
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally induced intermetallic Rh1Zn1 nanoparticles with high phase-purity for highly selective hydrogenation of acetylene†
Xiaocheng
Lan‡
,
Yu
Wang‡
 ,
Boyang
Liu
,
Zhenyu
Kang
and
Tiefeng
Wang
*
,
Boyang
Liu
,
Zhenyu
Kang
and
Tiefeng
Wang
*
Beijing Key Laboratory of Green Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: wangtf@tsinghua.edu.cn
First published on 23rd December 2023
Abstract
Ordered M1Zn1 intermetallic phases with structurally isolated atom sites offer unique electronic and geometric structures for catalytic applications, but lack reliable industrial synthesis methods that avoid forming a disordered alloy with ill-defined composition. We developed a facile strategy for preparing well-defined M1Zn1 intermetallic nanoparticle (i-NP) catalysts from physical mixtures of monometallic M/SiO2 (M = Rh, Pd, Pt) and ZnO. The Rh1Zn1 i-NPs with structurally isolated Rh atom sites had a high intrinsic selectivity to ethylene (91%) with extremely low C4 and oligomer formation, outperforming the reported intermetallic and alloy catalysts in acetylene semihydrogenation. Further studies revealed that the M1Zn1 phases were formed in situ in a reducing atmosphere at 400 °C by a Zn atom emitting–trapping–ordering (Zn-ETO) mechanism, which ensures the high phase-purity of i-NPs. This study provides a scalable and practical solution for further exploration of Zn-based intermetallic phases and a new strategy for designing Zn-containing catalysts.
Introduction
The intermetallic phase, with a definite crystal structure and well-defined atom composition, is an emerging and promising catalyst in various catalytic fields.1,2 The intermetallic phase exhibits an ordered surface atomic arrangement to regulate a specific adsorption mode and configuration that modulates certain reaction pathways and controls the reaction performance.3–6 Taking the intermetallic phase of M1Zn1 (M = Pd, Pt, Rh; molar ratio of M/Zn = 1) as an example, the active metal M1 and the metal Zn stack in an orderly manner to form a body-centered structure. Regarding the specific atomic geometry of the ordered M1Zn1 intermetallic phase, the distance between adjacent M atoms within M1Zn1 is enlarged, as M atoms are segregated by a Zn atom into isolated ensembles. The M1Zn1 phase provides a naturally high surface density of structurally isolated active ensembles with an identical coordination environment. Compared with single atom alloys (SAAs) and single atom catalysts (SACs), the isolated M1 site is in a long-range order structure with fixed atom positions and site occupancies, which offers consistent site isolation throughout the catalyst, as well as unique electronic properties of the M1 site modified by the neighbouring Zn atom.5,7,8 The ordered atomic arrangement in intermetallic phases successfully realizes the “site-isolation” concept that could control the reaction performance according to the structure–performance relationship.However, for the preparation of supported i-NP catalysts, harsh synthesis conditions such as high temperature annealing (usually >500 °C) is often required, which often leads to a sintering problem, a wide distribution of ensemble size, and ill-defined composition.1,5,9,10 Alternative approaches, such as the colloidal method and atomic layer deposition (ALD), are developed to achieve atom level control of the structure. However, these methods are still challenging in some aspects, for example the colloidal method generally requires special ligands that need to be removed to activate the catalyst, and ALD requires typical metal organic precursors and a specific instrument with complex processes.11–13 The absence of a facile and controllable synthesis method that attains high phase-purity and specific composition on each active ensemble has restricted the broad application of supported i-NP catalysts.5,14 Therefore, an industrially feasible, controllable, and scalable synthesis method for supported i-NP catalysts is desired.
We have developed a facile synthesis of well-defined M1Zn1 (M = Rh, Pd, Pt) i-NPs from simple physical mixtures of monometallic M/SiO2 and ZnO at a temperature of 400 °C and under a reducing atmosphere. Extensive study indicates that the formation of the intermetallic M1Zn1 phase follows a novel Zn emitting–trapping–ordering (Zn-ETO) mechanism, where Zn atoms were emitted from the ZnO surface with the assistance of H2, then trapped by the vicinal monometallic nanoparticles and finally rearranged into well-defined M1Zn1 intermetallic phases. The thermodynamic driving force of the disorder-to-order transition imparts high phase-purity to the M1Zn1 i-NPs, independent of the particle size distribution of the starting monometallic NPs.
Intermetallic compound catalysts exhibit distinct catalytic properties for alkyne semihydrogenation. Previous studies mainly focused on Pd-based3–5,15,16 and Ni-based6,17–19 intermetallic phases, where the primary active metal was alloyed with a second metal of Ag, Ga, Zn, etc., to improve the selectivity to ethylene. The enhanced selectivity is attributed to the isolated active-site ensembles in a specific intermetallic phase.5,6,17,20–22 Recently, Rioux et al.5 demonstrated that Pd8Zn44 consisting of only Pd1 monomers on the surface showed higher selectivity towards ethylene than Pd9Zn43 consisting of Pd3 sites. However, it is still challenging to prevent the side reaction of C–C bond coupling to oligomers. It is believed that the specific active-site ensembles in i-NPs are critical to highly selective hydrogenation. Thus, improving the understanding of the catalytic structure–performance relationship is facilitated by the phase-purity of the desired intermetallic phase.
Here the proposed Zn-ETO strategy provides a platform for designing desired intermetallic phase M1Zn1 (M = Rh, Pd, Pt) i-NPs. Specific emphasis was placed on Rh to study the reaction mechanism with specific active sites. The reduction time during Zn-ETO directly controls the composition of the catalytic active sites within the Rh–Zn phase, which tunes the product distribution of acetylene hydrogenation. The characterization results (HAADF-STEM, EDS, in situ XRD, XPS and CO-FTIR) verified the formation of Rh1Zn1 i-NPs. The crystal structure of the intermetallic phase provided a specific ensemble of surface atoms, where the Rh atoms were structurally isolated by Zn atoms. Catalytic evaluation and DFT calculations further reveal that Rh1Zn1 i-NPs with structurally isolated Rh atom sites efficiently suppress the C–C bond coupling reaction that forms C4 and green oil (GO) side products. The RhZn/SiO2-10 catalyst with high phase-purity of Rh1Zn1 had a high intrinsic selectivity to ethylene (91%) at a high acetylene conversion (nearly 100%), which outperforms the reported intermetallic and alloy catalysts for acetylene semihydrogenation.
Results and discussion
Synthesis and characterization of intermetallic M1Zn1 i-NPs with isolated atom M sites
The details of the Zn-ETO strategy are illustrated in Fig. 1a. We used one-step reduction for physically mixed monometallic M/SiO2 (prepared by impregnation) and ZnO. The physically mixed catalyst (PMC) was treated by a temperature-programmed reduction (TPR) procedure (Fig. S1†). The obtained M–Zn/SiO2 intermetallic compound catalysts were characterized by HAADF-STEM and EDS (Fig. 1b–g and S2†). EDS elemental mapping showed that the Zn element signal uniformly distributed over the region of element M. For Rh–Zn/SiO2, two perpendicular interplanar distances of 3.01 Å and 2.13 Å were found, corresponding to the (001) and (110) planes of Rh1Zn1, respectively (Fig. 1e), indicating this image shows the RhZn [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] zone axis. The atomic image matched well with the top view of the standard RhZn crystal (PDF#53-0470) from the [10] zone axis, which confirmed the existence of the Rh1Zn1 intermetallic compound. For Pd–Zn/SiO2, we also observed the PdZn [10] zone axis in the corresponding sample from the matched simulated atomic projection (Fig. 1f). The RhZn and PdZn intermetallic phase displayed the [10] zone axis, which showed the 2D projection of a line of Rh or Pd atoms and a line of Zn atoms packed alternatively. For Pt–Zn/SiO2, the [001] zone axis was observed (Fig. 1g), which showed the 2D projection of a Zn atom in the center of four Pd atoms in a square arrangement. The electron microscopy results verified the successful synthesis of an evenly distributed M1Zn1 intermetallic phase. The atomic images of the exposed surfaces of M1Zn1 also showed the formation of M sites that were isolated by the particular geometry and composition of the intermetallic phase crystal structure.
0] zone axis. The atomic image matched well with the top view of the standard RhZn crystal (PDF#53-0470) from the [10] zone axis, which confirmed the existence of the Rh1Zn1 intermetallic compound. For Pd–Zn/SiO2, we also observed the PdZn [10] zone axis in the corresponding sample from the matched simulated atomic projection (Fig. 1f). The RhZn and PdZn intermetallic phase displayed the [10] zone axis, which showed the 2D projection of a line of Rh or Pd atoms and a line of Zn atoms packed alternatively. For Pt–Zn/SiO2, the [001] zone axis was observed (Fig. 1g), which showed the 2D projection of a Zn atom in the center of four Pd atoms in a square arrangement. The electron microscopy results verified the successful synthesis of an evenly distributed M1Zn1 intermetallic phase. The atomic images of the exposed surfaces of M1Zn1 also showed the formation of M sites that were isolated by the particular geometry and composition of the intermetallic phase crystal structure.
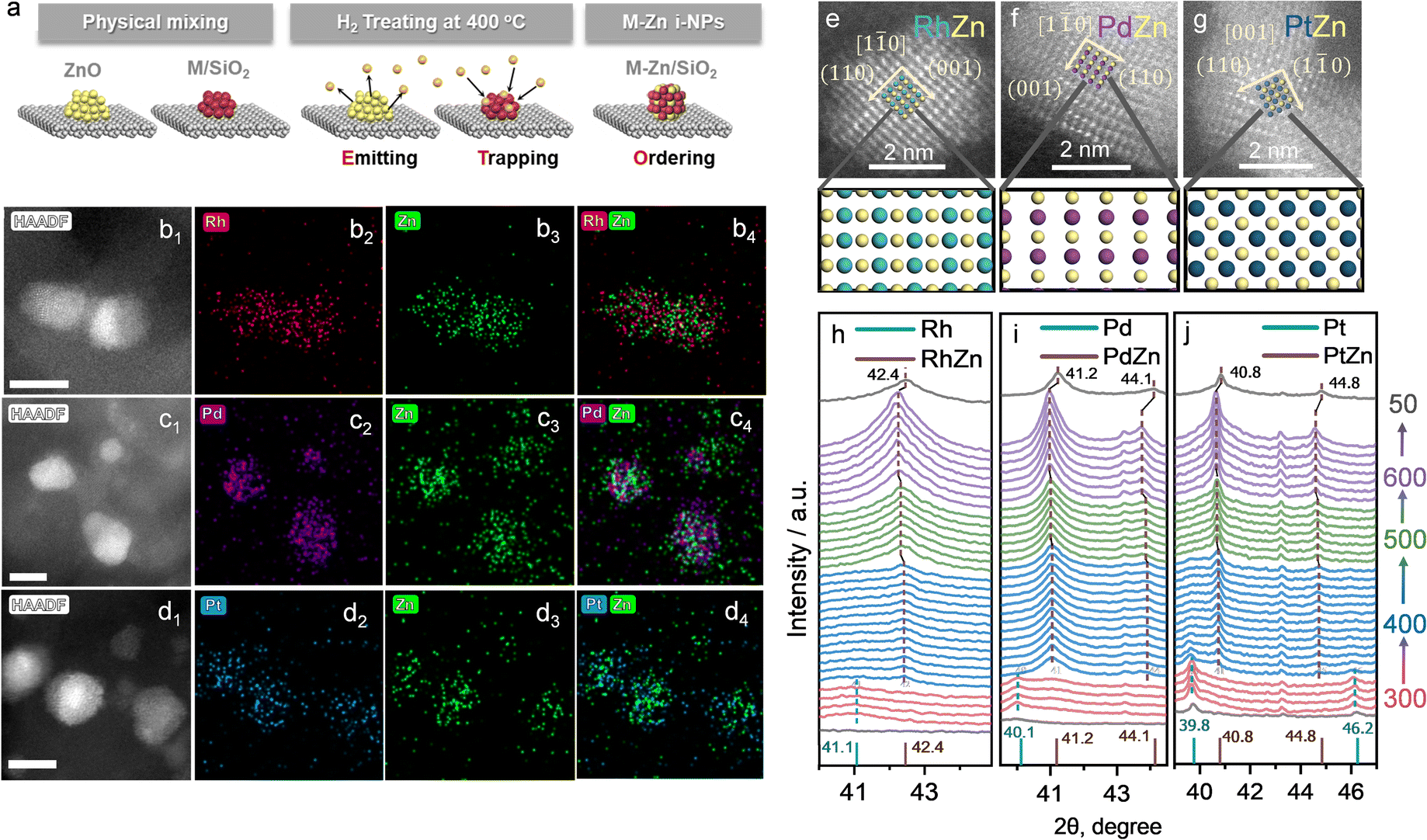 | ||
| Fig. 1 (a) Scheme of the Zn-ETO strategy. EDS mappings of (b) Rh–Zn/SiO2, (c) Pd–Zn/SiO2, and (d) Pt–Zn/SiO2 with a reduction time of 4 h. HAADF-STEM images of M1Zn1 i-NP catalysts (e) Rh–Zn/SiO2, (f) Pd–Zn/SiO2, and (g) Pt–Zn/SiO2 with a reduction time of 4 h. Rh, Pd, Pt and Zn atoms are shown in cyan, purple, blue and yellow, respectively. In situ XRD patterns of (h) Rh–Zn/SiO2, (i) Pd–Zn/SiO2, and (j) Pt–Zn/SiO2 during TPR. The TPR temperatures are 300 °C (red), 400 °C (blue), 500 °C (green), 600 °C (purple) and 50 °C (black) from bottom to top. The standard peaks of the monometallic (cyan sticks) and intermetallic phases (brown sticks) are also provided for reference (Rh: PDF#05-0685, RhZn: PDF#53-0470, Pd: PDF#46-1043, PdZn: PDF#06-0620, Pt: PDF#04-0802, PtZn: PDF#06-0604). | ||
The XRD patterns of the prepared catalysts are shown in Fig. S3.† The M/SiO2 without mixing ZnO showed corresponding peaks to the metal phases of M (Pd, PDF#46-1043, Fig. S3a;† Pt, PDF#04-0802, Fig. S3d;† Rh, PDF#05-0685, Fig. S3g†). For the sample of M/SiO2 physically mixed with ZnO (PMC), the XRD patterns were obtained after a reduction treatment at 400 °C for 4 h. All of the PMC samples showed the typical diffraction peaks of physically mixed ZnO (PDF#36-1451). In addition, the corresponding peaks of M1Zn1 phases (Pd1Zn1, PDF#06-0620; Pt1Zn1, PDF#06-0604; Rh1Zn1, PDF#53-0470) were also detected. For example, Pd1Zn1 (Fig. S3b and c†) showed the main peaks of (111), (200) and (311), Pt1Zn1 (Fig. S3e and f†) showed the main peaks of (111), (200), (002) and (311), and Rh1Zn1 (Fig. S3h and i†) showed the main peaks of (110) and (211).
To understand this transformation, in situ XRD was used to identify the diffraction peaks shift of the physical mixture of MOx/SiO2 (M = Rh, Pd, Pt) and ZnO during TPR (Fig. 1e–g). The peaks of M and M1Zn1 were emphasized with a dashed line going across the entirety of the XRD patterns. In the first stage (temperature kept at 300 °C for 1 h), the noble metals were reduced to their metallic state as the peaks of Rh, Pd and Pt were observed (corresponding to the metal phases of PDF#05-0685, PDF#46-1043, and PDF#04-0802). When the temperature increased to 400 °C and kept for 4 h, the diffraction peaks of intermetallic compounds dominated (brown dashed line) and the diffraction peaks of the mono metal (cyan dashed line) disappeared within the first hour. This phenomenon corresponded to the transformation from mono metals into M1Zn1 intermetallic phases. As the temperature increased to 500 °C and 600 °C, the diffraction peaks of the intermetallic compounds slightly shifted to a lower theta angle, which was due to the lattice expansion induced by the higher temperature. When the sample was cooled to 50 °C, the diffraction peak at 42.4° corresponded well to the main peak of the RhZn intermetallic phase (PDF#53-0470). For Pd–Zn/SiO2, the diffraction peaks at 41.2° and 44.1° correspond to the main peaks of the PdZn intermetallic phase (PDF#06-0620). For Pt–Zn/SiO2, the diffraction peaks at 40.8° and 44.8° correspond to the main peaks of the PtZn intermetallic phase (PDF#06-0604). The in situ XRD results clearly depicted the phase transformation from monometallic NPs to the bulk intermetallic phase during the TPR procedure.
To further identify this phase evolution of the supported NPs during TPR, HAADF-STEM and energy dispersive X-ray spectroscopy (EDS) were employed to monitor the elemental distribution. Specific emphasis was placed on Rh–Zn/SiO2 catalysts with different reduction time (the PMC with a reduction time of t (h) at 400 °C is denoted as Rh–Zn/SiO2-t). The mean metal particle size of Rh–Zn/SiO2-0 was 1.82 nm, which increased to 2.41 nm after trapping Zn atoms during a reduction time of 10 h (Fig. S4†). The elemental mappings of Rh–Zn/SiO2-t are shown in Fig. 2a–d. The Zn/Rh ratio (Fig. S5–S7†) was 0.3 over Rh–Zn/SiO2-0.5 and increased to 0.7–1.0 when the reduction time was prolonged to 4 and 10 h, which was approximately equal to the nominal value of the Rh1Zn1 intermetallic phase.
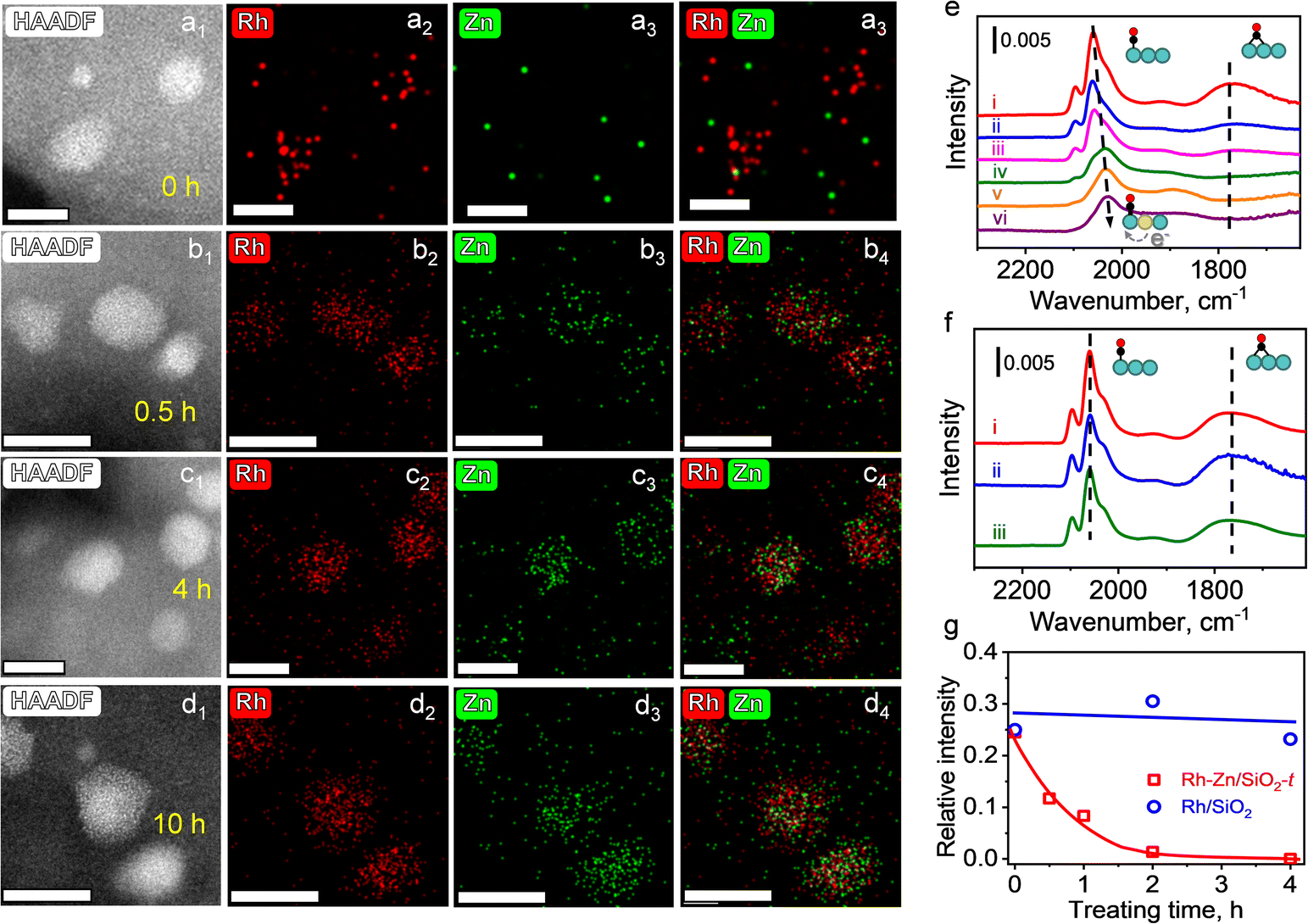 | ||
| Fig. 2 HAADF-STEM images and EDS mappings of Rh–Zn/SiO2-t catalysts with different reduction time of (a) 0 h, (b) 0.5 h, (c) 4 h, and (d) 10 h. (Scale bar: 5 nm). (e) CO adsorption on the Rh–Zn/SiO2-t catalysts with reduction time of (i) 0 h, (ii) 0.5 h, (iii) 1 h, (iv) 2 h, (v) 4 h, and (vi) 10 h. (f) CO adsorption on Rh/SiO2-t with reduction time of (i) 0 h, (ii) 2 h, and (iii) 4 h. (g) Relationship between reduction time and relative intensity of bridged CO to linear CO. The samples were pretreated by TPR and then transferred to the IR cell. CO-FTIR spectra were recorded at room temperature. | ||
With an increase in reduction time, the Zn element signal over the region of Rh element was steadily enhanced, providing evidence that Zn species migrated to the Rh NPs. It is also possible that Rh species migrated to Zn species, while the steadily enhanced Zn/Rh ratio suggested the migration of Zn to Rh but not the migration of Rh to Zn. The EDS elemental mapping also demonstrated the uniform distribution of Rh and Zn on individual NPs, that is, there was no segregation into core–shell or other hetero-structures. In addition, the Zn/Rh ratio in the NPs (Fig. S5–S7†) showed that a longer reduction time was beneficial for converting NPs with an ill-defined composition to uniform i-NPs.
In contrast, the co-impregnation method generally produces a mixture of monometallic and bimetallic NPs with ill-defined compositions.23,24 The bimetallic catalyst was prepared by conventional co-impregnation (denoted as RhZn/SiO2-IMP) for comparison. As shown in Fig. S8 and S9,† Rh and Zn were found to have different structures that ranged from completely segregated monometallic Rh NPs to well-mixed RhZn alloy NPs. Compared with the conventional impregnation method, the TPR of the PMC provides an efficient approach to preparing catalysts with uniform M1Zn1 i-NPs.
CO-FTIR experiments were performed to further examine the surface structure of the Rh–Zn/SiO2-t catalysts. The Rh–Zn/SiO2-0 catalyst (Fig. S10a†) mainly showed a linear CO peak at 2060 cm−1 and a broad bridged CO peak in the 1850–1700 cm−1 region. In addition, geminal dicarbonyl species at 2097 cm−1 and 2021 cm−1 and Rh2(CO)3 species at 1920 cm−1 were also observed.25,26 The CO-FTIR spectra of Rh–Zn/SiO2-t are shown in Fig. 2e. With increasing reduction time, the linear CO peak gradually shifted from 2060 to 2030 cm−1, and the bridged CO peak decreased. No bridged CO peak was observed at t = 4 and 10 h. For comparison, the RhOx/SiO2 sample with the same reduction procedure as Rh–Zn/SiO2-t but without physically mixing with ZnO/SiO2 (denoted as Rh/SiO2-t) was examined by CO-FTIR (Fig. 2f). With this sample, no redshift of linear CO and no decrease of bridged CO were observed. A quantitative analysis of the relative intensity (RI) of bridged CO to linear CO showed that the RI value remained at 0.25–0.3 over Rh/SiO2-t but decreased from 0.25 to 0 over Rh–Zn/SiO2-t with increasing reduction time (Fig. 2g). The above variation of CO adsorption over Rh–Zn/SiO2-t was ascribed to the formation of the Rh1Zn1 intermetallic phase. It has been reported that the electron donation from high electronegative metals (such as Sn and Zn) to noble metals leads to the red-shift of the linear CO peak.27–29 Similar changes of CO-FTIR were also observed in Pt1Zn1 and Pd1Zn1 i-NPs (Fig. S11†).
The charge transfer from Zn to Rh over Rh1Zn1 was verified by X-ray photoelectron spectroscopy (XPS). The Rh/SiO2 sample was physically mixed with excess ZnO for the Zn-ETO process. Most Zn species remained as ZnO and only a part of the Zn species was emitted and trapped by Rh NPs to form the Rh1Zn1 intermetallic phase at 400 °C. When prolonging the Zn-ETO reduction time from 0 to 4 h, the binding energy of Rh 3d5/2 shifted from 307.8 eV to 306.9 eV (Fig. S12a†). The Zn 2p3/2 binding energy of metallic Zn only shifts 0.25 eV to lower binding energy compared with that of ZnO.30 As shown in Fig. S12b,† the Zn 2p3/2 peak observed at 1022.3–1022.7 eV was the convolution of Zn species for ZnO and RhZn. When prolonging the Zn-ETO reduction time from 0.5 to 4 h, the EDS results (Fig. S5 and S6†) showed that the Zn/Rh ratio of RhZn NPs increased from 0.3 to 0.9, which indicated that more metallic Zn was formed in RhZn NPs, and thus the Zn 2p3/2 binding energy of Rh–Zn/SiO2-4 shifted to 1022.45 eV compared with that of Rh–Zn/SiO2-0.5 (1022.7 eV).
Bader charge analysis confirmed the charge transfer from Zn to Rh with an average of 0.39e over the Rh1Zn1 intermetallic phase. The d-band model was used to further analyze the CO adsorption over the corresponding surface. According to this model, the closer the d-band center value is to the Fermi level, the more unfilled the anti-bonding states are in the metal-adsorbate molecular orbital, and the stronger the adsorption of the adsorbate on the metal surface.31,32 In a simplified frontier molecular orbital description, the surface bond arises from the charge transfer from the 5σ orbital of CO to the metal, and the concurrent donation of charge from the metal to the 2π* orbital of CO (back-donation). The d-band center values calculated for Rh(111) and RhZn(110) are −1.68 and −0.85 eV, respectively (Fig. S12c†). This indicates a stronger adsorption of CO on the metal surface and weaker C–O bond vibration frequency. The calculated linear CO adsorption wavenumbers on the Rh atom were 1971.9 cm−1 and 1964.6 cm−1 over Rh(111) and Rh1Zn1(110), respectively. The DFT calculated IR shift and d-band center value change follow the same trend of the CO-FTIR experiment.
It is reported that the incorporation of metal M′ like Sn and Zn into the noble metal M like Pd, Pt, and Rh prevents the CO adsorption in the bridging site and threefold hollow site.33–35 The ensemble hollow site of M was separated by M′, while CO interacted much more weakly with the Zn and Sn site compared with the noble metal site, and favored the bonding configuration of CO on the top site of M.34,35 Herein, a well ordered Rh1Zn1 phase was formed by prolonging the reduction time. Compared with the Rh–Rh distance over Rh(111), the Rh–Rh distance was enlarged to 3.00 Å and 4.24 Å over Rh1Zn1(110) (Fig. S13†). CO is mainly adsorbed on Rh atoms but not on Zn atoms, and thus the bridged CO peak gradually disappeared and the linear CO peak became dominant.
The red-shift of the linear CO peak was ascribed to the charge transfer by alloying Rh with Zn. When increasing the reduction time from 0 to 10 h, the degree of alloying gradually increases from zero in monometallic Rh to a maximum in the intermetallic Rh1Zn1 phase, and thus the linear CO peak gradually shifts from 2060 to 2030 cm−1. The RhZn/SiO2-IMP catalysts with different reduction time were also characterized by CO-FTIR (Fig. S10b†). RhZn/SiO2-IMP contained heterogeneous structures varying from segregated monometallic Rh NPs to well-mixed RhZn alloy (as shown in Fig. S8 and S9†). The higher linear CO peak over RhZn/SiO2-IMP (2039 cm−1) than that over Rh–Zn/SiO2-10 (2030 cm−1) verified that RhZn/SiO2-IMP had a lower degree of RhZn alloying than Rh–Zn/SiO2-10. In addition, all the samples ((i)–(iii) in Fig. S10b†) showed similar spectra of CO adsorption and no shift of the linear CO peak was observed. These results indicated that a longer reduction time was ineffective for annealing RhZn/SiO2-IMP into an intermetallic phase.
Enhanced catalytic performance of the Rh1Zn1 phase and its dependence on reduction time
The selective hydrogenation of acetylene is a fundamental model reaction that focuses on appropriate hydrogenation reactivity to control overhydrogenation and leave the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond intact.21,24,36 Moreover, oligomerization produces C4 and GO by-products, which could be solved by the “site-isolation” concept to isolate adjacent adsorbate molecules to suppress oligomerization. The obtained Rh–Zn/SiO2-t catalysts were evaluated by acetylene hydrogenation (Fig. 3 and S14†). The HAADF-STEM, EDS, in situ XRD and CO-FTIR results confirmed the formation of Rh1Zn1 intermetallic phases over the PMC with increasing the reduction time. The relation between the denoted catalyst with the active phase is summarized in Fig. 3f. The obtained Rh1Zn1 catalysts showed enhanced selectivity and stability with an interesting reduction time dependence on catalytic performance.
C bond intact.21,24,36 Moreover, oligomerization produces C4 and GO by-products, which could be solved by the “site-isolation” concept to isolate adjacent adsorbate molecules to suppress oligomerization. The obtained Rh–Zn/SiO2-t catalysts were evaluated by acetylene hydrogenation (Fig. 3 and S14†). The HAADF-STEM, EDS, in situ XRD and CO-FTIR results confirmed the formation of Rh1Zn1 intermetallic phases over the PMC with increasing the reduction time. The relation between the denoted catalyst with the active phase is summarized in Fig. 3f. The obtained Rh1Zn1 catalysts showed enhanced selectivity and stability with an interesting reduction time dependence on catalytic performance.
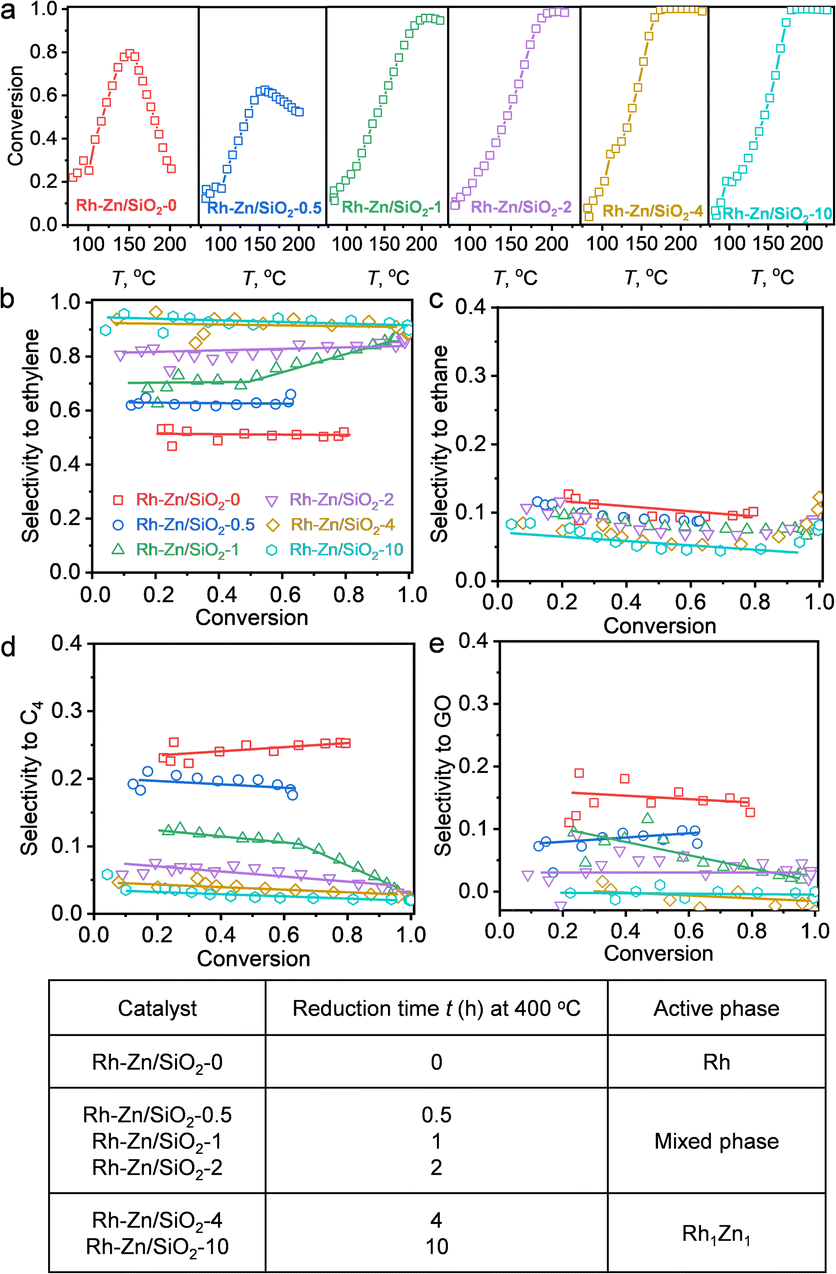 | ||
| Fig. 3 Reduction time dependent catalytic performance of Rh–Zn/SiO2-t catalysts in acetylene hydrogenation. (a) Acetylene conversion over Rh–Zn/SiO2-t catalysts during temperature programmed reaction. Selectivity to (b) ethylene, (c) ethane, (d) C4 and (e) GO versus acetylene conversion. (f) Relation between the denoted catalyst with the active phase. The product quantification was completed using GC (details of the calculation method are provided in the ESI†). | ||
With Rh–Zn/SiO2-0, the acetylene conversion first increased with increasing reaction temperature but started to decrease at 150 °C. This deactivation of the catalyst during the temperature programmed reaction was gradually suppressed by prolonging the catalyst reduction time and vanished with a reduction time of 4 and 10 h (Fig. 3a). The product distribution also varied significantly with the reduction time. With Rh–Zn/SiO2-0, the selectivity to ethylene was ∼50%, and ∼40% of acetylene was converted to C4 and GO. With Rh–Zn/SiO2-10, the selectivity to C4 was less than 3% and GO was almost undetectable, and thus the ethylene selectivity was enhanced to >90% (Fig. 3b–e). C4 is the precursor of oligomers and Rh NPs favored the C–C coupling reaction to C4 and further reaction to GO. These oligomers blocked the Rh active sites and deactivated the catalyst. The product distribution results showed that prolonging the catalyst reduction time significantly reduced the selectivity to C4 and GO. Thermogravimetric analysis (TGA) on spent Rh–Zn/SiO2-0 showed a mass loss at 200–400 °C, while spent Rh–Zn/SiO2-10 showed almost the same TGA curve as fresh Rh–Zn/SiO2 (Fig. S14†), that is, almost no accumulated oligomers. The spent Rh–Zn/SiO2-10 catalyst was further characterized by STEM-EDS and CO-FTIR (Fig. S15 and S16†). The individual NPs in the spent catalyst showed the signal of Rh and Zn elements. The CO-FTIR results showed that for Rh1Zn1 the linear CO peak was shifted to ∼2030 cm−1 with an undetectable broad bridged CO peak. The deactivation of the catalyst was mainly due to the formation of an oligomer.20,37,38 The Rh1Zn1 intermetallic phase was generated at 400 °C which could be thermally stable under the lower reaction temperature. The STEM-EDS and CO-FTIR also showed that the spent catalyst retained the morphology and surface structure of the intermetallic phase. RhZn/SiO2-IMP was used for comparison. It showed a slight deactivation during the temperature programmed reaction. Its selectivities to C4 and GO were both ∼10%, which were much higher than those over Rh–Zn/SiO2-4 and Rh–Zn/SiO2-10 (Fig. S17†). Long-term reaction evaluation of catalyst stability (Fig. S18†) showed that the conversion decreased from 100% to 40% in the first 4 h over RhZn/SiO2-IMP while it remained >99% over Rh–Zn/SiO2-10 for 12 h. These reduction time dependent results showed that the PMC with a long reduction time had decreased C4 and GO formation that significantly improved catalyst selectivity and stability.
The catalytic performance of Rh1Zn1 was compared with the reported intermetallic and alloy catalysts (Table S1†). Recently, many efforts have been made to design intermetallic catalysts. Rioux et al.5 designed Pd8Zn44 consisting of only Pd1 monomers on the surface, resulting in 85% selectivity to C2H4. Chen et al.3 developed a complex palladium hydride catalyst, CaPdH2, with a selectivity of 80%. The side reaction of C–C bond coupling to C4 and GO led to the low intrinsic selectivity to ethylene. For example, the PdCu with a B2 or fcc crystal phase showed 4–5% selectivity to ethane but almost 20% selectivity to C4, resulting in an ethylene selectivity of <80%.15 Herein we proposed a novel Zn-ETO strategy to design M1Zn1 intermetallic phases, and found that Rh1Zn1 was a new promising active site for suppressing the C–C bond coupling reaction and obtaining high intrinsic selectivity to ethylene. The Rh–Zn/SiO2-4 was further evaluated under the tail-end acetylene hydrogenation conditions with excess ethylene in the feed (Fig. S19†). A high selectivity to ethylene (94%) was obtained with a low selectivity to C4, which was consistent with the results in pure acetylene hydrogenation.
To further compare the Rh1Zn1 with the Pd-based and Pt-based catalysts, Pd1Zn1 and Pt1Zn1 catalysts prepared by Zn-ETO were also evaluated by acetylene hydrogenation (Fig. S20 and Table S3†). With the Pd–Zn/SiO2-0 catalyst, the selectivities to ethylene, ethane, C4, and GO were 55%, 13%, 16%, and 16%, respectively. By prolonging the catalyst reduction time of Zn-ETO, the selectivity to ethylene with the Pd–Zn/SiO2-4 catalyst was enhanced to 81% with the decrease of SC2H6 and SGO. Pt–Zn/SiO2-0 showed a very low selectivity to ethylene (<30%) and the main product is ethane. With the formation of the Pt1Zn1 intermetallic phase (Pt–Zn/SiO2-4), the over-hydrogenation of ethylene was significantly suppressed, giving a 60–70% selectivity to ethylene. The characterization results (Fig. 1 and S3†) indicated that Pd1Zn1 and Pt1Zn1 intermetallic phases were formed with a reduction time of 4 h (Pd–Zn/SiO2-4 and Pt–Zn/SiO2-4). The product selectivity over Pd–Zn/SiO2-4 and Pt–Zn/SiO2-4 indicated that Pd1Zn1 and Pt1Zn1 mainly suppress the over-hydrogenation of ethylene. Compared with Pd1Zn1, Pt1Zn1 and the catalyst in previous work, Rh1Zn1 had a higher intrinsic selectivity to ethylene (>91%) and much lower selectivity to C4 (2.2%) and GO (<0.5%), which outperforms the reported intermetallic and alloy catalysts.
The isolated Rh atom ensembles on the well-defined Rh1Zn1 intermetallic phase facilitate a theoretical study of how the C–C coupling reaction is suppressed. We calculated the reaction pathways of butadiene formation on Rh(111) and RhZn(110) (Fig. 4). The Rh3 site in the Rh(111) surface was assigned to be the starting adsorption site for further calculation. Adjacent Rh3 sites (Rh–Rh: 2.68 Å) on the Rh(111) surface give spatial proximity to the reaction intermediates (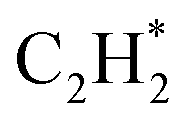 and H*), which facilitates C–C and C–H bond formation between the two intermediates adsorbed on the same NP. In contrast, on the RhZn(110) surface, the elongated Rh2 bridge sites (Rh–Rh: 3.00 Å and 4.24 Å), linearly distributed on the NPs, restrict the adsorption of adjacent reaction intermediates. In addition, the position of the H atom is restricted on the Rh2 bridge site to be on the left or right or the next line of hydrocarbons on RhZn(110). For the final step of butadiene formation, a spatially separated H needs to climb over a zinc atom to form the final C–H bond, which significantly slows down the formation of butadiene. The reaction trajectory (Fig. 4 and Table S4†) indicates that on Rh(111), the rate determining step of butadiene formation is C–C bond formation with an activation energy of 0.58 eV, while on RhZn(110), it is the third step with an activation energy of 1.40 eV, which is due to the structure-induced difficulty in C–H bond formation. It should be noted that the C–C coupling reaction on both Rh(111) and RhZn(110) follows the same pattern: C2H2 and
and H*), which facilitates C–C and C–H bond formation between the two intermediates adsorbed on the same NP. In contrast, on the RhZn(110) surface, the elongated Rh2 bridge sites (Rh–Rh: 3.00 Å and 4.24 Å), linearly distributed on the NPs, restrict the adsorption of adjacent reaction intermediates. In addition, the position of the H atom is restricted on the Rh2 bridge site to be on the left or right or the next line of hydrocarbons on RhZn(110). For the final step of butadiene formation, a spatially separated H needs to climb over a zinc atom to form the final C–H bond, which significantly slows down the formation of butadiene. The reaction trajectory (Fig. 4 and Table S4†) indicates that on Rh(111), the rate determining step of butadiene formation is C–C bond formation with an activation energy of 0.58 eV, while on RhZn(110), it is the third step with an activation energy of 1.40 eV, which is due to the structure-induced difficulty in C–H bond formation. It should be noted that the C–C coupling reaction on both Rh(111) and RhZn(110) follows the same pattern: C2H2 and 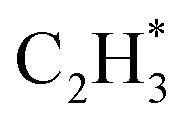 adsorbed on a shared Rh atom and the C–C bond is formed on and above that Rh atom. This leads to the similar activation energies for the C–C coupling reaction on these two surfaces. The DFT calculation results confirmed that the C–C coupling reaction to C4 was suppressed on the intermetallic Rh1Zn1 surface.
adsorbed on a shared Rh atom and the C–C bond is formed on and above that Rh atom. This leads to the similar activation energies for the C–C coupling reaction on these two surfaces. The DFT calculation results confirmed that the C–C coupling reaction to C4 was suppressed on the intermetallic Rh1Zn1 surface.
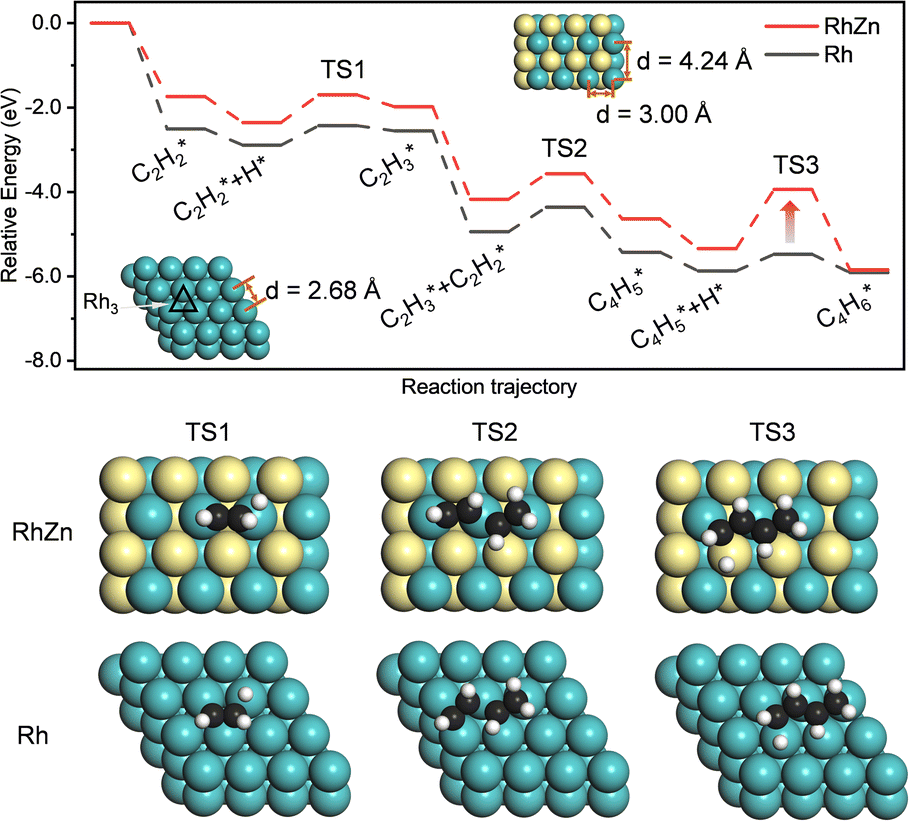 | ||
| Fig. 4 Reaction pathway and key structures in the transitional steps of butadiene formation on Rh(111) and RhZn(110) surfaces. Rh, Zn, C, and H atoms are in cyan, yellow, black, and white, respectively. | ||
The mechanism of intermetallic M1Zn1 phase formation in Zn-ETO
The reaction and characterization results above showed that the PMC underwent a migration of Zn to form intermetallic M1Zn1 phases, and its transformation was kinetically controlled by the reduction time and atmosphere. We further carried out the following experiment (4-packing mode experiment) to reveal the Zn-ETO mechanism and the apparent Zn transformation distance (Fig. 5). Four packing modes were used to represent different mixing degrees of Rh/SiO2 and ZnO/SiO2 (Fig. 5a). Mode 1 was a completely separate mode with ZnO/SiO2 located upstream and RhOx/SiO2 located downstream. The same amount of precursor powder was equally divided into 4, 8 and 16 portions and packed as modes 2, 3 and 4, respectively.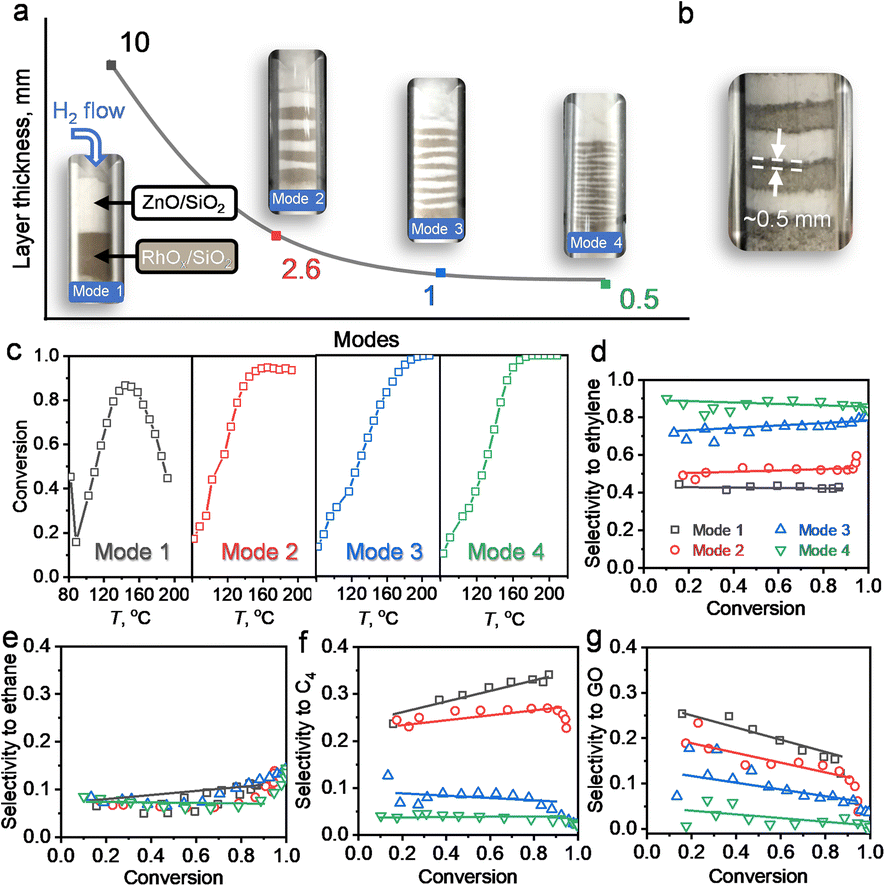 | ||
| Fig. 5 Catalytic performance using different packing modes for the temperature programmed reduction. (a) Different packing modes and layer thickness of ZnO/SiO2 and RhOx/SiO2. The photos were taken before catalyst reduction. (b) Photo of mode 2 after TPR treatment. (c) Acetylene conversion over the catalysts with different packing modes during temperature programmed reaction. Selectivity to (d) ethylene, (e) ethane, (f) C4 and (g) GO versus acetylene conversion. To divide the samples into several layers, 0.1 g ZnO/SiO2 and 0.1 g RhOx/SiO2 were separately pre-mixed with 0.4 g quartz sand of 80–120 mesh. All packing modes were treated by TPR with a reduction time of 10 h before reaction evaluation. | ||
Upon treatment by the TPR process, the mixed samples of modes 1 and 4 were recollected and characterized by EDS and CO-FTIR (Fig. S21 and S22†). For mode 1, the nanoparticles only showed the Rh element signal and the CO-FTIR spectrum showed the typical peaks of mono-Rh metal with a linear CO peak at 2060 cm−1 and a broad bridged CO peak in the 1850–1700 cm−1 region. For mode 4, the individual NPs showed the signal of Rh and Zn elements, and the CO-FTIR results indicated that the linear CO peak was shifted to 2035 cm−1 and the broad bridged CO peak was almost undetectable. The reaction evaluation indicated that mode 1 showed rapid catalyst deactivation during the reaction and the selectivities to C4 and GO both reached ∼20%, which was similar to that over Rh–Zn/SiO2-0. In modes 2 and 3, catalyst deactivation was suppressed and ethylene selectivity was enhanced to ∼50% and ∼80%, respectively. Mode 4 exhibited the same excellent catalytic performance as Rh–Zn/SiO2-10, enhancing ethylene selectivity to ∼90% with minor C4 and GO formation.
The characterization and reaction results showed that the atmosphere in the catalyst bed during TPR was similar in mode 4 and PMC, indicating that the maximum diffusion distance between the catalyst components is ∼0.5 mm under the current reduction treatment. The observed darker layer of ∼0.5 mm in the Rh/SiO2 layer after the TPR process also confirmed this distance (Fig. 5b). For packing modes 1–3, the Rh1Zn1 i-NPs were only formed in the partial Rh/SiO2 layer with a thickness of ∼0.5 mm which is directly connected to the downstream ZnO layer. For packing mode 4, the Rh/SiO2 sample was divided into 16 portions with each layer thickness of ∼0.5 mm. Most of the Rh NPs in mode 4 were ordering into the Rh1Zn1 phase, and thus the catalyst in mode 4 showed similar reaction performance to Rh–Zn/SiO2-10.
The reaction and characterization results indicated that the PMC underwent a migration of Zn to form intermetallic M1Zn1 phases. The migration of metal species was also reported in the re-dispersion of metal nanoparticles into single atoms.39–41 Zhang et al.42 indicated that the thermal aging dispersion of RuO2 powders into Ru single atoms underwent an anti-Ostwald ripening which was promoted by a strong covalent metal support interaction. Herein, the characterization indicated that the Zn was selectively trapped by M metal NPs rather than dispersed into single atoms. In addition, the anti-Ostwald ripening generally required a much higher aging temperature of >900 °C and a strong interaction support like FeOx. The 4-packing mode experiment indicated that the Zn migration distance was in the mm level. Therefore, the formation of the M1Zn1 phase for the PMC could undergo a gas phase migration rather than anti-Ostwald ripening.
Several works indicated that the decomposition of metal nanoparticles into single atoms underwent gas phase transfer. The volatile metal oxide species was generated from metal NPs at high temperature (∼800 °C) under an oxidizing environment and was further deposited on the specific supports.41,43 ZnO decomposes into Zn2+ and O2− at 1350 °C in a vacuum environment44,45 or to Zn atoms by thermal reduction at ≥550 °C with H2.46,47 Here, we found that Zn could transfer from ZnO at a lower temperature of 400 °C under H2 flow. In order to identify the Zn species for gas phase transformation, we carried out DFT calculations to study the thermodynamics. The DFT calculation results (Tables S5 and S6†) indicated that in the inert environment, a ZnO molecule evaporates from a ZnO(221) step surface with a high energy requirement of >3 eV at a temperature below 500 °C ((i) in Fig. 6a and b). In a reducing environment, however, a free Zn atom evaporates with the simultaneous formation of a H2O molecule and the energy required is only −0.31 eV at 500 °C ((ii) in Fig. 6a and b). Thus the Zn-ETO mechanism in which Zn atom emission is the first step is thermodynamically favored by coupling with water formation. In addition, temperature plays a critical role in Zn-ETO. The evaporation of Zn atoms is endothermic (0.18 eV) at 300 °C but is slightly exothermic (−0.06 eV) at 400 °C on a ZnO(221) step, which is consistent with the experimental results. The 4-packing mode experiments indicated that the apparent Zn transformation distance was ∼0.5 mm. It is suggested that only a small fraction of Zn atoms were emitted from ZnO at the critical temperature of 400 °C, which generated a constant Zn atom atmosphere over a local area.
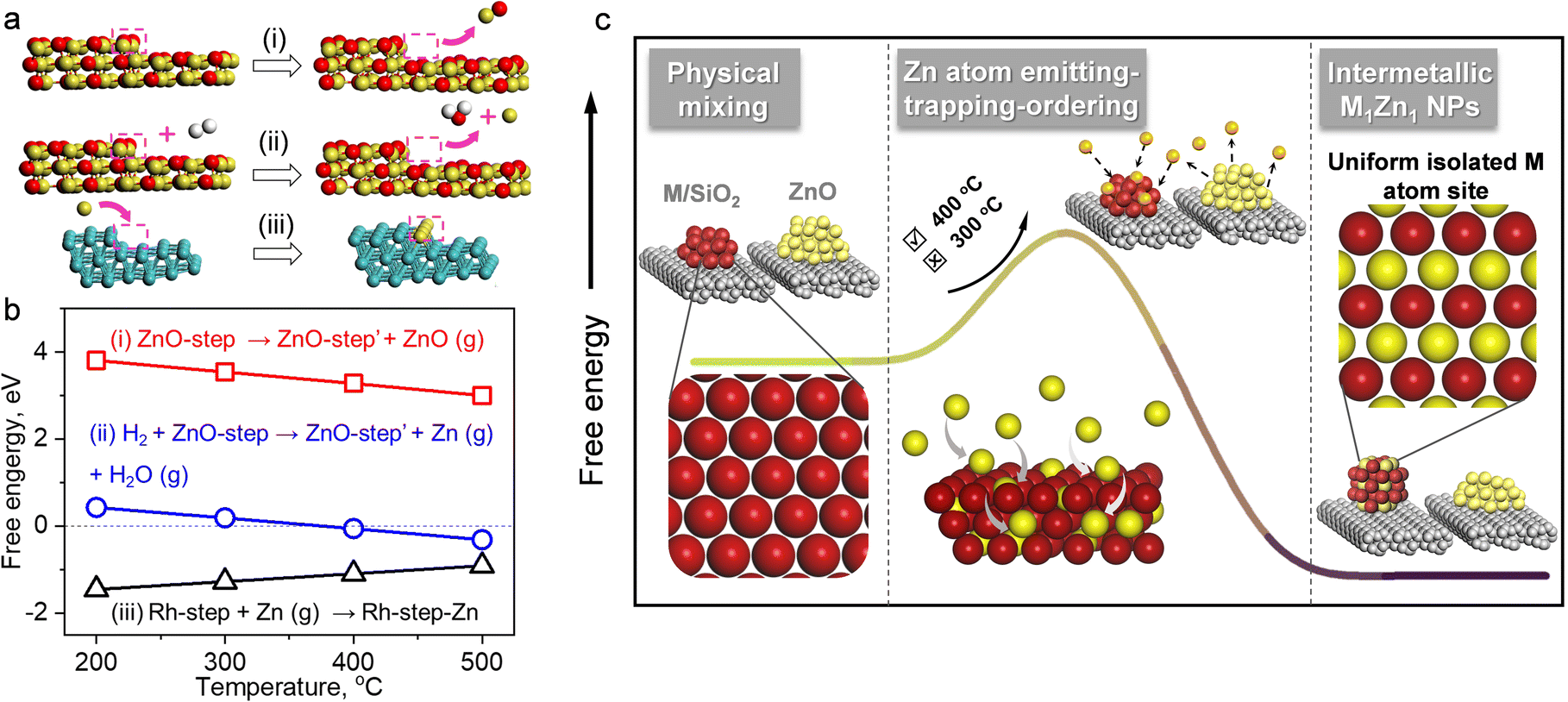 | ||
| Fig. 6 (a) Optimized structures from DFT calculations and (b) Gibbs free energy of (i) direct emission of ZnO, (ii) Zn atom emission with the assistance of H2O formation and (iii) trapping of Zn atoms on Rh step sites. Rh, Zn, O, and H atoms are in cyan, yellow, red, and white, respectively. (c) Schematic of intermetallic M1Zn1 phase formation by the Zn-ETO mechanism. | ||
The emitted Zn atoms are adsorbed and trapped by Rh NPs. We confirmed by DFT calculations that trapping a Zn atom on a Rh(221) step surface is exothermic with a low barrier ((iii) in Fig. 6a and b). This mild temperature is essential for preparing M1Zn1 i-NP catalysts because a high temperature would cause Ostwald ripening of metal NPs. The formation of an intermetallic phase from a bimetallic random alloy is driven by both thermodynamic and kinetic factors.1 For the thermodynamic factor, the intermetallic phase is energetically favored over the disordered alloy at low temperatures. For the kinetic factor, the thermal emission of Zn atoms from the ZnO surface generated a Zn atmosphere over a local area of the NPs for the trapping and ordering steps, which provided an effective driving force to access the intermetallic phase at lower temperature. In conventional co-impregnation, the pre-nucleation step inevitably generates various NPs with compositions ranging from a segregated mono-metal to a well-mixed bimetallic alloy, which required a much higher temperature to overcome the barrier for redistribution. The high temperature annealing would cause Ostwald ripening of metal NPs.
With the above discussion, we then proposed a Zn emitting–trapping–ordering (Zn-ETO) mechanism in which Zn atoms were emitted from ZnO into the gas phase with the thermodynamic assistance of H2 oxidation to H2O. These Zn atoms were adsorbed and trapped by noble metal NPs. The trapped Zn atoms diffused into the NPs, and finally the Zn and M atoms rearranged to form M1Zn1 i-NPs.
In the Zn-ETO approach (Fig. 6c), the monometallic NPs were used as the starting phase. The thermal emission of Zn atoms from the ZnO surface provides a Zn atmosphere over a local area of the NPs for the trapping and ordering steps. That is, each monometallic NP was in contact with the same and constant Zn atom atmosphere, unrestricted by the predetermined M/Zn ratio. The thermodynamic driving force of the disorder-to-order transition resulted in the formation of stable intermetallic compound NPs, independent of the particle size distribution of the starting monometallic NPs. The strong affinity between the host metal atoms and Zn atoms dominates the thermodynamic stability of the M1Zn1 phase and the kinetics of the penetration of Zn atoms into the bulk, which is the key to the success of the Zn-ETO mechanism.
For the proposed Zn-ETO strategy, the starting monometallic NPs are prepared by traditional impregnation and only need to be physically mixed with ZnO species for further reduction by H2 flow. All of the steps are suitable for scalable synthesis. As a future perspective, it is feasible to directly use cadmium under the same conditions as in Zn-ETO with the assistance of H2 flow. For other metals with higher boiling point, it is possible to emit the metal species with the assistance of other gas flow like CO or NH3, considering that the formation of a carbonyl metal compound or amino metal compound is feasible in thermodynamics.
Conclusions
In summary, we developed an industrially feasible and controllable strategy for preparing M1Zn1 (M = Rh, Pd, Pt) intermetallic phase catalysts by reduction of a physical mixture of monometallic M/SiO2 and ZnO. HAADF-STEM, in situ XRD, EDS and CO-FTIR results confirm that the obtained M1Zn1 i-NP catalysts have well-defined isolated M sites dictated by the crystal geometry and composition. Experiments and DFT calculations indicate that the isolated Rh atom sites on the Rh1Zn1 i-NPs realize the catalytic “site-isolation” concept for acetylene hydrogenation, that is, these sites significantly suppress C–C coupling to C4 and GO and thus greatly enhance ethylene selectivity to ∼90% and catalyst stability, outperforming other reported intermetallic compounds. Further DFT calculations and condition experiments demonstrate that this synthesis strategy follows a Zn-ETO mechanism, in which Zn atoms are emitted from ZnO and trapped on monometallic NPs in a reducing environment, and the NPs are ordered into an intermetallic M1Zn1 phase. The reducing temperature of 400 °C is a sufficiently high temperature for emitting Zn atoms from ZnO with the assistance of H2O formation. This temperature thermodynamically favors the M1Zn1 intermetallic phase, and is low enough to avoid i-NP sintering. This study provides an understanding of the formation mechanism of the M1Zn1 intermetallic compound structure, and would open fresh avenues for designing Zn-containing multi-component catalysts.Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
T. W. and X. L. directed the project. Y. W. and B. L. conducted DFT calculations. X. L., Y. W. and Z. K. carried out catalyst synthesis, characterization and catalytic tests. X. L., Y. W., B. L. and T. W. contributed to data analysis and wrote the manuscript. X. L. and Y. W. contributed equally to this work. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22108148 and 22178195). DFT calculations in this work are supported by Tsinghua National Laboratory for Information Science and Technology.References
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Intermetallic nanocrystals: syntheses and catalytic applications, Adv. Mater., 2017, 29(14), 1605997 CrossRef PubMed.
- S. Furukawa and T. Komatsu, Intermetallic compounds: promising inorganic materials for well-structured and electronically modified reaction environments for efficient catalysis, ACS Catal., 2017, 7(1), 735–765 CrossRef CAS.
- Q. Guo, R. Chen, J. Guo, C. Qin, Z. Xiong, H. Yan, W. Gao, Q. Pei, A. Wu and P. Chen, Enabling semihydrogenation of alkynes to alkenes by using a calcium palladium complex hydride, J. Am. Chem. Soc., 2021, 143(49), 20891–20897 CrossRef CAS PubMed.
- X.-T. Li, L. Chen, C. Shang and Z.-P. Liu, In situ surface structures of pdag catalyst and their influence on acetylene semihydrogenation revealed by machine learning and experiment, J. Am. Chem. Soc., 2021, 143(16), 6281–6292 CrossRef CAS PubMed.
- A. Dasgupta, H. He, R. Gong, S.-L. Shang, E. K. Zimmerer, R. J. Meyer, Z.-K. Liu, M. J. Janik and R. M. Rioux, Atomic control of active-site ensembles in ordered alloys to enhance hydrogenation selectivity, Nat. Chem., 2022, 14(5), 523–529 CrossRef CAS.
- X. Ge, M. Dou, Y. Cao, X. Liu, Q. Yuwen, J. Zhang, G. Qian, X. Gong, X. Zhou, L. Chen, W. Yuan and X. Duan, Mechanism driven design of trimer Ni1Sb2 site delivering superior hydrogenation selectivity to ethylene, Nat. Commun., 2022, 13(1), 5534 CrossRef CAS PubMed.
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, PdZn intermetallic nanostructure with Pd–Zn–Pd ensembles for highly active and chemoselective semi-hydrogenation of acetylene, ACS Catal., 2016, 6(2), 1054–1061 CrossRef CAS.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, Sub-4 nm PtZn intermetallic nanoparticles for enhanced mass and specific activities in catalytic electrooxidation reaction, J. Am. Chem. Soc., 2017, 139(13), 4762–4768 CrossRef CAS PubMed.
- A. Miura, H. Wang, B. M. Leonard, H. D. Abruña and F. J. DiSalvo, Synthesis of intermetallic PtZn nanoparticles by reaction of Pt nanoparticles with Zn vapor and their application as fuel cell catalysts, Chem. Mater., 2009, 21(13), 2661–2667 CrossRef CAS.
- C. L. Yang, L. N. Wang, P. Yin, J. Liu, M. X. Chen, Q. Q. Yan, Z. S. Wang, S. L. Xu, S. Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H. W. Liang, Sulfur-anchoring synthesis of platinum intermetallic nanoparticle catalysts for fuel cells, Science, 2021, 374(6566), 459–464 CrossRef CAS.
- J. Lu, K. B. Low, Y. Lei, J. A. Libera and J. W. Elam, Toward atomically-precise synthesis of supported bimetallic nanoparticles using atomic layer deposition, Nat. Commun., 2014, 5(1), 3264 CrossRef.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Bimetallic nanocrystals: syntheses, properties, and applications, Chem. Rev., 2016, 116(18), 10414–10472 CrossRef CAS PubMed.
- M. Cargnello, Colloidal nanocrystals as building blocks for well-defined heterogeneous catalysts, Chem. Mater., 2019, 31(3), 576–596 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem, 2018, 2(6), 65–81 CrossRef CAS.
- S. Liu, Y. Li, X. Yu, S. Han, Y. Zhou, Y. Yang, H. Zhang, Z. Jiang, C. Zhu, W.-X. Li, C. Wöll, Y. Wang and W. Shen, Tuning crystal-phase of bimetallic single-nanoparticle for catalytic hydrogenation, Nat. Commun., 2022, 13(1), 4559 CrossRef CAS PubMed.
- W. Zhang, X. Zhang, J. Wang, A. Ghosh, J. Zhu, N. J. LiBretto, G. Zhang, A. K. Datye, W. Liu and J. T. Miller, Bismuth-modulated surface structural evolution of Pd3Bi intermetallic alloy catalysts for selective propane dehydrogenation and acetylene semihydrogenation, ACS Catal., 2022, 12(17), 10531–10545 CrossRef CAS.
- Y. Cao, H. Zhang, S. Ji, Z. Sui, Z. Jiang, D. Wang, F. Zaera, X. Zhou, X. Duan and Y. Li, Adsorption site regulation to guide atomic design of Ni–Ga catalysts for acetylene semi-hydrogenation, Angew. Chem., Int. Ed., 2020, 59(28), 11647–11652 CrossRef CAS.
- Y. Niu, X. Huang, Y. Wang, M. Xu, J. Chen, S. Xu, M.-G. Willinger, W. Zhang, M. Wei and B. Zhang, Manipulating interstitial carbon atoms in the nickel octahedral site for highly efficient hydrogenation of alkyne, Nat. Commun., 2020, 11(1), 3324 CrossRef CAS PubMed.
- X. Ge, Z. Ren, Y. Cao, X. Liu, J. Zhang, G. Qian, X. Gong, L. Chen, X. Zhou, W. Yuan and X. Duan, Enhanced acetylene semi-hydrogenation on a subsurface carbon tailored Ni–Ga intermetallic catalyst, J. Mater. Chem. A, 2022, 10(37), 19722–19731 RSC.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, Pd−Ga intermetallic compounds as highly selective semihydrogenation catalysts, J. Am. Chem. Soc., 2010, 132(42), 14745–14747 CrossRef PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Srensen and J. K. Nrskov, Identification of non-precious metal alloy catalysts for selective hydrogenation of acetylene, Science, 2008, 320(5881), 1320–1322 CrossRef CAS PubMed.
- A. Pachulski, R. Schödel and P. Claus, Performance and regeneration studies of Pd–Ag/Al2O3 catalysts for the selective hydrogenation of acetylene, Appl. Catal., A, 2011, 400(1), 14–24 CrossRef CAS.
- A. Wong, Q. Lin, S. Griffin, A. Nicholls and J. R. Regalbute, Synthesis of ultrasmall, homogeneously alloyed, bimetallic nanoparticles on silica supports, Science, 2017, 358(6369), eaao6538 CrossRef PubMed.
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, A general synthesis approach for supported bimetallic nanoparticles via surface inorganometallic chemistry, Science, 2018, 362(6414), 560–564 CrossRef CAS PubMed.
- I. A. Fisher and A. T. Bell, A Comparative study of CO and CO2 hydrogenation over Rh/SiO2, J. Catal., 1996, 162(1), 54–65 CrossRef CAS.
- H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, CO2 hydrogenation reactivity and structure of Rh/SiO2 catalysts prepared from acetate, chloride and nitrate precursors, Appl. Catal., A, 2001, 205(1), 285–294 CrossRef CAS.
- Y. Pei, Z. Qi, T. W. Goh, L.-L. Wang, R. V. Maligal-Ganesh, H. L. MacMurdo, S. Zhang, C. Xiao, X. Li, F. Tao, D. D. Johnson and W. Huang, Intermetallic structures with atomic precision for selective hydrogenation of nitroarenes, J. Catal., 2017, 356, 307–314 CrossRef CAS.
- J. Camacho-Bunquin, M. S. Ferrandon, H. Sohn, A. J. Kropf, C. Yang, J. Wen, R. A. Hackler, C. Liu, G. Celik, C. L. Marshall, P. C. Stair and M. Delferro, Atomically precise strategy to a PtZn alloy nanocluster catalyst for the deep dehydrogenation of n-butane to 1,3-butadiene, ACS Catal., 2018, 8(11), 10058–10063 CrossRef CAS.
- Y. Pei, B. Zhang, R. V. Maligal-Ganesh, P. J. Naik, T. W. Goh, H. L. MacMurdo, Z. Qi, M. Chen, R. K. Behera, I. I. Slowing and W. Huang, Catalytic properties of intermetallic platinum-tin nanoparticles with non-stoichiometric compositions, J. Catal., 2019, 374, 136–142 CrossRef CAS.
- C. D. Wagner and G. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer, 1979 Search PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Effect of Strain on the Reactivity of Metal Surfaces, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Effects of d-band shape on the surface reactivity of transition-metal alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 89, 115114 CrossRef.
- H. W. Jen, Y. Zheng, D. F. Shriver and W. M. H. Sachtler, Characterization of Zn-promoted Rh/SiO2 catalysts by chemisorption, infrared spectroscopy, and temperature-programmed desorption, J. Catal., 1989, 116(2), 361–372 CrossRef CAS.
- Z.-X. Chen, K. M. Neyman, K. H. Lim and N. Rösch, CH3O decomposition on PdZn(111), Pd(111), and Cu(111). a theoretical study, Langmuir, 2004, 20(19), 8068–8077 CrossRef CAS.
- E. Jeroro, V. Lebarbier, A. Datye, Y. Wang and J. M. Vohs, Interaction of CO with surface PdZn alloys, Surf. Sci., 2007, 601(23), 5546–5554 CrossRef CAS.
- P. Wu, S. Tan, J. Moon, Z. Yan, V. Fung, N. Li, S.-Z. Yang, Y. Cheng, C. W. Abney, Z. Wu, A. Savara, A. M. Momen, D.-e. Jiang, D. Su, H. Li, W. Zhu, S. Dai and H. Zhu, Harnessing strong metal–support interactions via a reverse route, Nat. Commun., 2020, 11(1), 3042 CrossRef CAS.
- A. Borodziński and G. C. Bond, Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts. Part 1. effect of changes to the catalyst during reaction, Catal. Rev., 2006, 48(2), 91–144 CrossRef.
- A. Borodziński and G. C. Bond, Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts, Part 2: steady-state kinetics and effects of palladium particle size, carbon monoxide, and promoters, Catal. Rev., 2008, 50(3), 379–469 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Thermally stable single-atom platinum-on-ceria catalysts via atom trapping, Science, 2016, 353(6295), 150–154 CrossRef CAS.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms, Nat. Catal., 2018, 1(10), 781–786 CrossRef CAS.
- E. D. Goodman, A. C. Johnston-Peck, E. M. Dietze, C. J. Wrasman, A. S. Hoffman, F. Abild-Pedersen, S. R. Bare, P. N. Plessow and M. Cargnello, Catalyst deactivation via decomposition into single atoms and the role of metal loading, Nat. Catal., 2019, 2(9), 748–755 CrossRef CAS.
- K. Liu, X. Zhao, G. Ren, T. Yang, Y. Ren, A. F. Lee, Y. Su, X. Pan, J. Zhang, Z. Chen, J. Yang, X. Liu, T. Zhou, W. Xi, J. Luo, C. Zeng, H. Matsumoto, W. Liu, Q. Jiang, K. Wilson, A. Wang, B. Qiao, W. Li and T. Zhang, Strong metal-support interaction promoted scalable production of thermally stable single-atom catalysts, Nat. Commun., 2020, 11(1), 1263 CrossRef CAS PubMed.
- R. Lang, W. Xi, J.-C. Liu, Y.-T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A.-Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Non defect-stabilized thermally stable single-atom catalyst, Nat. Commun., 2019, 10(1), 234 CrossRef PubMed.
- D. Kohl, M. Henzler and G. Heiland, Low temperature sublimation processes from clean cleaved polar surfaces of zinc oxide crystals during first heating, Surf. Sci., 1974, 41(2), 403–411 CrossRef.
- X. Y. Kong and Z. L. Wang, Spontaneous polarization-induced nanohelixes, nanosprings, and nanorings of piezoelectric nanobelts, Nano Lett., 2003, 3(12), 1625–1631 CrossRef CAS.
- J. Goldberger, R. He, Y. Zhang, S. Lee, H. Yan, H.-J. Choi and P. Yang, Single-crystal gallium nitride nanotubes, Nature, 2003, 422(6932), 599–602 CrossRef CAS.
- D. Zhao, X. Tian, D. E. Doronkin, S. Han, V. A. Kondratenko, J.-D. Grunwaldt, A. Perechodjuk, T. H. Vuong, J. Rabeah, R. Eckelt, U. Rodemerck, D. Linke, G. Jiang, H. Jiao and E. V. Kondratenko, In situ formation of ZnOx species for efficient propane dehydrogenation, Nature, 2021, 599(7884), 234–238 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: TPR procedure, experimental details, HAADF-STEM details and DFT calculation details. See DOI: https://doi.org/10.1039/d3sc05460h |
| ‡ X. Lan and Y. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |