrossRef CAS.
- J. A. Fournier, W. B. Carpenter, N. H. C. Lewis and A. Tokmakoff, Nat. Chem., 2018, 10, 932–937 CrossRef CAS PubMed.
- C. T. Wolke, J. A. Fournier, L. C. Dzugan, M. R. Fagiani, T. T. Odbadrakh, H. Knorke, K. D. Jordan, A. B. McCoy, K. R. Asmis and M. A. Johnson, Science, 2016, 354, 1131–1135 CrossRef CAS PubMed.
- T. M. Chang, J. S. Prell, E. R. Warrick and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 15805–15813 CrossRef CAS.
- K. Akasaka, K. Hirata, F. Haddad, O. Dopfer, S. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2023, 25, 4481–4488 RSC.
- K. Chatterjee and O. Dopfer, J. Phys. Chem. A, 2020, 124, 1134–1151 CrossRef CAS PubMed.
- N. Takeda, K. Hirata, K. Tsuruta, G. D. Santis, S. S. Xantheas, S. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2022, 24, 5786–5793 RSC.
- G. D. Santis, N. Takeda, K. Hirata, K. Tsuruta, S. Ishiuchi, S. S. Xantheas and M. Fujii, J. Am. Chem. Soc., 2022, 144, 16698–16702 CrossRef CAS PubMed.
- M.-P. Gaigeot, A. Cimas, M. Seydou, J.-Y. Kim, S. Lee and J.-P. Schermann, J. Am. Chem. Soc., 2010, 132, 18067–18077 CrossRef CAS.
- B. Ucur, O. J. Shiels, S. J. Blanksby and A. J. Trevitt, J. Am. Soc. Mass Spectrom., 2023, 34, 1428–1435 CrossRef CAS.
- K. Ohshimo, S. Miyazaki, K. Hattori and F. Misaizu, Phys. Chem. Chem. Phys., 2020, 22, 8164–8170 RSC.
- K. Ohshimo, R. Sato, Y. Takasaki, K. Tsunoda, R. Ito, M. Kanno and F. Misaizu, J. Phys. Chem. Lett., 2023, 8281–8288 CrossRef CAS PubMed.
- Z. Tian and S. R. Kass, Angew. Chem., Int. Ed., 2009, 48, 1321–1323 CrossRef CAS PubMed.
- J. L. Campbell, J. C. Y. Le Blanc and B. B. Schneider, Anal. Chem., 2012, 84, 7857–7864 CrossRef CAS PubMed.
- P. R. Batista, T. C. Penna, L. C. Ducati and T. C. Correra, Phys. Chem. Chem. Phys., 2021, 23, 19659–19672 RSC.
- J. Seo, S. Warnke, S. Gewinner, W. Schöllkopf, M. T. Bowers, K. Pagel and G. von Helden, Phys. Chem. Chem. Phys., 2016, 18, 25474–25482 RSC.
- S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf and K. Pagel, et al. , J. Am. Chem. Soc., 2015, 137, 4236–4242 CrossRef CAS PubMed.
- M. J. Hebert and D. H. Russell, J. Phys. Chem. B, 2020, 124, 2081–2087 CrossRef CAS PubMed.
- S. Ishiuchi, H. Wako, D. Kato and M. Fujii, J. Mol. Spectrosc., 2017, 332, 45–51 CrossRef CAS.
- B. M. Marsh, J. M. Voss and E. Garand, J. Chem. Phys., 2015, 143, 204201 CrossRef PubMed.
- E. Sato, K. Hirata, J. M. Lisy, S. Ishiuchi and M. Fujii, J. Phys. Chem. Lett., 2021, 12, 1754–1758 CrossRef CAS PubMed.
- O. V. Boyarkin, S. R. Mercier, A. Kamariotis and T. R. Rizzo, J. Am. Chem. Soc., 2006, 128, 2816–2817 CrossRef CAS PubMed.
- J. G. Redwine, Z. A. Davis, N. L. Burke, R. A. Oglesbee, S. A. McLuckey and T. S. Zwier, Int. J. Mass Spectrom., 2013, 348, 9–14 CrossRef CAS.
- K. Hirata, F. Haddad, O. Dopfer, S. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2022, 24, 5774–5779 RSC.
- T. Khuu, N. Yang and M. A. Johnson, Int. J. Mass Spectrom., 2020, 457, 116427 CrossRef CAS PubMed.
- J. M. Voss, K. C. Fischer and E. Garand, J. Phys. Chem. Lett., 2018, 9, 2246–2250 CrossRef CAS PubMed.
- T. Khuu, S. J. Stropoli, K. Greis, N. Yang and M. A. Johnson, J. Chem. Phys., 2022, 157, 131102 CrossRef CAS PubMed.
- H. Ōkawa, A. Shigematsu, M. Sadakiyo, T. Miyagawa, K. Yoneda, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 13516–13522 CrossRef PubMed.
- M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472–5475 CrossRef CAS PubMed.
- H. Xia and A. B. Attygalle, J. Mass Spectrom., 2018, 53, 353–360 CrossRef CAS PubMed.
- J. L. Campbell, A. M.-C. Yang, L. R. Melo and W. S. Hopkins, J. Am. Soc. Mass Spectrom., 2016, 27, 1277–1284 CrossRef CAS PubMed.
- Y. Matsuda, A. Yamada, K. Hanaue, N. Mikami and A. Fujii, Angew. Chem., Int. Ed., 2010, 49, 4898–4901 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05455a |
| This journal is © The Royal Society of Chemistry 2024 |