
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pyridinium-based strategy for lysine-selective protein modification and chemoproteomic profiling in live cells†
Chuan
Wan‡
c,
Dongyan
Yang‡
d,
Chunli
Song
b,
Mingchan
Liang
b,
Yuhao
An
 b,
Chenshan
Lian
b,
Chuan
Dai
b,
Yuxin
Ye
b,
Feng
Yin
*b,
Rui
Wang
*b and
Zigang
Li
*ab
b,
Chenshan
Lian
b,
Chuan
Dai
b,
Yuxin
Ye
b,
Feng
Yin
*b,
Rui
Wang
*b and
Zigang
Li
*ab
aState Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, P. R. China. E-mail: lizg.sz@pku.edu.cn
bPingshan Translational Medicine Center, Shenzhen Bay Laboratory, Shenzhen, 518118, P. R. China. E-mail: yinfeng@szbl.ac.cn; wangrui@szbl.ac.cn; lizg@szbl.ac.cn
cCollege of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen, 518118, P. R. China
dCollege of Chemistry and Chemical Engineering, Zhongkai University of Agriculture and Engineering, Guangzhou, 510225, P. R. China
First published on 13th March 2024
Abstract
Protein active states are dynamically regulated by various modifications; thus, endogenous protein modification is an important tool for understanding protein functions and networks in complicated biological systems. Here we developed a new pyridinium-based approach to label lysine residues under physiological conditions that is low-toxicity, efficient, and lysine-selective. Furthermore, we performed a large-scale analysis of the ∼70% lysine-selective proteome in MCF-7 cells using activity-based protein profiling (ABPP). We quantifically assessed 1216 lysine-labeled peptides in cell lysates and identified 386 modified lysine sites including 43 mitochondrial-localized proteins in live MCF-7 cells. Labeled proteins significantly preferred the mitochondria. This pyridinium-based methodology demonstrates the importance of analyzing endogenous proteins under native conditions and provides a robust chemical strategy utilizing either lysine-selective protein labeling or spatiotemporal profiling in a living system.
Introduction
Site-selective protein modification under native, complex conditions is useful for both biological and pharmaceutical sciences. A facile and biocompatible method to incorporate synthetic molecules onto proteins could enable direct protein identification, providing information about protein function and structure relevant for disease diagnosis and drug development.1–5 Several methods have been developed for selective protein labeling in living systems. Among them, genetically encodable strategies using unnatural amino acids have been the most widely used in a live cell environment.6,7 However, some chemical methods for protein modification are often limited by high toxicity, poor uptake/distribution, and the complicated operation of many conventional methods.8,9 Creating further complexity, protein functions are regulated by a variety of subcellular compartments within their native habitats. As a result, few chemical methods are suitable for labeling native/endogenous proteins for live cell-based proteomics.10–14 Ideally, combining site-selective modifications and protein feature profiling would also directly exploit the subcellular organelle compartments in live cells.Lysine makes up 5.7% of the human proteome, is typically found on protein surfaces (8.9% of surface residues15), and is a weak nucleophile at physiological pH,16,17 making it the third most popular target in the CovPDB database.18 Although it has great promise, only one FDA approved drug “vigabatrin17” targets a lysine site. Thus, generating a broader lysine targeting toolbox is of great interest to current drug discovery.19,20
Recently, state-of-art methodologies were developed for lysine-selective modification and active-lysine protein profiling.21–25 Among them, an activated ester amine transesterification reaction is one of the most practical and robust methodologies (Fig. 1a).26–28 For example, a sulfotetrafluorophenyl (STP) ester probe was utilized by the Cravatt group for global lysine reactivity and ligandability profiling in the human proteome.29,30 Each of these activated esters has relative advantages and disadvantages, but a method that enables efficient proteome labeling and profiling in live cells is missing from the current toolbox.31–33
 | ||
| Fig. 1 (a) Examples of classical activated esters for ∼70% lysine-selective bioconjugation. (b) Design concept of pyridinium-based lysine-selective modification and profiling. | ||
To overcome these live system application limitations, we recently investigated using sulfonium probes.20,34–38 We achieved several successful applications and found that the probes had good aqueous solubility, good cellular uptake, and outstanding nucleophilicity from their positive charge.39–41 However, sulfonium is a soft acid, making it relatively unstable in the reductive cellular environment.42 Thus, we envisioned that another positively charged compound and harder acid, quaternary pyridinium, which has been widely used in mitochondria-targeting moieties, would be more applicable to live systems.43,44 Several excellent bioconjugate methods were previously reported that used pyridinium substrates, including cysteine conjugation45 and elimination to dehydroalanine (Dha),46 protein N-terminal amine transamination,47,48 and photoinduced tryptophan-selective modification in peptides and proteins49 (see Fig. S1†). Considering this, we expect that the exceedingly electron deficient quaternizated N atom would act as a carboxyl activating group that could be utilized in an efficient amine transesterification reaction for quick, clean, and selective lysine modification. Actually, Mukaiyama's reagent (2-halogenated pyridinium) has been extensively used for amidation and esterification in organic chemistry via a 2-acyloxy-N-methylpyridinium intermediate,50 which provides a solid foundation for our proposition. However, direct application of Mukaiyama's reagent would not achieve chemo-selective peptide and protein amidation.51 Therefore, revisiting pyridinium activated esters is an opportunity to assess a highly reactive methodology together with optimized biophysical properties and mitochondrial enrichment for endogenous protein modification.
Here we report a facile lysine-selective protein modification using a cationic pyridinium activated ester (Fig. 1b). The esters could be readily prepared, were bench stable for months, and had high amino reactivity and a 70% lysine-selective labeling. We then performed activity-based protein profiling (ABPP) for active lysines in cells. In total, we quantitatively identified 350 high-reactive lysine-labeled peptides in 250 proteins in MCF-7 cell lysates. In addition, we achieved labeling 248 proteins containing 386 modified lysine residues in live cells, confirmed by a certain mitochondrial colocalization imaging, suggesting the mitochondrial targeting was due to the positively charged esters.52 Consequently, pyridinium activated esters provide a promising toolbox to further facilitate spatiotemporal proteome investigation and genetic manipulation.
Results and discussion
Design and identification of pyridinium activated esters
We initially examined the direct aromatic nucleophilic substitution (SNAr) of carboxylic acid by Mukaiyama's reagent but obtained complex and inseparable mixtures. Alternately, we tested 4-pyridinol ester 1a methylation by methyl iodide (MeI) or methyl trifluoromethanesulfonate (MeOTf) and obtained pyridinium pellets with nearly quantitative yields (Fig. 2a). In addition, we established that the pyridinium ester water solubility was around 10 mM, and the pyridinium esters were satisfactorily hydrolysis-resistant in different aqueous solutions (see the preparation of pyridinium activated esters in Fig. S2†). In the metabolic properties of pyridinium ester probes, we performed a hydrolysis assay with carboxylesterase enzyme (using carboxylesterase 1 (CE1) in vitro and Chang liver cells (rich in carboxylesterases) in cells). We observed no obvious hydrolysis with carboxylesterase enzyme and found a satisfactorily effective pyridinium ester concentration within live cells (Fig. S2b†). In contrast, 2-pyridinol ester methylation led to a highly unstable pyridinium. | ||
| Fig. 2 Developing of pyridinium activated esters enable amidation. (a) Postulated mechanism of amidation with pyridinium esters. (b) Kinetic study between activated esters and Boc-Lys-OH. Conditions: activated ester 1 (20 mM), Boc-Lys-OH 2 (10 mM) in NaPi (pH 7, in D2O with 40% CD3OD) at room temperature. Two replicates were conducted for the calculation of observed rate constants (k2, M−1 s−1). | ||
Having successfully synthesized aqueous-stable pyridinium activated esters, we examined the amidation of 1b, 1c, and control reagents (pyridinol ester 1a and NHS ester 1d) with Boc-Lys-OH via1H NMR spectroscopy (in sodium phosphate (NaPi, pH 7.0, in D2O with 40% CD3OD) solution for 1 hour) (see more esters in Fig. S3†). We added CD3OD to increase solubility, as substrates other than pyridinium esters are highly hydrophobic. Preliminary activating group screening demonstrated that the pyridinium esters' reactivities were comparable to NHS ester (80–90% yields). Interestingly, we detected a prototropic rearrangement of the leaving enol to the more stable keto form (Fig. S4†), which may provide additional driving force for the transesterification (Fig. 1a).53 We conducted a detailed kinetic study to gain more insight into the pyridinium esters' reactivities (Fig. 2b and S4†). The results demonstrated that pyridinium esters 1b and 1c reacted with lysine significantly faster (observed rate constant kobs = 0.50 M−1 s−1) than pyridinol ester 1a (kobs = 0.16 M−1 s−1) and NHS ester 1d (kobs = 0.15 M−1 s−1). Moreover, the excellent efficiency in the aqueous solvent suggested that the pyridinium activated esters would be physiologically compatible.
We further investigated the pyridinium esters to comprehensively determine their reactivity, solubility, and stability. We conducted peptide and protein amidation to demonstrate the pyridinium esters' reactivity and chemo-selectivity. We prepared lysine (K)-reactive probes (KP1 and KP2, 10 mM), the alkynyl derivatives of 1b and 1c, and reacted them with model peptides in PBS (pH 7.4, 50% CH3CN was added to dissolve peptides) (Fig. 3). Product 4-a, with dual amidation at the N-terminus and lysine side chain, was observed with 99% conversion after 1 hour of the KP1/KP2 and peptide 4 reaction. Note that we used KP2 as a substrate for the following reactions due to the iodine anion's potential reducibility. The kinetic study demonstrated that the reaction was finished within 10 min (inset chart in Fig. 3a and Data S1†). To further illustrate their reactivity and chemo-selectivity, we conducted a kinetic comparison between KP2 and NHS ester KP11 (Fig. 4) in pH 6.0–8.0 buffers. KP2 maintained a high reactivity (99% conversion for 4-a) even at a pH as low as 6.5, but the NHS probe KP11 could barely complete the reaction (83% conversion for 4-a) at pH 8.0 after 1 hour (see ESI Tables S1 and S2† for additional data). Interestingly the pyridinium ester exhibited apparent selectivity at the N-terminus in low pH 6.0 buffer. In contrast, the NHS ester only slightly improved the N-terminal reaction selectivity at low pH 6.0 to 6.5. Thus, we performed an in-depth study of the N-terminal-selective peptide and protein modification with pyridinium esters. We found that cyclic peptide 5, which contains only one N-terminal amino group, yielded 96% amidated product 5-a at pH 6.5, indicating that the pyridinium ester has the potential to be N-terminal-selective in a weakly acidic environment. Moreover, we obtained LC traces by directly analyzing reaction mixtures (Fig. 3a) and found that the reactions typically proceeded with high chemo-selectivity. We also detected the remaining excess KP2 in the LC traces.
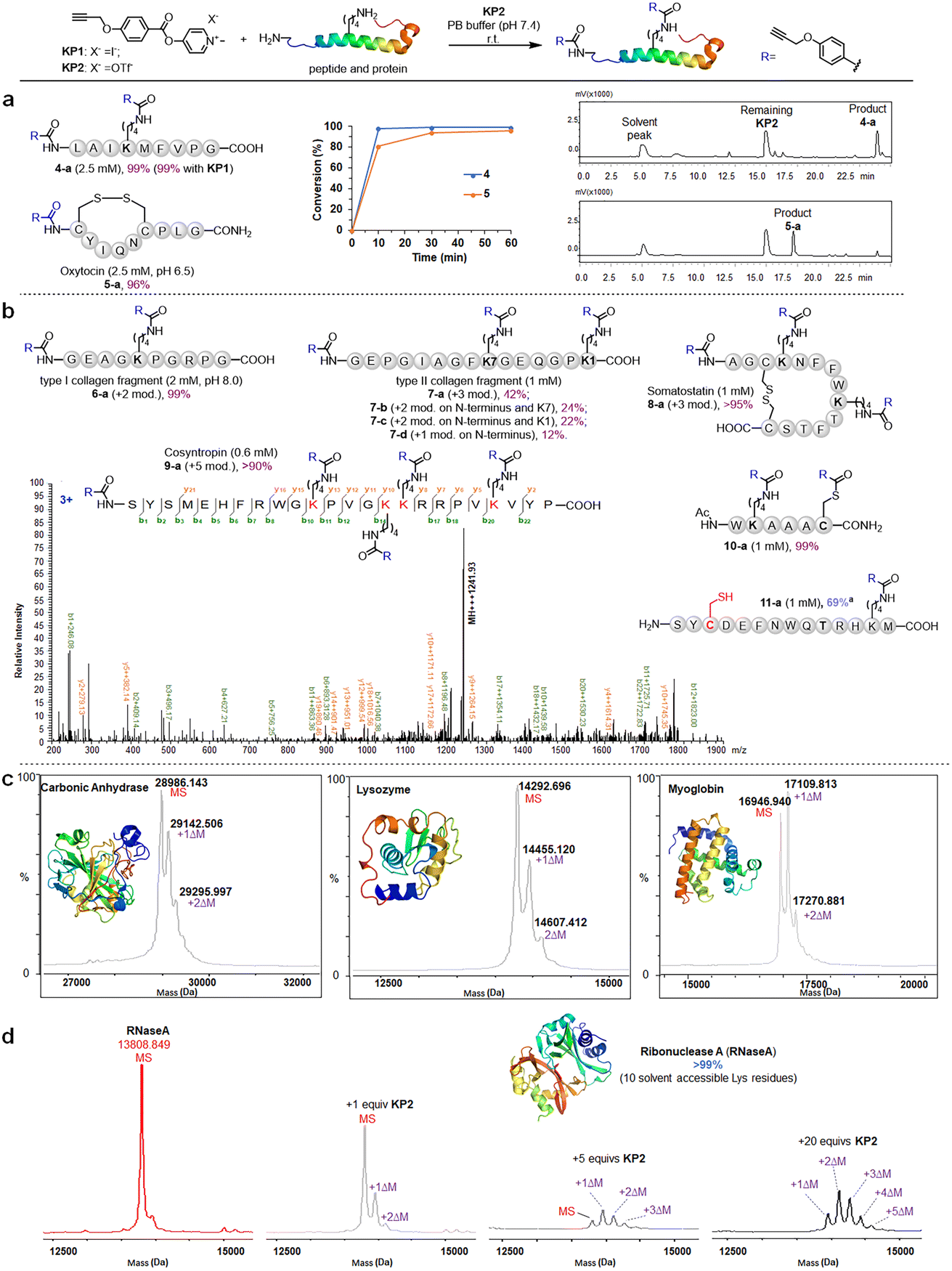 | ||
| Fig. 3 Chemoselective amidation of pyridinium esters on lysine residue sites in peptides and proteins. (a) Model reactions between KP2 and peptides, the inserted chart exhibits HPLC determined conversion versus time plot, and the LC traces of directly analyzing of reaction mixtures of peptide 4 and 5. Standard conditions for peptide: KP2 (10 mM), peptide (indicated concentration in parenthesis) and in PB buffer (pH 7.4, 50% MeCN) for 1 hour at rt. (b) Range of bioactive peptides and protein fragments. “+n mod.” refers to the number of modifications on the amino groups. (c and d) Modification efficiency of KP2 on proteins by MALDI-TOF analysis. ΔM = 158 Da. Standard conditions for protein: KP2 (50 μM, 250 μM and 1000 μM, respectively), protein (50 μM) and in PBS (pH 7.4) for 1 hour at 37 °C. a 20 mM DTT was added after amidation. | ||
 | ||
| Fig. 4 Qualitative assessment of respective modification performance of probes (KP1–KP13). (a) Modification of BSA (10 μg) with KP1–KP13 (100 μM). (b) Dose-dependent modification of BSA protein with KP2. (c) Competitive modification of BSA protein with KP2 (100 μM) in the increased dose of IAM, NHS-Ac and KP-B. (d) Labeling performance of probes in cell lysates. (e) KP2 preferentially labels lysine residues in MCF-7 cell proteomes. FL, in-gel fluorescence scanning. CBB, Coomassie gel. | ||
Pyridinium activated ester probe labels lysine containing proteins
We next moved to more complex bioactive peptides and protein fragments. KP2 reacted well with all investigated 10-mer to 24-mer peptides (Fig. 3b) that contained a free N-terminal amine and at least one lysine residue. For peptides 6–8 (10–15 amino acid (AA) resides), we conducted the type I and type II collagen fragment (6 and 7) and peptide 8 reactions in a different manner. The peptide 6 reaction resulted in 99% conversion of bisamidated product 6-a at a slightly higher pH (8.0), and we isolated four products from the peptide 7 reaction comprising triple modification (42% of 7-a), double modification (24% of 7-b, 22% of 7-c), and single modification on the N-terminus (12% of 7-d) at standard conditions (pH 7.4). Note that a trial reaction at pH 8.0 caused significant degradation of peptide 7. The relatively low reactivity may be due to the helical conformation and multiple exposed carboxyl groups in collagen fragments 6 and 7, which may cause lysine residue pKa changes and/or pH changes in the peptide microenvironment. Reacting the cyclic peptide somatostatin 8 (a human hormone analogue) resulted in excellent conversion (>95% for 8-a) for amine group triple modification at standard conditions. We further demonstrated KP2's reactivity and selectivity during the late stage amidation of a 24 AA-containing peptide. Peptide 9 (cosyntropin, a synthetic adrenocorticotropic hormone) was reacted with KP2 and resulted in a +5 modification product 9-a with excellent conversion (>90%). To investigate the chemo-selectivity between amino and thiol groups, we conducted a reaction at standard conditions using lysine and cysteine residue-containing peptide 10. We found double modification on both 10-a lysine and cysteine sites, and we observed no other side reactions between the pyridinium salt and cysteine. Additionally, we found that cysteine contributed few side effects in the following profiling studies, which may be attributed to thioesters' instability under physiological conditions. To validate the hypothesis regarding thioester and to conduct a more in-depth exploration of the chemical selectivity of this reaction, we designed a model peptide 11 containing all nucleophilic amino acids. Following the reaction, the addition of dithiothreitol (DTT) facilitated the successful acquisition of the amidation product 11a, achieving a conversion of 69%. This result further demonstrates the chemical selectivity of the peptide amidation method based on pyridinium ester. We analyzed all peptide products using tandem MS/MS to confirm the reactions' site selectivity (Fig. 3b and ESI†).We next evaluated the amidation reaction of KP2 with lysine-containing proteins at high dilution. We used ribonuclease A (RNaseA), carbonic anhydrase (CA), lysozyme, and myoglobin as model proteins, and we conducted equimolar reactions between model proteins (50 μM) and KP2 (50 μM) under physiological conditions (PBS, pH 7.4, 37 °C for 1 hour). We analyzed the resulting KP2-labeled protein products using MALDI-TOF (Fig. 3c). We clearly observed one or two modifications for all four proteins at this dilute concentration. Furthermore, we found that the RNaseA amidation was KP2 dose-dependent over concentrations ranging from 0 to 1 mM (Fig. 3d). We increased the conversions by increasing ester equivalents. With 20 equiv. of KP2, we observed quantitative conversion with one to five amidated modifications. Collectively, we revealed that peptide and protein amidation reactions with pyridinium esters had high reactivity and chemo-selectivity, suggesting a potential pyridinium-based tool to be further validated for protein labeling and profiling in more complex systems.
We next prepared a series of alkyne-tagged ester probes (KP1–KP13), including pyridinium esters and control reagents (Fig. 4a), and utilized them to investigate bovine serum albumin (BSA) labeling profiles after copper-catalyzed azide alkyne cycloaddition (CuAAC) coupling with a fluorescent dye (TAMRA-PEG3-N3). We incubated the probes (100 μM) with BSA under physiological conditions (PBS, pH 7.4, 37 °C for 1 hour) and found that KP2 performed comparable well. Furthermore, the reaction between KP2 and BSA proceeded in a dose-dependent manner (Fig. 4b). We also performed competition reactions using lysine- and cysteine-reactive reagents and found that excess NHS-Ac pretreatment decreased the fluorescence intensity, but iodoacetamide (IAM) pretreatment had negligible influence (Fig. 4c), indicating that KP2 predominantly labeled BSA protein lysine residues but not cysteine residues. We further confirmed this using LC-MS/MS (ESI Table S3†). We consistently found weaker fluorescence intensity after pretreatment with excess pyridinium ester competitor (KP-B).
In vitro labeling lysine residues containing proteome
Inspired by pyridinium ester's high protein-labeling efficiency, we next evaluated the human proteome lysine reactivity profile. We first established the KP1–KP13 (100 μM, room temperature (RT) for 1 hour) labeling profiles in MCF-7 cell lysates using in-gel fluorescence scanning. Similar labeling intensity was observed for 100 μM KP2 compared to 100 μM of KP11 and KP12 (NHS esters), and KP13 (STP) (Fig. 4d). Also, KP2 labeling appeared to be dose-dependent and time-dependent (Fig. S5a and b†). In a competitive labeling experiment, the fluorescent bands became weaker after pretreatment with NHS-Ac or competitor (KP-B) but not IAM, suggesting that KP2 labeled proteins in a lysine-/KP-specific manner (Fig. S5c of in-gel fluorescence imaging and Fig. S7c of cellular imaging in cells†). Next we incorporated KP2-labeled fragments into photolytic tags (PC-biotin-N3) to enrich for the labeled MCF-7 cell lysate proteome using streptavidin beads. The photo-released labeled peptides were then analysed using LC-MS/MS. The hydroxyl-containing AAs tyrosine (9.5%) and serine (8.3%) were partially labeled, and the majority of KP2-labeled sites were lysine residues (67.9% on average) (Fig. 4e and ESI Table S4†), highlighting a certain lysine-selectivity of pyridinium esters comparing with the NHS and STP esters (Fig. S6†). To visualize the statistical significance of the frequency of modified-lysines containing peptides (Tables S4 and S5d†), we further performed a pLogo54 analysis to show the KP2 labeling pattern. Argine (R), glycine (G) and alaine (A) residues were significantly enriched at −1/+3/−6 positions around lysines, respectively. Next, we quantitatively assessed the active lysine residues labeled by KP2 using tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) with label-free quantification (LFQ) without normalization (see ESI†) when a large-scale change in the global modification occurs.55 We prepared two pairwise KP2 concentrations (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 μM and 100:10 μM) in two parallel experiments with MCF-7 cells (Fig. 5a and ESI Table S5†). Highly reactive lysines (387 KP2-labeled peptides) could be enriched with low-concentration KP2, yielding nearly equivalent intensities as that of high-dose KP2 (0 < R100:10 ≤ 2) (Fig. 5b). The KP2-labeled peptide spectra that met our quality control confidence criteria with quantified values in all samples were selected for analysis. The 350 potential high-reactive lysine-labeled peptides (at both ratios of 0.67 ≤ R10:10 ≤ 1.5 and 0 < R100:10 ≤ 2.0) in 250 proteins that spanned protein classes, including kinases, chaperones, and structural proteins (Fig. 5c).
:10 μM and 100:10 μM) in two parallel experiments with MCF-7 cells (Fig. 5a and ESI Table S5†). Highly reactive lysines (387 KP2-labeled peptides) could be enriched with low-concentration KP2, yielding nearly equivalent intensities as that of high-dose KP2 (0 < R100:10 ≤ 2) (Fig. 5b). The KP2-labeled peptide spectra that met our quality control confidence criteria with quantified values in all samples were selected for analysis. The 350 potential high-reactive lysine-labeled peptides (at both ratios of 0.67 ≤ R10:10 ≤ 1.5 and 0 < R100:10 ≤ 2.0) in 250 proteins that spanned protein classes, including kinases, chaperones, and structural proteins (Fig. 5c).
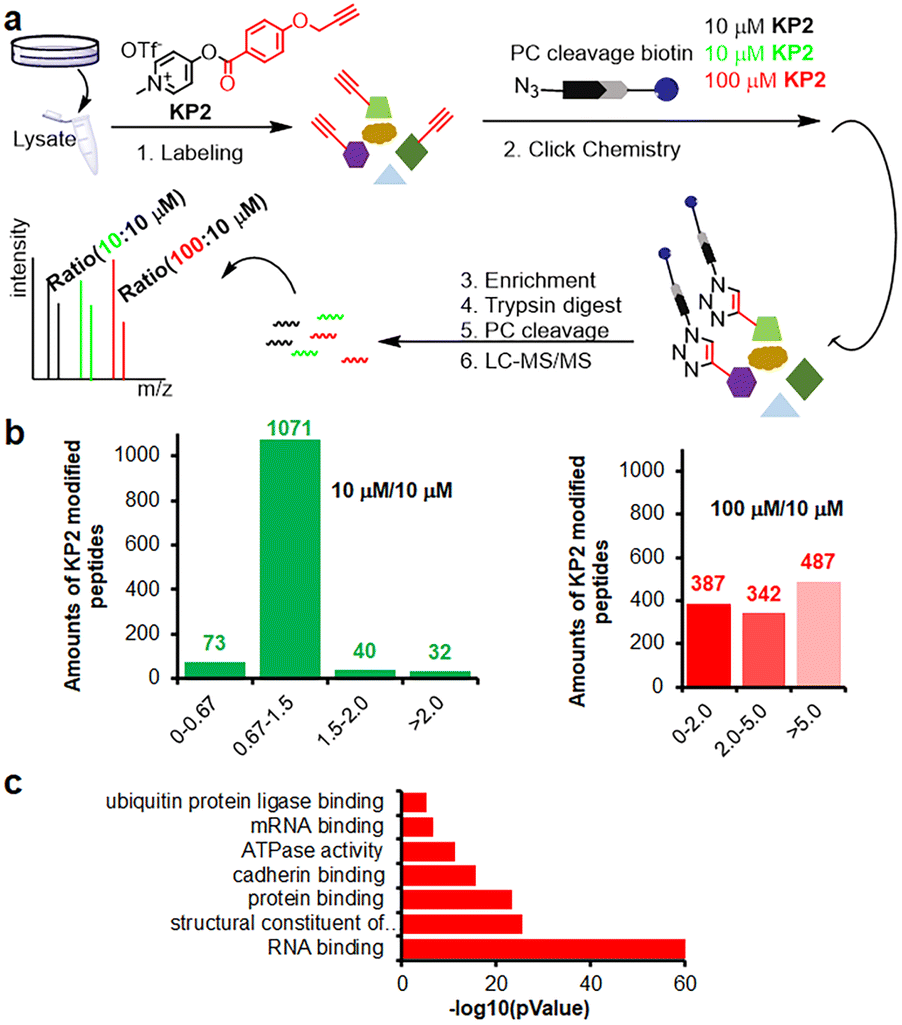 | ||
| Fig. 5 Chemoproteomic lysine profiling with KP2 in MCF-7 cells. (a) The workflow of tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) combined with label-free quantification. (b) Identified LFQ TOP-ABPP ratios for peptides from MCF-7 cells labeled with two KP2 concentrations (100:10 μM in red color) and (10:10 μM in green color) (ESI Table S5†). (c) MF (molecular function) analysis for 350 high-reactive lysine containing proteins (at both ratios of 0.67 ≤ R10:10 ≤ 1.5 and 0 < R100:10 ≤ 2.0). | ||
Mitochondrial-labeled proteome profiling in live cells
We next evaluated KP2 biocompatibility in live cells. We measured IC50 values in MCF-7 cells and found that 500 μM KP2 showed almost no toxicity, much less than NHS and STP esters (Fig. 6a and S7a†). To perform an in situ labeling of KP2, we first used in-gel fluorescent signals to preliminarily optimize the labeling conditions. Notably, cell culture medium ingredients consume KP2; thus, it was necessary to conduct the live cell labeling in PBS buffer for 1 hour. As additionally shown in Fig. 6b, labeling proteins with 500 μM KP2 in cells yielded the lowest signals but no toxicity to MCF-7 live cells. The cellular imaging was then performed to assess KP2 labeling56 (Fig. 6c and S7b†), in which 2.5 μM KP2 was incubated in live MCF-7/HeLa cells for 1 hour, then fixed and permeabilized, followed by TAMRA-N3 click. It is noteworthy that a lower concentration of 2.5 μM KP2 was utilized in the cell imaging assays to mitigate the detrimental effects of excessive fluorescence probe on imaging outcomes. Since macrophage inflammatory responses are important during immune responses,57 we also applied KP2 labeling to RAW264.7 macrophages. Interestingly, we discovered that KP2 appeared to be a partially mitochondrial location, possibly because of its positive charge. Therefore, we examined KP2 labeling colocalization with two mitochondrial markers in live MCF-7/HeLa/RAW264.7 cells, heat shock protein 60 (HSP60, blue) and cytochrome c oxidase subunit 4 isoform 1 (COX4, green) (Fig. 6d and S7b†). | ||
| Fig. 6 In situ profiling human proteome by KP2. (a) IC50 values of the probes (KP2, KP12, KP13) against MCF-7 cell. Data represent means ± standard deviation for three experiments. (b) KP2 probe labeling proteins in MCF-7 live cells, by ABPP in-gel fluorescence scanning and CBB, treated with KP2 probe 1 hour followed conjugation of TAMRA-N3 by CuAAC click-chemistry. FL, in-gel. (c) Cellular imaging of KP2 (2.5 μM) with live MCF-7 cells. KP2 exhibited the strongest in situ labeling in live MCF-7 cells after 1 hour of treatment, colocalized with that of MitoTracker green FM. Scale bar = 10 μm. Correlation was determined by Manders overlap coefficient (MOC = 0.94529). (d) Immunofluorescent analysis of MCF-7 live cells after KP2 labeling validated the labeled mitochondria proteins. The colocalization of TAMRA immunofluorescence (red) conjugated by KP2 with two mitochondrial markers cytochrome c oxidase subunit 4 isoform 1 (COX4, green) and Heat Shock Protein 60 (HSP60, blue). Scale bar = 10 μm. (e) Cellular component (CC) analysis for KP2 labeled lysines containing proteins labeled by 500 μM KP2. | ||
Finally, based on above optimized conditions (Fig. 6a and b), we used 500 μM KP2 to label MCF-7 live cell in culture dish replaced with PBS buffer for one hour to successfully collect 386 modified lysine sites in 248 proteins (ESI Table S6†). Distinguishable KP2-labeled proteins were further explored and systematically compared (Fig. 6e). These proteins showed a diverse cellular components (CC) as annotated in the GO database.58 CC analysis in live cells revealed 43 unique proteins in mitochondrion, consistent with our findings in imaging. Taken together, we revealed that to a certain extent pyridinium-based KP2 labeling was lysine-selective and mitochondria-labeling. Thus, KP2 can potentially be used to detect endogenous lysine-specific sites both in vitro and in live cells. In the future, it would be valuable to derive more pyridinium-based strategies for spatiotemporal changes in living systems.
Conclusions
Here we developed a cationic pyridinium activated ester to label and profile active lysine proteins. The pyridinium ester exhibited effective lysine reactivity and ∼70% selectivity. We applied KP2 to human proteome ABPP and quantitatively identified 350 high-reactive lysine-labeled peptides by LFQ. Remarkably, we also used KP2 for lysine-selective proteome profiling in live cells and identified 386 lysine sites in 248 proteins. To the best of our knowledge, this is a rare, successful case of lysine selective ABPP in a complex system. Additionally, the cationic pyridinium ester demonstrated the potential for mitochondrial-localized labeling in live cells. In future, we will continue to improve KP2's lysine selectivity, organelle-specificity, and metabolic features. In conclusion, we developed a pyridinium-based activated ester with good reactivity, a ∼70% lysine selectivity, and biocompatibility that provides a complimentary alternative for lysine-selective modification and organelle-targeted chemoproteomic profiling.Data availability
Materials, protocols, and data characterizations for all chemical and biological experiments are provided in the ESI.† Protein lists from mass spectrometry identification (XLSX), proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository59 with the project ID IPX0004016001. The data that support the findings of this study are available.Author contributions
Research was conceived by FY, RW and ZL. Experiments were designed by CW, FY and ZL. Chemical experiments were conducted by CW and CD. Biochemical assays and chemo-proteomic experiments were performed by DY, CS, ML, CL, XY, YA and RW. The manuscript was written and proofread by all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge financial support from the National Key Research and Development Program “Synthetic Biology” Key Special Project of China, 2021YFC2103902 and 2021YFA0910803; Natural Science Foundation of China grants 22307081, 32302420, 22307084, 21977010, 21977011 and 22107045; the Guangdong Basic and Applied Basic Research Foundation, 2022A1515010996; the Shenzhen Science and Technology Program, JCYJ20230807151059004, JCYJ20220818095808019, RCJC20200714114433053, JCYJ201805081522131455, JCYJ20200109140406047 and JCYJ20180507182427559; Tian Fu Jin Cheng Laboratory (Advanced Medical Center) Group Racing Project, TFJC2023010008; Science and Technology Program in Guangzhou 202102020214; the Shenzhen People’s Hospital funds SYKYPY201909. We acknowledge financial support from Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2023SHIBS0004). Shenzhen High-tech Zone Development Special Plan Pingshan District Innovation Platform Construction Project (29853MKCJ202300208). Technical support from the Mass Spectrometry Core Facility of Shenzhen Bay Laboratory and Dr Fan Yang of Peking University are gratefully acknowledged.Notes and references
- T. Tamura and I. Hamachi, Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Contemporary approaches to site-selective protein modification, Nat. Rev. Chem, 2019, 3, 147–171 CrossRef CAS.
- O. Koniev and A. Wagner, Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- C. G. Parker and M. R. Pratt, Click Chemistry in Proteomic Investigations, Cell, 2020, 180, 605–632 CrossRef CAS PubMed.
- C. Kwak, C. Park, M. Ko, C. Y. Im, H. Moon, Y. H. Park, S. Y. Kim, S. Lee, M. G. Kang, H. J. Kwon, E. Hong, J. K. Seo and H. W. Rhee, Identification of proteomic landscape of drug-binding proteins in live cells by proximity-dependent target ID, Cell Chem. Biol., 2022, 29(12), 1739–1753 CrossRef CAS PubMed.
- Y. Ge, X. Fan and P. R. Chen, A genetically encoded multifunctional unnatural amino acid for versatile protein manipulations in living cells, Chem. Sci., 2016, 7, 7055–7060 RSC.
- R. Zhu, G. Zhang, M. Jing, Y. Han, J. Li, J. Zhao, Y. Li and P. R. Chen, Genetically encoded formaldehyde sensors inspired by a protein intra-helical crosslinking reaction, Nat. Commun., 2021, 12, 581 CrossRef CAS PubMed.
- A. E. Speers and B. F. Cravatt, Chemical strategies for activity-based proteomics, Chembiochem, 2004, 5, 41–47 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Bioorthogonal chemistry: fishing for selectivity in a sea of functionality, Angew Chem. Int. Ed. Engl., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. Gonzalez-Paez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Proteome-wide covalent ligand discovery in native biological systems, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Ligand and Target Discovery by Fragment-Based Screening in Human Cells, Cell, 2017, 168, 527–541 CrossRef CAS PubMed.
- A. E. Speers, G. C. Adam and B. F. Cravatt, Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS PubMed.
- R. M. Reja, W. Wang, Y. Lyu, F. Haeffner and J. Gao, Lysine-Targeting Reversible Covalent Inhibitors with Long Residence Time, J. Am. Chem. Soc., 2022, 144, 1152–1157 CrossRef CAS PubMed.
- M. Tan, J. Ma, X. Yang, Q. You, X. Guo, Y. Li, R. Wang, G. Han, Y. Chen, X. Qiu, X. Wang and L. Zhang, Quantitative proteomics reveals differential immunoglobulin-associated proteome (IgAP) in patients of acute myocardial infarction and chronic coronary syndromes, J. Proteomics, 2022, 252, 104449 CrossRef CAS PubMed.
- F. Tekaia, E. Yeramian and B. Dujon, Amino acid composition of genomes, lifestyles of organisms, and evolutionary trends: a global picture with correspondence analysis, Gene, 2002, 297, 51–60 CrossRef CAS PubMed.
- G. Platzer, M. Okon and L. P. McIntosh, pH-dependent random coil (1)H, (13)C, and (15)N chemical shifts of the ionizable amino acids: a guide for protein pK a measurements, J. Biomol. NMR, 2014, 60, 109–129 CrossRef CAS PubMed.
- J. W. Wheless, R. E. Ramsay and S. D. Collins, Vigabatrin, Neurotherapeutics, 2007, 4, 163–172 CrossRef CAS PubMed.
- M. Gao, A. F. A. Moumbock, A. Qaseem, Q. Xu and S. Gunther, CovPDB: a high-resolution coverage of the covalent protein-ligand interactome, Nucleic Acids Res., 2022, 50, D445–D450 CrossRef CAS PubMed.
- J. Ha, H. Park, J. Park and S. B. Park, Recent advances in identifying protein targets in drug discovery, Cell Chem. Biol., 2021, 28, 394–423 CrossRef CAS PubMed.
- Y. Xie, S. Du, Z. Liu, M. Liu, Z. Xu, X. Wang, J. X. Kee, F. Yi, H. Sun and S. Q. Yao, Chemical Biology Tools for Protein Lysine Acylation, Angew Chem. Int. Ed. Engl., 2022, 61, e202200303 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, K. Thakur, L. Purushottam, N. C. Reddy, N. Kalra, S. Shukla and V. Rai, Chemoselective and Site-Selective Lysine-Directed Lysine Modification Enables Single-Site Labeling of Native Proteins, Angew. Chem., 2020, 132, 10418–10422 CrossRef.
- T. Tamura, T. Ueda, T. Goto, T. Tsukidate, Y. Shapira, Y. Nishikawa, A. Fujisawa and I. Hamachi, Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide, Nat. Commun., 2018, 9, 1870 CrossRef PubMed.
- M. J. Matos, B. L. Oliveira, N. Martinez-Saez, A. Guerreiro, P. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jimenez-Oses and G. J. L. Bernardes, Chemo- and Regioselective Lysine Modification on Native Proteins, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- D. A. Shannon, R. Banerjee, E. R. Webster, D. W. Bak, C. Wang and E. Weerapana, Investigating the proteome reactivity and selectivity of aryl halides, J. Am. Chem. Soc., 2014, 136, 3330–3333 CrossRef CAS PubMed.
- P. M. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. Gois, Iminoboronates: a new strategy for reversible protein modification, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed.
- Y. Yasueda, T. Tamura, A. Fujisawa, K. Kuwata, S. Tsukiji, S. Kiyonaka and I. Hamachi, A Set of Organelle-Localizable Reactive Molecules for Mitochondrial Chemical Proteomics in Living Cells and Brain Tissues, J. Am. Chem. Soc., 2016, 138, 7592–7602 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T. A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. Am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, A proteome-wide atlas of lysine-reactive chemistry, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- S. Choi, S. Connelly, N. Reixach, I. A. Wilson and J. W. Kelly, Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma, Nat. Chem. Biol., 2010, 6, 133–139 CrossRef CAS PubMed.
- S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Global profiling of lysine reactivity and ligandability in the human proteome, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T. A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. Am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, A proteome-wide atlas of lysine-reactive chemistry, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Chemistry in living systems, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- D. L. Fortin, M. R. Banghart, T. W. Dunn, K. Borges, D. A. Wagenaar, Q. Gaudry, M. H. Karakossian, T. S. Otis, W. B. Kristan, D. Trauner and R. H. Kramer, Photochemical control of endogenous ion channels and cellular excitability, Nat. Methods, 2008, 5, 331–338 CrossRef CAS PubMed.
- Z. Huang, Z. Liu, X. Xie, R. Zeng, Z. Chen, L. Kong, X. Fan and P. R. Chen, Bioorthogonal Photocatalytic Decaging-Enabled Mitochondrial Proteomics, J. Am. Chem. Soc., 2021, 143, 18714–18720 CrossRef CAS PubMed.
- D. Wang, M. Yu, N. Liu, C. Lian, Z. Hou, R. Wang, R. Zhao, W. Li, Y. Jiang, X. Shi, S. Li, F. Yin and Z. Li, A sulfonium tethered peptide ligand rapidly and selectively modifies protein cysteine in vicinity, Chem. Sci., 2019, 10, 4966–4972 RSC.
- A. D. Guo, D. Wei, H. J. Nie, H. Hu, C. Peng, S. T. Li, K. N. Yan, B. S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X. H. Chen, Light-induced primary amines and o-nitrobenzyl alcohols cyclization as a versatile photoclick reaction for modular conjugation, Nat. Commun., 2020, 11, 5472–5485 CrossRef CAS PubMed.
- R. Griffiths, F. R. Smith, J. E. Long, H. Williams, R. Layfield and N. Mitchell, Site-Selective Modification of Peptides and Proteins via Interception of Free Radical-Mediated Dechalcogenation, Angew Chem. Int. Ed. Engl., 2020, 59(52), 23659–23667 CrossRef CAS PubMed.
- Z. Hou, Y. Wang, C. Wan, L. Song, R. Wang, X. Guo, D. Yang, Y. Zhang, X. Qin, Z. Zhou, X. Zhang, F. Yin and Z. Li, Sulfonium Triggered Alkyne-Azide Click Cycloaddition, Org. Lett., 2022, 24, 1448–1453 CrossRef CAS PubMed.
- C. Wan, Y. Zhang, J. Wang, Y. Xing, D. Yang, Q. Luo, J. Liu, Y. Ye, Z. Liu, F. Yin, R. Wang and Z. Li, Traceless Peptide and Protein Modification via Rational Tuning of Pyridiniums, J. Am. Chem. Soc., 2024, 146(4), 2624–2633 CrossRef CAS PubMed.
- X. Shi, R. Zhao, Y. Jiang, H. Zhao, Y. Tian, Y. Jiang, J. Li, W. Qin, F. Yin and Z. Li, Reversible stapling of unprotected peptides via chemoselective methionine bis-alkylation/dealkylation, Chem. Sci., 2018, 9, 3227–3232 RSC.
- W. Li, D. Wang, X. Shi, J. Li, Y. Ma, Y. Wang, T. Li, J. Zhang, R. Zhao, Z. Yu, F. Yin and Z. Li, A siRNA-induced peptide co-assembly system as a peptide-based siRNA nanocarrier for cancer therapy, Mater. Horiz., 2018, 5, 745–752 RSC.
- Y. Y. Li, X. J. Wang, Y. L. Su, Q. Wang, S. W. Huang, Z. F. Pan, Y. P. Chen, J. J. Liang, M. L. Zhang, X. Q. Xie, Z. Y. Wu, J. Y. Chen, L. Zhou and X. Luo, Baicalein ameliorates ulcerative colitis by improving intestinal epithelial barrier via AhR/IL-22 pathway in ILC3s, Acta Pharmacol. Sin., 2022, 43, 1495–1507 CrossRef CAS PubMed.
- T.-L. Ho, Hard soft acids bases (HSAB) principle and organic chemistry, Chem. Rev., 1975, 75, 1–20 CrossRef CAS.
- Y. Huang, G. Zhang, R. Zhao and D. Zhang, Aggregation-Induced Emission Luminogens for Mitochondria-Targeted Cancer Therapy, ChemMedChem, 2020, 15, 2220–2227 CrossRef CAS PubMed.
- M. Tian, J. Zhan and W. Lin, Single fluorescent probes enabling simultaneous visualization of duple organelles: Design principles, mechanisms, and applications, Coord. Chem. Rev., 2022, 451, 214266 CrossRef CAS.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Companon, F. Corzana, T. P. J. Knowles, G. Jimenez-Oses and G. J. L. Bernardes, Quaternization of Vinyl/Alkynyl Pyridine Enables Ultrafast Cysteine-Selective Protein Modification and Charge Modulation, Angew Chem. Int. Ed. Engl., 2019, 58, 6640–6644 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Methods for converting cysteine to dehydroalanine on peptides and proteins, Chem. Sci., 2011, 2, 1666 RSC.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C. H. Weng, A. T. Iavarone and M. B. Francis, Site-specific protein transamination using N-methylpyridinium-4-carboxaldehyde, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- K. S. Palla, L. S. Witus, K. J. Mackenzie, C. Netirojjanakul and M. B. Francis, Optimization and expansion of a site-selective N-methylpyridinium-4-carboxaldehyde-mediated transamination for bacterially expressed proteins, J. Am. Chem. Soc., 2015, 137, 1123–1129 CrossRef CAS PubMed.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, Selective Modification of Tryptophan Residues in Peptides and Proteins Using a Biomimetic Electron Transfer Process, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- T. Mukaiyama, New Synthetic Reactions Based on the Onium Salts of Aza-Arenes [New synthetic methods (29)], Angew Chem. Int. Ed. Engl., 1979, 18, 707–721 CrossRef.
- D. Crich and I. Sharma, Epimerization-free block synthesis of peptides from thioacids and amines with the Sanger and Mukaiyama reagents, Angew Chem. Int. Ed. Engl., 2009, 48, 2355–2358 CrossRef CAS PubMed.
- F. N. Vogtle, J. M. Burkhart, H. Gonczarowska-Jorge, C. Kucukkose, A. A. Taskin, D. Kopczynski, R. Ahrends, D. Mossmann, A. Sickmann, R. P. Zahedi and C. Meisinger, Landscape of submitochondrial protein distribution, Nat. Commun., 2017, 8, 290 CrossRef PubMed.
- Z. J. Kaminski, B. Kolesinska, J. Kolesinska, G. Sabatino, M. Chelli, P. Rovero, M. Blaszczyk, M. L. Glowka and A. M. Papini, N-triazinylammonium tetrafluoroborates. A new generation of efficient coupling reagents useful for peptide synthesis, J. Am. Chem. Soc., 2005, 127, 16912–16920 CrossRef CAS PubMed.
- J. P. O'Shea, M. F. Chou, S. A. Quader, J. K. Ryan, G. M. Church and D. Schwartz, pLogo: a probabilistic approach to visualizing sequence motifs, Nat. Methods, 2013, 10, 1211–1212 CrossRef PubMed.
- O. Kauko, T. D. Laajala, M. Jumppanen, P. Hintsanen, V. Suni, P. Haapaniemi, G. Corthals, T. Aittokallio, J. Westermarck and S. Y. Imanishi, Label-free quantitative phosphoproteomics with novel pairwise abundance normalization reveals synergistic RAS and CIP2A signaling, Sci. Rep., 2015, 5, 13099 CrossRef CAS PubMed.
- D. Zhu, H. Guo, Y. Chang, Y. Ni, L. Li, Z.-M. Zhang, P. Hao, Y. Xu, K. Ding and Z. Li, Cell- and Tissue-Based Proteome Profiling and Dual Imaging of Apoptosis Markers with Probes Derived from Venetoclax and Idasanutlin, Angew. Chem., Int. Ed., 2018, 57, 9284–9289 CrossRef CAS PubMed.
- R. Medzhitov, Origin and physiological roles of inflammation, Nature, 2008, 454, 428–435 CrossRef CAS PubMed.
- D. N. Itzhak, S. Tyanova, J. Cox and G. H. Borner, Global, quantitative and dynamic mapping of protein subcellular localization, Elife, 2016, 5, e16950 CrossRef PubMed.
- J. Ma, T. Chen, S. Wu, C. Yang, M. Bai, K. Shu, K. Li, G. Zhang, Z. Jin, F. He, H. Hermjakob and Y. Zhu, iProX: an integrated proteome resource, Nucleic Acids Res., 2019, 47, D1211–D1217 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05766f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |