
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal-free strategy to construct fluoroalkyl–olefin linkages using fluoroalkanes†
Kaushik
Chakrabarti
a,
Michael M.
Wade Wolfe
a,
Shuo
Guo
 b,
Joseph W.
Tucker
c,
Jisun
Lee
c and
Nathaniel K.
Szymczak
*a
b,
Joseph W.
Tucker
c,
Jisun
Lee
c and
Nathaniel K.
Szymczak
*a
aDepartment of Chemistry, University of Michigan, 930 N. University, Ann Arbor, Michigan 48109, USA. E-mail: nszym@umich.edu
bCollege of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot 010021, P.R. China
cMedicine Design, Pfizer Inc., Eastern Point Rd, Groton, CT 06340, USA
First published on 22nd December 2023
Abstract
We present a metal-free strategy to access fluoroalkyl–olefin linkages from fluoroalkane precursors and vinyl-pinacol boronic ester (BPin) reagents. This reaction sequence is templated by the boron reagent, which induces C–C bond formation upon oxidation. We developed this strategy into a one-pot synthetic protocol using RCF2H precursors directly with vinyl-BPin reagents in the presence of a Brønsted base, which tolerated oxygen- and nitrogen-containing heterocycles, and aryl halogens. We also found that HCF3 (HCF-23; a byproduct of the Teflon industry) and CH2F2 (HCF-32; a low-cost refrigerant) are amenable to this protocol, representing distinct strategies to generate RCF2H and RCF3 molecules. Finally, we demonstrate that the vinyldifluoromethylene products can be readily derivatized, representing an avenue for late-stage modification after installing the fluoroalkyl unit.
Introduction
Fluorine-containing functional groups are often incorporated within medicinally-relevant compounds to modulate molecular properties, including metabolic stability, lipophilicity, stereoelectronics, and affinity to biological targets.1,2 In addition to these general features, difluoromethylene units (–CF2–) are considered as bioisosteres of oxygen atoms in ether, carbonyl, and sulfonyl groups.3 For example, within a series of ledipasvir (GS-5885) variants that feature an Ar–X–Ar linkage, highest activity was found when X = CF2, compared to –O– or –CH2– analogues.4 Despite their potential advantages, molecules containing Ar–CF2–R and Ar–CF2–Ar linkages are not common in commercial pharmaceuticals, likely due to limited synthetic pathways for direct –CF2–R incorporation.Metal-free routes to prepare Ar–CF2–alkyl and Ar–CF2–Ar linkages include radical difluorination of C–H bonds using fluorine atom transfer reagents (e.g. Selectfluor),5,6 or fluorodeoxygenation of ketones using trifluorosulfuranes (e.g. DAST; Et2NSF3)7,8 and defluorinative functionalization.9–13 Many of these strategies require reagents that are toxic, explosive,14 and of limited scope, which lessen their synthetic utility.15 Metal-catalyzed routes include reactions of aryl/alkyl boronic acids with ArCF2X; (X = halide, amides),16–19 and recently, reactions of ArCF2SiMe3 with aryl halides.20 However, limitations of these approaches include: (a) low availability of ArCF2X electrophiles and pronucleophiles, and (b) low compatibility of procedures needed to prepare the requisite reagents.9,21–24
Difluoromethanes (RCF2H; R = H, R = Ar) are widely available pronucleophiles25 that can be unmasked by deprotonation,26 and represent an attractive entry point to access R–CF2–R′ molecules. A key challenge to broad use of RCF2H compounds as synthons is that upon deprotonation, RCF2− anions decompose to fluorocarbenes via fluoride elimination, even at ambient temperatures.27 To overcome this deleterious pathway, our group developed a strategy to use Lewis acids (e.g. hexamethylborazine; B3N3Me6) to stabilize RCF2− anions against defluorination.28–33
One class of fluorinated compounds that is underdeveloped is aryl/alkyl difluoromethylene containing internal olefins.34 These moieties are an attractive functionality in medicinal chemistry, and are present within marketed drugs such as tafluprost and glecaprevir (Fig. 1a).35,36 Such motifs are particularly desirable not only because fluorine atoms may improve metabolic stability and/or binding affinity with the target of interest,37 but also because they are amenable to late-stage functionalization.38,39 This latter feature exploits an olefin as a functional handle to build molecular complexity and/or as a branch point for structure–activity relationship studies in drug discovery. Unfortunately, there are limited synthetic strategies to access vinyldifluoromethylene linkages and most proceed by coupling RCF2Br (R = aryl, alkyl, ester) electrophiles with either alkane, alkyne, or aryl nucleophiles (Fig. 1b).40–49 Alternatively, RCF2-internal olefins have been prepared photochemically via either Ru or Ni based catalytic systems.50,51 Although promising, all these strategies require electrophilic RCF2Br as RCF2– sources, whose preparation by radical bromination is incompatible with many substrate classes.21,22 Defluoroalkylation of trifluoromethyl alkenes is an alternative approach to access vinyldifluormethylene linkages.52–56
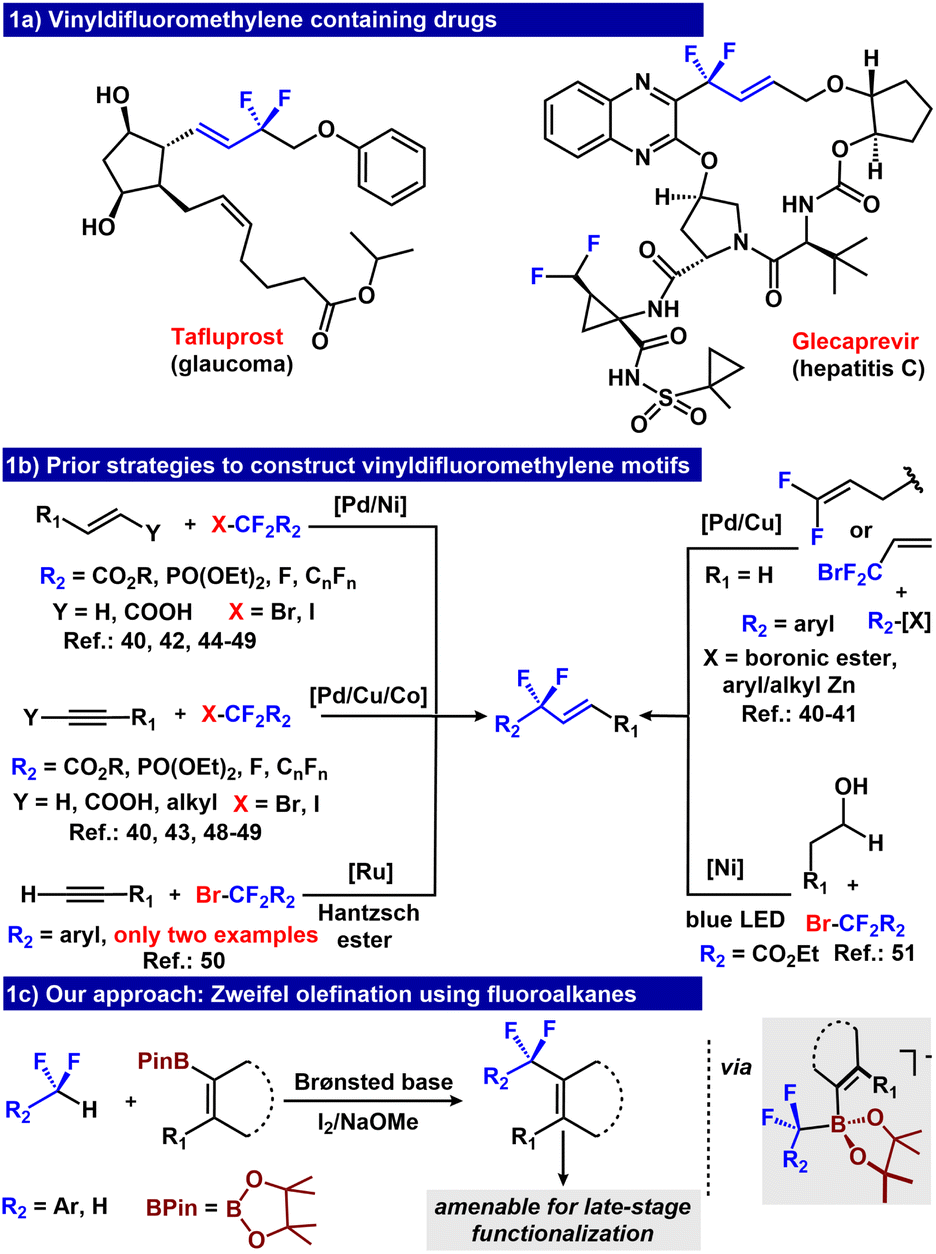 | ||
| Fig. 1 (a) Vinyldifluoromethylene containing drugs. (b) Prior strategies to construct vinyldifluoromethylene motifs. (c) Our approach: Zweifel olefination using fluoroalkanes. | ||
Zweifel olefination represents an attractive metal-free route to construct a vinylic C–C bond.57–59 This reaction sequence proceeds through an anionic vinyl borate intermediate. Oxidation induces 1,2-rearrangement, and addition of an alkoxide base leads to a product containing a new C–C bond. We hypothesized that if the key anionic vinyl borate intermediate could be accessed by deprotonating RCF2H molecules, subsequent oxidation would provide α,α-difluoroalkylated vinyl compounds. Our group previously established that fluoroalkyl B3N3Me6 adducts [RCF2B3N3Me6]− can transfer the RCF2− group to stronger Lewis acids such as B(OMe)3.26,29,30,33 Given the similar Lewis acidity of R-BPin to B(OMe)3,60,61 we hypothesized that [(R)(ArCF2)BPin]− could be formed analogously,62 and if R = vinyl, the borate intermediate would be uniquely situated for a net Zweifel olefination reaction to form α,α-difluoroalkylated olefin products (Fig. 1c).
Results and discussion
To establish feasibility of fluoroalkyl transfer to R-BPin, we allowed an equimolar mixture of [K(18-crown-6)(B3N3Me6-CF2Ph)] (1) and Ph-BPin (2a) to react in tetrahydrofuran solvent for 1.5 h at 50 °C. The reaction furnished [K(18-c-6)(Ph-BPin-CF2Ph)] (2b) quantitatively (Fig. 2) as assessed by 1H, 13C, 19F, 11B NMR and ESI-MS analyses (11B: 3.95 ppm and 19F: −104.21 ppm). The clean transfer of PhCF2− from B3N3Me6 to Ph-BPin demonstrates feasibility of the approach. To evaluate the Zweifel olefination sequence, we introduced 1 to vinyl-BPin 2c. The reaction afforded adduct 2d with 98% conversion, as assessed by 19F and 11B NMR spectroscopy (11B: 3.67 ppm and 19F: −105.66 ppm), and was isolated as a white powder in 88% yield.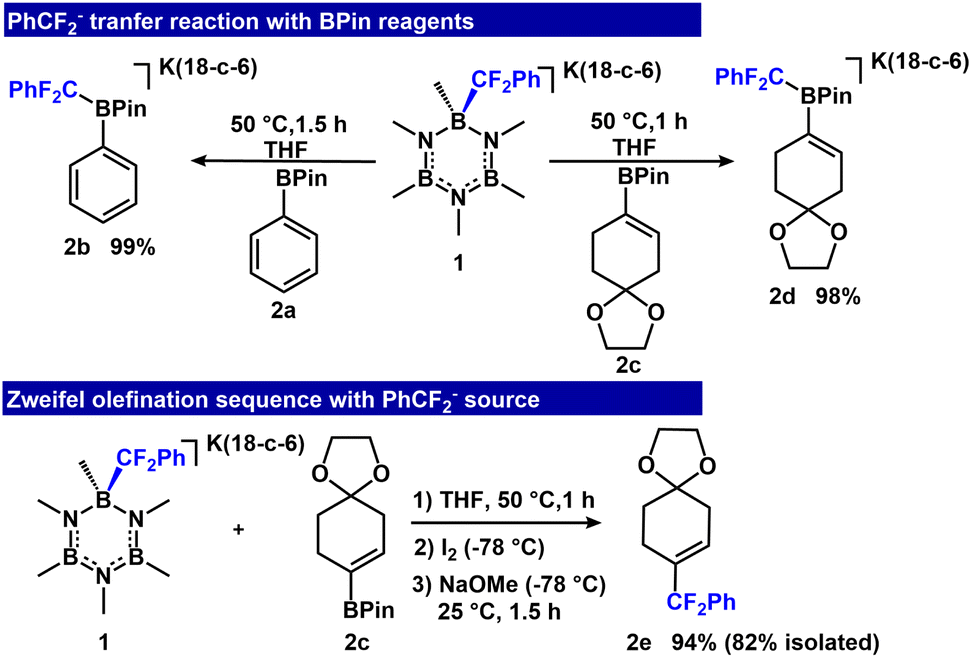 | ||
| Fig. 2 PhCF2− transfer reactions to aryl and vinyl-BPin (top) and Zweifel olefination (bottom). | ||
After achieving clean conversion to 2d above, we evaluated a one-pot Zweifel olefination sequence to form α,α-difluoroalkylated olefin (2e). Initial conditions using 1 equiv. I2 and NaOMe at room temperature afforded 2e in 26% yield (ESI, Table S1,† entry 1). The identity of 2e was established by 1H and 19F NMR spectroscopy (1H NMR: vinyl hydrogen at 5.79–5.82 ppm (m) and 19F NMR: −95.41 ppm). The yield was increased to 40% by lowering the temperature to −78 °C prior to adding I2 and NaOMe (ESI, Table S1,† entry 2). Importantly, increasing the stoichiometry of I2 and NaOMe to 2 equiv. further improved the conversion to 94% (Fig. 2).
We assessed the reaction scope by varying the vinyl-BPin derivatives (Fig. 3, entries 3a–3i). Aliphatic vinyl-BPin reagents generally responded well, with good yields (73–81%; Fig. 3, entries 3a–3c). Dihydronaphthalene and 2H-chromene derivatives furnished 89 and 84% isolated yields for 3e and 3g, respectively. The latter substrate is of particular interest because chromene derivatives are an important class of heterocycles used in cosmetic agents, food additives, and biodegradable agrochemicals.63,64 Acyclic vinyl-BPin reagents were also tolerated, affording 3d (77%), 3f (89%) and 3i (94%) in good yields.
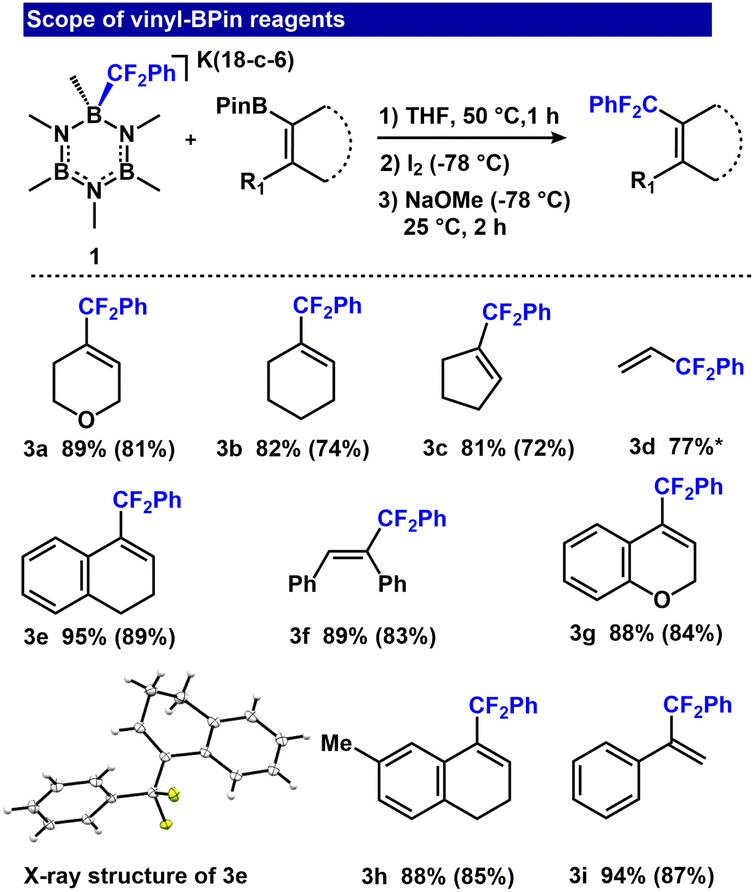 | ||
| Fig. 3 Zweifel olefination scope by varying the vinyl-BPin reagents. R-BPin (0.1 mmol), [K(18-c-6)(B3N3Me6-CF2Ph)] (0.1 mmol), I2 (0.2 mmol), NaOMe (0.2 mmol), THF (1.8 mL). 19F NMR yields (isolated yields in parentheses).*Only 19F NMR yield acquired due to product volatility. | ||
Although the reaction sequence provides high yields of PhCF2–olefin products, one limitation to broad scope adoption is the requirement of B3N3Me6. To overcome this requirement, we evaluated whether the use of B3N3Me6 could be eliminated in the Zweifel olefination sequence. Importantly, because 2c is more Lewis acidic than B3N3Me6, we hypothesized that it could effect the direct capture/olefination of PhCF2− following deprotonation of PhCF2H. To examine the feasibility of the one-pot Zweifel olefination strategy, we evaluated a series of bases to deprotonate ArCF2H in the presence of 2c. We optimized the formation of 2d in a single step starting from 2c, PhCF2H and base. In the absence of 18-crown-6, deprotonation of PhCF2H with 1.5 equiv. KN(iPr)2 gave 95% conversion to 2d, but for the tandem Zweifel reaction, only 27% yield of 2e (Fig. 4, ESI Table S2†). However, adding 18-crown-6 after deprotonation enabled subsequent conversion to 2e in 64% yield over two steps.
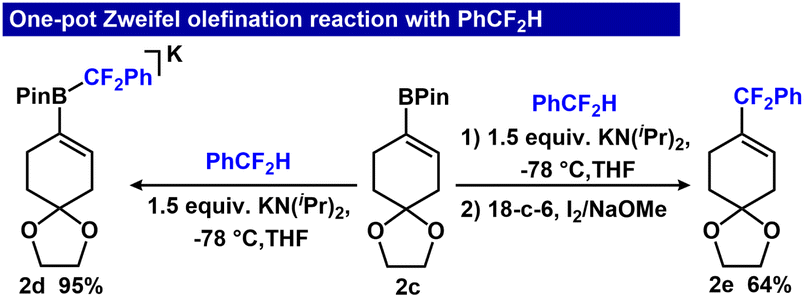 | ||
| Fig. 4 Strategy for the one-pot Zweifel olefination reaction by in situ deprotonation of PhCF2H with KN(iPr)2. 2c (0.06 mmol), KN(iPr)2 (0.09 mmol), 18-c-6 (0.06 mmol), I2 (0.12 mmol), NaOMe (0.12 mmol), THF (1 mL). 19F NMR yields (PhOCF3 used as internal standard). | ||
After demonstrating feasibility of the one-pot methodology, we assessed scope in both the vinyl-BPin, and fluoroalkyl component (Fig. 5). Although most of the previous scope in vinyl-BPin (Fig. 3) translated to the one-pot method, two substrates were incompatible (3f and 3a), which we attribute to deleterious reactivity with KN(iPr)2. Dihydronaphthalene and the 2H-chromene derivative responded well, providing isolated yields of 71% (3e) and 51% (3g). Vinyl-BPin reagents containing aliphatic functionality afforded moderate isolated yields (2e (56%) and 3b (40%)). Styrenyl-BPin similarly afforded 3i in 72% isolated yield.
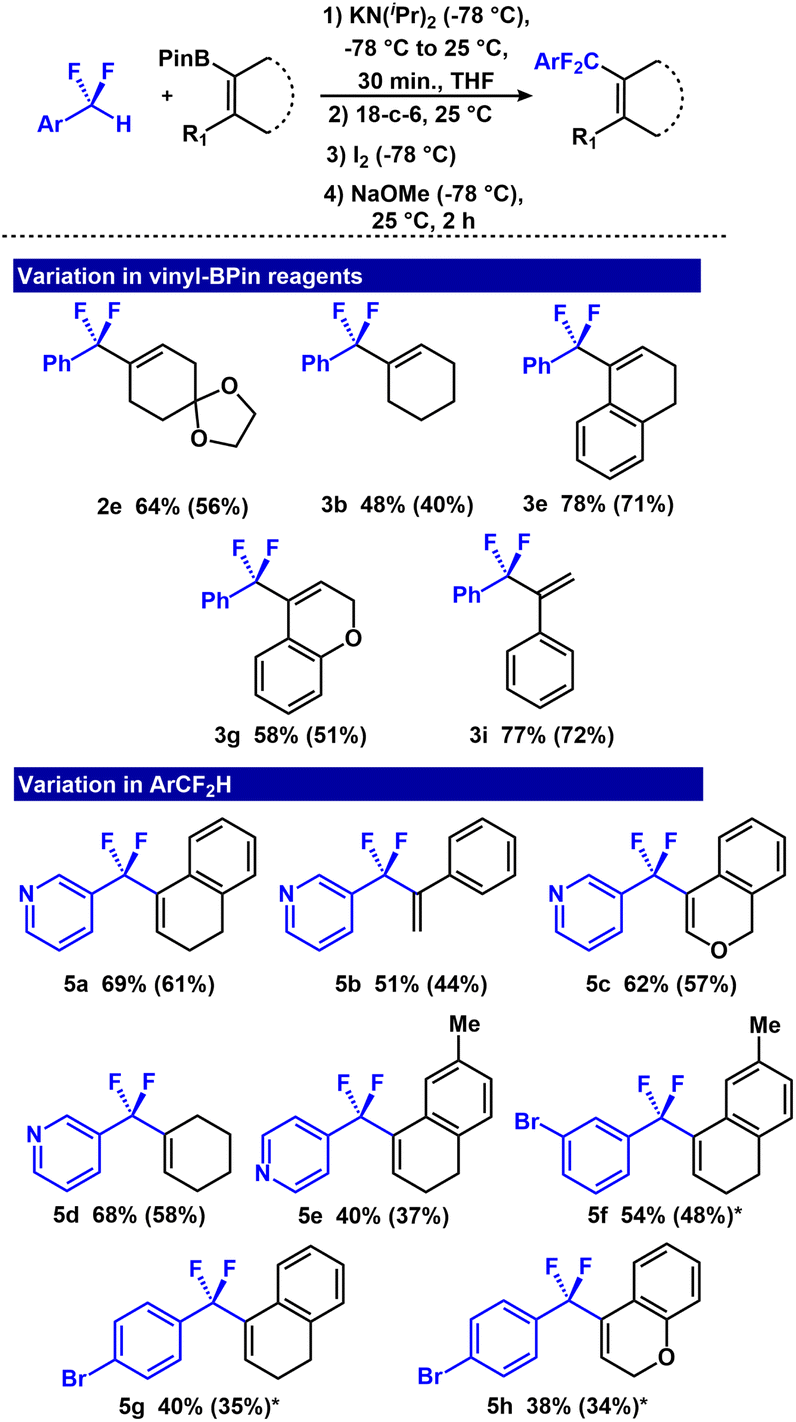 | ||
| Fig. 5 Scope of one-pot Zweifel olefination reaction. BPin derivatives (0.26 mmol), KN(iPr)2 (0.39 mmol), 18-c-6 (0.26 mmol) I2 (0.52 mmol), NaOMe (0.52 mmol), THF (3 mL). 19F NMR yields (PhOCF3 used as internal standard). Isolated yields are in parentheses. *5–8% hydrodebromination also observed. | ||
Next, we explored the scope in pronucleophile by investigating difluoromethyl pyridine and difluoromethyl aryl bromide substrates. Pyridine units are an important structural motif in pharmaceutical and medicinal chemistry.65,66 We evaluated the reactivity of 3-(difluoromethyl)pyridine and 4-(difluoromethyl)pyridine with four representative vinyl-BPin substrates: (dihydronaphthalene, 2H-chromene, cyclohexenyl and styrenyl) (Fig. 5). They all furnished the Py–CF2–olefin products in 44–61% isolated yields (5a–5e). Finally, although aryl bromides generally have low compatibility with electrochemical and photoredox fluorination reactions,67,68 they were compatible with the one-pot Zweifel protocol. For example, 1-bromo-4-(difluoromethyl)benzene and 1-bromo-3-(difluoromethyl)benzene substrates provided their respective products in 34–48% (5f–5h) isolated yield.
Our group previously demonstrated that hydrofluorocarbons (HFCs) can be used as low cost –CF3, and –CF2H sources,28–30 representing a strategy to repurpose refrigerants or fluorinated waste products that are produced on a >15 kt scale.69,70 Importantly, the use of CH2F2 (HCF-32) as a fluoroalkyl precursor affords products that are also pro-nucleophiles, primed for subsequent functionalization. We evaluated the feasibility of a one-pot difluoromethylation reaction using [K(18-c-6)(B3N3Me6-CF2H)] (6a) and vinyl-BPin derivatives. Although higher temperature (80 °C) was required for –CF2H transfer, the previously optimized conditions for Zweifel olefination afforded 6b in 76% yield (Fig. 6, ESI Table S3†). We also evaluated the protocol using dihydronaphthalene and styrenyl BPin derivatives, which provided conversions of 72% and 68% for 6c and 6d, respectively (Fig. 6). We directly compared this approach to Me3Si-CF2H, a common –CF2H pronucleophile that requires activation by F− or OH−.71–73 Addition of vinyl-BPin (2c) to a solution of Me3Si-CF2H activated with either [Me4N][OH] or [nBu4N][F] did not afford 19F NMR resonances consistent with the –CF2H analogue of 2d (see ESI, S75†), under similar conditions used with 6a (see ESI, Fig. S114 and S115†). These results highlight the challenge of selective nucleophilic fluoroalkyl transfer from R-SiMe3 reagents that contain more than one nucleophilic sites, and demonstrate a clear advantage for using –CF2R transfer reagents that do not need an activator.
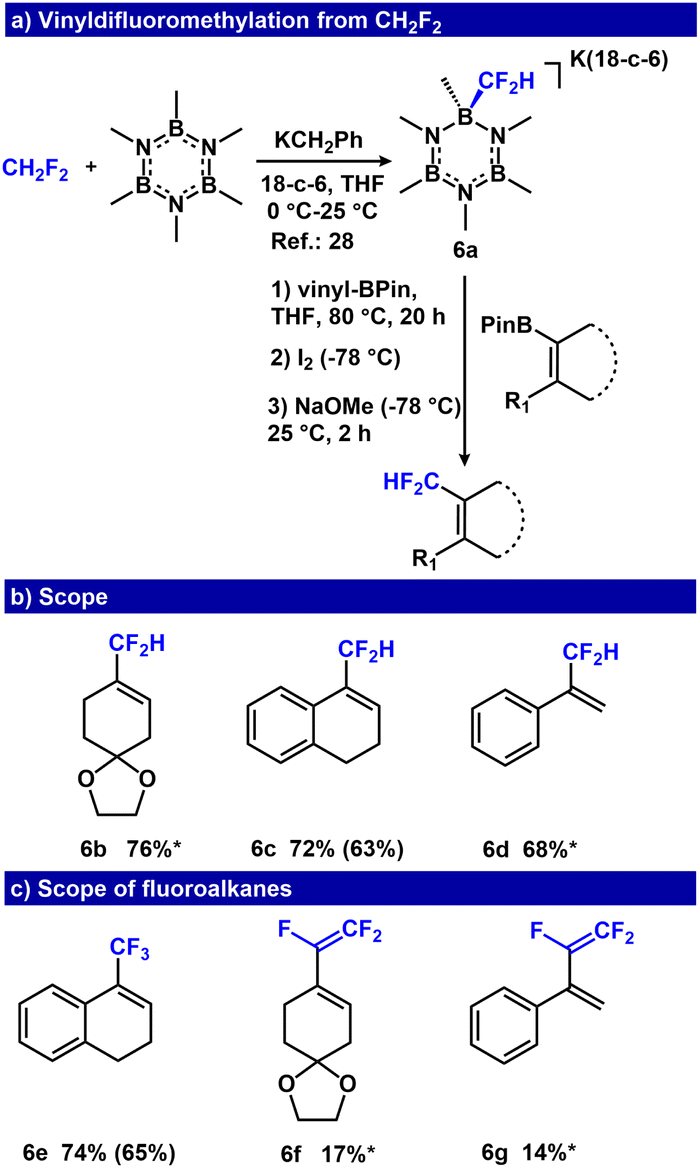 | ||
| Fig. 6 (a) Zweifel olefination for the synthesis of vinyldifluoromethane molecules from CH2F2. (b) Scope of vinyldifluoromethanes, conditions: R-BPin (0.2 mmol), [K(18-c-6)(B3N3Me6-CF2H)] (0.2 mmol), I2 (0.4 mmol), NaOMe (0.4 mmol), THF (4.0 mL). (c) Scope in fluoroalkanes. *Only 19F NMR yield acquired due to product volatility. 19F NMR yields (isolated yields are in parentheses). | ||
Next, we examined the feasibility of the Zweifel olefination using other fluoroalkanes (HCF3, CH3CF2H, CF3CFH2 and CF3CH3). We found that the HCF3-derived reagent, [(B3N3Me6-CF3)(K(18-c-6))], was readily adapted, affording the trifluoromethyl containing product, 6e, in 65% isolated yield (Fig. 6c). When translating these conditions to fluoroethanes, we found that although CH3CF2H and CF3CH3 did not afford the olefination products, CF3CFH2 furnished 6f and 6g with 17% and 14% yield as assessed by 19F NMR spectroscopy. In these cases, β-fluoride elimination occurred at the first step (generating [(B3N3Me6-CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2)(K(18-c-6))]), a species competent for the net Zweifel olefination (Fig. 6c, ESI S78–S80†). Finally, we attempted the direct capture/olefination of HCF2− following deprotonation of CF2H2. Deprotonation of CF2H2 with 5 equiv. KN(iPr)2 gave 32% conversion to the vinyl boronate intermediate, that when subjected to NaOMe/I2, afforded 9% yield of 6d over two steps (ESI S77†).
CF2)(K(18-c-6))]), a species competent for the net Zweifel olefination (Fig. 6c, ESI S78–S80†). Finally, we attempted the direct capture/olefination of HCF2− following deprotonation of CF2H2. Deprotonation of CF2H2 with 5 equiv. KN(iPr)2 gave 32% conversion to the vinyl boronate intermediate, that when subjected to NaOMe/I2, afforded 9% yield of 6d over two steps (ESI S77†).
Given previous data showing rapid transfer of the RCF2− unit to carbonyl electrophiles,26 we examined whether transfer to vinyl BPin may occur in the presence of another, competitive electrophile. We performed a series of competition experiments to examine reaction tolerance to carbonyl electrophiles as additives. The addition of esters and a ketone (benzyl benzoate, phenyl benzoate and benzophenone) provided low conversion to the final product (Fig. 7, 8%, 5% and 10%, respectively). However, the reaction tolerated an amide electrophile, N,N-dimethylbenzamide, and we observed only 3% reduction in product formation (92% yield of 3e) (Fig. 7, ESI S78†). These results demonstrate the feasibility of the reaction sequence with select competitive electrophiles.
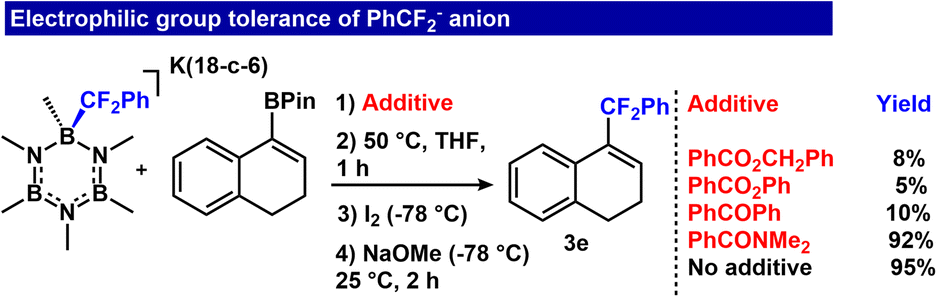 | ||
| Fig. 7 Tolerance to ester, ketone and amide additives. | ||
We next investigated the feasibility of late-stage modifications of three representative Ar–CF2–olefins formed from the Zweifel olefination sequence (Fig. 8). Hydrogenation of 3e using Pd/C at 25 psi H2 furnished 8a in 84% isolated yield. A net dehydrogenation reaction was effected by treating 3e with AIBN/NBS at room temperature, affording 8b in 72% isolated yield. An epoxidation reaction of 3i with m-CPBA in CH2Cl2 at 60 °C provided 8c in 74% isolated yield. Reduction of 3fvia BH3 hydroboration followed by oxidation furnished 8d in 56% isolated yield. Finally, a defluorinative SN2′ reaction with nBuLi yielded the E isomer 8e with 73% isolated yield (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity of E/Z alkenes).
:1 selectivity of E/Z alkenes).
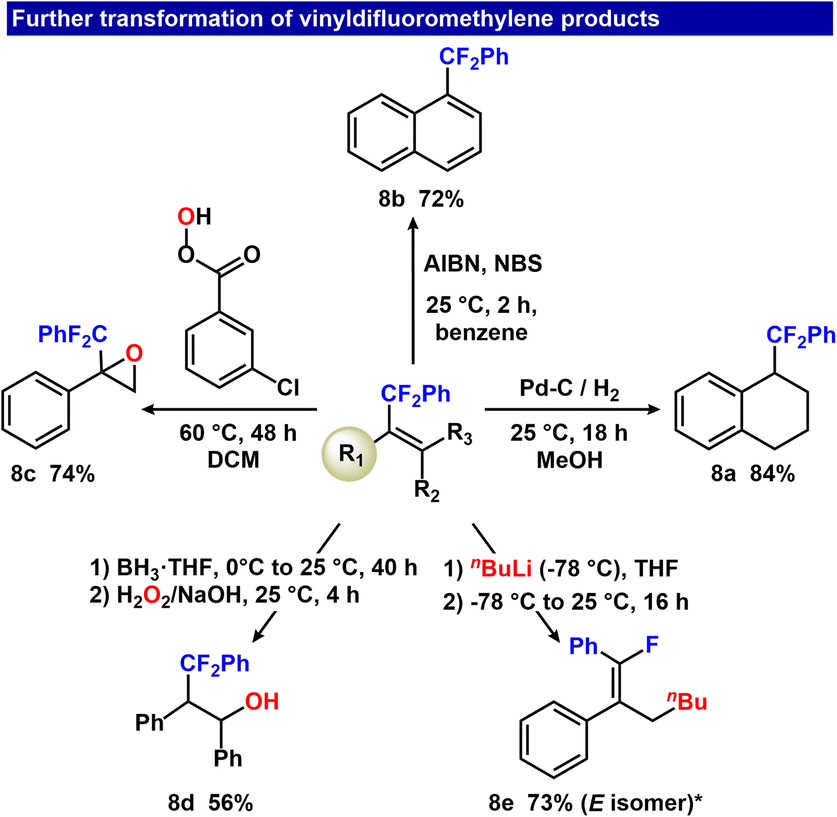 | ||
| Fig. 8 Further transformation of vinyldifluoromethylene products spanning oxidative, reductive and substitution chemistry. *10% Z isomer also formed. | ||
Conclusion
In summary, we have developed a metal-free strategy to access vinyldifluoromethylene molecules from fluoroalkane precursors and vinyl-BPin reagents. Lewis acidic boranes stabilize and template α,α-difluoroalkyl- and vinylic-moieties, ultimately enabling C–C bond formation. This strategy was extended to a straightforward one-pot synthesis using RCF2H precursors and vinyl-BPin reagents. We showed that this reaction sequence can be used to generate RCF2H precursors directly from inexpensive CH2F2 (HCF-32), which represents a new strategy to repurpose this underutilized synthon. Importantly, we demonstrated that this CF2H− reactivity is a unique application of the deprotonation approach, and not possible using the most common –CF2H transfer reagent: Me3Si-CF2H. Finally, we established that the vinyldifluoromethylene products can be readily derivatized, representing an avenue for late-stage modification of fluoroalkylated compounds.Data availability
All relevant experimental data and characterization details are provided in the ESI.† Crystallographic data for compound 3e has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under access number 2299679.Author contributions
The manuscript was written through contributions of all authors. The project was designed by K. Chakrabarti, M. M. Wade Wolfe, S. Guo, and N. K. Szymczak. All optimizations, syntheses and characterizations were performed by K. Chakrabarti. J. W. Tucker and J. Lee provided insights related to project development. All authors have given approval to the final version of the manuscript.Conflicts of interest
NKS holds a patent relating to fluoroalkyl transfer reagents.Acknowledgements
This work was supported by the NSF (CHE 1955284). We thank Dr Fengrui Qu for acquiring data for X-ray crystallography and Dr Russell Bornschein for Mass Spectrometry assistance. We thank Dr Daniel Beagan for assistance solving the crystal structure.Notes and references
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- J. O. Link, J. G. Taylor, L. Xu, M. Mitchell, H. Guo, H. Liu, D. Kato, T. Kirschberg, J. Sun, N. Squires, J. Parrish, T. Keller, Z.-Y. Yang, C. Yang, M. Matles, Y. Wang, K. Wang, G. Cheng, Y. Tian, E. Mogalian, E. Mondou, M. Cornpropst, J. Perry and M. C. Desai, J. Med. Chem., 2014, 57, 2033–2046 CrossRef CAS PubMed.
- S. P. Vincent, M. D. Burkart, C.-Y. Tsai, Z. Zhang and C.-H. Wong, J. Org. Chem., 1999, 64, 5264–5279 CrossRef CAS PubMed.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- L. N. Markovskij, V. E. Pashinnik and A. V. Kirsanov, Synthesis, 1973, 1973, 787–789 CrossRef.
- J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC.
- K. Aikawa, K. Maruyama, J. Nitta, R. Hashimoto and K. Mikami, Org. Lett., 2016, 18, 3354–3357 CrossRef CAS PubMed.
- V. V. Levin, A. A. Zemtsov, M. I. Struchkova and A. D. Dilman, Org. Lett., 2013, 15, 917–919 CrossRef CAS PubMed.
- R. Gupta, A. K. Jaiswal, D. Mandal and R. D. Young, Synlett, 2020, 31, 933–937 CrossRef CAS.
- D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572–2578 CrossRef CAS PubMed.
- D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682–10686 CrossRef CAS PubMed.
- H. A. Tennekes, Toxicology, 2010, 276, 1–4 Search PubMed.
- F. S. Fawcett, C. W. Tullock and D. D. Coffman, J. Am. Chem. Soc., 1962, 84, 4275–4285 CrossRef CAS.
- Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS PubMed.
- J.-W. Gu, W.-H. Guo and X. Zhang, Org. Chem. Front., 2015, 2, 38–41 RSC.
- Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed.
- A. Bunnell, N. Lalloo, C. Brigham and M. S. Sanford, Org. Lett., 2023, 25, 7584–7588 CrossRef CAS PubMed.
- K. Choi, M. G. Mormino, E. D. Kalkman, J. Park and J. F. Hartwig, Angew. Chem., Int. Ed., 2022, 61, e202208204 CrossRef CAS PubMed.
- J. He and C. U. Fittman, Synth. Commun., 1999, 29, 855–862 CrossRef CAS.
- Y. Masato, S. Daiki and I. Masahiko, Chem. Lett., 1994, 23, 2357–2360 CrossRef.
- L. Santos, A. Panossian, M. Donnard, J.-P. Vors, S. Pazenok, D. Bernier and F. R. Leroux, Org. Lett., 2020, 22, 8741–8745 CrossRef CAS PubMed.
- P. Clavel, M. P. Léger-Lambert, C. Biran, F. Serein-Spirau, M. Bordeau, N. Roques and H. Marzouk, Synthesis, 1999, 1999, 829–834 CrossRef.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- J. B. Geri, M. M. Wade Wolfe and N. K. Szymczak, J. Am. Chem. Soc., 2018, 140, 9404–9408 CrossRef CAS PubMed.
- A. Streitwieser Jr and F. Mares, J. Am. Chem. Soc., 1968, 90, 2444–2445 CrossRef.
- J. B. Geri, E. Y. Aguilera and N. K. Szymczak, Chem. Commun., 2019, 55, 5119–5122 RSC.
- J. B. Geri, M. M. Wade Wolfe and N. K. Szymczak, Angew. Chem., Int. Ed., 2018, 57, 1381–1385 CrossRef CAS PubMed.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 9811–9814 CrossRef CAS PubMed.
- S. Guo, W. Sun, J. W. Tucker, K. D. Hesp and N. K. Szymczak, Chem.–Eur. J., 2023, 29, e202203578 CrossRef CAS PubMed.
- M. M. Wade Wolfe, S. Guo, L. S. Yu, T. R. Vogel, J. W. Tucker and N. K. Szymczak, Chem. Commun., 2022, 58, 11705–11708 RSC.
- M. M. Wade Wolfe, J. P. Shanahan, J. W. Kampf and N. K. Szymczak, J. Am. Chem. Soc., 2020, 142, 18698–18705 CrossRef CAS PubMed.
- D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587–1595 CrossRef.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- Y. N. Lamb, Drugs, 2017, 77, 1797–1804 CrossRef CAS PubMed.
- Y. Matsumura, in Fluorine in Medicinal Chemistry and Chemical Biology, 2009, pp. 47–66 Search PubMed.
- S. Woo and R. A. Shenvi, Acc. Chem. Res., 2021, 54, 1157–1167 CrossRef CAS PubMed.
- M. D. Levin, T. Q. Chen, M. E. Neubig, C. M. Hong, C. A. Theulier, I. J. Kobylianskii, M. Janabi, J. P. O'Neil and F. D. Toste, Science, 2017, 356, 1272–1276 CrossRef CAS PubMed.
- Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed.
- Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230–1233 CrossRef CAS PubMed.
- X.-T. Feng, J.-X. Ren, X. Gao, Q.-Q. Min and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210103 CrossRef CAS PubMed.
- L. Liao, R. An, H. Li, Y. Xu, J.-J. Wu and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11010–11019 CrossRef CAS PubMed.
- G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS PubMed.
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270–1274 CrossRef CAS PubMed.
- F. Ke, C. Yu, X. Li, H. Sheng and Q. Song, Org. Lett., 2023, 25, 2733–2738 CrossRef CAS PubMed.
- G. Wu and A. Jacobi von Wangelin, Chem. Sci., 2018, 9, 1795–1802 RSC.
- E. Zhu, X.-X. Liu, A.-J. Wang, T. Mao, L. Zhao, X. Zhang and C.-Y. He, Chem. Commun., 2019, 55, 12259–12262 RSC.
- M. Ke, Q. Feng, K. Yang and Q. Song, Org. Chem. Front., 2016, 3, 150–155 RSC.
- S. Sumino, M. Uno, T. Fukuyama, I. Ryu, M. Matsuura, A. Yamamoto and Y. Kishikawa, J. Org. Chem., 2017, 82, 5469–5474 CrossRef CAS PubMed.
- Y. Bai, L. Cao, S. Li, G. a. Zhang, Y. Liu, F. Zhao and J. Wu, Org. Lett., 2023, 25, 6511–6516 CrossRef CAS PubMed.
- N. A. Phillips, G. J. Coates, A. J. P. White and M. R. Crimmin, Chem.–Eur. J., 2020, 26, 5365–5368 CrossRef CAS PubMed.
- C. Zhu, M.-M. Sun, K. Chen, H. Liu and C. Feng, Angew. Chem., Int. Ed., 2021, 60, 20237–20242 CrossRef CAS PubMed.
- L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701–8705 RSC.
- S.-S. Yan, D.-S. Wu, J.-H. Ye, L. Gong, X. Zeng, C.-K. Ran, Y.-Y. Gui, J. Li and D.-G. Yu, ACS Catal., 2019, 9, 6987–6992 CrossRef CAS.
- F. Ye, Y. Ge, A. Spannenberg, H. Neumann, L.-W. Xu and M. Beller, Nat. Commun., 2021, 12, 3257 CrossRef CAS PubMed.
- G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS.
- D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS.
- R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 49, 3323–3336 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- A. Yokawa and S. Ito, Chem.–Asian J., 2020, 15, 3432–3436 CrossRef CAS PubMed.
- N. Kaur, in Metal and Nonmetal Assisted Synthesis of Six-Membered Heterocycles, ed. N. Kaur, Elsevier, 2020, pp. 351–412 Search PubMed.
- A. Chaudhary, K. Singh, N. Verma, S. Kumar, D. Kumar and P. P. Sharma, Mini-Rev. Med. Chem., 2022, 22, 2736–2751 CrossRef CAS PubMed.
- E. Khan, ChemistrySelect, 2021, 6, 3041–3064 CrossRef CAS.
- S. De, A. Kumar S K, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, RSC Adv., 2022, 12, 15385–15406 RSC.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed.
- Difluoromethane (HFC-32) CAS No. 75-10-5, European Centre for Ecotoxicology and Toxicology of Chemicals, Brussels, 2nd edn, 2008 Search PubMed.
- D. J. Sheldon and M. R. Crimmin, Chem. Soc. Rev., 2022, 51, 4977–4995 RSC.
- M. Miele and V. Pace, Aust. J. Chem., 2021, 74, 623–625 CrossRef CAS.
- X. Ispizua-Rodriguez, C. Barrett, V. Krishamurti and G. K. Surya Prakash, in The Curious World of Fluorinated Molecules, ed. K. Seppelt, Elsevier, 2021, vol. 6, pp. 117–218 Search PubMed.
- R. Britton, V. Gouverneur, J. H. Lin, M. Meanwell, C. Ni, G. Pupo, J. C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 48 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2299679. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05616c |
| This journal is © The Royal Society of Chemistry 2024 |