
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic ethane conversion on rutile TiO2(110): identifying the role of the ethyl radical†
Fangliang
Li‡
 a,
Yuemiao
Lai‡
a,
Yi
Zeng‡
a,
Xiao
Chen
a,
Tao
Wang
a,
Xueming
Yang
abc and
Qing
Guo
*a
a,
Yuemiao
Lai‡
a,
Yi
Zeng‡
a,
Xiao
Chen
a,
Tao
Wang
a,
Xueming
Yang
abc and
Qing
Guo
*a
aShenzhen Key Laboratory of Energy Chemistry & Department of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, PR China. E-mail: guoq@sustech.edu.cn
bState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, Liaoning 116023, PR China
cHefei National Laboratory, Hefei 230088, PR China
First published on 27th November 2023
Abstract
Oxidative dehydrogenation of ethane (C2H6, ODHE) is a promising approach to producing ethene (C2H4) in the chemical industry. However, the ODHE needs to be operated at a high temperature, and realizing the ODHE under mild conditions is still a big challenge. Herein, using photocatalytic ODHE to obtain C2H4 has been achieved successfully on a model rutile(R)-TiO2(110) surface with high selectivity. Initially, the C2H6 reacts with hole trapped OTi− centers to produce ethyl radicals 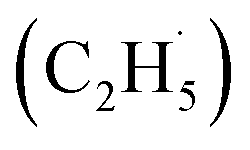 , which can be precisely detected by a sensitive TOF method, and then the majority of the
, which can be precisely detected by a sensitive TOF method, and then the majority of the 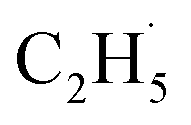 radicals spontaneously dehydrogenate into C2H4 without another photo-generated hole. In addition, parts of the
radicals spontaneously dehydrogenate into C2H4 without another photo-generated hole. In addition, parts of the 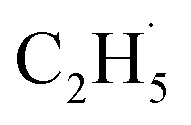 radicals rebound with diversified surface sites to produce C2 products via migration along the surface. The mechanistic model built in this work not only advances our knowledge of the C–H bond activation and low temperature C2H6 conversion, but also provides new opportunities for realizing the ODHE with high C2H4 efficiency under mild conditions.
radicals rebound with diversified surface sites to produce C2 products via migration along the surface. The mechanistic model built in this work not only advances our knowledge of the C–H bond activation and low temperature C2H6 conversion, but also provides new opportunities for realizing the ODHE with high C2H4 efficiency under mild conditions.
Introduction
Ethene (C2H4), as an important basic material for manufacturing diverse consumer products, accounts for about 75% of petrochemical products.1,2 With the increase of global C2H4 demand, environmental and economic issues have become serious problems facing the world because of the energy- and emission-intensive activities for C2H4 production (e.g., naphtha steam cracking,3–5 fluidized catalytic cracking (FCC),6–8 methanol-to-olefins (MTO)2 and Fischer–Tropsch to olefins (FTO)2). The vigorous exploitation of shale gas containing abundant light alkanes has promoted the development of the direct dehydrogenation of ethane (C2H6) to C2H4. However, compared with the non-oxidative dehydrogenation of the C2H6 technique, which is thermodynamically limited with highly endothermic properties, the selective oxidative dehydrogenation (ODH) of C2H6 is a promising alternative route for C2H4 production due to its autothermal conditions.1,2,9,11Although the ODH of C2H6 (ODHE) is thermodynamically favored, it is still often conducted under harsh conditions (high temperature and pressure) because of the high chemical stability of the C–H bonds (414 kJ mol−1), resulting in high energy consumption, catalyst deactivation, and over oxidation.1,2,10 Therefore, various new catalysts (such as boron nitride (BN) based catalysts,12,13 metal dopants14,15) and approaches (including CO2-assisted oxidation,16,17 chemical looping oxidative dehydrogenation (CL-ODH),18,19 and so on) were developed to achieve the ODHE process with high selectivity and high efficiency under mild conditions. Among them, photocatalysis, as an emerging technology, can efficiently utilize clean solar energy for the C–H bond activation under mild conditions. Recently, both theoretical and experimental results have shown that TiO2-based catalysts have potential for C–H bond activation of light alkanes,20–25 indicating that the photocatalytic ODHE may achieve selective C2H4 production under mild conditions. Although both theoretical and experimental studies claimed that alkyl radical intermediates may be formed in the photocatalytic conversion of light alkanes,20–25 the formation of alkyl radicals is rarely identified due to the sensitivity of the experimental methods, which have confused the fundamental understandings of these reactions. Therefore, illustrating the formation of the ethyl 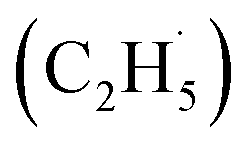 radical in photocatalytic ODHE could play a vital role in understanding the microkinetic mechanisms underlying the reaction.
radical in photocatalytic ODHE could play a vital role in understanding the microkinetic mechanisms underlying the reaction.
Herein, we systematically investigated the use of photocatalytic ODHE with rutile-TiO2(110) using temperature-programmed desorption (TPD), photo stimulated desorption (PSD), and time-of-flight (TOF) methods. The results demonstrate that using photocatalytic ODHE to obtain C2H4 can be achieved efficiently on the O atom covered R-TiO2(110) surface, and the 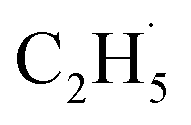 radical intermediate is captured very sensitively. Furthermore, a clear insight into the microkinetic mechanism of the photocatalytic ODHE has been explored.
radical intermediate is captured very sensitively. Furthermore, a clear insight into the microkinetic mechanism of the photocatalytic ODHE has been explored.
Experimental
All the TPD experiments were performed with a home-built apparatus, which has been described in detail elsewhere.26 The preparation of the well-ordered R-TiO2(110) crystal surfaces (Princeton Scientific, 10 mm × 10 mm × 1 mm) was accomplished using cycles of Ar+ sputtering and annealing at 850 K in an ultra-high vacuum (UHV). The characterization of the ordering and cleanness of the R-TiO2(110) surfaces was conducted using low-energy electron diffraction (LEED) and Auger electron spectroscopy (AES), respectively. The density of the oxygen vacancies (Ov) on the surface was measured using H2O TPD, and was determined to be about 6–7%. The purity of the C2H6 and O2 gases used in the experiment was ≥99.99%. The 355 nm light was produced using a fiber laser (Braze Laser UV), and the pulse time and repetition rate of the laser were ≤15 ps and 400 kHz, respectively. The third harmonic (343 nm) output was produced from a 1030 nm laser (Flare NX laser, Coherent), and the pulse time and repetition rate of the UV laser were 1.5 ns and 200 Hz, respectively. To minimize the increase of surface temperature by the UV irradiation, the maximum power of the laser is 5 mW, corresponding to a flux of 2.1 × 1016 photons per cm2 per s at 355 nm, and 2.0 × 1016 photons per cm2 per s at 343 nm. During the UV light irradiation process, the temperature increase of the surface was less than 2 K. The previously described laser (Braze Laser UV) was used in photocatalytic reactions and the PSD measurements. In order to improve the signal-to-background ratio for sensitively detecting trace signals of the photo-desorbed products, the Flare NX laser (Coherent) was used in the TOF measurements.For the PSD measurements, the time resolution was set to 0.5 s. If we assume that the ionization of the background residual gases under vacuum will produce 1 × 106 counts per second (cps) at m/z = 29, and the ionization of the 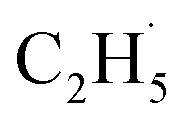 radical product will produce 1 × 105 cps for C2H5+ (m/z = 29), and the signal-to-noise ratio (SNR) the PSD method used here was 10
radical product will produce 1 × 105 cps for C2H5+ (m/z = 29), and the signal-to-noise ratio (SNR) the PSD method used here was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. If the fluctuation of the signal of m/z = 29 from the background was about 10%, it was hard to determine whether the
:1. If the fluctuation of the signal of m/z = 29 from the background was about 10%, it was hard to determine whether the 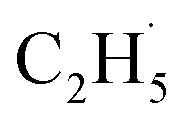 radical had been produced. However, when the 200 Hz light was used for the experiment with a TOF analyzer, the moment that the laser light arrived at the surface was set as time zero, and the C2H5+ signal arriving at the detector could be precisely counted in an exact relationship to their arrival time in each pulse. The time interval between every laser shot was 5 ms, and the time resolution was set to 256 ns for the TOF measurements. The background signal of the C2H5+ signal was produced randomly, and then the background signal collected in each frequency bin was (1 × 106 ÷ 200) ÷ (5 ms ÷ 256 ns) ≈ 0.256 count per 256 ns. However, the production of
radical had been produced. However, when the 200 Hz light was used for the experiment with a TOF analyzer, the moment that the laser light arrived at the surface was set as time zero, and the C2H5+ signal arriving at the detector could be precisely counted in an exact relationship to their arrival time in each pulse. The time interval between every laser shot was 5 ms, and the time resolution was set to 256 ns for the TOF measurements. The background signal of the C2H5+ signal was produced randomly, and then the background signal collected in each frequency bin was (1 × 106 ÷ 200) ÷ (5 ms ÷ 256 ns) ≈ 0.256 count per 256 ns. However, the production of 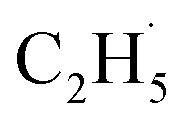 radical was not random, and it was produced at the time scale of 0.1 ms (see below). Correspondingly, the C2H5+ product signal collected in each frequency bin is (1 × 105 ÷ 200) ÷ (0.1 ms ÷ 256 ns) ≈ 1.28 count per 256 ns, and the SNR of the TOF method was 1:5. As a result, the sensitivity of the TOF method was much higher than that of the PSD method (10:1).
radical was not random, and it was produced at the time scale of 0.1 ms (see below). Correspondingly, the C2H5+ product signal collected in each frequency bin is (1 × 105 ÷ 200) ÷ (0.1 ms ÷ 256 ns) ≈ 1.28 count per 256 ns, and the SNR of the TOF method was 1:5. As a result, the sensitivity of the TOF method was much higher than that of the PSD method (10:1).
Results
The TPD results of the C2H6 conversion into C2H4
Fig. 1 shows the typical TPD spectra of the mass-to-charge ratios (m/z) of 15 (CH3+), 18 (H2O+), 26 (C2H2+), 27 (C2H3+), 28 (C2H4+ and CO+), 29 (C2H5+ and CHO+), 30 (C2H6+), 31 (CH2OH+) and 43 (CH3CO+) collected on the oxidized R-TiO2(110) surfaces after adsorbing 0.28 ML (1 ML = 5.2 × 1014 molecules per cm2) of C2H6 followed by 355 nm irradiation for 0 (black lines) and 10 min (red lines). The oxidized R-TiO2(110) surfaces were prepared by exposing the reduced surfaces to 200 L of O2 at 300 K.25,27,28 After surface oxidation, the bridging oxygen vacancies (Ov) will be healed, leaving oxygen atoms on the five coordinated Ti4+ sites (Ti5c, OTi).25 Before irradiation, only one desorption peak at 129 K appeared in the TPD traces of m/z = 15, 26, 27, 28, 29, 30, and 31, which was attributed to the desorption of the molecular C2H6 on the Ti5c sites (C2H6(Ti)).29 No signals from thermocatalytic products suggested that the oxidized R-TiO2(110) surface was not thermally active for the C–H bond activation of C2H6.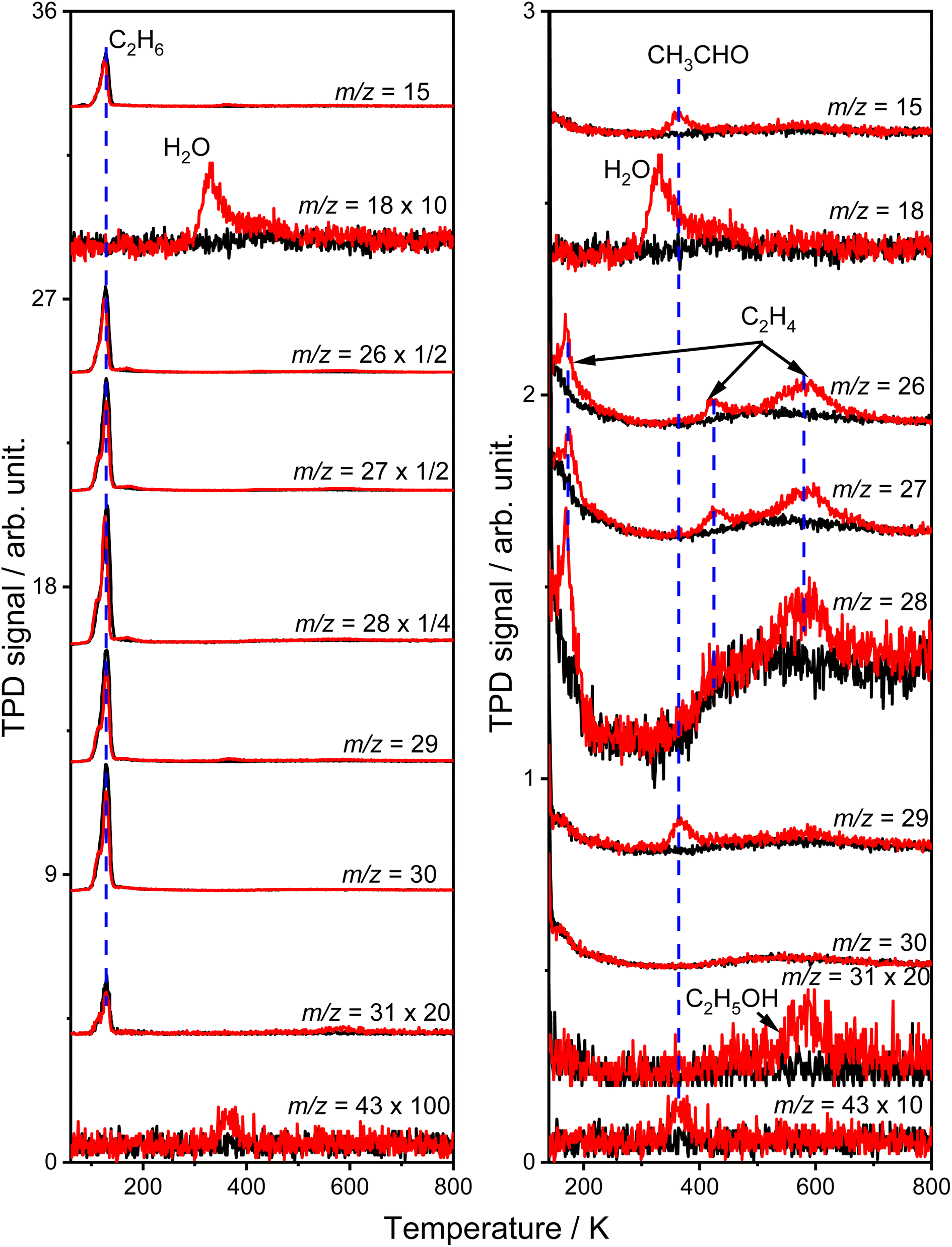 | ||
| Fig. 1 Left: Typical TPD spectra acquired at m/z = 15 (CH3+), 18 (H2O+), 26 (C2H2+), 27 (C2H3+), 28 (C2H4+ and CO+), 29 (C2H5+ and CHO+), 30 (C2H6+), 31 (CH2OH+), and 43 (CH3CO+) after irradiating the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces for 0 min (black lines) and 10 min (red lines) by 355 nm at 75 K, respectively. Right: The TPD spectra at the temperature range >140 K are highlighted. The oxidized R-TiO2(110) surfaces were prepared by exposing the reduced surfaces to 200 L of O2 at 300 K. The photon flux of 355 nm light was 2.1 × 1016 photons per cm2 per s. | ||
After 355 nm irradiation, a new desorption peak at 330 K was observed in the TPD spectra of m/z = 18 (Fig. 1a), which was contributed by the molecular H2O desorption on the Ti5c sites (H2OTi) or recombinational H2O desorption from the terminal OH groups on the Ti5c sites (OHTi).30 The H atoms of the H2O product could only be from C2H6(Ti), demonstrating that the photocatalytic dehydrogenation of C2H6(Ti) occurred on the OTi atom covered R-TiO2(110) surface. Conversely, the reduced R-TiO2(110) showed no photoactivity for the ODHE process (see Fig. S1, ESI†). Based on our previous results of the photocatalytic oxidative dehydrogenation of propane (C3H8, ODHP) on R-TiO2(110),25 the hole trapped OTi− centers rather than hole trapped bridging oxygen atoms (Ob−) were the active species for the initial C–H bond activation of C2H6, leading to the formation of H2OTi and OHTi (Fig. 1a).
The H2OTi formation was accompanied by several desorption features of carbon-containing products, which were observed at 168 K, 365 K, 423 K, 580 K, and 585 K (Fig. 1b). The broad peak (400–700 K) in the TPD traces of m/z = 15, 26, 27, 28, 29 and 30 was due to the desorption of C2H6 from the copper blocks, which were used for the mounting tantalum sample holder (Fig. S2, ESI†)The relative intensity ratio of the 365 K peak in the TPD traces of m/z = 15, 29, and 43 were calculated to be 0.80:1:0:14, respectively, which was very close to that of acetaldehyde (CH3CHO) measured by mass spectrometry (Fig. S3, ESI†). Therefore, this peak could be attributed to the formation of CH3CHO on the Ti5c sites.31 The tiny peak at 585 K (m/z = 31) was likely to be due to the formation of ethanol (C2H5OH).32 In addition, as shown in Fig. 1b, the relative intensities of the 168 K, 423 K, and 580 K peaks in the TPD traces of m/z = 26 and 27 were calculated to be 0.87:1 (168 K), 0.89:1 (423 K) and 0.88:1 (580 K), respectively, which were very close to that of the C2H4 sample (Fig. S4, ESI†), and very different from that of the alkanes and alkenes (CnH2n (3 ≤ n ≤ 10) and CnH2n+2 (2 ≤ n ≤ 10)) found in the NIST database. Therefore, all three peaks could be assigned to the C2H4 product, illustrating that the photocatalytic ODHE process to produce C2H4 could be realized on oxidized R-TiO2(110).
Although the structure of C2H6 was simpler than that of C3H8, and the initial C–H bond activation process for these two molecules was nearly the same, the pathways for C2H4 production from photocatalytic ODHE on oxidized R-TiO2(110) were more complicated than that of the photocatalytic ODHP.25 For the photocatalytic ODHP on R-TiO2(110), the majority of the propylene (C3H6) product can be formed efficiently under UV irradiation at 100 K. Only a tiny amount of the C3H6 product is formed at 340 K via the thermal dehydrogenation of the C3H7− groups on the Ti5c sites (C3H7(Ti)−), whereas, no oxygenated carbon products were produced.25 However, for methane (CH4) dehydrogenation via either thermocatalysis or photocatalysis,20,33 the 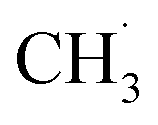 radicals are thought to be suspended above the TiO2 surface or to enter directly into the gas phase, showing a very high mobility. Therefore, once the
radicals are thought to be suspended above the TiO2 surface or to enter directly into the gas phase, showing a very high mobility. Therefore, once the 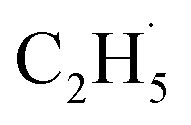 radical is produced via photocatalytic ODHE on R-TiO2(110), it may also migrate on the surface or enter directly into the gas phase, in a similar way to the
radical is produced via photocatalytic ODHE on R-TiO2(110), it may also migrate on the surface or enter directly into the gas phase, in a similar way to the 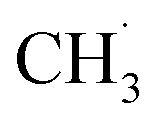 radical from CH4 conversion,20,33 leading to the complicated reaction pathways in the photocatalytic ODHE process.
radical from CH4 conversion,20,33 leading to the complicated reaction pathways in the photocatalytic ODHE process.
Evidence of 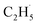 radical formation and the fate of
radical formation and the fate of 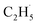 radical
radical
To confirm the formation of the 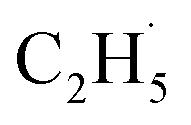
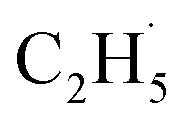
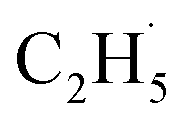 radical intermediate in photocatalytic ODHE on R-TiO2(110), the PSD signals were collected at m/z = 26, 27, 28, 29, and 30 from the 0.28 ML C2H6 covered oxidized R-TiO2(110) surface during the UV irradiation (Fig. 2). Upon irradiation, no obvious PSD signal was detected at m/z = 30, indicating that no photodesorption of C2H6 had occurred. Conversely, sharp increases of the desorption signals at m/z = 26, 27 and 28 were detected immediately when the UV light was on. The relative intensity of the PSD signals at m/z = 26 and 27 was 0.89:1, suggesting that the signals were contributed by photo-desorbed C2H4 molecules. In addition, a tiny PSD signal also appeared at m/z = 29, which may come from two sources. The first was due to the fragmentation of the C2H4 product at m/z = 29 (Fig. S4, ESI†). The other one was the
radical intermediate in photocatalytic ODHE on R-TiO2(110), the PSD signals were collected at m/z = 26, 27, 28, 29, and 30 from the 0.28 ML C2H6 covered oxidized R-TiO2(110) surface during the UV irradiation (Fig. 2). Upon irradiation, no obvious PSD signal was detected at m/z = 30, indicating that no photodesorption of C2H6 had occurred. Conversely, sharp increases of the desorption signals at m/z = 26, 27 and 28 were detected immediately when the UV light was on. The relative intensity of the PSD signals at m/z = 26 and 27 was 0.89:1, suggesting that the signals were contributed by photo-desorbed C2H4 molecules. In addition, a tiny PSD signal also appeared at m/z = 29, which may come from two sources. The first was due to the fragmentation of the C2H4 product at m/z = 29 (Fig. S4, ESI†). The other one was the 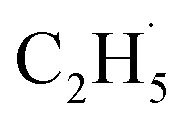 radical product. However, due to the small PSD signal at m/z = 29, it was hard to determine whether a
radical product. However, due to the small PSD signal at m/z = 29, it was hard to determine whether a 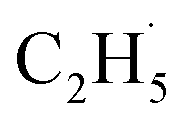 radical was formed.
radical was formed.
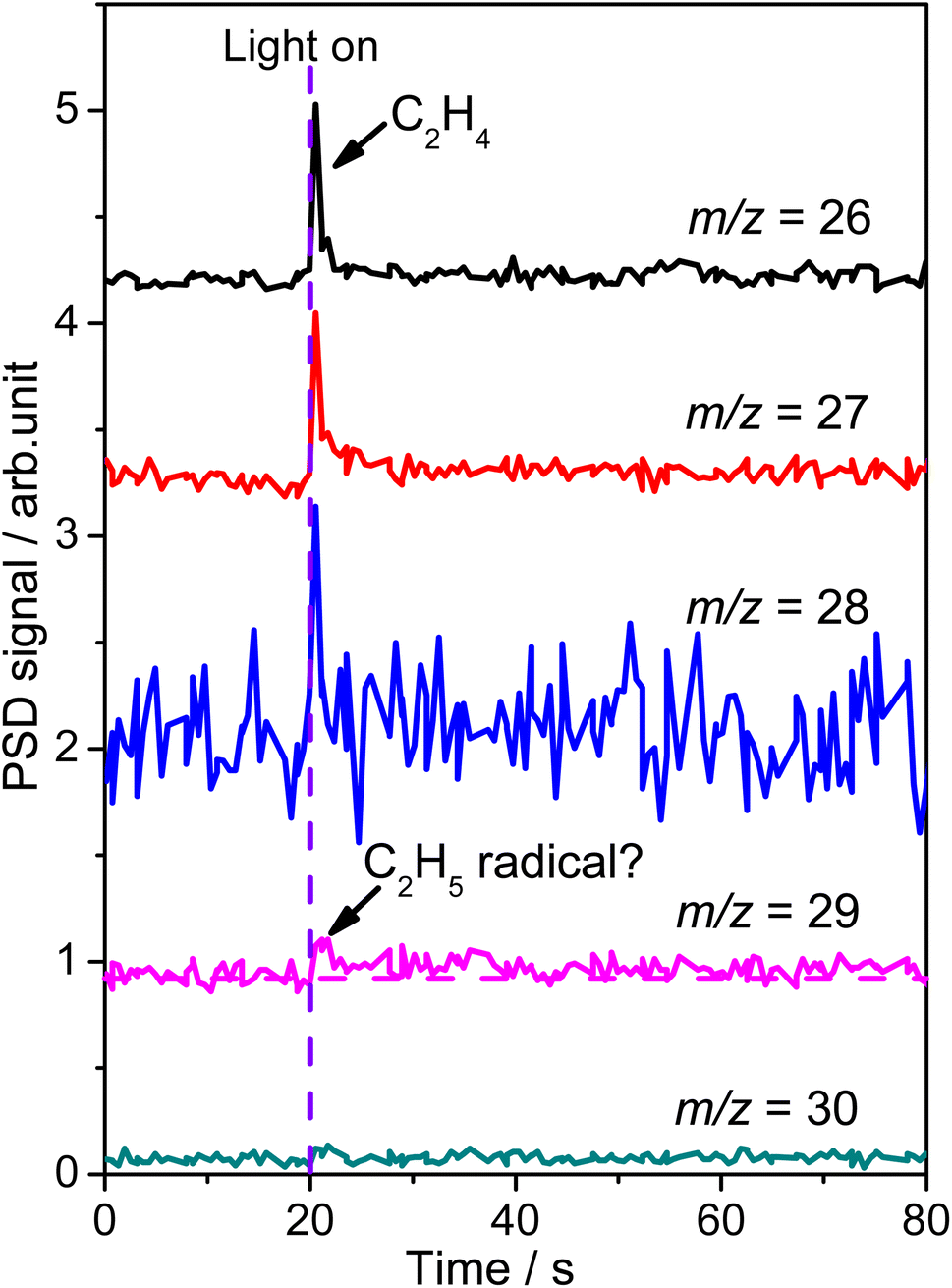 | ||
| Fig. 2 The PSD spectra acquired at m/z = 26 (C2H2+), 27 (C2H3+), 28 (C2H4+), 29 (C2H5+) and 30 (C2H6+) on the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces. The purple dashed line represents the moment when the light is turned on (t = 20 s). The photon flux of the 355 nm light is 2.4 × 1016 photons per cm2 per s. | ||
Subsequently, the TOF method, which can enhance the detection sensitivity significantly by improving the SNR,34 was used to monitor the desorbed products from the photocatalytic ODHE on R-TiO2(110) during the irradiation. As shown in Fig. 3, the TOF signals at m/z = 27 (C2H3+), 29 (C2H5+) and 30 (C2H6+) were collected. Obvious peaks appeared in the TOF spectra of m/z = 27 and 29. According to the result shown in Fig. 2, the TOF peak at m/z = 27 was due to the desorption of C2H4. Interestingly, the relative intensities of the TOF signal at m/z = 27 and 29 were about 3:1, which was much smaller than that of the C2H4 sample (Fig. S4, ESI†). However, no discernible TOF signal at m/z = 30 suggested that no photodesorption of C2H6 had occurred, and this was consistent with the results in Fig. 2. As a result, the large TOF signal at m/z = 29 could only be from the desorption of 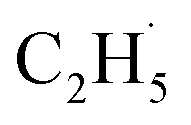 radical upon irradiation, illustrating that the initial photocatalytic C–H activation of C2H6 on R-TiO2(110) produced the
radical upon irradiation, illustrating that the initial photocatalytic C–H activation of C2H6 on R-TiO2(110) produced the 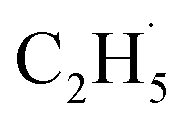 radical.
radical.
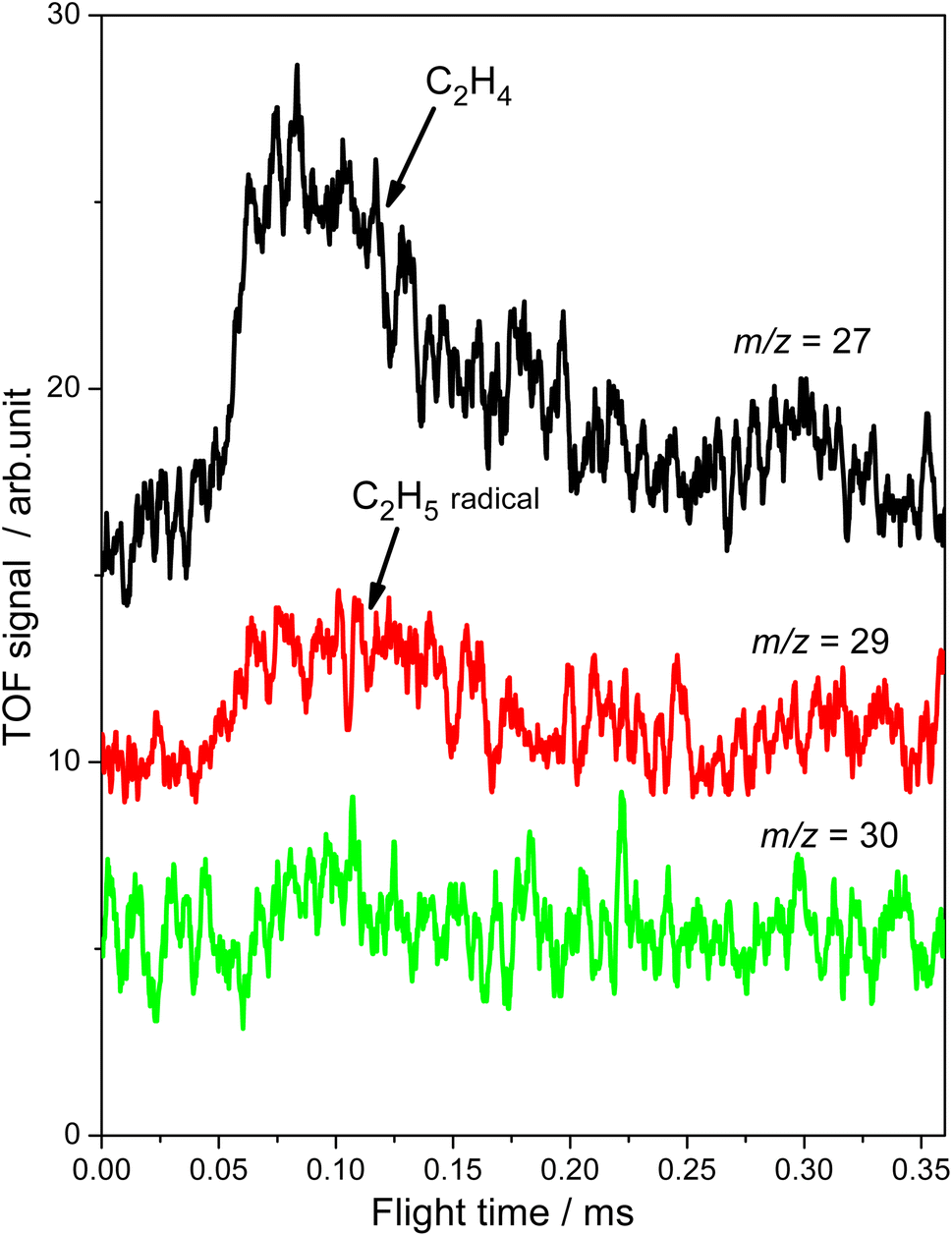 | ||
| Fig. 3 The TOF signals of C2H4 collected at m/z = 27 (C2H3+), 29 (C2H5+) and 30 (C2H6+) as a function of the flight time when irradiating the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces at 343 nm. The photon flux of the 343 nm light is 2.0 × 1016 photons per cm2 per s. | ||
Thus, after the OTi2− centers trap the photogenerated holes to form excited OTi− centers:
| TiO2 + hv → h+ + e− | (1) |
| OTi2− + h+ → [OTi−]* | (2) |
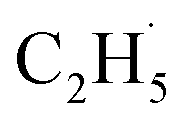 radical:
radical: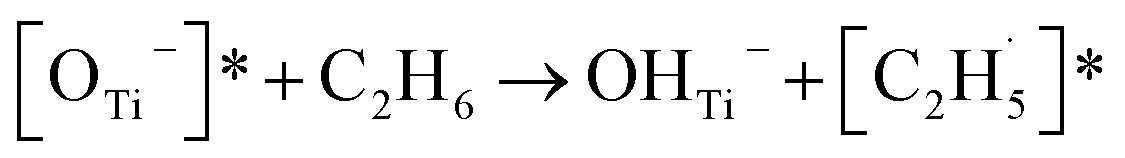 | (3) |
When the 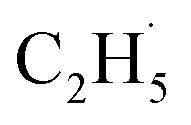 radical was produced, it may further dehydrogenate spontaneously into C2H4 and H atoms on the OTi2−/Ob2− sites or be ejected into the gas phase:21
radical was produced, it may further dehydrogenate spontaneously into C2H4 and H atoms on the OTi2−/Ob2− sites or be ejected into the gas phase:21
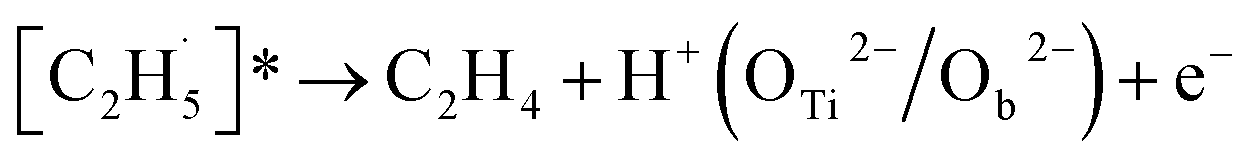 | (4) |
Here, it is not certain whether the second C–H bond cleavage needs another hole. The detection of the 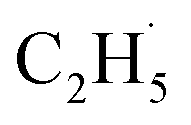 radical demonstrates that the interaction between the
radical demonstrates that the interaction between the 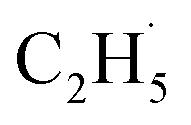 radical and the R-TiO2(110) was very weak. Therefore, the migration of the
radical and the R-TiO2(110) was very weak. Therefore, the migration of the 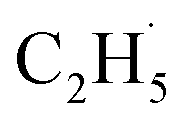 radical on the surface may occur easily. Once the
radical on the surface may occur easily. Once the 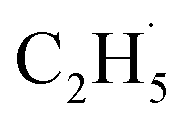 radicals migrate on the surface, the C2H5(Ti)− groups, ethoxy groups (C2H5Ob− and C2H5OTi−) and C2H5OHTi may be produced via the rebounding of the
radicals migrate on the surface, the C2H5(Ti)− groups, ethoxy groups (C2H5Ob− and C2H5OTi−) and C2H5OHTi may be produced via the rebounding of the 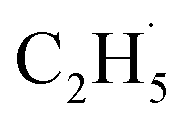 radicals to the Ti5c, Ob2−, OTi2− and OHTi− groups:35
radicals to the Ti5c, Ob2−, OTi2− and OHTi− groups:35
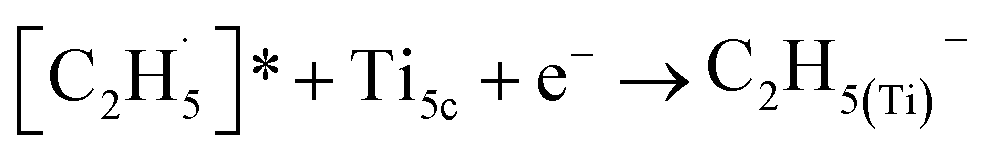 | (5) |
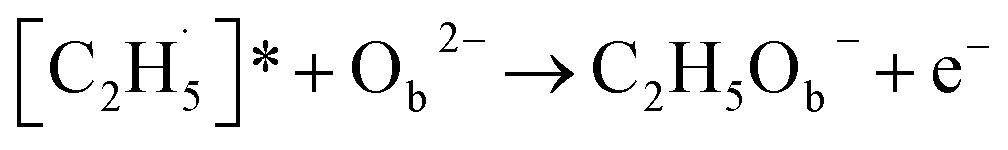 | (6) |
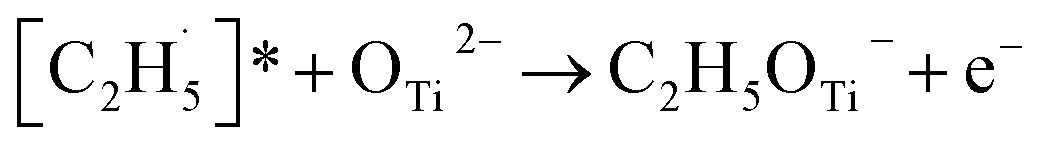 | (7) |
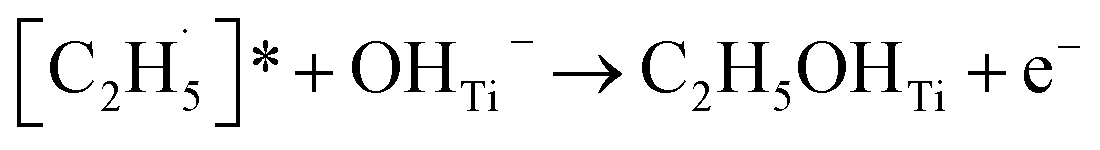 | (8) |
Among them, the C2H5(Ti)− groups would further dehydrogenate into C2H4via a similar thermocatalytic pathway used for the thermocatalytic C3H6 formation,25 giving a C2H4 desorption peak at 423 K. The C2H5OTi−/C2H5OHTi will decompose into CH3CHOTi easily upon irradiation:36,37
| C2H5OTi− + h+ → CH3CHOTi + H+ (at OTi2−/Ob2−) + e− | (9) |
| C2H5OH + h+ → CH3CHOTi + 2H+ (at OTi2−/Ob2−) + 2 e− | (10) |
During the reaction steps, most of the steps were hole induced half-reactions, which will leave electrons on the surface. It seems that the photocatalytic ODHE on R-TiO2(110) will produce excess electrons on the surface. In fact, when the reduced R-TiO2(110) surface is oxidized by O2 at room temperature to form the OTi covered surface, the surface Ob vacancies will be healed, the excess electrons of R-TiO2(110) contributed by the vacancies (Ov2−) and Ti interstitials (Ti3+) under the surface or in the bulk, will be trapped by the dissociated OTi atoms to form OTi2−.25,27,28 Upon irradiation, after the electron–hole separation, the holes will be trapped at the Ob2− and OTi2−, forming Ob− and OTi−. The photogenerated electrons will be trapped by the vacancies and Ti interstitials that gave the electrons to the surface of the OTi atoms before. Similarly, in the later reactions, even electrons are left behind, and most of them are probably trapped by vacancies and the Ti interstitials. As a result, although the excess electrons of R-TiO2(110) did not transfer to C2H6 and C2H5OTi−/C2H5OHTi during the photocatalytic ODHE process, they are trapped by OTi atoms initially to form OTi2−, and most of them go back to the surface after the reactions because the formation of low temperature C2H4 and CH3CHO are whole reactions, not half reactions. The overall reaction via photocatalysis follows eqn (11):
| C2H6 + OTi + hv → CH3CHOTi or C2H4 + H2OTi | (11) |
In addition, the minor reaction pathways of reactions in eqn (5) and (6) may produce excess electrons on the surface.
Furthermore, although the C2H5Ob− groups have little photo reactivity,36 the C2H5Ob− groups could dissociate to C2H4 with a small amount of C2H5OH product during the TPD process,38 which was consistent with our TPD result for the C2H5OH desorption on the Ov sites of R-TiO2(110) (Fig. S5, ESI†). Therefore, the C2H4 formation at 580 K was due to the thermocatalytic dehydrogenation of the C2H5Ob− groups, and the tiny peak at 585 K (m/z = 31) may be assigned to the recombinational C2H5OH desorption from the C2H5Ob− groups and dissociated protons (H+) during the TPD process.
To evaluate the importance of the C2H4 production via photocatalytic ODHE on R-TiO2(110), the formation of carbon containing products and H2O were monitored using the TPD traces of m/z = 18, 27, 29, and 31 collected on the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces as a function of irradiation time (Fig. 4). As the irradiation time increased, the signals of the thermocatalytic products (the 423 K (C2H4), 580 K (C2H4), and 585 K (C2H5OH) peaks) increased very fast and reached plateaus after approximately 60 s irradiation (the green traces). However, the signals of the 168 K (C2H4), 330 K (H2O), and 365 K peaks (CH3CHO) increased slowly and almost reached plateaus after 600 s irradiation. As discussed, previously, the OTi is involved in the formation of 168 K (C2H4), 330 K (H2O), and 365 K (CH3CHO) products. Here, the coverage of OTi atoms on the surface was only about 0.06–0.07 ML, which was strongly dependent on the concentration of the Ov sites.27,28 In contrast, the coverages of Ti5c and Ob sites on the oxidized R-TiO2(110) were nearly 1.0 ML. As a result, the possibility for the C2H5 moieties bonding to the Ti5c and Ob sites to produce C2H5(Ti)− and C2H5Ob− groups would be much higher than that for the C2H5 moieties bonding to OTi2−/OHTi− to produce C2H5OTi−/C2H5OHTi, resulting in the formation of C2H5(Ti)− and C2H5Ob− groups much faster than the C2H5OTi−/C2H5OHTi.
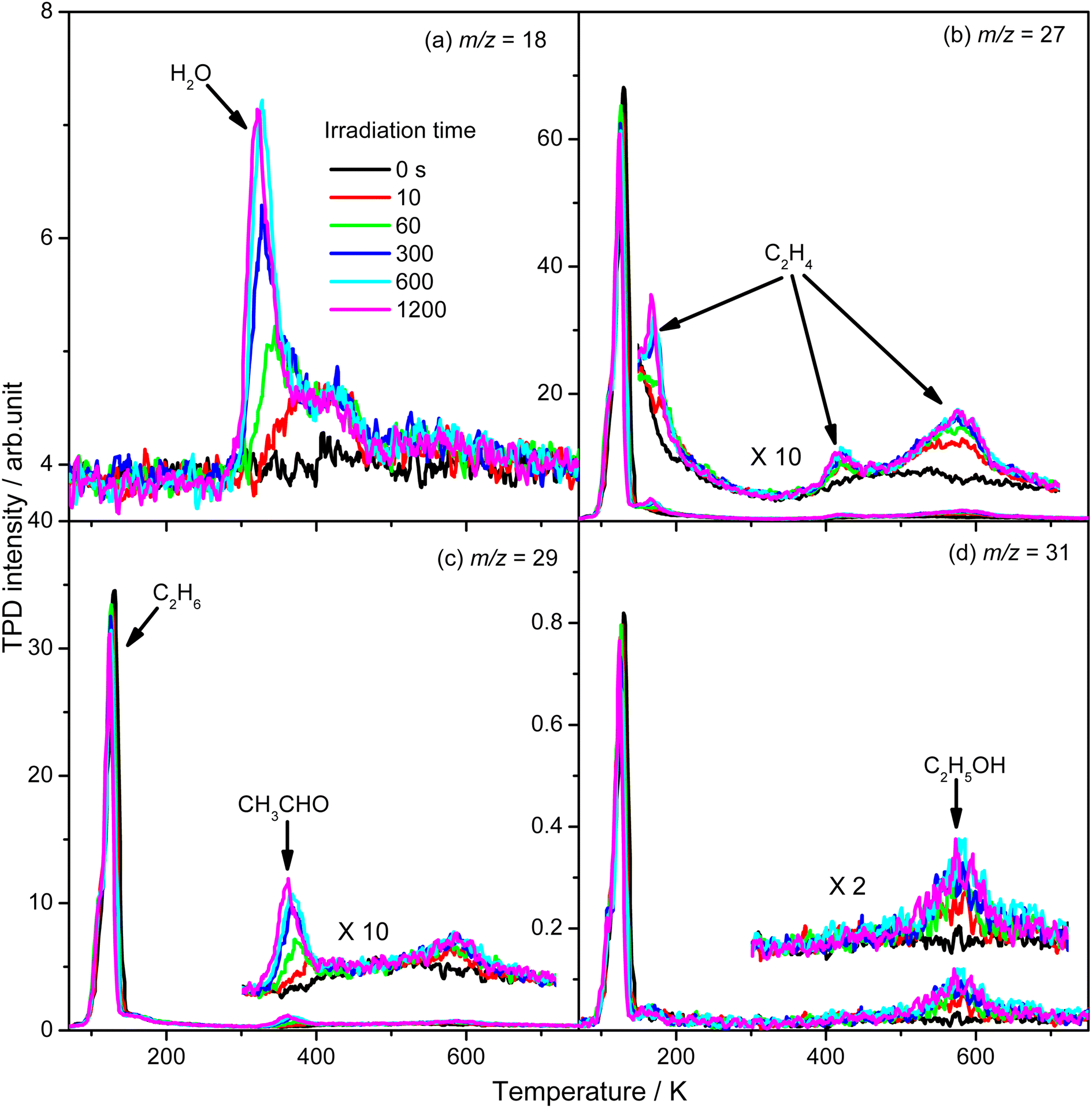 | ||
| Fig. 4 The TPD spectra acquired at m/z = 18 (H2O+) (a), 27 (C2H3+) (b), 29 (C2H5+) (c), and 31 (CH3O+) (d) on the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces as a function of irradiation time. The desorption peaks of the carbon containing products are been highlighted in the inserts of (b–d). | ||
From Fig. 4, the yields of H2O and carbon containing products (C2H4, CH3CHO, and C2H5OH) were derived and are plotted in Fig. 5. The total yield of C2H4 contained the 168 K, 423 K, and 580 K peaks. With an increasing irradiation time, the difference between the yield of H2O and carbon containing products became larger and larger. At 20 min irradiation, about 0.041 ML of H2O was produced, and the yields of C2H5OH, CH3CHO, and C2H4 are about 0.004 ML, 0.0075 ML, and 0.02 ML, respectively. Combining the results from Fig. 2 and 3, it was seen that the big difference between the yields of H2O and carbon containing products was due to the photo-desorbed C2H4 and 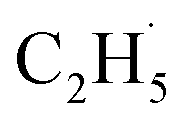 radicals. Therefore, the C2H4 product (including the 168 K, 423 K, and 580 K peaks) was the main product in photocatalytic ODHE on R-TiO2(110).
radicals. Therefore, the C2H4 product (including the 168 K, 423 K, and 580 K peaks) was the main product in photocatalytic ODHE on R-TiO2(110).
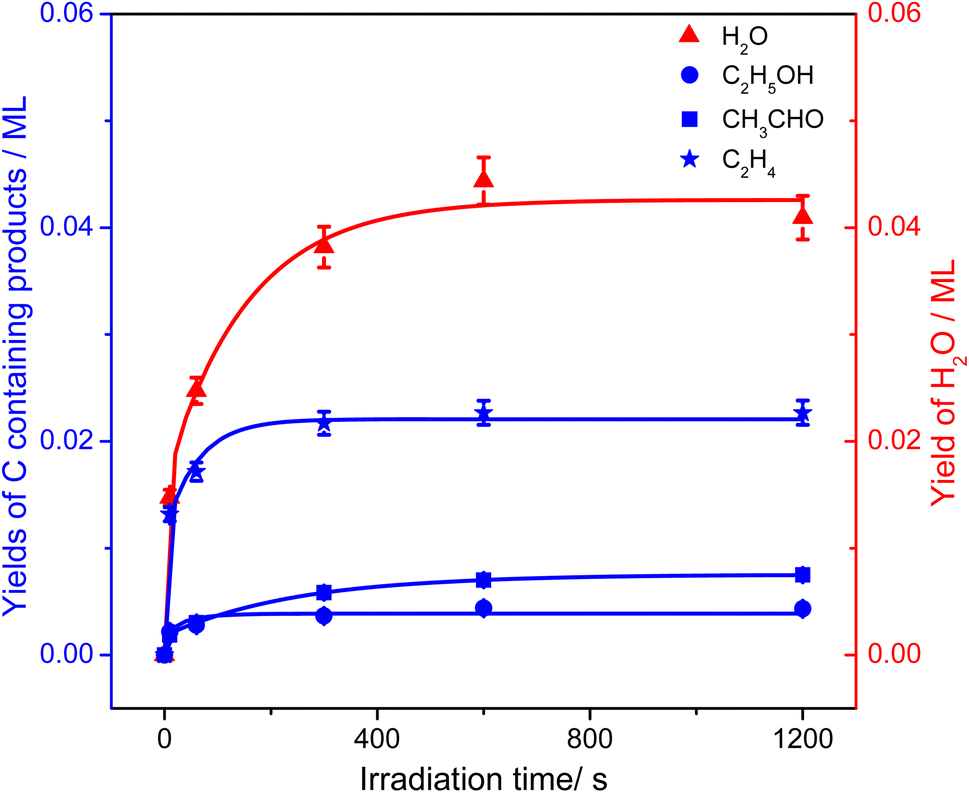 | ||
| Fig. 5 The yields of H2O (red triangles) and carbon containing products (C2H5OH (blue circles), CH3CHO (blue squares), and C2H4 (blue stars)) in photocatalytic ODHE on oxidized R-TiO2(110) as a function of irradiation time, derived from Fig. 4. All the plotted lines are only to guide the eye. | ||
Microscopic kinetics of the C2H4 formation
Similar to the case of the other hydrocarbons (C3H8, ethylbenzene (C6H5C2H5, EB)),25,39 the initial C–H bond activation of C2H6 could be induced by hole trapped OTi−. However, the second C–H bond cleavage in the C2H6 conversion on R-TiO2(110) was still not clear. On the one hand, the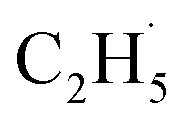 radicals may further dehydrogenate into C2H4 directly,21 suggesting that the whole process only needs one hole. On the other hand, because the
radicals may further dehydrogenate into C2H4 directly,21 suggesting that the whole process only needs one hole. On the other hand, because the 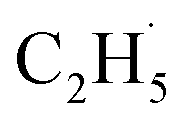 radicals can decay into C2H5(Ti)− groups via the de-excitation process, the C2H5(Ti)− group may also trap another hole to form a
radicals can decay into C2H5(Ti)− groups via the de-excitation process, the C2H5(Ti)− group may also trap another hole to form a 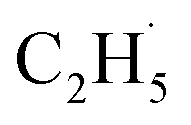 radical again for C2H4 production. For this process, a total of two holes are consumed for the stepwise C2H4 production. Although the involvement of C2H5(Ti)− in photocatalytic ODHE does not affect C2H4 production macroscopically, the microkinetic mechanism is very distinct.
radical again for C2H4 production. For this process, a total of two holes are consumed for the stepwise C2H4 production. Although the involvement of C2H5(Ti)− in photocatalytic ODHE does not affect C2H4 production macroscopically, the microkinetic mechanism is very distinct.
To confirm whether C2H5(Ti)− is involved in photocatalytic ODHE into C2H4 on R-TiO2(110), the PSD signals of C2H4 at m/z = 27 were collected from the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces as a function of the laser power. As the laser power increased, the intensity of the C2H4 PSD signal (the initial data point in each photodesorption experiment) increased significantly. More importantly, the intensity of the PSD signal of the C2H4 scaled linearly with the square root of the photon flux (Fhν1/2) (see the inset of Fig. 6). According to the results of previous work on O2 photodesorption on R-TiO2(110)40,41 and C2H5OH photodecomposition on R-TiO2(110),42 such a linear relationship illustrates that the photocatalytic ODHE to C2H4 on oxidized R-TiO2(110) was governed by the second-order electron–hole (h+/e−) pair recombination kinetics, and only one hole (or one photon) was involved in the complicated process. Furthermore, the pathway of C2H4 production from the photocatalytic ODHE on oxidized R-TiO2(110) with the involvement of C2H5(Ti)− could be ruled out, because it needs two holes (or photons). Therefore, the C2H4 formation from photocatalytic ODHE on oxidized R-TiO2(110) occurs in a stepwise manner, in which the C2H6 first undergoes the initial C–H bond cleavage to form 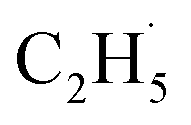 radicals with the help of hole trapped OTi− centers, and is then followed by further spontaneous dehydrogenation of the
radicals with the help of hole trapped OTi− centers, and is then followed by further spontaneous dehydrogenation of the 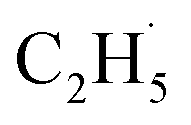 radicals into C2H4 without the involvement of an extra photon or hole. During the C2H4 formation process, the initial C–H bond activation is the rate-limiting step.
radicals into C2H4 without the involvement of an extra photon or hole. During the C2H4 formation process, the initial C–H bond activation is the rate-limiting step.
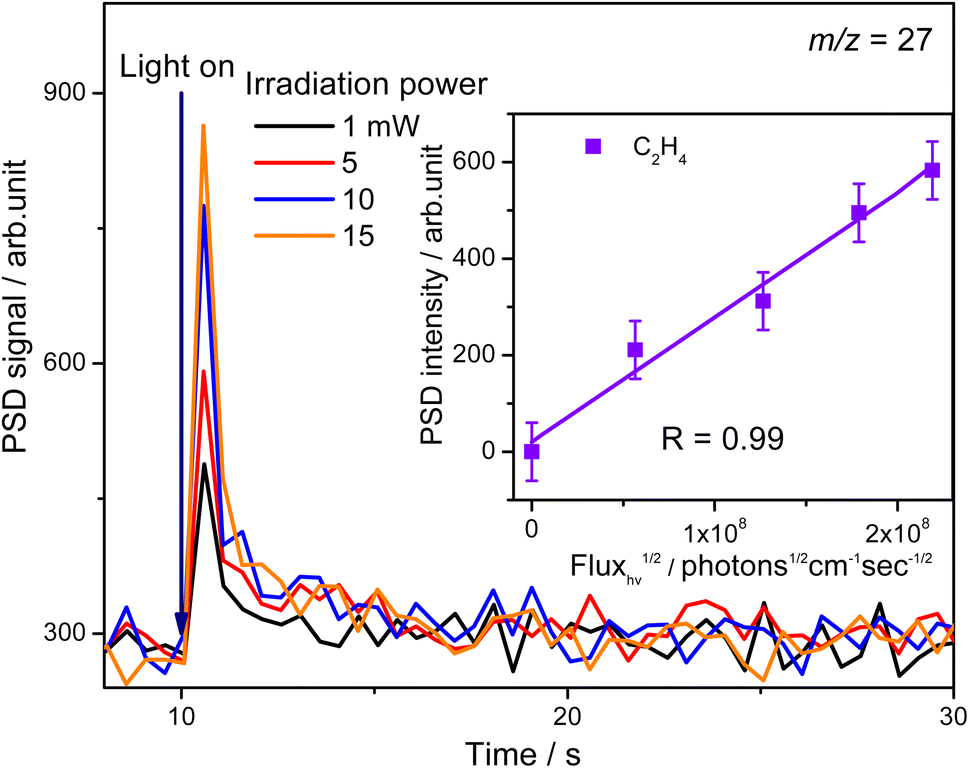 | ||
| Fig. 6 The PSD signals of C2H4 collected from the 0.28 ML C2H6 covered oxidized R-TiO2(110) surfaces as a function of the laser irradiation power. The intensity of the PSD signals (the initial point when the UV light is turned on) of C2H4 has a linear relationship with the square root of the incident light flux (Fhν1/2), as shown in the inset of this figure. | ||
In addition, as shown in Fig. S6 (ESI),† C2H4 can be photo-desorbed on the C2H4 covered R-TiO2(110), indicating that the desorption of C2H4 was induced by the photogenerated charge carriers (electron or hole). If the C2H4 product from the photocatalytic ODHE prefers to adsorb on the R-TiO2(110) surface first, and then be photo-desorbed from the surface, at least two photons are consumed for the C2H4 formation and desorption processes. However, the whole process was accomplished by one hole (or one photon), thus, once C2H4 was formed by further C–H bond cleavage of the 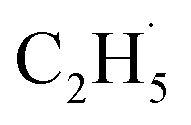 radicals, it was preferentially ejected directly into the vacuum rather than being adsorbed on the surface followed by photoinduced desorption. Therefore, only a tiny signal for the C2H4(Ti) desorption can be observed during the TPD process.
radicals, it was preferentially ejected directly into the vacuum rather than being adsorbed on the surface followed by photoinduced desorption. Therefore, only a tiny signal for the C2H4(Ti) desorption can be observed during the TPD process.
Discussion
As shown previously, both reduced and oxidized R-TiO2(110) surfaces do not show thermocatalytic reactivities for C2H6 activation. This was very different from the results of the C–H activation of the light alkanes on the PdO(101), RuO2(110), and IrO2(110) surfaces,43–46 in which alkanes adsorb on these surfaces stably by forming strongly adsorbed σ-complex species as the precursor, leading to a weakening of the C–H bond in the alkanes. Similarly, Yue and co-workers proposed that the initial C–H bond activation of C2H6 on the M/TiO2 (M = Pd and Cu) surfaces was realized via the interaction of C2H6 with the surface sites to produce surface C2H5− groups, and then the C2H5− groups trapped the photogenerated holes to form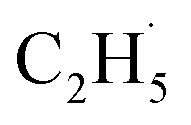 radicals, which converted rapidly into the C2H4 product.22,24 However, although the R-TiO2(110) has the same surface structure with RuO2(110) and IrO2(110), the weak interaction between the C2H6 and Ti5c sites of R-TiO2(110) inhibit the formation of the C2H6 σ-complex. As a result, the H atom abstraction from C2H6 by the Ob and OTi atoms (or Ob2− and OTi2−) following the Langmuir–Hinshelwood (L–H) mechanism is difficult. Namely, the initial C–H bond activation of C2H6 on R-TiO2(110) seems to occur ineffectively at the ground state via thermocatalysis.21,22,24
radicals, which converted rapidly into the C2H4 product.22,24 However, although the R-TiO2(110) has the same surface structure with RuO2(110) and IrO2(110), the weak interaction between the C2H6 and Ti5c sites of R-TiO2(110) inhibit the formation of the C2H6 σ-complex. As a result, the H atom abstraction from C2H6 by the Ob and OTi atoms (or Ob2− and OTi2−) following the Langmuir–Hinshelwood (L–H) mechanism is difficult. Namely, the initial C–H bond activation of C2H6 on R-TiO2(110) seems to occur ineffectively at the ground state via thermocatalysis.21,22,24
However, when the Ob2− and OTi2− centers trap photogenerated holes, the nucleophilic Ob2− and OTi2− convert into electrophilic Ob− and OTi− centers,47,48 which have a stronger ability than Ob2− and OTi2− to abstract the H atoms of the small alkanes.20,21,25 Correspondingly, the study of the photocatalytic EB dehydrogenation on R-TiO2(110)39 demonstrated that both the Ob− and OTi− centers produced by trapping the holes can activate the α-C–H bond of the side chain alkyl groups of EB. In addition, theoretical works also suggest that the Ob− centers formed upon the UV irradiation play a vital role in the C–H bond activation of CH4 and C2H6 on R-TiO2(110).20,21 Unfortunately, no product signal of photocatalytic ODHE was detected on reduced R-TiO2(110) (Fig. S1, ESI†) under 355 nm irradiation, indicating that the Ob− center produced with the 355 nm irradiation finds it difficult to activate the inert C–H bond of C2H6 under the current conditions.
In contrast, the EB can be regarded as one H atom of the C2H6 molecule substituted by a phenyl group (C6H5), in which the C–H bond of the ethyl group can be activated efficiently by the hole trapped Ob− center.39 The difference in the initial C–H bond activation of C2H6 and EB by the Ob− center on R-TiO2(110) may be because of two possible reasons. Firstly, compared with the H atom, the phenyl group as an electron withdrawing group will decrease the electron density of the α-C in the C2H5 group via a σ–π hyperconjugation, resulting in weakening of the α-C–H bond. As a result, the α-C–H bond will be activated more easily than the C–H bond of C2H6. Secondly, the desorption temperature of EB on R-TiO2(110)39 was much higher than that of C2H6 by about 120 K, indicating that the former has a stronger interaction between the aromatic ring and the surface, which may be more beneficial for energy and charge transfer between adsorbates with the surface, leading to the second C–H bond cleavage of EB.
In addition, for C3H8, one H atom of the C2H6 molecule substituted by a methyl group (CH3–), contained two types of C–H bonds (1° and 2°), and its structure was more complicated than that of C2H6. However, the C3H6 was produced with a high selectivity via photocatalytic ODHP on R-TiO2(110), and no oxygenates were produced,25 indicating that the reaction intermediates in the photocatalytic ODHP did not show high mobility on the surface. This was most likely due to the different interaction strengths between different intermediates with R-TiO2(110). According to the desorption temperature of C2H6 (Fig. 1) and C3H8,25,29 both C2H6 and C3H8 were weakly bound to the surface, causing both C2H6 and C3H8 to easily migrate on the surface. As a result, the initial C–H bond activation of C2H6 on R-TiO2(110) was more likely to follow the OTi atom mediated Eley–Rideal (E–R) mechanism,1,25 forming a movable 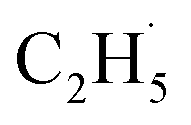 radical by the abstraction of an H atom from C2H6 by an excited OTi− center, which may significantly affect the selectivity of the products. The interaction between the
radical by the abstraction of an H atom from C2H6 by an excited OTi− center, which may significantly affect the selectivity of the products. The interaction between the 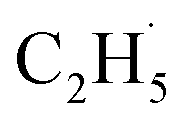 radicals, which were worse electron donors, with the Ti5c sites should be weaker than that of the possible
radicals, which were worse electron donors, with the Ti5c sites should be weaker than that of the possible 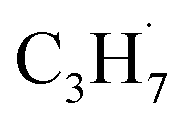 radicals produced in the photocatalytic ODHP on R-TiO2(110), which tended to form an allyl σ–p hyperconjugation configuration (CH3CH˙CH3) with a stronger electron-donating ability.25,49 Then, the
radicals produced in the photocatalytic ODHP on R-TiO2(110), which tended to form an allyl σ–p hyperconjugation configuration (CH3CH˙CH3) with a stronger electron-donating ability.25,49 Then, the 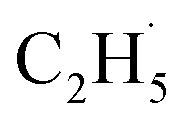 radicals may migrate on the surface more easily, resulting in the formation of additional byproducts via the diffusion and rebounding processes.
radicals may migrate on the surface more easily, resulting in the formation of additional byproducts via the diffusion and rebounding processes.
Similarly, previous research on the photooxidation of tert-butanol and ketones on R-TiO2(110) also observed 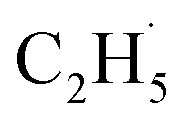 radical ejection,50–52 which was associated with hole-induced chemistry.20,21,25,50,52 According to the work on
radical ejection,50–52 which was associated with hole-induced chemistry.20,21,25,50,52 According to the work on 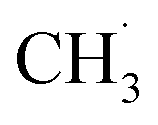 radical formation from ketone photooxidation on R-TiO2(110),52 two dissociation channels (“fast” and “slow” channels) of the
radical formation from ketone photooxidation on R-TiO2(110),52 two dissociation channels (“fast” and “slow” channels) of the 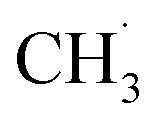 radical desorption were detected. The ‘‘fast’’
radical desorption were detected. The ‘‘fast’’ 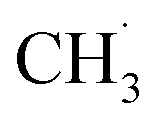 radical production was attributed to the prompt dissociation of an internally “hot’’ acetone–oxygen complex (intermediates at the excited state), and the acetone–oxygen complex weakly coupled to the surface. However, the ‘‘slow’’
radical production was attributed to the prompt dissociation of an internally “hot’’ acetone–oxygen complex (intermediates at the excited state), and the acetone–oxygen complex weakly coupled to the surface. However, the ‘‘slow’’ 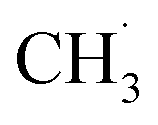 radical production was assigned to the dissociation of a relaxed acetone–oxygen complex formed via internal vibrational redistribution (IVR), which consumed the available energy for the C–C bond cleavage. In the case of the
radical production was assigned to the dissociation of a relaxed acetone–oxygen complex formed via internal vibrational redistribution (IVR), which consumed the available energy for the C–C bond cleavage. In the case of the 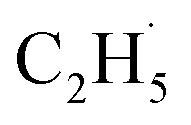 radical ejection from 2-butanone photooxidation on R-TiO2(110), the ‘‘slow’’ channel dominated.50 As a result, no obvious C2H4 product obtained from
radical ejection from 2-butanone photooxidation on R-TiO2(110), the ‘‘slow’’ channel dominated.50 As a result, no obvious C2H4 product obtained from 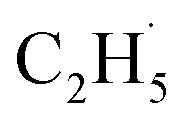 radical dehydrogenation was detected.50
radical dehydrogenation was detected.50
However, for the photocatalytic ODHE on the R-TiO2 (110) surface, C2H6 was also weakly adsorbed on the surface and its structure was very simple without a π-conjugated system, and the IVR process will not occur to make the “hot” 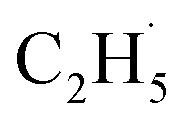 radical relax efficiently, thus resulting in the further dehydrogenation of the excited
radical relax efficiently, thus resulting in the further dehydrogenation of the excited 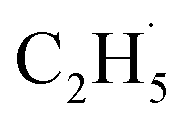 radical into C2H4. Compared with these results,50–52 it was not too difficult to conclude that the IVR process can affect the energy relaxation in excited molecules or ions on the R-TiO2(110) surface, which can further affect the bond breaking and product formation. The smaller molecules (such as C2H6) may inhibit the IVR process in photocatalytic reactions due to having fewer vibrational energy levels than complicated molecules, leading to the high efficiency of bond breaking.
radical into C2H4. Compared with these results,50–52 it was not too difficult to conclude that the IVR process can affect the energy relaxation in excited molecules or ions on the R-TiO2(110) surface, which can further affect the bond breaking and product formation. The smaller molecules (such as C2H6) may inhibit the IVR process in photocatalytic reactions due to having fewer vibrational energy levels than complicated molecules, leading to the high efficiency of bond breaking.
Due to the formation of weakly bonded 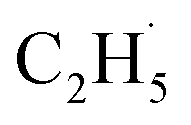 radical intermediates, it is reasonable that oxygen-containing species are formed via the rebounding between the
radical intermediates, it is reasonable that oxygen-containing species are formed via the rebounding between the 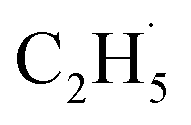 radicals and the surface O atoms (Ob2− and OTi2−). However, based on previous research about ODHE over vanadium oxides,53–55 terminal M
radicals and the surface O atoms (Ob2− and OTi2−). However, based on previous research about ODHE over vanadium oxides,53–55 terminal M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species (terminal metal oxo, VO) are the active sites for ODHE, in which either the direct insertion of the C–H bond across the MO bond resulted in C2H5O group formation or C–H bond activation by H abstraction to form M–OH and a transient alkyl radical product
O species (terminal metal oxo, VO) are the active sites for ODHE, in which either the direct insertion of the C–H bond across the MO bond resulted in C2H5O group formation or C–H bond activation by H abstraction to form M–OH and a transient alkyl radical product 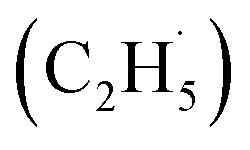 may occur. If the C2H5Ob− formation occurs via the Ob2− insertion pathway, the C2H4 product formed at 580 K by the dehydrogenation of C2H5Ob− groups should be observed on both the reduced and oxidized R-TiO2(110) surfaces. However, the formation of the C2H5Ob− groups was only detected on the oxidized surface, suggesting that the C2H5Ob− groups were produced via the recombination of the
may occur. If the C2H5Ob− formation occurs via the Ob2− insertion pathway, the C2H4 product formed at 580 K by the dehydrogenation of C2H5Ob− groups should be observed on both the reduced and oxidized R-TiO2(110) surfaces. However, the formation of the C2H5Ob− groups was only detected on the oxidized surface, suggesting that the C2H5Ob− groups were produced via the recombination of the 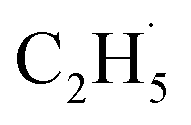 radical and Ob2− rather than the Ob2− insertion pathway. In addition, the direct insertion generally has a high barrier,54,55 which is difficult for C2H6 activation on TiO2 following the L–H mechanism due to weak adsorption energy. Despite all this, the existence of a direct heterolytic insertion of OTi− to C2H6 for C2H5OTi−/C2H5OHTi formation cannot be completely ruled out.
radical and Ob2− rather than the Ob2− insertion pathway. In addition, the direct insertion generally has a high barrier,54,55 which is difficult for C2H6 activation on TiO2 following the L–H mechanism due to weak adsorption energy. Despite all this, the existence of a direct heterolytic insertion of OTi− to C2H6 for C2H5OTi−/C2H5OHTi formation cannot be completely ruled out.
Interestingly, although the initial C–H bond cleavage of EB on R-TiO2(110) occurred more easily than that of C2H6, the possible radical intermediate 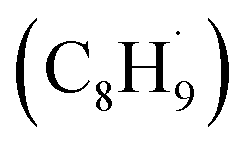 produced from the initial C–H bond cleavage of EB under UV irradiation preferred to decay to C8H9− rather than further dehydrogenate into styrene directly, leading to a low yield of low temperature styrene production.39 In contrast, in the case of C2H6 and C3H8 activation25 on R-TiO2(110), the intermediates mainly dehydrogenated into C2H4 and C3H6 spontaneously, whereas only tiny radicals decay to alkyl groups adsorbed on the Ti5c sites. This suggested that the photon energy for the C–H bond activation of small alkanes into alkenes via TiO2 photocatalysis may be utilized more efficiently than that of aromatic EB.39,56 Furthermore, due to phenyl group substitution, the rate-determining step of the photocatalytic dehydrogenation of hydrocarbons into corresponding alkenes shifts from the initial C–H bond activation to the second further dehydrogenation.25,39 This result may be more evidence that the IVR process in the larger molecule reduces the available energy for bond breaking. In order to overcome the consumption of available energy by the IVR process, excitation with higher energy photons may be a feasible way. Referring to the results of the recent photocatalytic conversion of EB into styrene on R-TiO2(100),56 the efficiency of the initial α-C–H bond activation is nearly the same at 257 nm and 343 nm, whereas the rate of the β-C–H bond cleavage was strongly enhanced with the photon energy. In contrast, for C2H6 and C3H8,21,25 once the initial C–H bond cleavage was activated by the hole derived from the 355 nm photoexcitation with a lower photon energy, the second dehydrogenation was still accomplished quite easily. However, the mobility of the intermediates determined the complexity of the reactions.
produced from the initial C–H bond cleavage of EB under UV irradiation preferred to decay to C8H9− rather than further dehydrogenate into styrene directly, leading to a low yield of low temperature styrene production.39 In contrast, in the case of C2H6 and C3H8 activation25 on R-TiO2(110), the intermediates mainly dehydrogenated into C2H4 and C3H6 spontaneously, whereas only tiny radicals decay to alkyl groups adsorbed on the Ti5c sites. This suggested that the photon energy for the C–H bond activation of small alkanes into alkenes via TiO2 photocatalysis may be utilized more efficiently than that of aromatic EB.39,56 Furthermore, due to phenyl group substitution, the rate-determining step of the photocatalytic dehydrogenation of hydrocarbons into corresponding alkenes shifts from the initial C–H bond activation to the second further dehydrogenation.25,39 This result may be more evidence that the IVR process in the larger molecule reduces the available energy for bond breaking. In order to overcome the consumption of available energy by the IVR process, excitation with higher energy photons may be a feasible way. Referring to the results of the recent photocatalytic conversion of EB into styrene on R-TiO2(100),56 the efficiency of the initial α-C–H bond activation is nearly the same at 257 nm and 343 nm, whereas the rate of the β-C–H bond cleavage was strongly enhanced with the photon energy. In contrast, for C2H6 and C3H8,21,25 once the initial C–H bond cleavage was activated by the hole derived from the 355 nm photoexcitation with a lower photon energy, the second dehydrogenation was still accomplished quite easily. However, the mobility of the intermediates determined the complexity of the reactions.
Conclusion
In summary, photocatalytic ODHE on the model R-TiO2(110) surface has been systematically investigated to determine the mechanism of the process. The C2H4 formation for the photocatalytic ODHE on the oxidized surface can be achieved via a stepwise manner at 75 K, in which the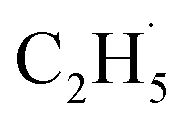 radical intermediate is detected directly. The hole trapped OTi− centers play a crucial role in the initial C–H bond activation of C2H6, and only one hole is involved in the cleavage of the two C–H bonds to produce C2H4. This result not only illustrates that photocatalysis is very suitable for the inert C–H bond activation of small alkanes, but also provides a novel mechanistic insight into photocatalytic ODHE, which offers new opportunities for the development of novel ODHE pathways with high C2H4 selectivity under mild conditions.
radical intermediate is detected directly. The hole trapped OTi− centers play a crucial role in the initial C–H bond activation of C2H6, and only one hole is involved in the cleavage of the two C–H bonds to produce C2H4. This result not only illustrates that photocatalysis is very suitable for the inert C–H bond activation of small alkanes, but also provides a novel mechanistic insight into photocatalytic ODHE, which offers new opportunities for the development of novel ODHE pathways with high C2H4 selectivity under mild conditions.
Data availability
The data supporting this study is available within the main text and the associated ESI.†Author contributions
YL and YZ conducted the TPD experiments and analysis. XC and TW helped to conduct the TPD experiments, and to revise the manuscript. FL and QG conceived the idea, designed the experiment, analyzed the data, and co-wrote the paper. XY supervised the research. All the authors discussed the results and commented on the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2022YFA1503102, 2018YFE0203002), the National Natural Science Foundation of China (NSFC Center for Chemical Dynamics, Grant No. 22173041, 22103033, 22103031, 22173042, 21973037), the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB17000000), the Shenzhen Science and Technology Innovation Committee (Grant No. 20220814164755002, ZDSYS20200421111001787), the Guangdong Innovative & Entrepreneurial Research Team Program (Grant No. 2019ZT08L455, 2019JC01X091), the International Partnership Program of Chinese Academy of Sciences (Grant No. 121421KYSB20170012), and the Innovation Program for Quantum Science and Technology (Grant No. 2021ZD0303304).References
- S. Najari, S. Saeidi, P. Concepcion, D. D. Dionysiou, S. K. Bhargava, A. F. Lee and K. Wilson, Chem. Soc. Rev., 2021, 50, 4564–4605 RSC.
- Y. Dai, X. Gao, Q. Wang, X. Wan, C. Zhou and Y. Yang, Chem. Soc. Rev., 2021, 50, 5590–5630 RSC.
- R. Gudgila and C. A. Leclerc, Ind. Eng. Chem. Res., 2011, 50, 8438–8443 CrossRef CAS.
- P. Sun, G. Siddiqi, W. C. Vining, M. Chi and A. T. Bell, J. Catal., 2011, 282, 165–174 CrossRef CAS.
- J.-P. Lange, R. Schoonebeek, P. Mercera and F. Van Breukelen, Appl. Catal., A, 2005, 283, 243–253 CrossRef CAS.
- V. Zacharopoulou and A. A. Lemonidou, Catalysts, 2017, 8, 2 CrossRef.
- S. Najari, S. S. Hosseini, M. Omidkhah and N. R. Tan, RSC Adv., 2015, 5, 47199–47215 RSC.
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. Al-Ghrami and A. Bourane, Catal. Sci. Technol., 2017, 7, 12–46 RSC.
- M. Sun, J. Zhang, P. Putaj, V. Caps, F. d. r. Lefebvre, J. Pelletier and J.-M. Basset, Chem. Rev., 2014, 114, 981–1019 CrossRef CAS.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2017, 118, 2769–2815 CrossRef.
- C. A. Gärtner, A. C. van Veen and J. A. Lercher, ChemCatChem, 2013, 5, 3196–3217 CrossRef.
- Y. Zhou, J. Lin, L. Li, X. Pan, X. Sun and X. Wang, J. Catal., 2018, 365, 14–23 CrossRef CAS.
- Y. Honda, A. Takagaki, R. Kikuchi and S. T. Oyama, Chem. Lett., 2018, 47, 1090–1093 CrossRef CAS.
- T.-K. Cheung and B. C. Gates, J. Catal., 1997, 168, 522–531 CrossRef CAS.
- S. Wang, K. Murata, T. Hayakawa, S. Hamakawa and K. Suzuki, Chem. Commun., 1999, 103–104 RSC.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem, 2019, 3, 638–649 CrossRef CAS.
- S. Deng, S. Li, H. Li and Y. Zhang, Ind. Eng. Chem. Res., 2009, 48, 7561–7566 CrossRef CAS.
- S. Yusuf, L. Neal, V. Haribal, M. Baldwin, H. H. Lamb and F. Li, Appl. Catal., B, 2018, 232, 77–85 CrossRef CAS.
- Y. Gao, F. Haeri, F. He and F. Li, ACS Catal., 2018, 8, 1757–1766 CrossRef CAS.
- M. Zhou and H. Wang, JACS Au, 2022, 2, 188–196 CrossRef CAS.
- X. Wang, L. Wan, Z. Wang, X. Liu, Y. Gao, L. Wang, J. Liu, Q. Guo, W. Hu and J. Yang, J. Phys. Chem. Lett., 2022, 13, 6532–6540 CrossRef CAS PubMed.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280–9286 CrossRef CAS.
- Y. Jiang, W. Zhao, S. Li, S. Wang, Y. Fan, F. Wang, X. Qiu, Y. Zhu, Y. Zhang and C. Long, J. Am. Chem. Soc., 2022, 144, 15977–15987 CrossRef CAS PubMed.
- L. Song, R. Zhang, C. Zhou, G. Shu, K. Ma and H. Yue, Chem. Commun., 2023, 59, 478–481 RSC.
- F. Li, B. Wang, X. Chen, Y. Lai, T. Wang, H. Fan, X. Yang and Q. Guo, JACS Au, 2022, 2, 2607–2616 CrossRef CAS.
- Z. Ren, Q. Guo, C. Xu, W. Yang, C. Xiao, D. Dai and X. Yang, Chin. J. Chem. Phys., 2012, 25, 507–512 CrossRef.
- E. Lira, J. Ø. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Lægsgaard, B. Hammer, S. Wendt and F. Besenbacher, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- I. Sokolović, M. Reticcioli, M. Čalkovský, M. Wagner, M. Schmid, C. Franchini, U. Diebold and M. Setvín, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14827–14837 CrossRef.
- L. Chen, R. S. Smith, B. D. Kay and Z. Dohnálek, Surf. Sci., 2016, 650, 83–92 CrossRef CAS.
- M. A. Henderson, W. S. Epling, C. H. Peden and C. L. Perkins, J. Phys. Chem. B, 2003, 107, 534–545 CrossRef CAS.
- R. T. Zehr and M. A. Henderson, Surf. Sci., 2008, 602, 2238–2249 CrossRef CAS.
- R. Sun, X. Liu, X. Chen, L. Che, X. Yang and Q. Guo, J. Phys. Chem. Lett., 2022, 13, 801–807 CrossRef CAS PubMed.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar and D. Ma, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- J. J. Lin, D. W. Hwang, S. Harich, Y. T. Lee and X. Yang, Rev. Sci. Instrum., 1998, 69, 1642–1646 CrossRef CAS.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. Specht, W. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS.
- J. Ø. Hansen, R. Bebensee, U. Martinez, S. Porsgaard, E. Lira, Y. Wei, L. Lammich, Z. Li, H. Idriss and F. Besenbacher, Sci. Rep., 2016, 6, 21990 CrossRef CAS PubMed.
- Z. Ma, Q. Guo, X. Mao, Z. Ren, X. Wang, C. Xu, W. Yang, D. Dai, C. Zhou and H. Fan, J. Phys. Chem. C, 2013, 117, 10336–10344 CrossRef CAS.
- Y. K. Kim, B. D. Kay, J. White and Z. Dohnálek, Catal. Lett., 2007, 119, 1–4 CrossRef CAS.
- F. Li, X. Chen, Y. Lai, T. Wang, X. Yang and Q. Guo, J. Phys. Chem. Lett., 2022, 13, 9186–9194 CrossRef CAS PubMed.
- T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2005, 109, 18230–18236 CrossRef CAS.
- T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2006, 110, 7431–7435 CrossRef CAS PubMed.
- Y. Lai, Y. Zeng, X. Chen, T. Wang, X. Yang and Q. Guo, J. Phys. Chem. C, 2023, 127, 1863–1869 CrossRef CAS.
- J. F. Weaver, C. Hakanoglu, A. Antony and A. Asthagiri, Chem. Soc. Rev., 2014, 43, 7536–7547 RSC.
- V. Fung, F. F. Tao and D.-e. Jiang, Phys. Chem. Chem. Phys., 2018, 20, 22909–22914 RSC.
- C.-C. Wang, S. S. Siao and J.-C. Jiang, J. Phys. Chem. C, 2012, 116, 6367–6370 CrossRef CAS.
- V. Fung, G. Hu, F. Tao and D. E. Jiang, ChemPhysChem, 2019, 20, 2217–2220 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS.
- Q. Zhang, Y. J. Li, H. F. Wen, Y. Adachi, M. Miyazaki, Y. Sugawara, R. Xu, Z. H. Cheng, J. N. Brndiar and L. Kantorovich, J. Am. Chem. Soc., 2018, 140, 15668–15674 CrossRef CAS PubMed.
- J. Mullins, J. Chem. Educ., 2012, 89, 834–836 CrossRef CAS.
- D. Wilson, D. Sporleder and M. White, J. Phys. Chem. C, 2013, 117(18), 9290–9300 CrossRef CAS.
- C. Walenta, S. Kollmannsberger, C. Courtois, M. Tschurl and U. Heiz, Phys. Chem. Chem. Phys., 2018, 20, 7105–7111 RSC.
- D. Wilson, D. Sporleder and M. White, Phys. Chem. Chem. Phys., 2012, 14, 13630–13637 RSC.
- X. Rozanska and J. Sauer, Int. J. Quantum Chem., 2008, 108, 2223–2229 CrossRef CAS.
- C. Coperet, Chem. Rev., 2010, 110, 656–680 CrossRef CAS.
- G.-L. Dai, Z.-P. Liu, W.-N. Wang, J. Lu and K.-N. Fan, J. Phys. Chem. C, 2008, 112, 3719–3725 CrossRef CAS.
- Y. Lai, Y. Zeng, F. Li, X. Chen, T. Wang, X. Yang and Q. Guo, J. Phys. Chem. Lett., 2023, 14, 6286–6294 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05623f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |