
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThia[n]helicenes with long persistent phosphorescence†
Zhen
Sun‡
,
Wan
Xu‡
,
Shuai
Qiu
,
Zhiying
Ma
 ,
Chunli
Li
,
Sheng
Zhang
* and
Hua
Wang
*
,
Chunli
Li
,
Sheng
Zhang
* and
Hua
Wang
*
Institute of Nanoscience and Engineering, Henan University, Kaifeng 475004, Henan, China. E-mail: zhang.sheng.2022@henu.edu.cn; hwang@henu.edu.cn
First published on 8th December 2023
Abstract
Helicenes with persistent luminescence have received relatively little attention, despite their demonstrated highly efficient intersystem crossing (ISC) from the excited singlet to the triplet state. Herein, we designed a series of ortho-fused aromatics by combining dithieno[2,3-b:3′,2′-d]thiophene (DTT) with annulated benzene fragments, denoted as TB[n]H (n = 3–8), to achieve persistent luminescence. Wherein, thia[n]helicenes (n = 5–8) exhibited intense phosphorescence with millisecond-range lifetimes (τp) at 77 K. Particularly interesting was the observation that the odd-numbered ring helicenes displayed longer τp values than their neighboring even-numbered counterparts. Notably, TB[7]H showcased the longest τp of 628 ms. This phenomenon can be attributed to the more favorable ISC channels and stronger spin-orbital coupling (SOC) of old-numbered helicenes than even-numbered ones. Furthermore, both conformers of TB[7]H exhibited significant circularly polarized phosphorescent (CPP) responses, with luminescence dissymmetry factors (glum) of 0.015 and −0.014. These discoveries suggest that thiahelicenes may be a specific class of organic phosphorescent and CPP materials.
Introduction
Helicenes are a class of π-conjugated compounds with screw configuration, consisting of ortho-fused aromatic rings. This unique geometry, along with their simple structural modifiability and distinctive electronic properties, has made helicenes a subject of continuous research interest in the field of polycyclic aromatic hydrocarbons for the past century.1–5 Among their various attributes, the optical features of helicenes have gained significant attention in recent years, particularly in the field of chiroptics. The chiroptical response, encompassing optical rotation, circular dichroism (CD), and circularly polarized luminescence (CPL) properties, positions helicenes as promising materials for applications such as 3D displays, information encryption, spintronics, optoelectronic devices, and more.6,7 Numerous strategies for structural modification have been proposed to fine-tune the luminescent properties of helicenes. These strategies encompass polyploidization of the helicene skeleton, the extension of π-conjugation within the helicene backbone, heteroatom doping (e.g., N, B, Si, and P), the creation of helicenes by combining electron-donating and accepting moieties, and the formation of helicenoids through the annulation of aromatic and nonaromatic rings, among various other approaches.7–14 However, it is worth noting that the predominant focus of research has been on prompt fluorescence, while the development of persistent luminescence, such as phosphorescence, has lagged behind, despite the high estimated ISC efficiency of helicenes.15It is widely recognized that an efficient ISC process leads to the population of triplet states, which is the prerequisite to obtain long-lasting phosphorescence.16 The twisted configuration of helicenes induces out-of-plane vibrations, a phenomenon that has been demonstrated to significantly enhance ISC efficiency.16–21 Building on this effect, several classical structural modifications of helicenes have been explored to enhance their phosphorescent properties, with relevant studies summarized in Fig. 1a. Earlier investigations revealed that increasing the torsion of the helical skeleton, achieved by incorporating more ortho-fused rings, led to faster ISC in carbohelicenes.15,17 Another effective modification involves the introduction of heteroatoms (such as N and O) onto the helicene skeleton, which strengthens SOC (S1–Tn) through an n–π* admixture into the π–π* singlet state, consequently promoting ISC efficiency.16,22 Among heteroatom-containing helicenes, metal–helicene complexes, featuring metals such as Pt and Ir, exhibited notably higher ISC efficiency. The strong SOC and charge transfer between metal centers and helicene ligands facilitated the generation of phosphorescence, making metallohelicenes popular as candidates for various applications, including circularly polarized luminescent materials.6,9,23 Additionally, organic phosphorescence devoid of metals can be achieved through strategic substitutions on helicenes.24–26 For instance, Hariharan et al. introduced bromine atoms to a planar perylene diimide (PDI), resulting in an octabrominated derivative. The steric effects of these substitutions induced significant out-of-plane distortion in the aromatic core of PDI, yielding a double[3]helicene. This alteration in the perylene structure moderately reduced the energy gap between singlet and triplet states compared to unsubstituted PDI. Moreover, the heavy-atom effect of bromine amplified the strength of SOC compared to pristine PDI. The combined impact of these factors led to ultrafast ISC and a nearly complete population of the triplet state.24 More recently, Crassous et al. reported the photophysical and chiroptical properties of [6]helicene-based emitters bearing electron-donating (TMS, –NMe2, and –NH2) and electron-accepting (–CN, pyridyl, and –NO2) substituents. All tested samples exhibited robust circularly polarized phosphorescence in a frozen state, owing to efficient ISC.25 Despite some progress in helicene-based emitters, phosphorescent helicenes remain relatively scarce, especially helicenes with exceptionally long τp. Leveraging their unique electronic properties and intrinsic chirality, the development of helicene-based phosphors with ultra-long τp promises to not only broaden the scope of helicene applications but also enrich the palette of organic phosphorescent materials suitable for diverse uses, including room temperature applications (such as encryption and biological imaging),27–30 temperature-dependent applications (luminescence thermometry and surface icing detection), and chiroptical applications.31–37
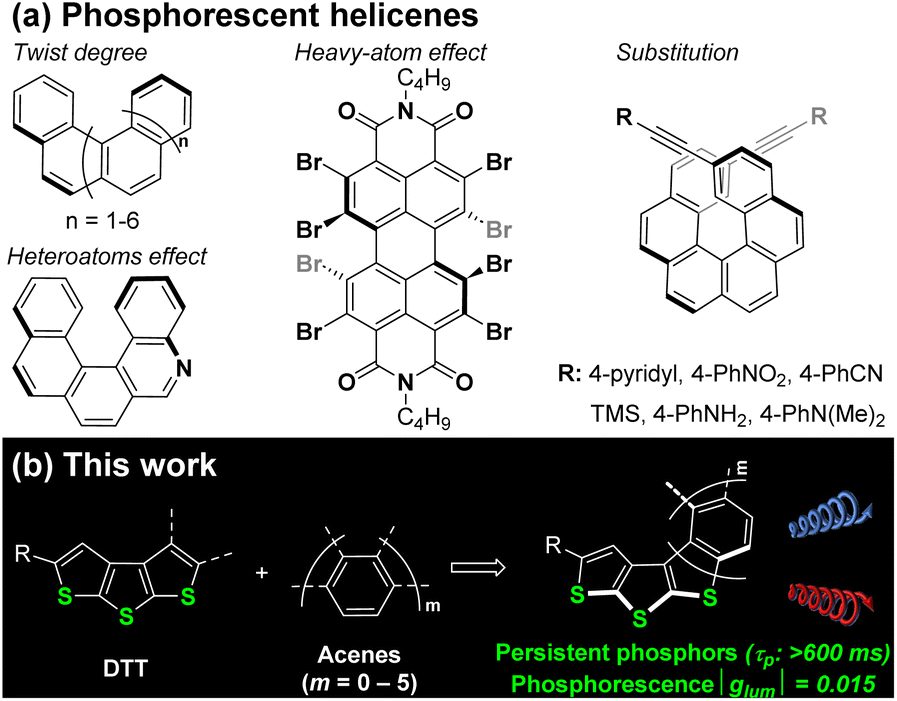 | ||
| Fig. 1 Strategies for the population of triplets and promotion of phosphorescence of helicenes. (a) The reported strategies. (b) Design of thia[n]helicenes having long-lived phosphorescence and chiroptical properties in this work. | ||
In light of these considerations, we have undertaken the design of a series of helicenes featuring the DTT fragment annulated with varying numbers of benzene rings (Fig. 1b). Among the diverse heteroatom-containing helicenes,9,38–40 thiahelicene emerges as a noteworthy category.41–43 The presence of a sulfur atom has been shown to have a favorable impact on the promotion of SOC via n–π* transitions, thereby paving the way to induce robust phosphorescence through SOC-triggered ISC.44,45 Remarkably, this unique feature has been underutilized in helicene design for enhancing phosphorescence. Despite a synthesis by Bossi et al. of fluorinated thiahelicenes exhibiting phosphorescence, they attributed this behavior to the inductive effect of fluorine rather than sulfur.26 Typically, thiophene rings serve as building blocks for constructing thiahelicenes due to their excellent optoelectronic properties.46 As a component of π-conjugation, sulfur (S) atoms are anticipated to facilitate n–π* transitions more readily, thus leading to a stronger SOC compared to thioethers. The presence of multiple S atoms is believed to amplify this effect. Guided by this rationale, we have designed a series of thia[n]helicenes incorporating a DTT47 moiety. The fused thiophene moieties not only contribute a surplus of n-electrons for engaging in n–π* transitions but also impart excellent electron-donating properties to modulate the energy gap between singlet and triplet states. Importantly, all sulfur atoms within the DTT unit are positioned opposite to carbon, a configuration that facilitates helicene formation upon annulation of aromatic rings.47 In this context, we have introduced different numbers of fused benzene rings (ranging from 1 to 5) to modulate the extent of π-conjugation and twist in the helicenes, thereby fine-tuning their τp and achieving ultra-long phosphorescence. We conducted a comprehensive study of photoluminescence, encompassing both fluorescence and phosphorescence, employing a range of spectroscopic techniques, including steady-state, time-resolved, and transient-state analyses. All helicenes exhibited ultra-long phosphorescence with τp exceeding 290 ms at the low temperature. Interestingly, the odd-numbered-ring helicenes exhibited more persistent phosphorescence compared to their even-numbered counterparts. Notably, thia[7]helicene demonstrated the most impressive phosphorescent performance, with a remarkable τp exceeding 600 milliseconds. Computational calculations were employed to elucidate the difference in τp between odd- and even-numbered-ring thiahelicenes. Additionally, we selected thia[7]helicene for an in-depth investigation of its chiroptical properties. Both enantiomers (P and M) exhibited robust CPP at low temperatures, with respective glum values of 0.015 and −0.014.
Results and discussion
Syntheses
According to the molecular design in the current work, a series of DTT-containing polyaromatic hydrocarbons would be synthesized including the two planar-shaped molecules (TB[3]H and TB[4]H) as reference compounds. TB[3]H (Scheme 1a) and the starting material 3a were previously reported in our work.47 The synthesis of TB[4]H involved a sequence of steps, beginning with Suzuki coupling of 1 and 2-trimethylsilyl-4-Bpin-thiophene, followed by n-BuLi-induced deprotonation, and culminating in vulcanizing cyclization with (PhSO2)2S (Scheme 1b). The target sulfur-doped helicenes, denoted as TB[n]H (n = 5–8), were synthesized through the dehydrogenative photocyclization of diarylethene derivatives containing DTT and ortho-fused benzene rings (Scheme 1c and the ESI†). Except that 5d was produced by alkyne reduction (3c with 6),49 other diarylethene species (5a–5c50) were synthesized by the Wittig reaction of 3b (described in previous work48) with phosphorous ylide salts (4a–4c). All dehydrogenated photocyclizations were performed under the irradiation of 500-watt mercury lamps. Notably, TB[8]H was obtained in a lower yield (approximately 36%) compared to the other thiahelicenes (68–76%), even with extended reaction times. This observation can be attributed to the significant steric hindrance between the two terminal rings, leading to a higher cyclization barrier than in the case of other compounds. Interestingly, in the 1H NMR spectra of TB[n]H (n = 5–8), the singlet proton signals on the terminal thiophene shifted toward a higher magnetic field (at 8.17, 6.78, 5.26, and 5.04 ppm) with an increasing number of rings. This shift indicates enhanced overlap of the terminal rings, resulting in a stronger magnetic shielding effect.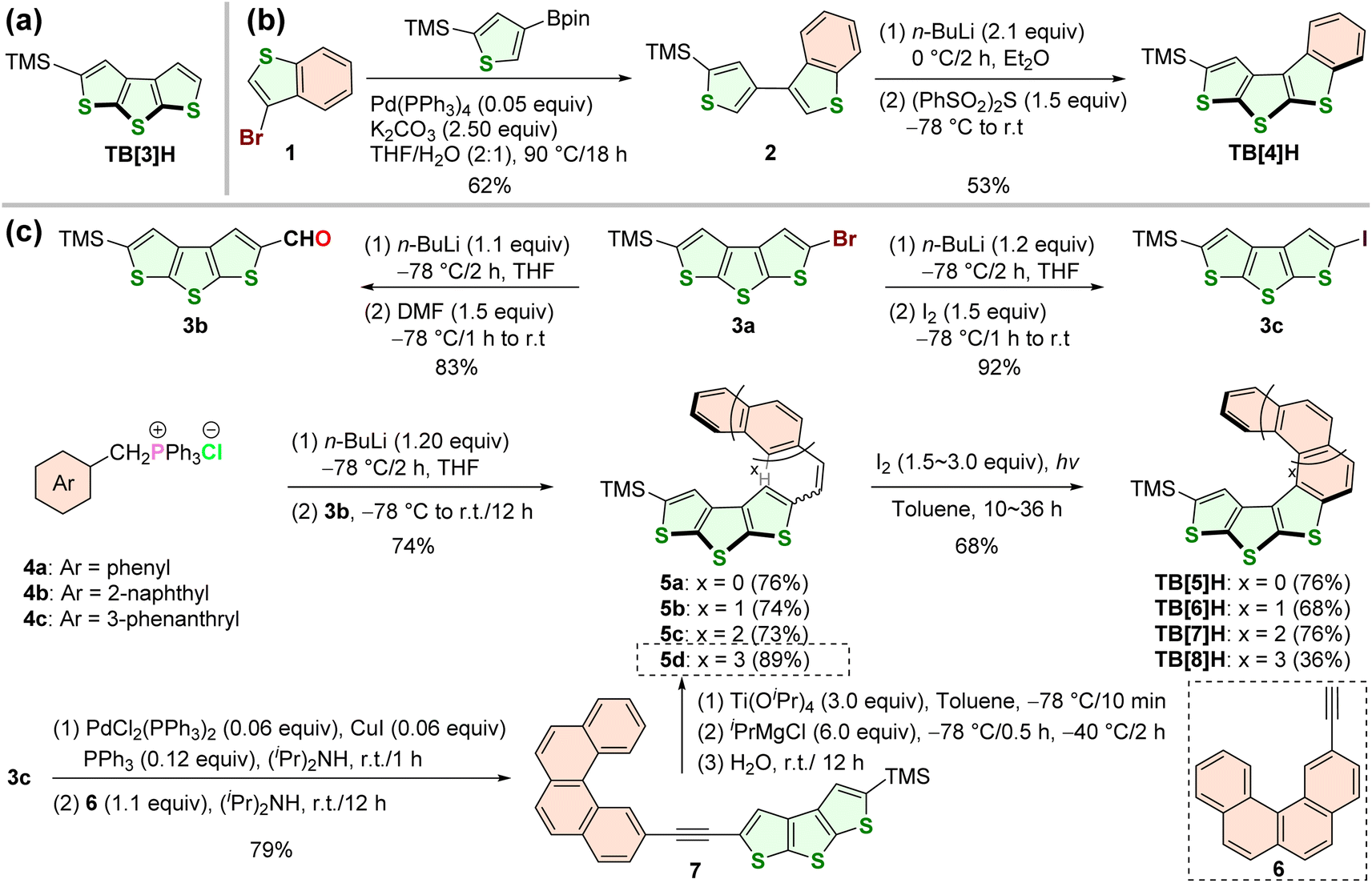 | ||
| Scheme 1 Molecular structures and syntheses of TH[n]H. (a) Chemical structure of TB[3]H. (b) Synthesis of TB[4]Hvia vulcanizing cyclization of dianionic 2. (c) Photocyclization syntheses of TB[n]H (n = 5–8). TMS: trimethylsilyl. DMF: N, N-dimethyl formaldehyde. Pr: propyl. | ||
Single crystal X-ray structures
The structures of TB[4]H, TB[5]H, TB[6]H, and TB[7]H were further validated through X-ray crystallographic diffraction (Fig. 2). Single crystals were obtained by gradually introducing methanol vapor into chloroform solutions of the compounds. The data from the crystallographic analysis have been deposited at the Cambridge Crystallography Data Centre, with assigned CCDC numbers as follows: 2292750 (TB[4]H), 2225686 (TB[5]H), 2225687 (TB[6]H), and 2225691 (TB[7]H). Crystals of TB[n]H (n = 4–7) exhibit triclinic, orthorhombic, orthorhombic, and monoclinic crystal systems with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Pbca, Pbca, and P21/c space groups, respectively. Crystallography reveals that TB[4]H exhibits a planar configuration (Fig. 2a), while the other three molecules exhibit helical scaffolds. However, there is a notable difference in their helicity, especially with TB[5]H displaying a slight deviation of the benzene ring from the DTT plane due to reduced steric hindrance between the two terminal rings (Fig. 2b and S41b†). Several parameters related to the helicity and distortion of the helicenes are schematically presented in Fig. 2e, with TB[7]H serving as an example. These parameters include the average angle between the least-squares planes of neighboring rings (θL), the dihedral angle of the two terminal rings (θD), turns in-plane (θT), and helix climbs (hH).47 As shown in Table 1, it's observed that TB[5]H has a θL value of 4.1°, which is smaller than that of TB[6]H (10.0°) and TB[7]H (9.9°). It's noteworthy that the θL values of the fused thiophene moiety (ranging from 4.8° to 8.7°) were smaller than those of the ortho-fused benzene fragments (ranging from 8.1° to 14.8°) in TB[6]H and TB[7]H. This difference suggests that the helicity is likely governed by the fused benzene rings. Additionally, the θD values (0°, 15.7°, 47.8°, and 51.8°) progressively increase with an increase in the number of rings. Both θT and hH were calculated using the carbon atoms in the inner rim (as labeled in Fig. 2a–d) of helicene and the middle ring plane. The θT and hH were calculated to be 230.8° and 0.56 Å (C1–C6) for TB[5]H, 265.0° and 2.19 Å (C1–C7) for TB[6]H, and 315.6° and 3.08 Å (C1–C8) for TB[7]H. These values indicate an increasing tendency along with the molecular length due to the progressively enhanced steric hindrance between the two terminal rings (Fig. S41†). When compared to related carbohelicenes51–53 and carbon–sulfur helicenes,47,54,55TB[n]H (n = 4–7) exhibit smaller helical parameters than former helicenes but larger than the latter, indicating their medium helicity. Furthermore, in the packing state of all thiahelicenes TB[n]H (n = 5–7), two enantiomers (P and M conformers) were identified (Fig. S41†). These enantiomers tend to assemble together through multiple intermolecular interactions (C⋯C, S⋯C, S⋯H, C⋯H, and H⋯H), while fewer interactions exist between homo conformers. Moreover, the magnitude of π-plane overlaps between two conformers was found to decrease with an increase in the number of rings, reflecting the increasingly distorted configuration of higher thia[n]helicenes.
, Pbca, Pbca, and P21/c space groups, respectively. Crystallography reveals that TB[4]H exhibits a planar configuration (Fig. 2a), while the other three molecules exhibit helical scaffolds. However, there is a notable difference in their helicity, especially with TB[5]H displaying a slight deviation of the benzene ring from the DTT plane due to reduced steric hindrance between the two terminal rings (Fig. 2b and S41b†). Several parameters related to the helicity and distortion of the helicenes are schematically presented in Fig. 2e, with TB[7]H serving as an example. These parameters include the average angle between the least-squares planes of neighboring rings (θL), the dihedral angle of the two terminal rings (θD), turns in-plane (θT), and helix climbs (hH).47 As shown in Table 1, it's observed that TB[5]H has a θL value of 4.1°, which is smaller than that of TB[6]H (10.0°) and TB[7]H (9.9°). It's noteworthy that the θL values of the fused thiophene moiety (ranging from 4.8° to 8.7°) were smaller than those of the ortho-fused benzene fragments (ranging from 8.1° to 14.8°) in TB[6]H and TB[7]H. This difference suggests that the helicity is likely governed by the fused benzene rings. Additionally, the θD values (0°, 15.7°, 47.8°, and 51.8°) progressively increase with an increase in the number of rings. Both θT and hH were calculated using the carbon atoms in the inner rim (as labeled in Fig. 2a–d) of helicene and the middle ring plane. The θT and hH were calculated to be 230.8° and 0.56 Å (C1–C6) for TB[5]H, 265.0° and 2.19 Å (C1–C7) for TB[6]H, and 315.6° and 3.08 Å (C1–C8) for TB[7]H. These values indicate an increasing tendency along with the molecular length due to the progressively enhanced steric hindrance between the two terminal rings (Fig. S41†). When compared to related carbohelicenes51–53 and carbon–sulfur helicenes,47,54,55TB[n]H (n = 4–7) exhibit smaller helical parameters than former helicenes but larger than the latter, indicating their medium helicity. Furthermore, in the packing state of all thiahelicenes TB[n]H (n = 5–7), two enantiomers (P and M conformers) were identified (Fig. S41†). These enantiomers tend to assemble together through multiple intermolecular interactions (C⋯C, S⋯C, S⋯H, C⋯H, and H⋯H), while fewer interactions exist between homo conformers. Moreover, the magnitude of π-plane overlaps between two conformers was found to decrease with an increase in the number of rings, reflecting the increasingly distorted configuration of higher thia[n]helicenes.
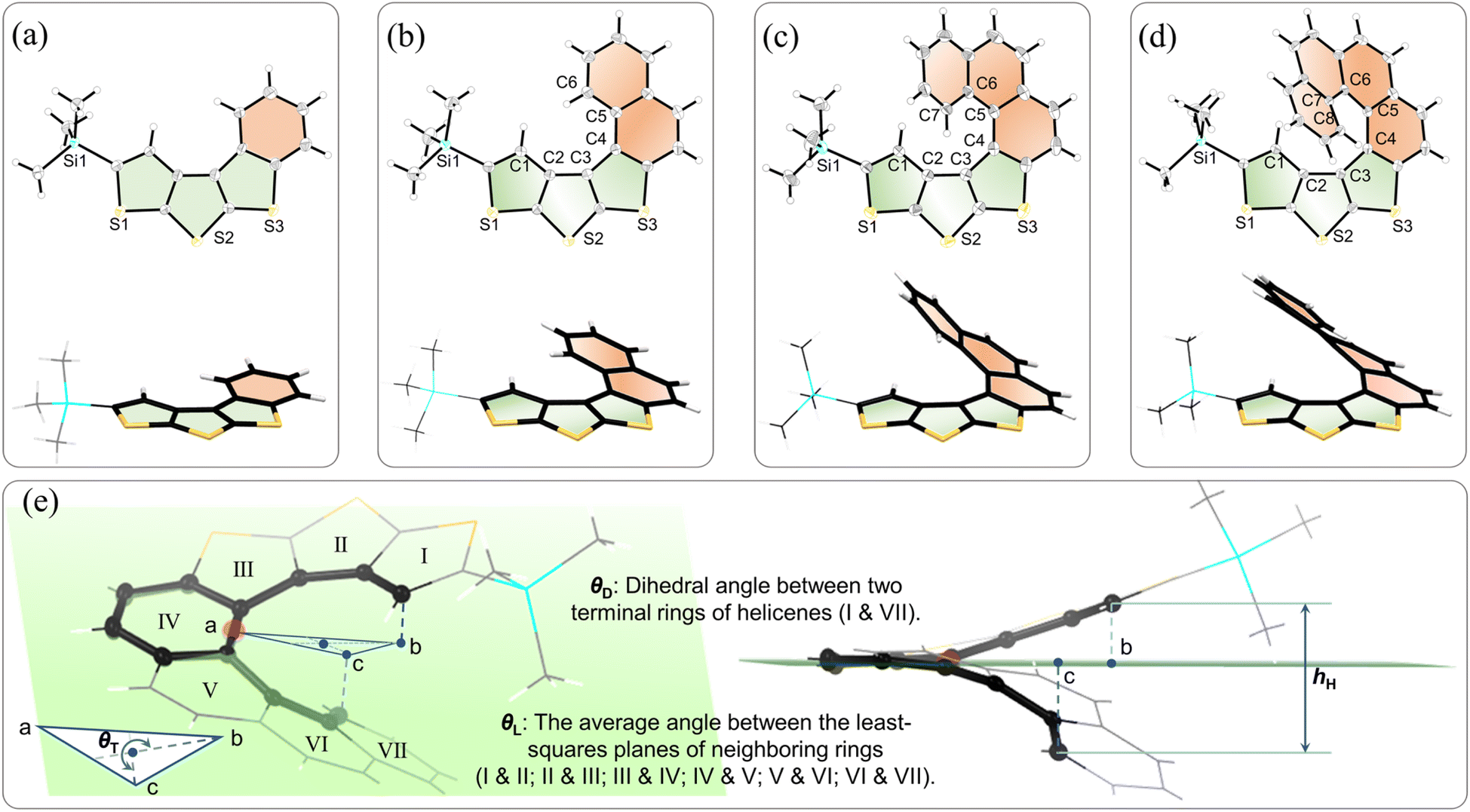 | ||
| Fig. 2 Crystal structures of TH[n]H (n = 4–7). The top-view (ellipsoid mode with 50% probability levels) and side-view structures of (a) TH[4]H; (b) TH[5]H; (c) TH[6]H; (d) TH[7]H. (e) Thia[7]helicene (TB[7]H) was selected as an example to present schematic representations of the dihedral angle between two terminal rings (θD), the average angle between the least-squares of neighboring rings (θL), the turns in-plane (θT), and helix climb (hH). | ||
| TB[n]H | θ L (°) | θ D (°) | θ T (°) | h H (Å) | Crystal system/space group |
|---|---|---|---|---|---|
| a The angles between the least-squares planes of neighboring rings. b Dihedral angle between two terminal rings of helicenes. c Turns angles in-plane. d Helical climbs. | |||||
| TB[4]H | — | 0 | — | — | Triclinic/P |
| TB[5]H | 4.1 | 15.7 | 230.8 | 0.56 | Orthorhombic/Pbca |
| TB[6]H | 10.0 | 47.8 | 265.0 | 2.19 | Orthorhombic/Pbca |
| TB[7]H | 9.9 | 51.8 | 315.6 | 3.08 | Monoclinic/P21/c |
The molecular system under investigation encompasses a range of structures exhibiting varying degrees of nonplanarity, leading to distinct out-of-plane vibrations. These structural variations have the potential to influence electronic properties in both the ground state and the excited state. Consequently, our primary research objective centers on elucidating the structure–property relationship within this series of thiahelicenes using various photophysical techniques.
UV-vis absorption
The UV-vis spectra of TB[n]H (where n = 3–8) are depicted in Fig. 3a (indicated by the orange curves), and pertinent photophysical data are presented in Table 2. It is observed that the maximum absorption wavelength of TB[n]H exhibits a bathochromic shift as the number of benzene rings increases (TB[3]H: 257 nm; TB[4]H: 264 nm and 308 nm; TB[5]H: 261 nm and 319 nm; TB[6]H: 295 nm and 339 nm; TB[7]H: 316 nm and 360 nm; TB[8]H: 337 nm and 376 nm). This shift underscores the progressive extension of π-conjugation within the molecular structures. To gain deeper insights into the changes in absorption across this series of molecules, we conducted time-dependent density functional theory (TD-DFT) calculations. These calculations revealed a reduction in excitation energies with an increase in the number of benzene rings, indicative of extended conjugation facilitating electron excitations. Furthermore, the presence of multiple excitation modes with low oscillator strength helps elucidate the complex shoulder band observed in the UV-vis spectra of the higher thia[n]helicenes (Fig. S45†).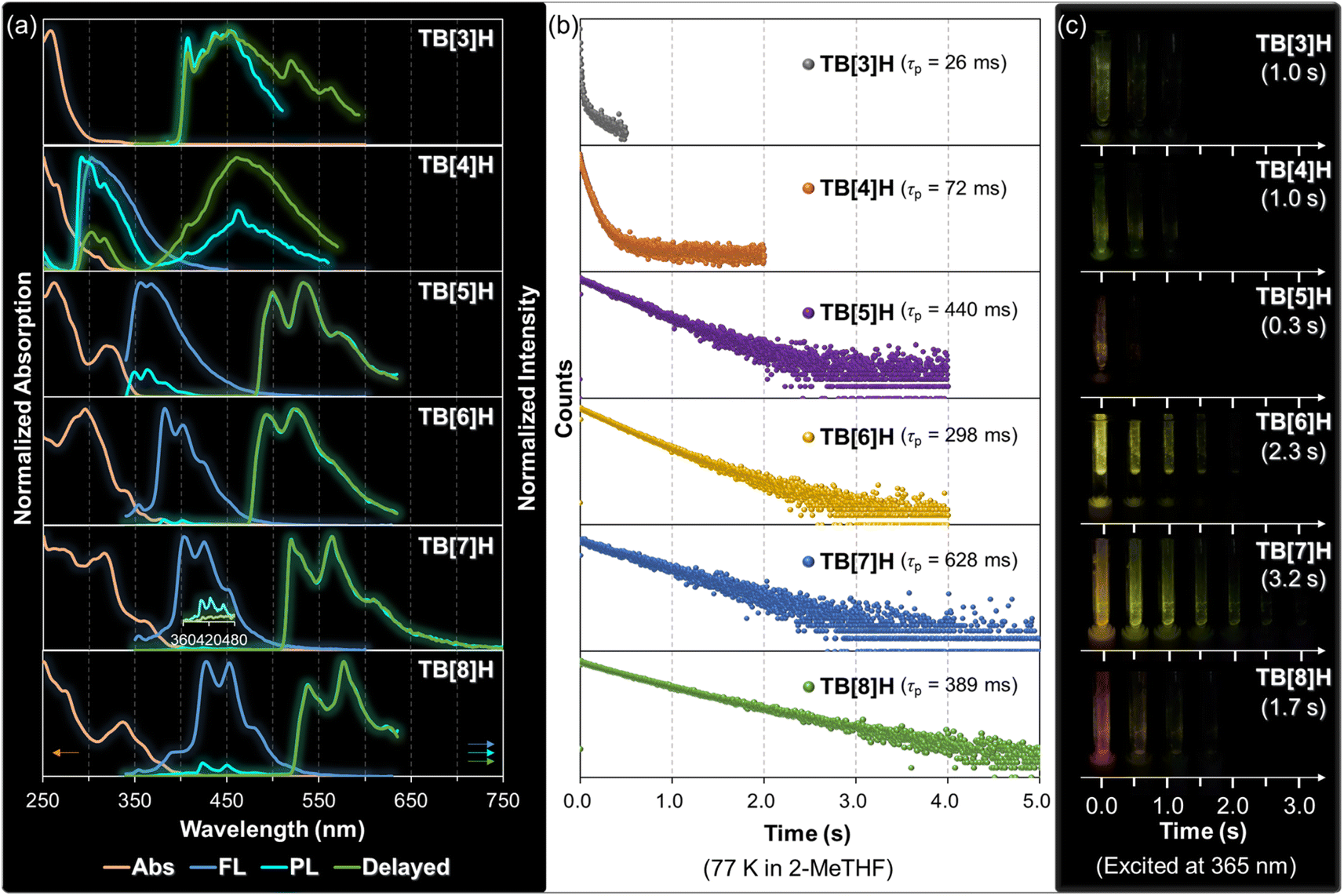 | ||
| Fig. 3 Photophysical properties. (a) UV-vis absorption spectra (orange curves), fluorescence spectra at r.t. (blue curves), emission spectra at 77 K (cyan curves), and the delayed emission spectra at 77 K (5 ms, delayed, green curves) of TB[n]H (n = 3–8). (b) Phosphorescence decays of TB[n]H at 77 K with the emission wavelength at 410 nm for TB[3]H, 460 nm for TB[4]H, 498 nm for TB[5]H, 492 nm for TB[6]H, 518 nm for TB[7]H, and 534 nm for TB[8]H. The UV-vis spectra were recorded in dichloromethane at room temperature with a concentration of [C] = 1 × 10−5 M. All emission spectra were recorded in 2-MeTHF with a concentration of [C] = 5 × 10−5 M. The excitation wavelength (λex) for TB[3]H and TB[4]H were set at 230 nm and 320 nm for other TB[n]H (n = 5–8). (c) Solid-state phosphorescence (77 K) evolution over time and the duration of time. The solids for all compounds were loaded on the inside wall of the quartz tube by evaporation of the sample solution. A UV light with a wavelength of 365 nm was used to excite all samples. | ||
| TB[n]H | λ ABS (nm) | λ FL (nm) | λ PL (nm) | Φ FL (%) | τ f (ns) | k r (s−1) | k nr (kISC + kIC, s−1) | τ p (ms) |
|---|---|---|---|---|---|---|---|---|
| a Recorded in the solution of dichloromethane [1 × 10−5 M]. b Recorded in the solution of 2-MeTHF [5 × 10−5 M] at r.t. c Recorded in the solution of 2-MeTHF [5 × 10−5 M] at 77 K with a delayed time of 5 ms. | ||||||||
| TB[3]H | 257 | — | 400–500 | — | — | — | — | 26 |
| TB[4]H | 264, 308 | 302 | 460 | 1.21 | 1.03 | 1.17 × 107 | 9.59 × 108 | 72 |
| TB[5]H | 261, 319 | 356, 367 | 498, 530 | 0.23 | 1.93 | 1.19 × 106 | 5.17 × 108 | 440 |
| TB[6]H | 295, 339 | 382, 402 | 492, 520 | 0.45 | 1.09 | 4.13 × 106 | 8.34 × 108 | 298 |
| TB[7]H | 316, 360 | 403, 425 | 518, 560 | 0.19 | 0.95 | 2.00 × 106 | 1.05 × 108 | 628 |
| TB[8]H | 337, 376 | 427, 452 | 534, 574 | 0.51 | 1.15 | 4.43 × 106 | 8.65 × 108 | 389 |
Room temperature emission
In Fig. 4a (blue curve), it is evident that, except for the reference molecule TB[3]H, compounds TB[n]H (where n = 4–8) exhibit fluorescent luminescence (FL) at room temperature. This highlights the significant role played by the benzene rings in governing radiative transitions. The fluorescence spectra of TB[n]H (n = 4–8) display emission bands with vibrational structures, indicative of structural rigidity. The fluorescence bands exhibit a bathochromic shift (302 nm for TB[4]H, 356 and 367 nm for TB[5]H, 382 and 402 nm for TB[6]H, 403 and 425 nm for TB[7]H, and 427 and 452 nm for TB[8]H) as the number of benzene rings increases, owing to the gradual extension of the molecular conjugation system. Fluorescence quantum yields (ΦFL) of TB[n]H (n = 4–8) were quantified using quinine sulfate as the standard.56 The resulting ΦFL values for TB[n]H (n = 4–8) were 1.26%, 0.23%, 0.45%, 0.19%, and 0.51%, respectively (Table 2). In comparison to the planar structure of TB[4]H, the lower ΦFL observed in thiahelicenes TB[n]H (n = 5–8) may be attributed to radiationless deactivation induced by their screw configuration. The fluorescence lifetimes (τf) for TB[n]H (n = 4–8) were determined to be 1.03, 1.93, 1.09, 0.95, and 1.15 ns, respectively. Subsequently, radiative (kr) and radiationless (knr) rates were derived from ΦFL and τf (ESI†). All molecules exhibit low kr values at the level of 106–107 s−1, considerably lower than their knr. Consequently, the dominant transition process from the S1 to S0 state is mainly governed by radiationless pathways, including ISC and internal conversion (IC). The sulfur atom in thiophene has been shown to make a substantial contribution to the triplet spin density, and the low-lying n electrons of sulfur atoms play a crucial role in enhancing ISC.26 Additionally, based on the research conducted by Hasobe et al., the ISC rate constant (kISC) was found to be over 4 times larger than the IC rate constant (kIC) in the multiple sulfur-containing helicene.57 Consequently, although the combined kISC and kIC (knr = kISC + kIC) collectively determine the radiationless deactivation, the ISC process was speculated to be the dominant factor, suggesting efficient population of the triplet state for TB[n]H (n = 4–8). However, no phosphorescence was observed at room temperature. This could be attributed to the low stability of the triplet state, which is more susceptible to quenching by molecular vibration and collisions with other media, such as solvents.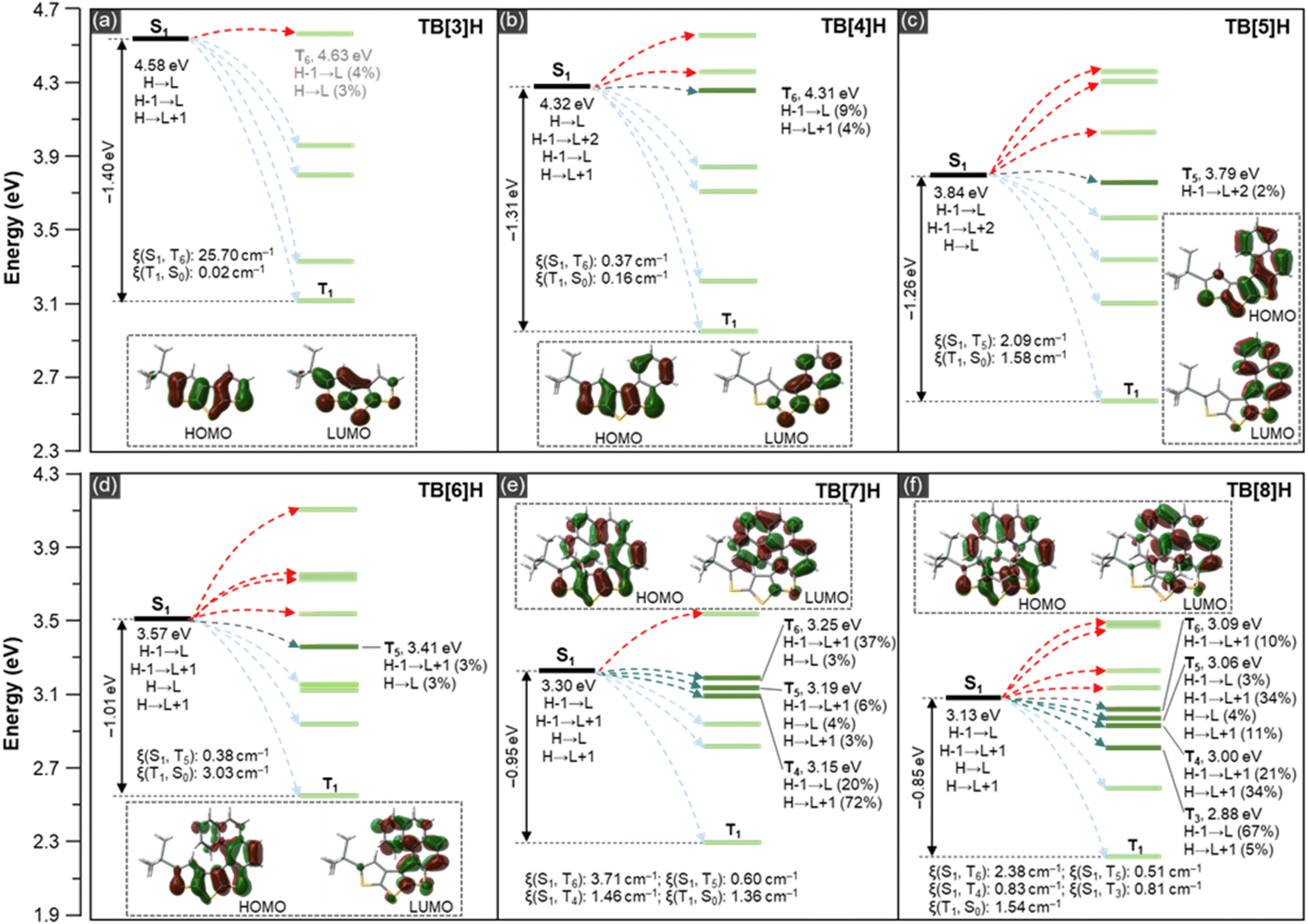 | ||
| Fig. 4 Theoretical calculations of ISC channels, SOC matrix elements, and frontier molecular orbitals. (a) TB[3]H, (b) TB[4]H, (c) TB[5]H, (d) TB[6]H, (e) TB[7]H, and (f) TB[8]H. TD-DFT calculated (B3LYP/6-31G*) energy levels, the possible ISC channels (blue arrow and dark green line), other ISC channels with small possibility (red arrow marked high-lying triplets, light blue arrow marked low-lying triplets, and triplet state marked as a light green line), SOC matrix elements (ξ), and DFT calculated frontier molecular orbitals (HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital) are involved in each figure. | ||
Cryogenic emission
Given that sulfur atoms can enhance ISC through n–π* transitions, these thia[n]helicenes hold promise for potential applications as phosphorescent materials. Nevertheless, it is crucial to implement measures to suppress the pronounced radiationless process from T1 to S0 state. Considering that molecular motion is restricted at low temperatures, effectively suppressing radiationless decay, we conducted steady-state emission spectroscopy under cryogenic conditions (77 K). As exhibited in Fig. 3a (cyan curves), it is evident that the reference compound TB[3]H exhibits a complex vibrational emission band in the wavelength range of 400 to 500 nm. This emission has been assigned as phosphorescence, demonstrating that the annulated thiophenes, DTT, can serve as an excellent building block for constructing phosphorescent molecules. Following the annulation of benzene rings onto DTT, TB[n]H (n = 4–8) exhibited dual emission under the same experimental conditions. The emission bands in the short wavelength region (291 nm for TB[4]H, 349 and 363 nm for TB[5]H, 381 and 402 nm for TB[6]H, 402 and 423 nm for TB[7]H, and 423 and 450 nm for TB[8]H) correspond to FL, coinciding with the FL observed at room temperature. In contrast, the emission bands in the long wavelength region (460 nm for TB[4]H, 498 and 530 nm for TB[5]H, 492 and 520 nm for TB[6]H, 518 and 560 nm for TB[7]H, and 534 and 574 nm for TB[8]H) are attributed to steady-state phosphorescent luminescence (PL). It's noteworthy that both the FL and PL of TB[n]H (n = 3–8) undergo a gradual bathochromic shift as the number of benzene units increases due to the extension of molecular conjugation. Additionally, the PL/FL ratio (49%/51% for TB[4]H, 90%/10% for TB[5]H, 98%/2% for TB[6]H, 99%/1% for TB[7]H, and 92%/8% for TB[8]H) increases progressively. The higher PL/FL ratio in twisted structures highlights the efficiency of ISC and the T1 → S0 transition. The singlet and triplet energies (ES1/ET1) were calculated based on the cryogenic spectra, yielding values of 4.36/3.19 eV (TB[4]H), 3.69/2.57 eV (TB[5]H), 3.47/2.62 eV (TB[6]H), 3.19/2.42 eV (TB[7]H), and 2.99/2.39 eV (TB[8]H), respectively. The decreasing energy gap between these states suggests a more efficient ISC in twister helicenes. Beyond these aspects, the heavy-atom effect of sulfur and helical molecular distortion is also inferred as a contributing factor to ISC since it can influence the energy levels and orbital distributions of both singlet and triplet states.19,58Furthermore, these thiahelicenes also exhibited remarkable afterglow luminescence in the solid state at 77 K following excitation with 365 nm UV light. As illustrated in Fig. 3c and S44,† the emission color transitioned from light yellow to orange, which may be attributed to variations in their structures and packing arrangements. The emission persisted for a duration ranging from 0.3–3.2 s across this series of compounds. Notably, the twister structures TB[n]H (n = 6–8) exhibited longer durations compared to the less distorted TB[n]H (n = 3–5), suggesting that the extent of distortion plays a crucial role in modulating the duration of solid-state phosphorescence. In line with the findings in the frozen state of solution, TB[7]H exhibited the longest afterglow duration of 3.2 s. This suggests that TB[7]H holds potential as a cryogenic phosphorescent material, suitable for application in both liquid and solid states. Under the cryogenic conditions, molecular motion is restricted, efficiently suppressing nonradiative decay and facilitating persistent emission. Additionally, it is speculated that molecular structures and intermolecular interactions (as discussed in the Single crystal X-ray structure section) contribute to the solid-state emission and its duration, resulting in distinct afterglow colors and times. The favorable photoluminescent properties observed in both molecular and solid states indicate that the designed molecules in this study hold promise as long-lived phosphorescent materials.
Delayed emission
Time-resolved PL measurements were conducted with a delayed time of 5 ms (Fig. 3a, green curves) at 77 K. Given that FL typically exhibits nanosecond-level lifetimes, this technique effectively eliminates the initial FL, making it a standard approach for investigating long-lived luminescence. As depicted in Fig. 3a, TB[3]H displayed a profile nearly identical to the non-delayed spectrum. This further underscores that this molecule exclusively exhibits phosphorescence in the frozen state. Unexpectedly, TB[4]H continued to exhibit dual emission even with the extended delay time. This FL may arise from certain photon upconversion processes, such as triplet–triplet annihilation (TTA).59,60 In contrast, all thiahelicenes TB[n]H (n = 5–8) exclusively displayed PL under the same conditions when compared to TB[4]H. The higher PL/FL ratio observed in helical structures in the non-time-resolved emission spectra suggested that the upconversion process is less likely to occur. Consequently, the time-resolved technique efficiently screens out FL efficiently in the helicene structures, providing pure PL spectra.Phosphorescence decay
To explore the lifetimes of PL in TB[n]H, transient PL decays were measured at 77 K with a delayed time of 5 ms, as depicted in Fig. 3b. The reference molecule TB[3]H exhibited a rapid decay process, resulting in a relatively short lifetime of 26 ms, while TB[4]H displayed a somewhat longer lifetime of 72 ms. With the annulation of additional benzene rings, the decay process gradually slowed down and the lifetimes extended to hundreds of milliseconds. Notably, the PL lifetime did not exhibit a direct positive correlation with the extension of the π-system. Instead, an alternative trend in τp emerged in relation to the ring number, with odd-numbered-ring molecules having larger τp values (440 ms for TB[5]H and 628 ms for TB[7]H) than their neighboring even-number-ring counterparts (298 ms for TB[6]H and 389 ms for TB[8]H). Furthermore, τp for both odd and even-number-ring molecules increased with the accumulation of ring numbers, respectively. Consequently, it can be inferred that odd-number-ring thiahelicenes may function as more persistent phosphors than their even-number-ring counterparts. From a structural similarity perspective, we speculate that this odd–even effect might be applicable to other types of helicenes, such as heterohelicenes, helicenoids, and multiple helicenes. This observed phenomenon could potentially serve as a referential rule for designing high-performance phosphorescent helicenes.Under frozen conditions, molecular movement and vibration are typically constrained, thereby suppressing various nonradiative decay pathways during the transition from the T1 to S0 state, such as the collision with solvent molecules. However, it should be noted that this frozen state would play a similar role for all molecules in the current study due to their similar elemental composition. Therefore, the differential phosphorescence behavior observed in TB[n]H (n = 3–8) might be attributed to their varying π-conjugated lengths and helical configurations.18–22,24,58 Both of these factors have the potential to reduce the singlet–triplet energy gap and enhance the SOC, thereby facilitating the ISC process.16 Consequently, ISC was presumed to dominate the decay process in the current molecular series, resulting in stronger phosphorescent luminescence and longer lifetimes for the higher thiahelicenes. While low-temperature phosphorescence is a common occurrence in many organic molecules, the exploration of long-persistent phosphorescence has not garnered attention from researchers specializing in helicenes. This has led to an unclear understanding of the relationship between structure and phosphorescence properties in this context. The findings in this work aim to address this gap and provide new insights into the matter.
Theoretical calculations
In order to gain insight into the reasons behind the distinct PL behaviours observed in the current series of molecules, we conducted TD-DFT calculations at the B3LYP/6-31G* level of theory. Given that the ISC process might govern the phosphorescent properties of the TB[n]H (n = 3–8), the computational calculations primarily focused on investigating the key factors influencing ISC. In accordance with perturbation theory, two critical factors, namely the energy gap between singlet and triplet states (ΔEST) and SOC, impact the kISC. This relationship can be described as the following expression:where ξ (Sn, Tn) represents the SOC matrix element.16 As per this expression, there is a negative correlation between ΔEST and the ISC rate, whereas the magnitude of SOC exhibits a positive correlation with kISC. The quantum-chemical calculation results for ΔEST and ξ (Sn, Tn) are illustrated in Fig. 4.
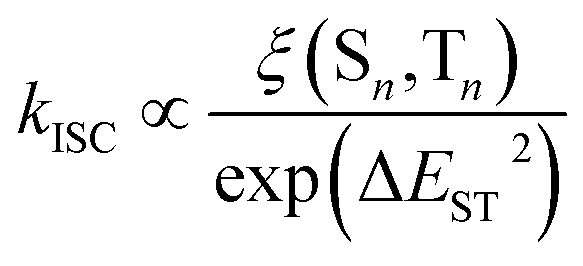
The energy gap between S1 and T1 of all molecules (1.40 eV for TB[3]H, 1.31 eV for TB[4]H, 1.26 eV for TB[5]H, 1.01 eV for TB[6]H, 0.95 eV for TB[7]H, and 0.85 eV for TB[8]H) gradually decreases with the increase in the ring number. The trend in ΔEST aligns well with the experimental data and also explains the bathochromic shift observed in PL spectra. For possible ISC to occur, it is necessary for the S1 and Tn states to have matching transition orbital compositions, with a triplet energy level falling within the energy range of ±0.3 eV around ES1 (the energy level of the S1 state). In the case of TB[3]H, although it has four low-lying triplet levels with the same transition configuration as the S1 state (Fig. S48†), the energy gaps between the S1 and Tn states in all channels exceed 0.3 eV. This large energy gap results in inefficient ISC. In contrast, TB[n]H (n = 4–8) all exhibit multiple transition-identical triplets as S1 states with sufficient ISC channels. TB[4]H, TB[5]H, and TH[6]H each have one ISC-favorable triplet level (T6, T5, and T5) with energy gaps (EISC: energy gap between S1 and the most favorable ISC-actionable highest Tn) of 0.02, 0.05, and 0.16 eV, respectively. TB[7]H and TH[8]H feature three (S1 → T6, S1 → T5, and S1 → T4) and four (S1 → T6, S1 → T5, S1 → T4, and S1 → T3) energy-favorable ISC channels with an EISC of 0.05 and 0.04 eV, respectively. The presence of these accessible triplets coupled with low EISC values imparts intensive and long-lived phosphorescence to these molecules. Additionally, molecules with more rings, which have more ISC channels, should exhibit superior PL. However, it's worth noting that these simulated results do not fully explain the observed τp differences between odd and even-number-ring helicenes. Thus, we believe that another crucial factor, namely SOC, may simultaneously play a key role in ISC, in addition to the ISC channels.
The SOC matrix element (ξ) was calculated by employing TD-DFT in conjugation with the PySOC program.61,62 As depicted in Fig. 4, SOC between T1 and S0 for TB[3]H and TB[4]H exhibited relatively small matrix elements of 0.02 cm−1 and 0.16 cm−1, respectively. These values indicate that the SOC is not sufficiently strong enough to support efficient PL with a long lifetime, which is in agreement with the experimental findings. In contrast, the helicenes TB[n]H (n = 5–8) displayed relatively larger ξ (T1, S0) values (1.58, 3.03, 1.36, and 1.54 cm−1) compared to the two reference species. This suggests that the T1 → S0 transitions are more favorable in thia[n]helicenes than in planar species. The most favorable ISC channels of helicenes TB[n]H (n = 5–8) exhibit ξ (S1, T5) of 2.09 cm−1 for TB[5]H, ξ (S1, T5) of 0.38 cm−1 for TB[6]H, ξ (S1, T6) of 3.71 cm−1 for TB[7]H, and ξ (S1, T6) of 2.38 cm−1 for TB[8]H, respectively. This trend closely aligns with the experimental τp value, suggesting that the ISC process follows this pattern. Furthermore, other accessible ISC channels of TB[7]H demonstrated stronger spin–orbital coupling than those of TB[8]H, providing further evidence that the former exhibits more efficient ISC than the latter. Given the close correlation between SOC and τp,29,63 the ξ values of these thiahelicenes efficiently explain the observed trend in their PL lifetimes.
The results presented above collectively underscore the pivotal role of spin–orbit interactions in the generation of PL through ISC. The presence of sulfur atoms in all molecules supplied sufficient n orbitals which can form good conjugation with π bonds on molecules providing an ample source of n orbitals, which can readily form effective conjugation with π bonds on the molecular backbones, as evidenced by the frontier molecular orbitals depicted in each figure. This heteroatom effect significantly enhances the SOC between singlet and triplet states. Consequently, the initially excited singlet 1nπ* state is believed to facilitate ISC. The spin density distribution of T1 further confirms that the spin is predominantly localized in the angular acene moieties of TB[n]H (n = 4–8), indicating that the triplets are predominantly of the 3ππ* state (Fig. S47†). According to EI-Sayed rules, the combination of the 1nπ* state (S1) with 3ππ* state (T1) significantly accelerates ISC and dramatically enhances PL.64,65 Beyond the aforementioned influences, the twisted molecular configuration may also contribute to the strong PL observed in higher helicenes. This configuration is speculated to narrow the singlet–triplet energy gap and intensify SOC through out-of-plane C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H vibrations,20,66 which aligns with the speculation in the single crystal section.
C and C–H vibrations,20,66 which aligns with the speculation in the single crystal section.
Hence, the combined influence of potential ISC channels and robust SOC could account for the longer-lasting PL observed in odd-numbered helicenes compared to their even-numbered counterparts. This insight could serve as a fundamental guideline for the molecular design of helicenes with superior optical performance.
Chiroptical properties
The chiroptical properties of phosphorescence were subsequently investigated using CD and CPL spectroscopy. Since TB[7]H displayed robust phosphorescent behavior and a longer lifetime compared to other structures in this work, it was selected for further research on chiroptical natures. The racemization barrier for the two TB[7]H conformers was determined to be 43.2 kcal mol−1 through DFT calculations at the B3LYP/6-31G* level of theory. This barrier demonstrates sufficient stability, allowing the existence, separation, and storage of the two enantiomers, consistent with findings in prior studies.41 The racemic mixture was chirally resolved through preparative high-performance liquid chromatography (HPLC, EnantioPak®Y1) using a supercritical fluid chromatographic technique with a CO2/isopropanol mixture (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35, v/v) as the mobile phase, leading to the efficient separation of two fractions with retention times (tR) of 6.49 min and 7.67 min, respectively (Fig. S49†).
:35, v/v) as the mobile phase, leading to the efficient separation of two fractions with retention times (tR) of 6.49 min and 7.67 min, respectively (Fig. S49†).
The two separated compounds exhibited distinct optical responses and mirror image profiles in CD spectra (Fig. 5, dashed curves), indicating their enantiomeric relationship. Both enantiomers displayed Cotton effects at 289, 319, and 363 nm with |gabs| values of 3.0 × 10−3, 2.9 × 10−3, and 1.8 × 10−3, respectively. Notably, the first HPLC-eluted enantiomer displayed a negative Cotton effect, while the second one exhibited a positive CD response. Based on TD-DFT calculations, the (M)-enantiomer ((M)-TB[7]H) was predicted to have a negative Cotton effect at the wavelengths of 270–300 nm and 350–400 nm (Fig. S50†), which closely matched the experimental CD spectra of the first HPLC-eluted enantiomer. Consequently, the absolute configurations of the two enantiomers were confidently assigned based on the comparison of experimental results and theoretical simulations. Specifically, the component with the shorter retention time was identified as the M-enantiomer ((M)-TB[7]H), while the other component with a longer retention time was assigned as the P-enantiomer ((P)-TB[7]H).
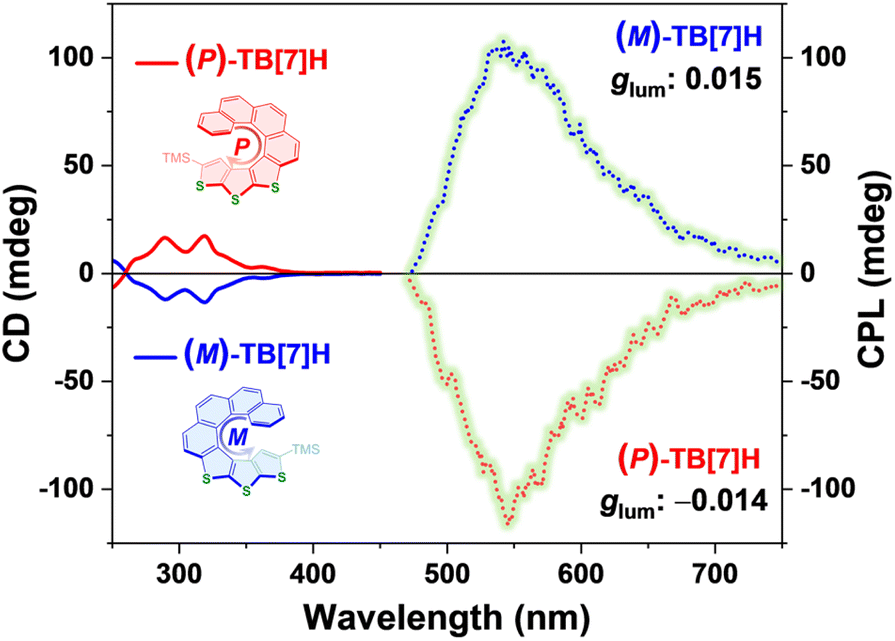 | ||
| Fig. 5 Chiroptical properties. CD (dashed curves) and CPL (dot curves) spectra of enantiomers (M)-TB[7]H and (M)-TB[7]H. Both samples were characterized in the solution of 2-MeTHF with a concentration of 5 × 10−5 M. The CD spectra were recorded at room temperature, while the CPL spectra were detected at 77 K. | ||
Armed with the two configurationally distinct samples, we delved into their CPL properties. Initially, we assessed the CPL characteristics at room temperature, but no CPL response was observed, likely due to their low fluorescence quantum yield. As discussed earlier, the sulfur heteroatom effect and the twisted configuration both enhance the ISC process, leading to inefficient fluorescent luminescence. However, when the solution was cooled to 77 K, both samples exhibited a robust CPL response, displaying a mirror CPL image profile at 470–750 nm (Fig. 5, dot curves). The emission wavelength aligned with the cryogenic PL spectra (Fig. S51†), confirming that the luminescence was circularly polarized phosphorescent luminescence. Notably, the CPL spectra exhibited opposite signs compared to CD spectra. (M)-TB[7]H exhibited a positive signal within 470–750 nm, while (P)-TB[7]H displayed a negative response in the same wavelength range. This discrepancy between CD and CPL spectra suggested significant geometric reorganization in the excited singlet or triplet state. The maximum glum values for the M and P- enantiomers were 0.015 (542 nm) and −0.014 (545 nm), respectively. In the realm of molecular CPP materials, these glum values represent a relatively high level.23,25 The substantial |glum| values indicate a high degree of polarization in the phosphorescence emitted by (P)- and (M)-TB[7]H, making them potential low-temperature CPP-active materials with prolonged luminescence lifetimes. Given their distinct CPL performances at room temperature and low temperature, thia[7]helicene enantiomers could find applications in temperature-dependent information encryption, spintronics, optoelectronic devices, and beyond.
Conclusion
In summary, we successfully synthesized a series of thia[n]helicenes, denoted as TB[n]H (n = 3–8), by employing a module-assembly strategy that incorporated DTT and ortho-fused benzene rings. The increasing helical character of this molecule series was unequivocally confirmed through X-ray crystallography. Our photophysical investigations revealed that thiahelicenes TB[n]H (n = 5–8) displayed notable luminescent properties, particularly exhibiting cryogenic phosphorescence. These impressive luminescent attributes on one hand can be attributed to the combined influence of the sulfur atom's heteroatom effect, the distorted molecular configuration, and the strong SOC, which collectively fostered efficient ISC processes, and on another hand can be attributed to the suppression of nonradiative decay under cryogenic conditions. Thiahelicenes TB[n]H (n = 5–8) exhibited millisecond-level PL lifetimes and persistent afterglow in the solid state. The intriguing alternation in phosphorescence lifetime between odd- and even-numbered ring thiahelicenes was observed and explained by the distinct ISC channels and SOC in the excited state. Notably, TB[7]H stood out with a remarkable τp of 628 ms and the longest solid-state afterglow luminescence lasting 3.2 s, highlighting its potential for application in organic optoelectronic materials. Furthermore, we discovered excellent low-temperature CPP with a |glum| of approximately 0.015. Consequently, our work suggests that rational molecular design can lead to heterohelicenes as a distinctive class of organic materials with persistent phosphorescence and CPP capabilities. These findings hold promise for cutting-edge applications in electronics and optical waveguides, particularly as organic persistent phosphors. Our future research endeavors will explore the use of this series of molecules as suitable chromophores for constructing ultra-long room-temperature phosphorescence (URTP) with CPP activity through crystallography or supramolecular polymerization. After designing rational strategies for suppression of nonradiative deactivation, we expect that the URTP of thiahelicenes with adjustable lifetime and emission band will be realized.Data availability
All experimental and characterization data are available in the ESI.†Author contributions
Z. Sun and W. Xu performed the syntheses, structural determinations, and partial physical characterization studies. C. Li and S. Qiu instructed some syntheses and physical characterization, respectively. Z. Ma analyzed single crystal X-ray structures and instructed the DFT calculations. S. Zhang performed partial physical characterization studies, DFT calculations, and data analysis and wrote the manuscript and ESI.† S. Zhang and H. Wang proposed the ideas and charged project administration. Z. Sun and W. Xu contributed equally to this paper.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. U2004213), Natural Science Foundation of Henan Province (general program, no. 232300421228), and Youth Natural Science Foundation of Henan Province (232300420373).References
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef PubMed.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–648 CrossRef CAS.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- Y. Zhu and J. Wang, Acc. Chem. Res., 2023, 56, 363–373 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. D. Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735–742 RSC.
- G. Farias, C. A. M. Salla, M. Aydemir, L. Sturm, P. Dechambenoit, F. Durola, B. de Souza, H. Bock, A. P. Monkman and I. H. Bechtold, Chem. Sci., 2021, 12, 15116–15127 RSC.
- I. Stary and I. G. Stara, Targets Heterocycl. Syst., 2017, 21, 23–53 Search PubMed.
- F. Dumitrascu, D. G. Dumitrescu and I. Aron, Arkivoc, 2010, 1–32 Search PubMed.
- F. Pop, N. Zigon and N. Avarvari, Chem. Rev., 2019, 119, 8435–8478 CrossRef PubMed.
- M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639–5643 CrossRef.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
- P. Ravat, T. Šolomek, M. Rickhaus, D. Hussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- C. A. M. Salla, G. Farias, M. RouziHres, P. Dechambenoit, F. Durola, H. Bock, B. de Souza and I. H. Bechtold, Angew. Chem., Int. Ed., 2019, 58, 6982–6986 CrossRef PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef PubMed.
- X. Wang, S. Ma, B. Zhao and J. Deng, Adv. Funct. Mater., 2023, 33, 2214364 CrossRef.
- A. Mohan, E. Sebastian, M. Gudem and M. Hariharan, J. Phys. Chem. B, 2020, 124, 6867–6874 CrossRef PubMed.
- K. Dhbaibi, P. Morgante, N. Vanthuyne, J. Autschbach, L. Favereau and J. Crassous, J. Phys. Chem. Lett., 2023, 14, 1073–1081 CrossRef PubMed.
- A. Bossi, P. Mussini, G. Farinola, M. Penconi, S. Cauteruccio, M. E. Thompson and E. Licandro, Chem. – Eur. J., 2023, 29, e202300339 CrossRef CAS PubMed.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS PubMed.
- Z. Wu, J. Nitsch and T. B. Marder, Adv. Opt. Mater., 2021, 9, 2100411 CrossRef CAS.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 1, 582–594 RSC.
- M. Mecklenburg, W. A. Hubbard, E. R. White, R. Dhall, S. B. Cronin, S. Aloni and B. C. Regan, Science, 2015, 347, 629–632 CrossRef CAS PubMed.
- X. Yang, L. Zhao, W. Chen, Z. Liu, X. Fan, S. Tian, X. Xu, J. Qiu and X. Yu, J. Lumin., 2019, 208, 290–295 CrossRef CAS.
- J. Du and D. Poelman, Adv. Opt. Mater., 2020, 8, 1901848 CrossRef CAS.
- L. Marciniak, K. Kniec, K. Elżbieciak-Piecka, K. Trejgis, J. Stefanska and M. Dramićanin, Coord. Chem. Rev., 2022, 469, 214671 CrossRef CAS.
- M. J. Kreder, J. Alvarenga, P. Kim and J. Aizenberg, Nat. Rev. Mater., 2016, 1, 15003 CrossRef CAS.
- H. Zhu, I. Badía-Domínguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. R. Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Angew. Chem., Int. Ed., 2022, 61, e20221310 Search PubMed.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef PubMed.
- E. Licandro, S. Cauteruccio and D. Dova, Adv. Heterocycl. Chem., 2016, 118, 1–46 CrossRef.
- S. K. Collins and M. P. Vachon, Org. Biomol. Chem., 2006, 4, 2518–2524 RSC.
- L. Zhou, H. Liu, J. Tan, C. Liu, X.-Y. Cao, A. Narita and Y. Hu, Chem.–Asian J., 2022, 17, e202200336 CrossRef CAS PubMed.
- J. Yang, X. Gao, Z. Xie, Y. Gong, M. Fang, Q. Peng, Z. Chi and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15299–15303 CrossRef CAS PubMed.
- G. Bergamini, A. Fermi, C. Botta, U. Giovanella, S. D. Motta, F. Negri, R. Peresutti, M. Gingras and P. Ceroni, J. Mater. Chem. C, 2013, 1, 2717–2724 RSC.
- A. Mishra, C.-Q. Ma and P. Baüerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- C. Li, J. Shi, L. Xu, Y. Wang, Y. Cheng and H. Wang, J. Org. Chem., 2009, 74, 408–411 CrossRef CAS PubMed.
- W. Xu, J. Shi, C. Li, D. Zhao, Z. Wang and H. Wang, Org. Chem. Front., 2022, 9, 5578–5585 RSC.
- R. E. Messersmith, M. A. Siegler and J. D. Tovar, J. Org. Chem., 2016, 81, 5595–5605 CrossRef CAS PubMed.
- S. Abbate, C. Bazzini, T. Caronna, F. Fontana, C. Gambarotti, F. Gangemi, G. Longhi, A. Mele, I. N. Sora and W. Panzeri, Tetrahedron, 2006, 62, 139–148 CrossRef CAS.
- A. C. Bédard, A. Vlassova, A. C. Hernandez-Perez, A. Bessette, G. S. Hanan, M. A. Heuft and S. K. Collins, Chem. – Eur. J., 2013, 19, 16295–16302 CrossRef PubMed.
- Y. Yoshida, U. Nakamura, H. Kishida, H. Hayama, Y. Nakano, H. Yamochi and G. Saito, CrystEngComm, 2017, 19, 3626–3632 RSC.
- C. Shen, G. Zhang, Y. Ding, N. Yang, F. Gan, J. Crassous and H. Qiu, Nat. Commun., 2021, 12, 2786 CrossRef PubMed.
- M. Miyasaka, A. Rajca, M. Pink and S. Rajca, J. Am. Chem. Soc., 2005, 127, 13806–13807 CrossRef PubMed.
- B. Wanno and V. Ruangpornvisuti, Struct. Chem., 2010, 21, 715–725 CrossRef.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- Y. Yamamoto, H. Sakai, J. Yuasa, Y. Araki, T. Wada, T. Sakanoue, T. Takanobu, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 7421–7427 CrossRef.
- P. Baronas, R. Komskis, E. Tankelevičiūtė, P. Adomėnas, O. Adomėnienė and S. Juršėnas, J. Phys. Chem. Lett., 2021, 12, 6827–6833 CrossRef CAS PubMed.
- R. Ieuji, K. Goushi and C. Adachi, Nat. Commun., 2019, 10, 5283 CrossRef PubMed.
- Y. Luo, K. Zhang, Z. Ding, P. Chen, X. Peng, Y. Zhao, K. Chen, C. Li, X. Zheng, Y. Huang, X. Pu, Y. Liu, S.-J. Su, X. Hou and Z. Lu, Nat. Commun., 2022, 13, 6892 CrossRef CAS PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- S. Hirata, J. Phys. Chem. Lett., 2018, 9, 4251–4259 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, synthesis, X-ray diffraction, photophysical properties, computational calculations, chiral resolution, and chiral spectra. CCDC 2292750, 2225686, 2225687 and 2225691. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05480b |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |