
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular modulation strategies for two-dimensional transition metal dichalcogenide-based high-performance electrodes for metal-ion batteries
Mingyuan
Gu
a,
Apparao M.
Rao
b,
Jiang
Zhou
 c and
Bingan
Lu
*a
c and
Bingan
Lu
*a
aSchool of Physics and Electronics, Hunan University, Changsha, P. R. China. E-mail: luba2012@hnu.edu.cn
bDepartment of Physics and Astronomy, Clemson Nanomaterials Institute, Clemson University, Clemson, SC 29634, USA
cSchool of Materials Science and Engineering, Central South University, Changsha, 410083, P. R. China
First published on 19th January 2024
Abstract
In the past few decades, great efforts have been made to develop advanced transition metal dichalcogenide (TMD) materials as metal-ion battery electrodes. However, due to existing conversion reactions, they still suffer from structural aggregation and restacking, unsatisfactory cycling reversibility, and limited ion storage dynamics during electrochemical cycling. To address these issues, extensive research has focused on molecular modulation strategies to optimize the physical and chemical properties of TMDs, including phase engineering, defect engineering, interlayer spacing expansion, heteroatom doping, alloy engineering, and bond modulation. A timely summary of these strategies can help deepen the understanding of their basic mechanisms and serve as a reference for future research. This review provides a comprehensive summary of recent advances in molecular modulation strategies for TMDs. A series of challenges and opportunities in the research field are also outlined. The basic mechanisms of different modulation strategies and their specific influences on the electrochemical performance of TMDs are highlighted.
 Mingyuan Gu | Mingyuan Gu received his bachelor's degree in electronic science and technology from Hunan University in 2012. His research interests include the design and preparation of advanced electrolytes and electrode materials for potassium ion batteries. |
 Bingan Lu | Bingan Lu, professor of Hunan University, obtained his PhD in physics from Lanzhou University in 2012. He has extensive experience in aqueous electrolyte batteries, potassium ion batteries and zinc ion batteries. He has published over 250 papers in many prestigious journals, including Nature, National Science Review, Nature Sustainability, and Nature Communications, with an H-index of 83 and other-citation of ∼20 |
1. Introduction
The enormous energy demand in modern society is causing excessive consumption of non-renewable energy sources and serious environment pollution. Energy resources, such as wind and solar energy, are clean and renewable, which are highly desirable for the long-term sustainable development of modern society. The main problem associated with these renewable clean energy sources is their intermittent characteristics, making energy storage systems (ESSs) necessary to store such intermittent energy for later use as a continuous and stable power. Due to their inherent advantages of low cost, long lifespan, and high efficiency, secondary batteries are one of the ideal candidates for storing energy.1 After decades of development, lithium-ion batteries (LIBs) have been successfully commercialized and are ubiquitous in our daily lives. However, the low abundance of lithium resources may restrict large-scale LIB-based ESSs.2,3 Therefore, alternate secondary metal-ion batteries with higher earth-abundance of the corresponding metals, such as sodium ion batteries (NIBs),4–6 potassium ion batteries (PIBs),7–10 and zinc ion batteries (ZIBs),11–13 have been extensively studied. Among various battery systems, electrode materials play an important role in the battery's electrochemical performance. Therefore, developing advanced electrode materials is desired for low cost, long lifespan, high efficiency, and large scale secondary electrochemical ESSs.Normally with a formula of MX2 (M is composed of transition metals in group 4 to group 7, and X = S, Se, Te), 2D transition metal dichalcogenides (TMDs) have attracted much attention in the field of energy storage due to their 2D structure and tunable physical and chemical properties.14–16 In monolayer MX2, X atoms are distributed in the outer layers, while the M atoms that form covalent bonds with X atoms are sandwiched between the two outer layers of X atoms. The adjacent MX2 layers in TMDs are coupled by weak van der Waals (vdW) forces, and the interlayers allow the intercalation of metal ions. For example, Whittingham demonstrated that Li+ can be inserted and extracted reversibly in the interlayer of TiS2 at a high speed in his pioneering work.17 In addition, the large interlayer spacing of MoS2 (0.62 nm) theoretically makes it easy to accommodate metal ions, such as alkali metal ions (Li+, Na+, and K+), Zn2+, etc.18–20 Furthermore, the conversion reaction of some TMDs in the deep discharge state provides them with a high theoretical specific capacity. However, structural aggregation, restacking, and limited cycling stability originating from the conversion reaction during cycling still exist.21,22 These negative factors cause TMD electrode materials to exhibit unsatisfactory electrochemical performance in metal ion batteries.
To address these issues, a large amount of research has been conducted to tune and optimize the properties of TMDs. Different morphologies of TMDs,23–25 such as nanoflowers,26 nanorods,27etc., were explored to increase the specific surface area and relieve the structural stress during cycling. The TMDs' larger specific surface area can increase the contact area with the electrolytes, which is beneficial for faster ion diffusion and better rate performance. The lower structural stress is beneficial for maintaining structural integrity and improving long-term cycling stability. Coupling with carbon materials is also one of the commonly used strategies, which can not only reduce the charge transfer impendence, but also alleviate the structural aggregation caused by vdW interaction.28–30 The above strategies effectively enhance TMDs' electrochemical performance, but they all modify TMDs from an ‘external’ perspective, and the basic properties of TMDs remain unchanged. In contrast, molecular modulation strategies modify the TMD molecules from an ‘internal’ perspective. Molecular modulation is mainly carried out at the molecular or atomic level, through the purposeful modulation of specific parts or regions of TMD molecules, so as to realize the effective modulation of TMDs' properties. Table 1 summarizes the cycling performance of some TMDs before and after molecular modulation, showing the advantages of molecular modulation.31–58 As a result, molecular modulation strategies are of great significance for the development of TMDs.
| Materials | Cycling performance | Current density | Systems | Ref. |
|---|---|---|---|---|
| Phase engineering | ||||
| 2H-MoS2 | 16.4 mA h g−1 after 300 cycles | 1 A g−1 | LIBs | 31 |
| 2H-MoS2/C | 350 mA h g−1 after 300 cycles | |||
| 1T-MoS2/C | 870 mA h g−1 after 300 cycles | |||
| 2H-MoS2@PNC | 369.9 mA h g−1 after 250 cycles | 0.5 A g−1 | NIBs | 32 |
| 1T/2H-MoS2@NC | 389.5 mA h g−1 after 250 cycles | |||
| 1T/2H-MoS2@PNC | 472.6 mA h g−1 after 250 cycles | |||
| MoS2-0 T | 182 mA h g−1 after 280 cycles | 1 A g−1 | NIBs | 33 |
| MoS2-9 T | 310 mA h g−1 after 280 cycles | |||
| 2H-MoS2 | 100 mA h g−1 after 50 cycles | 0.1 A g−1 | NIBs | 34 |
| 1T-MoS2 | 410 mA h g−1 after 150 cycles | |||
| 2H-MoS2NFs | 308 mA h g−1 after 50 cycles and 63.6 mA h g−1 after 415 cycles | 0.1 A g−1, 1 A g−1 | NIBs | 35 |
| TiO2–2H-MoS2NFs | 593 mA h g−1 after 50 cycles and 341 mA h g−1 after 700 cycles | |||
| TiO–1T-MoS2NFs | 676 mA h g−1 after 50 cycles and 501 mA h g−1 after 700 cycles | |||
| CNT@2H-MoS2 | 254.6 mA h g−1 after 50 cycles | 0.1 A g−1 | NIBs | 36 |
| 1T-MoS2 | 89.7 mA h g−1 after 50 cycles | |||
| CNT@1T-MoS2 | 542.3 mA h g−1 after 50 cycles | |||
| 2H-MoSe2 | ∼20 mA h g−1 after 50 cycles | 0.1 A g−1 | ZIBs | 37 |
| 1T-MoSe2 | 138.8 mA h g−1 after 50 cycles | |||
| 1T-MoSe2/CC | 265.1 mA h g−1 after 50 cycles | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Defect engineering | ||||
| MoS2 | 170 mA h g−1 after 220 cycles | 0.1 A g−1 | LIBs | 38 |
| MoS2/C-CPM | 831 mA h g−1 after 220 cycles | 0.2 A g−1 | ||
| WS2–SPAN-1 | 278 mA h g−1 after 450 cycles, 292 mA h g−1 after 1400 cycles | 0.5 A g−1, 2 A g−1 | NIBs | 39 |
| WS2–SPAN-2 | 464 mA h g−1 after 450 cycles, 354 mA h g−1 after 1400 cycles | |||
| WS2–SPAN-3 | 246 mA h g−1 after 450 cycles and 206 mA h g−1 after 1400 cycles | |||
| MoS2 | 45 mA h g−1 after 100 cycles | 0.5 A g−1 | NIBs | 40 |
| BD-MoS2 | 354 mA h g−1 after 100 cycles | |||
| NC@MoS2 | 103 mA h g−1 after 100 cycles | 0.1 A g−1 | NIBs | 41 |
| NC@MoS2–Ar | 430 mA h g−1 after 100 cycles | |||
| NC@MoS2–VS | 495 mA h g−1 after 100 cycles | |||
| VSe2 | 264 mA h g−1 after 100 cycles and 79 mA h g−1 after 500 cycles | 0.5 A g−1, 3 A g−1 | PIBs | 42 |
| P–VSe2−x | 351 mA h g−1 after 100 cycles and 143 mA h g−1 after 1000 cycles | |||
| Commercial WS2 | 85.6 mA h g−1 after 50 cycles, — | 0.1 A g−1, 2 A g−1 | PIBs | 43 |
| P–WS2 | 173.7 mA h g−1 after 50 cycles and 25.6 mA h g−1 after 50 cycles | |||
| Sv–WS2 | 230.8 mA h g−1 after 50 cycles and 93.2 mA h g−1 after 200 cycles | |||
| MoS2 750 | ∼5 mA h g−1 | 1 A g−1 | ZIBs | 44 |
| MoS2−x 250 | 88.6 mA h g−1 after 1000 cycles | |||
| VSe2-SS | 75.8 mA h g−1 after 1800 cycles | 4 A g−1 | ZIBs | 45 |
| VSe2−x-SS | 175.8 mA h g−1 after 1800 cycles | |||
|
||||
| Interlayer spacing expansion | ||||
| MoS2 | 248 mA h g−1 after 100 cycles | 0.2 A g−1 | LIBs | 46 |
| MoS2@PANI | 701 mA h g−1 after 100 cycles | |||
| MoS2/PANI | 1207 mA h g−1 after 100 cycles | |||
| MoS2 | ∼90 mA h g−1 after 100 cycles | 0.3 A g−1 | NIBs | |
| MoS2@PANI | ∼150 mA h g−1 after 100 cycles | |||
| MoS2/PANI | 456 mA h g−1 after 100 cycles | |||
| c-WSe2 | 61 mA h g−1 after 150 cycles | 0.1 A g−1 | PIBs | 47 |
| WSNC | 384 mA h g−1 after 200 cycles | |||
| ns-MoS2 | ∼200 mA h g−1 after 100 cycles | 0.5 A g−1 | PIBs | 48 |
| exp-MoS2 | ∼360 mA h g−1 after 100 cycles | |||
| p-MoS2 | 11.9 mA h g−1 after 1000 cycles | 1 A g−1 | ZIBs | 49 |
| MoS2/PANI-150 | 91.6 mA h g−1 after 1000 cycles | |||
|
||||
| Heteroatom doping | ||||
| Bulk WSe2 | 69 mA h g−1 after 50 cycles, — | 0.1 A g−1, 1 A g−1 | NIBs | 50 |
| 2H-WSe2–2 | 281 mA h g−1 after 50 cycles and 158 mA h g−1 after 900 cycles | |||
| 2H-WSe2–1 | 321 mA h g−1 after 50 cycles and 202 mA h g−1 after 900 cycles | |||
| 1T-WSe2–Sn | 460 mA h g−1 after 50 cycles and 285 mA h g−1 after 900 cycles | |||
| OMSCF-500 | 60 mA h g−1 after 100 cycles and 21 mA h g−1 after 100 cycles | 0.1 A g−1, 1 A g−1 | NIBs | 51 |
| OMSCF-300 | 221 mA h g−1 after 100 cycles and 79 mA h g−1 after 100 cycles | |||
| MSCF | 232 mA h g−1 after 100 cycles and 87 mA h g−1 after 100 cycles | |||
| OMSCF-400 | 330 mA h g−1 after 100 cycles and 181 mA h g−1 after 100 cycles | |||
| Pristine MoS2 | ∼47 mA h g−1 after 50 cycles | 1 A g−1 | ZIBs | 52 |
| D-MoS2–O | 281.9 mA h g−1 after 50 cycles | |||
|
||||
| Alloy engineering | ||||
| MoS2 | 228.9 mA h g−1 after 400 cycles | 1 A g−1 | LIBs | 53 |
| Mo0.5W0.5S2 | 271.9 mA h g−1 after 400 cycles | |||
| WS2 | 205.8 mA h g−1 after 400 cycles | |||
| MoS2-NC | 385.63 mA h g−1 after 1000 cycles | 0.2 A g−1 | PIBs | 54 |
| MoS1.5Se0.5-NC | 531.56 mA h g−1 after 1000 cycles | |||
| MoS2 | 18.3 mA h g−1 after 1000 cycles | 2 A g−1 | PIBs | 55 |
| MoSe2 | 114.3 mA h g−1 after 1000 cycles | |||
| MoSSe | 220.5 mA h g−1 after 1000 cycles | |||
| MoS2/rGO | 67.6 mA h g−1 after 150 cycles | 0.1 A g−1 | ZIBs | 56 |
| MoSe2/rGO | 44.5 mA h g−1 after 150 cycles | |||
| MoSSe/rGO | 210.3 mA h g−1 after 150 cycles | |||
|
||||
| Bond modulation | ||||
| MoSe2 | Almost no capacity after 20 cycles | 1 A g−1 | LIBs | 57 |
| Red MoSe2 | 1125.7 mA h g−1 after 500 cycles | |||
| MoSe–rGO | ∼100 mA h g−1 after 60 cycles | 0.1 A g−1 | PIBs | 58 |
| MoSe2+x–rGO | 168 mA h g−1 after 300 cycles | |||
In this review, we will introduce and summarize various molecular modulation strategies for TMD electrodes used in metal ion batteries, including phase engineering, defect engineering, interlayer spacing expansion, heteroatom doping, alloy engineering, and bond modulation. We begin by introducing the basic properties of different TMDs. Next, we focus on the mechanisms of different modulation methods and their specific influences on the TMDs' properties for metal-ion storage. Lastly, the challenges and perspectives of TMDs as metal-ion battery electrodes are outlined. We expect that this review will help improve the overall understanding of various molecular modulation mechanisms for TMDs in the application of electrochemical storage and serve as an excellent reference for the future development of TMD-based electrodes.
2. Basic properties of TMDs
2.1 Crystal structure
Due to the different coordination and stacking sequences of transition metals, TMDs exhibit polymorphs and stacking polytypes. For monolayer TMDs, two typical coordination structures exist for metal atoms: trigonal prismatic and octahedral coordination.59 As shown in Fig. 1a, the trigonal prismatic coordination can be expanded to hexagonal symmetry (belongs to the Oh (or D3h) point group) with Bernal stacking (AbA) (the upper and lower case letters represent chalcogen and metal atoms, respectively) in the monolayer, namely, the 1H phase.60 The octahedral coordination can be expanded to tetragonal symmetry (belongs to the D3d point group) with rhombohedral stacking (AbC), namely, the 1T phase (Fig. 1b). Due to distortions, other phases can also be found, such as the 1T′ phase (Fig. 1c). As shown in Fig. 1d–f, for bulk TMDs, the three most common polymorphs are 1T, 2H, and 3R, where the letters represent trigonal, hexagonal, and rhombohedral symmetry, and the digits indicate the number of MX2 units in the unit cell (in other words, the number of MX2 layers in the stacking sequences).61,62 Among them, 2H and 3R phases can both be formed by stacking the 1H phase in different ways with a stacking sequence of AbA BaB and AbA BcB CaC, respectively.63 A single TMD can also exist in the form of various polymorphs or stacking polytypes, depending on its synthesis method. For example, natural MoS2 usually exhibits the 2H phase. However, 1T or 3R phases are often found in synthesized MoS2.64,65 Among the above several phases, 1T and 2H have been widely studied in metal ion batteries.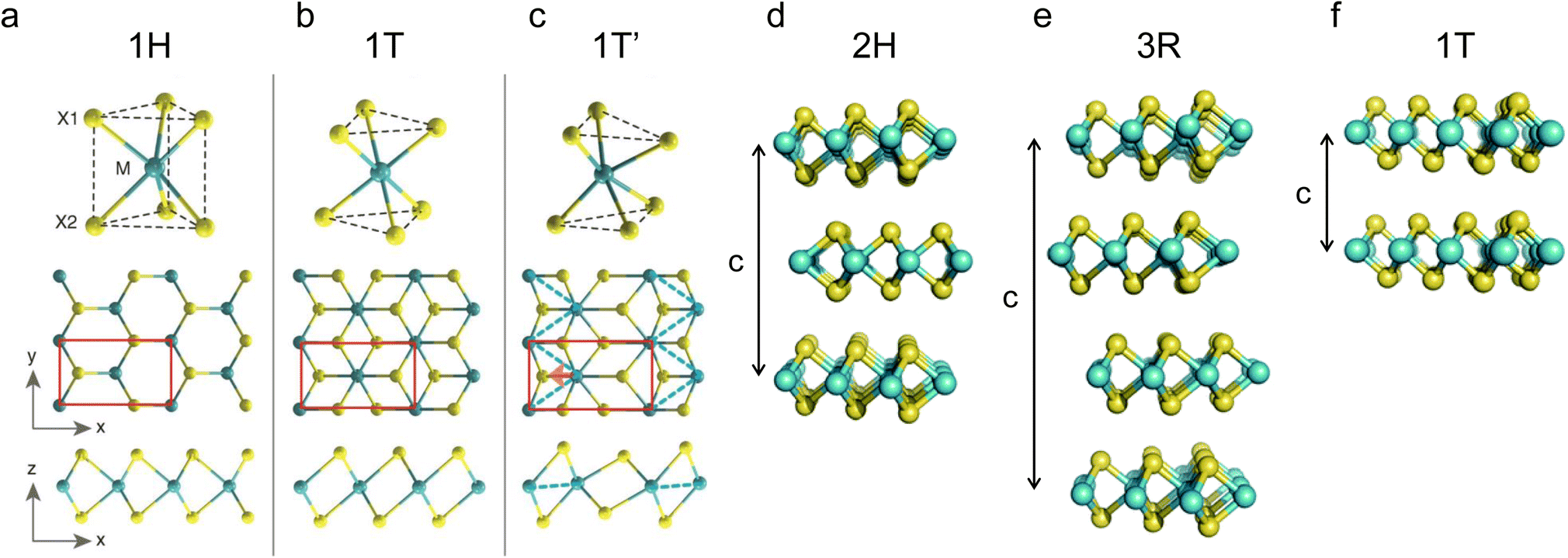 | ||
| Fig. 1 Schematics of the polymorphs and stacking polytypes: (a–f) 1H, 1T, 1T′, 2H, 3R, and 1T. Reproduced with permission from (a–c) ref. 60. Copyright 2014, Science; (d–f) ref. 62. Copyright 2015, Royal Society of Chemistry. | ||
2.2 Electronic structure
The electrical properties of TMDs depend on the transition metals' coordination environment and their d orbital filling states.62 In both 1H and 1T phases, the non-bonding d bands of TMDs are located within the gap between the bonding and antibonding bands of M–X bonds. Due to the influence of the lattice field, the d orbitals of transition metals with trigonal prismatic coordination (Oh) split into three groups, dz2(a1), dx2−y2,xy(e), and dxz,yz(e′), with a sizeable gap (∼1 eV) between the first two groups of orbitals. The d orbitals of transition metals with octahedral coordination (D3d) split into two groups, dz2,x2−y2(eg) and dyz,xz,xy(t2g).59 The filling states of non-bonding d bands play an important role in the electrical properties of TMDs. When the orbitals are fully occupied, such as in 2H-MoS2 and 2H-MoSe2,66,67 the materials exhibit semiconducting behaviour. When the orbitals are partially filled, such as in NbS2 and VS2,68,69 the materials show metallic behaviour. The phases and electrical properties of group 4–7 TMDs are summarized in Table 2.59,61,63,70–76 In addition, although the chalcogen atoms have a smaller influence on the electronic properties of TMDs, the broadening of d bands and the corresponding decrease in bandgap with increasing chalcogen atom number can still be observed. For example, the bandgap of 2H-MoS2, 2H-MoSe2, and 2H-MoTe2 decreases from 1.3 to 1.0 eV.77| Group | Metal | Chalcogen | Phases | Electrical properties | Ref. |
|---|---|---|---|---|---|
| 4 | Ti | S | 1T | Semiconducting | 31 and 70 |
| Se | |||||
| Te | |||||
| 5 | V | S | 1T | Metal | |
| Se | |||||
| Te | 71 and 72 | ||||
| 5 | Nb | S | 2H | Metal | 61 and 70 |
| Se | 70 and 73 | ||||
| Te | 1T | 61 and 70 | |||
| 6 | Mo | S | 2H | Semiconducting | 61 and 70 |
| Se | 70 and 73 | ||||
| Te | 61 and 70 | ||||
| 6 | W | S | 2H | Semiconducting | 61 and 70 |
| Se | 70 and 73 | ||||
| Te | 1T′ | 61 and 74 | |||
| 7 | Re | S | 1T′ | Semiconducting | 75 and 76 |
| Se | 61 and 63 | ||||
| Te | — | — | — |
The d electron counting of transition metals also plays a key role in selecting the preferred phase for TMDs. Group 4 TMDs with d0 transition metal centers all possess an octahedral structure. While in group 5 TMDs (d1), both the trigonal prismatic and octahedral structures exist. The trigonal prismatic phase accounts for the majority in group 6 TMDs (d2), and group 7 TMDs (d3) mainly consist of a distorted octahedral phase.59
2.3 Metal-ion storage mechanisms
When TMDs serve as alkali metal-ion battery electrodes, reversible intercalation and extraction reactions occur first at a relatively high cutoff voltage (∼1 V) with AxMX2 as the product (A represents alkali metal atoms).78 The M–X bonding interactions are larger than the A–MX2 interactions during these processes. When the discharge process continues, TiX2 and NbX2 still exhibit reversible intercalation and extraction processes due to the sufficiently strong M–X bonding. However, for the other TMDs whose M–X bonding interactions become smaller than the A–MX2 interactions at a low voltage, a conversion reaction occurs with the products of M and A2X. The reversibility of the conversion reactions is influenced by the dynamic properties of MX2,79 including the electrical properties, structural stability, and ion diffusion kinetics. The poor dynamic properties of MX2 result in irreversible conversion reactions and the oxidation products of M and X in the first cycle. In the subsequent cycles, the redox couple changes into X/A2X, which means that X replaces MX2 as the active material. Due to their poor dynamic properties, the semiconducting phases MoX2, WX2, and ReX2 exhibit irreversible conversion reactions.80–82 In contrast, the excellent dynamic properties of MX2 lead to reversible conversion reactions, where MX2 can be reformed after an oxidation reaction. i.e., MX2 is always the active material. The metal phase VX2 compounds have been demonstrated to exhibit partially reversible conversion reactions due to their better dynamic properties.83–85 In addition, the voltage at which the conversion reaction begins usually varies depending on the materials and the intercalated alkali metal ions. For example, commercial 2H-MoS2 begins to undergo a conversion reaction when discharged to 0.6 V in a LIB system. In NIB and PIB systems, however, the starting voltages decrease to 0.13 and 0.16 V, respectively.86When TMDs are used in multivalent-ion batteries, no ion storage mechanism associated with the conversion reaction has been reported so far.87 As a result, the main multivalent-ion storage mechanisms of TMDs are currently ion intercalation and extraction.
3. Molecular modulation strategies for TMDs in metal ion batteries
3.1 Phase engineering
The pristine electrical properties of different phases play a critical role in TMDs' electrochemical performance. For example, the conversion reaction is irreversible for the semiconducting phase Mo, W, and Re-based dichalcogenides due to their poor conductivities. Due to their much better conductivity, the conversion reaction becomes partially reversible in the case of metallic phase V-based dichalcogenides. As a result, converting semiconducting phase TMDs to the metallic phase could be an effective strategy to improve their electrical properties and electrochemical performance greatly. For example, Yao et al. prepared 1T-MoS2@GO for a PIB anode.88 As a comparison, 2H-MoS2@GO was also prepared. Due to the much improved electronic conductivity, a partially reversible conversion reaction is achieved when using 1T-phase MoS2, which is similar to the behaviour of metallic phase V-based dichalcogenides. In addition, 1T-MoS2@GO exhibits a better electrochemical performance than 2H-MoS2@GO at all the tested current densities. This section mainly discusses the phase engineering strategies of Mo and W-based dichalcogenides.As shown in Fig. 2a and b, under a trigonal prismatic crystal field, the Mo 4d orbitals of MoS2 are divided into dxy,x2−y2, dyz,xz, and dz2 groups, where dz2 is located at the lowest position of the orbitals.89 Therefore, the two 4d electrons fill the dz2 orbital in pairs, making 2H-MoS2 exhibit semiconducting behaviour. For 1T-MoS2, the octahedral crystal field makes the Mo 4d orbitals split into dxy,xz,yz and dz2,x2−y2 groups. In this case, the two 4d electrons tend to parallelly occupy two of the three orbitals in the lower group following the minimum Coulomb energy principle, resulting in metallic behaviour. Therefore, manipulating the electronic structures of the Mo 4d orbitals is the key to transitioning from the semiconducting phase to the metallic phase for Mo-based dichalcogenides. The partial density of states (PDOS) of Mo 4d in different phases also confirms the above conclusion (Fig. 2c and d).89
 | ||
| Fig. 2 Electronic structure manipulation of the Mo 4d orbitals as a key to transition from a semiconducting to a metallic phase for Mo-based dichalcogenides. (a and b) The occupation of electrons in Mo 4d orbitals under the crystal fields of the 1T phase and 2H phase. Calculated PDOSs of 4d states of Mo for the (c) 1T phase and (d) 2H phase. HRTEM images of the (e) 1T-MoS2/C hybrid and (f) 2H-MoS2/C hybrid. Water contact angle measurements of (g) 3DG/2H-MoS2 and (h) 3DG/1T-MoS2. Electrolyte contact angle measurements of (i) 3DG/2H-MoS2 and (j) 3DG/1T-MoS2. Calculated energy profiles of Zn2+ diffusion in (k) 2H phase and (l) 1T phase MoS2 with different interlayer distances. (m) Snapshots of trajectories for lithiated 2H-MoS2 monolayer, 2H-MoS2 bilayer, 1T-MoS2 monolayer, and 1T-MoS2 bilayer nanosheets following 6 ps ab initio molecular dynamics (AIMD) simulations at 300 K. (n) Digital photos of glass vials with the MXene/1T–2H MoS2–C–S and MXene/2H MoS2–C–S batteries as discharge time increases (0.05C). (o) The ΔG values per formula unit of the reaction between Na2Se/Mo and MoSe2 under strained and unstrained conditions. Reproduced with permission from: (a–d) ref. 88. Copyright 2015, AIP Publishing; (e and f) ref. 31. Copyright 2019, Wiley-VCH; (g–j) ref. 96. Copyright 2019, Royal Society of Chemistry; (k and l) ref. 97. Copyright 2022, Elsevier; (m) ref. 102. Copyright 2016, Royal Society of Chemistry; (n) ref. 103. Copyright 2018, Wiley-VCH; (o) ref. 113. Copyright 2022, Springer Nature. | ||
In addition to the greatly improved electrical conductivity, the 1T phase usually shows a larger interlayer spacing than the 2H phase, resulting in faster ion diffusion rates and smaller volume changes during cycling.90,91 Bai et al. prepared the 1T-MoS2/C composite for Li+ storage. The prepared 1T-MoS2 not only exhibits metallic conductivity but also shows an expanded interlayer spacing of 0.94 nm (Fig. 2e).31 The enlarged interlayer space of 1T-MoS2 compared to that of 2H-MoS2 (0.62 nm, Fig. 2f) enables rapid intercalation and extraction of Li+ and buffers the volume changes, leading to improved electrochemical performance. The 1T-WS2 nanoflowers (NFs) with an enlarged interlayer spacing of 0.67 nm (0.62 nm for 2H-WS2) also exhibit better electrochemical performance than the 2H-WS2 NFs and 2H-WS2 sheets.92
When used as aqueous metal-ion battery electrodes, 1T-MoS2 shows higher hydrophilicity than 2H-MoS2, which is beneficial for wetting the electrolytes and increasing the ion diffusion rate.93–95 The water contact angle measurements of three-dimensional graphene (3DG)/2H-MoS2 and 3DG/1T-MoS2 are shown in Fig. 2g and h.96 3DG/2H-MoS2 shows a contact angle of 97.2°, which is much higher than 37.2° of 3DG/1T-MoS2, suggesting that 1T-MoS2 is highly hydrophilic. Liu et al. synthesized MoS2 nanosheets with different 1T phase contents for aqueous zinc ion storage.97,98 Because of the high conductivity and hydrophilicity of 1T-MoS2, a tendency is observed that the electrochemical performance improves with increasing the content of the 1T phase, and the MoS2 nanosheets with the highest content of the 1T phase exhibit the best electrochemical performance. The density functional theory (DFT) calculations showed that 1T-MoS2 always exhibits lower Zn2+ diffusion energy barriers than 2H-MoS2 under the same interlayer spacing conditions (Fig. 2k and l).97 In addition to its high hydrophilicity, 1T-MoS2 also exhibits better organic electrolyte wettability (Fig. 2i) than 2H-MoS2 (Fig. 2j).96
Moreover, during the conversion reaction process, taking MoS2 as an example, the formation of polysulfides is unavoidable, resulting in the loss of active materials and the shuttle effect.34,99 This phenomenon can be mitigated by converting 2H-MoS2 to 1T-MoS2 due to the latter's strong adsorption toward polysulfides and higher chemical stability.100,101 As shown in Fig. 2m, the continuous release of S atoms from 2H-MoS2 and the formation of polysulfides within 6 ps are evident for both monolayer and bilayer nanosheets from the AMID simulations.102 In contrast, no S atoms are released from 1T-MoS2 within 6 ps for both monolayer and bilayer nanosheets, demonstrating the higher chemical stability of 1T-MoS2 compared to 2H-MoS2. Furthermore, a polysulfide adsorption experiment was carried out under continuous discharge conditions (Fig. 2n).103 For the MXene/1T–2H MoS2–C–S nanohybrids, almost no change in the color of the electrolyte is observed during the 12 h discharging process. However, in the case of MXene/2H MoS2–C–S nanohybrids, the color of the electrolyte changes from colorless to faint yellow within the first 3 h. Subsequently, the color of the electrolyte changes to dark yellow until the end of the discharge. Therefore, the strong adsorption of 1T-MoS2 toward polysulfides enables it to have excellent cycling stability and behave as a promising S host. Zhang et al. prepared MXene/1T–2H MoS2–C nanohybrids as an S host for Li–S batteries.103 Compared to the MXene/2H MoS2–C host, the MXene/1T–2H MoS2–C–S electrode exhibits better cycling stability.
Phase engineering is a very effective strategy to improve the dynamic properties and electrochemical performance of Mo and W-based dichalcogenides, and many methods have demonstrated the successful preparation of their 1T phases.104 The relatively lower temperature hydrothermal/solvothermal methods are often used to prepare 1T phase-containing Mo and W-based dichalcogenides.105,106 When increasing the reaction temperature or annealing the 1T phase at a high temperature, the 2H phase can be obtained. For example, 1T-MoS2 can be prepared by a hydrothermal reaction below 200 °C. When the reaction temperature is increased to 240 °C, only the 2H phase exists due to its higher thermodynamic stability.88 More specifically, when performing hydrothermal/solvothermal reactions or other phase engineering strategies, foreign species intercalation,107,108 vacancy and dopant introduction,109,110 exfoliation-restacking,111,112etc., are the main pathways for manipulating the Mo 4d electronic structures. Among these, foreign species (such as organic compounds) intercalation not only results in the formation of the 1T phase but can also regulate the Gibbs free energy of the redox reaction by using the strain. For example, Jiang et al. reported tensile-strained MoSe2 (TS-MoSe2) using 2-methylimidazole (2-MI) as a scaffold.113 The introduction of 2-MI not only results in the formation of 1T-MoSe2 but also reduces the Gibbs free energy of the redox chemistry (Fig. 2o), resulting in highly reversible sodium ion storage.
Since the 1T phase has many advantages, the phase transition ratio therefore becomes a very important parameter for the phase engineering strategy. From the viewpoint of thermodynamics, the 1T phase is metastable, and the 2H phase is more stable.114 Therefore, it is hard to achieve a 100% phase transition for Mo and W-based dichalcogenides. Currently, most TMDs constructed by phase engineering strategies contain around 70% of the 1T phase (Table 3).36,37,115,116 Although the electrochemical performance of these TMDs has been greatly improved, the construction of pure 1T phase Mo and W-based dichalcogenides is still desirable. He et al. reported that they induced a 100% phase transition of MoS2 from the 2H phase to the 1T phase by electron-injection-engineering.35 During the synthesis process, the reducing gases generated by the decomposition of melamine donate electrons to the MoS2 precursor, triggering the formation of pure 1T phase MoS2. Meanwhile, the TiO nanoparticles generated from the reduction of TiO2 are chemically bonded with 1T-MoS2, which ensures the stability of the 1T phase. In addition, a very interesting phenomenon was reported by Ding et al. where high magnetic fields can also induce the formation of pure 1T phase MoS2 and WS2.33 They found that 1T-MoS2 and 1T-WS2 can be prepared by magneto-hydrothermal processing under magnetic fields of 8 T and 9 T, respectively.
| Materials | Methods | 1T phase ratio (%) | Ref. |
|---|---|---|---|
| MoS2 | Hydrothermal method: 160 °C for 24 h | 51 | 97 |
| MoSe2 | Hydrothermal method: 200 °C for 12 h | 53.7 | 115 |
| MoS2 | Hydrothermal method: 180 °C for 24 h + solvothermal method: 200 °C for 8 h | 60 | 116 |
| MoSe2 | Solvothermal method: 220 °C for 24 h | 65 | 36 |
| MoS2 | Solvothermal method: 200 °C for 12 h | 62.5 | 106 |
| MoS2 | Solvothermal method: 180 °C for 20 h | 69.1 | 37 |
| MoS2 | Exfoliated and restacked method | 70 | 93 |
| MoS2 | Hydrothermal method: 160 °C for 24 h | 70 | 98 |
| MoSe2 | Solvothermal method: 220 °C for 48 h | 78.6 | 90 |
| MoSe2 | Plasma-assisted annealing method | 91 | 109 |
| MoS2 | Solvothermal method: 210 °C for 24 h + annealing method:700 °C for 2 h | 100 | 35 |
| MoS2 + WS2 | Magneto-hydrothermal method | 100 | 33 |
In short, phase engineering can improve the electrical conductivity of Mo and W-based TMDs. Additionally, a larger interlayer spacing, better electrolyte wettability, and stronger polysulfide adsorption are also beneficial for rapid ion storage and stable cycling performance.
3.2 Defect engineering
Generally, ions insert into defect-free TMDs from the edges and diffuse along the interlayers.117,118 The limited active sites and long diffusion paths are not beneficial for rapid ion storage, resulting in sluggish ion transport kinetics. Defects have been found to be able to increase the active adsorption sites, enhance the conductivity, improve the reactive kinetics, and create additional ion diffusion paths,55,119–121 which can effectively alleviate the problems faced by defect-free TMDs.According to the dimensions, the defects in TMDs can be divided into four main types: zero-dimensional (0D) point defects, one-dimensional (1D) line defects, two-dimensional (2D) planar defects, and three-dimensional (3D) volume defects.122 Among them, the 0D point defects are more favorable due to their positive effects on the electrochemical performance and their easy and controllable incorporation.123 Therefore, point defects will be mainly discussed in this section. For example, as shown in Fig. 3a–h,124 the most commonly observed point defects in monolayer MoS2 include the monosulfur vacancy (V1S), disulfur vacancy (V2S), Mo vacancy (VMo), vacancy complex VMoS3 (VMoS6) of Mo and nearby three (six) sulfur, and two types of antisite defects where the Mo atom substitutes an S2 column (MoS2) or an S2 column substitutes a Mo atom (S2Mo). The formation energies of various vacancies are listed in Table 2, from which it can be observed that the formation energies of V1S, V2S, and VMo are lower than those of the other vacancies, implying that they are easier to form and are more widely studied.122,123 In addition, it is worth pointing out that although the defect engineering strategies reported below claim that TMD electrodes containing anionic or cationic vacancies can be controllably synthesized, the preparation of TMD electrodes containing only specific types of defects has not yet been reported.
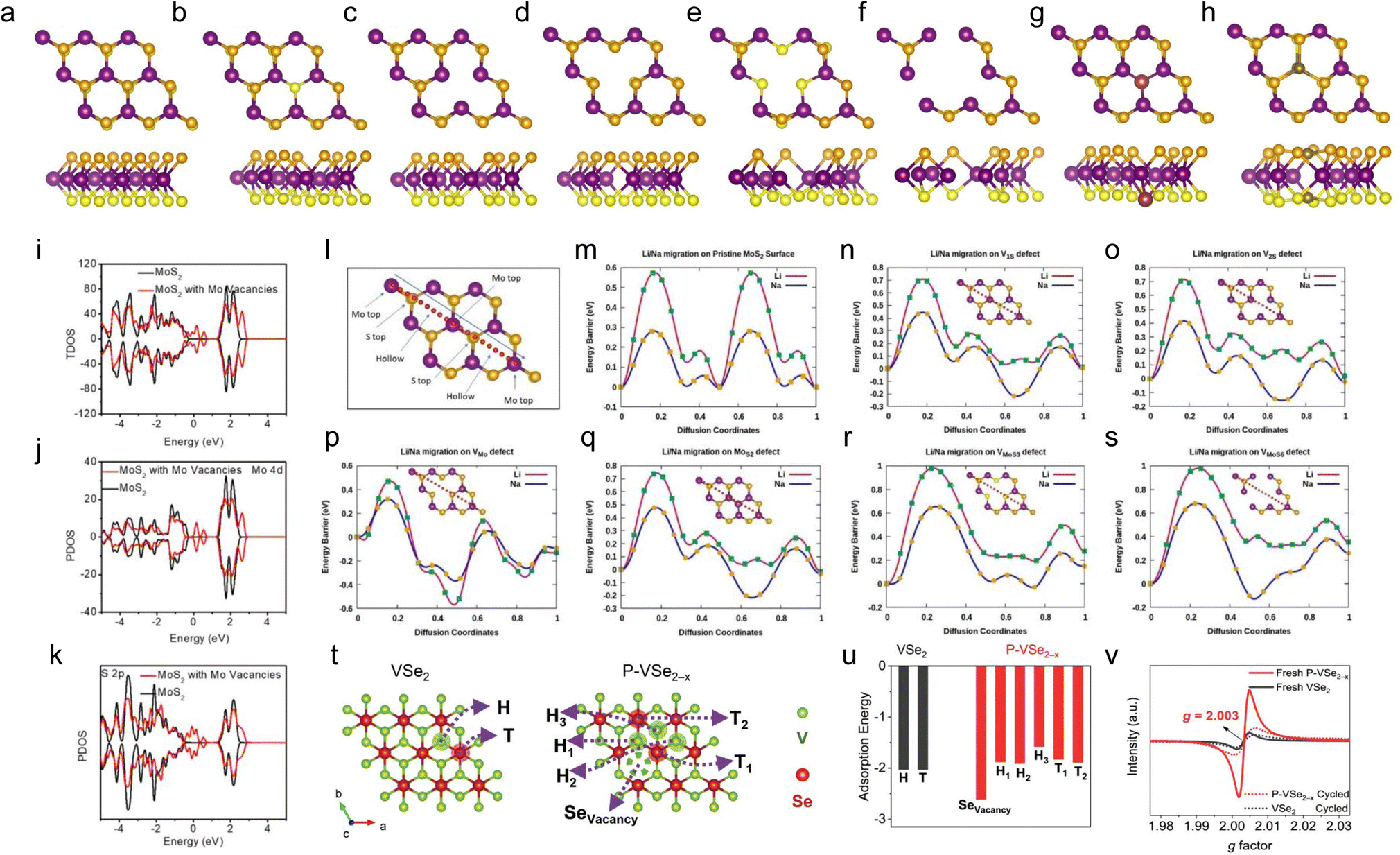 | ||
| Fig. 3 Optimized geometry of various defects in monolayer MoS2. (a) Pristine MoS2, (b) V1S, (c) V2S, (d) VMo, (e) VMoS3, (f) VMoS6, (g) MoS2, and (h) S2Mo vacancies. The purple and yellow colored balls denote Mo and S atoms, respectively. The top layer of S atoms is in golden yellow color, and the bottom layers are in light yellow color. In the MoS2 vacancy, the maroon-colored ball represents the antisite Mo substituting a sulfur atom. In the S2Mo vacancy, the gray ball represents the antisite S substituting a Mo atom. The diffusion path for Li/Na on MoS2 with defects. Total (i) and partial (j and k) charge density of states of MoS2 with Mo vacancies and pristine MoS2. Diffusion energy profiles for Li/Na diffusion barriers for (l) pristine MoS2, (m) Li/Na migration path on pristine MoS2, (n) monosulfur vacancy, (o) disulfur vacancy, (p) Mo vacancy, (q) MoS2 vacancy, (r) MoS3 vacancy, and (s) MoS6 vacancy. (t) K+ adsorption sites on VSe2 and P–VSe2−x, and (u) their corresponding adsorption energies. (v) EPR spectra of VSe2 and P–VSe2−x electrodes before and after one galvanostatic charge–discharge cycle. Reproduced with permission from (a–h and l–s) ref. 124. Copyright 2019, American Chemical Society; (i–k) ref. 126. Copyright 2019, American Chemical Society; (t–v) ref. 42. Copyright 2023, Wiley-VCH. | ||
By manipulating the electronic structures of the Mo 4d orbitals, the conversion of Mo-based TMDs from a semiconducting phase to a metallic phase was achieved via phase engineering, leading to a great improvement of conductivity and electrochemical performance. The presence of defects can also manipulate the electronic structures of the Mo 4d orbitals. Due to the presence of defects, the localized electrons generate midgap states within the valence or conduction bands, resulting in p-type or n-type conductors. The midgap states generate free charge carriers, thereby enhancing the conductivity.125 As shown in Table 4, pristine MoS2 exhibits a direct bandgap of around 1.67 eV. When different defects are created, the bandgap decreases to varying degrees, indicating enhanced conductivity. For example, Liang et al. reported nitrogen-doped carbon nanofiber@MoS2 nanosheet arrays with sulfur vacancies for Na+ storage.41 The introduction of sulfur vacancies enhances the conductivity of the materials and serves as the new active site for adsorbing Na+. Therefore, the charge transfer resistance is reduced, and the specific capacity and rate performance are improved. Li et al. developed a hollow microcube framework composed of ultrathin MoS2 nanosheets rich in Mo vacancies.126 Theoretical calculations reveal that the Mo vacancies can improve the electrical conductivity of MoS2 due to the narrowed bandgap (Fig. 3i–k) and the enhanced interaction between MoS2 and Na, resulting in rapid reaction kinetics and improved Na storage capacity.
| Vacancy | Vacancy formation energy (eV) | Energy gap Eg (eV) | Adsorption energy for Li Ead-Li (eV) | Adsorption energy for Na Ead-Na (eV) | Barrier height for Li (eV) | Barrier height for Na (eV) |
|---|---|---|---|---|---|---|
| Pristine MoS2 | — | 1.67 | −2.08 | −1.28 | 0.57 | 0.28 |
| V1S | 1.95 | 1.06 | −2.57 | −1.99 | 0.69 | 0.43 |
| V2S | 3.78 | 1.02 | −2.44 | −1.89 | 0.70 | 0.40 |
| VMo | 5.73 | 0.09 | −3.92 | −1.81 | 0.46 | 0.32 |
| VMoS3 | 7.90 | 0.64 | −2.83 | −2.09 | 0.91 | 0.65 |
| VMoS6 | 13.89 | 0.03 | −3.37 | −2.28 | 0.95 | 0.67 |
| MoS2 | 6.89 | 0.01 | −2.58 | −1.99 | 0.74 | 0.47 |
| S2Mo | 9.33 | 0.48 | −2.28 | −1.60 | — | — |
In addition, the presence of defects in TMDs is believed to be able to provide stronger binding sites for alkali metal atoms. The adsorption energies for Li and Na at the top site (above the top of a Mo atom) of pristine MoS2 and at the seven defective regions are listed in Table 4.124 It can be observed that pristine MoS2 shows a Li adsorption energy of −2.08 eV and a Na adsorption energy of −1.28 eV. When different defects are created, the adsorption energies for Li and Na are higher than that of pristine MoS2. The high adsorption energies of various defects indicate the easy binding and the favorable chemical interaction between Li/Na and defective MoS2. Besides the adsorption energies, the diffusion kinetics are also important for the electrochemical reaction rate. As shown in Fig. 3l–s, the associated activation energy barriers of pristine MoS2 and all types of defects were calculated with the fixed path.124 For the pristine MoS2 model, the path features two symmetrical absolute maxima, two symmetrical local maxima, and two local minima for the diffusion barriers when Li and Na pass through the two-unit cell (Fig. 3m). Consistent with the diffusion path of pristine MoS2, only one maximum is found in the vicinity of the defects (Fig. 3n–s), as the barrier height disappears in the defect regions. Thus, it is clear that the presence of vacancies reduces the barrier height in the defective regions, and Li+ and Na+ can diffuse efficiently into the vacancy regions and then diffuse across the vacancies to the top site of the next cell in the MoS2 plane (Fig. 3n–p). Although the introduction of vacancies enhances the barrier energies in the vicinity of the defects (Table 4), the barrier energies near 0.7 eV and 0.4 eV are believed to ensure the rate performance of LIBs and NIBs at room temperature. The enhanced adsorption energies of VMoS3 and VMoS6 essentially act as traps, which inhibits the atomic motion of Li+ and Na+ (Fig. 3r and s). The presence of antisite defects also promotes the migration of Li and Na atoms on the defective MoS2 surface (Fig. 3q). However, in the case of antisite defect S2Mo, the migration of both Li and Na ions ceases because the migrating Li+ and Na+ are repelled by the sulfur atoms replacing the Mo atoms. A systematic study is still lacking for the adsorption and diffusion properties of K+ in pristine and defective MoS2.
The presence of defects can enhance the conductivity and provide stronger and more binding sites, which is very beneficial for electrochemical performance. However, for those TMDs that undergo conversion reactions during cycling, the crystal structures experience a reconstruction process, which may be detrimental to the retention of defects. Sha et al. explored the role of anion vacancies of VSe2 in K+ storage.42 They found that the existence of anion vacancies in VSe2 can promote the electrochemical reaction kinetics. In addition, the defect regions provide more active sites (Fig. 3t), and they all exhibit large K+ adsorption energies (Fig. 3u). However, the anion vacancies in VSe2 are found to influence the first potassiation process primarily. In the subsequent cycles, they disappear and cannot be regenerated, which is demonstrated by electron paramagnetic resonance (EPR) (Fig. 3v). Although the vacancies in VSe2 disappear after the first cycle, the electrochemical performance of defective VSe2 is still much better than that of pristine VSe2, which is interesting and needs further exploration. In the case of those TMDs that only undergo intercalation and extraction reactions, the defects may be preserved well during cycling. Liu et al. reported a TiS2 anode rich in cation vacancies, which only experiences intercalation and extraction reactions during cycling.127Ex situ EPR spectra demonstrate that, even after 60 cycles, the EPR signal of defective TiS2 is almost unchanged, indicating that the defects were well retained. Still, much work is needed to determine the clear relationship between the reaction types and defect retention.
It is believed that introducing defects will bring new ion diffusion pathways, thus enhancing the electrochemical reaction kinetics. An experiment designed by Zhang et al. demonstrated that small-size ions can also intercalate through the top surface of few-layer defective MoS2 apart from the edges (Fig. 4a).128 The color changes of MoS2 flakes with sealed and open edges during cycling indicate that Li+ can both intercalate through the top surface and the edges (Fig. 4b), and the process of intercalating through the top surface of MoS2 flakes has much better reversibility. In the case of Na+ storage (Fig. 4c and d), the MoS2 flakes with sealed and open edges both exhibit excellent reversibility, although the ionic radius of Na+ is higher than that of Li+. This phenomenon is attributed to the relatively weak chemical binding between Na and MoS2.129 In contrast, no obvious change is observed when attempting to intercalate K+ into sealed MoS2, indicating that K+ cannot intercalate through the top surface of defective MoS2 flakes (Fig. 4e and f). The color changes of K+ intercalated into MoS2 flakes with open edges are similar to that of Li+, indicating that the reversibility of this process is also poor. These observations demonstrate that the defects on the top surface of MoS2 allow the intercalation of small-size ions (such as Li+ and Na+) but reject large-size ions (such as K+). Similarly, additional 3D ion diffusion pathways have also been explored through theoretical or experimental studies.40,42
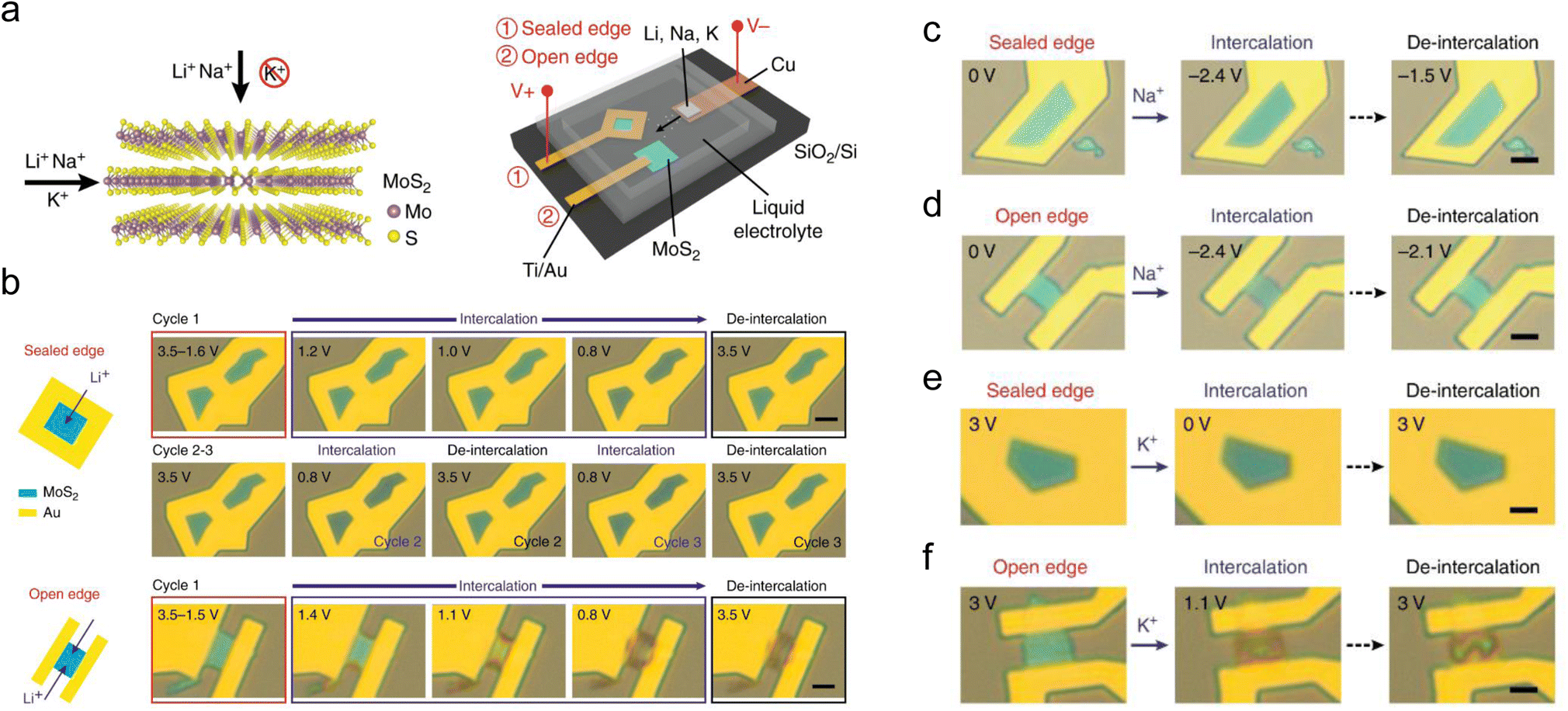 | ||
| Fig. 4 Schematic representation of MoS2 and of the experimental setup to study intercalation and de-intercalation. (a) Li+, Na+, and K+ intercalation into MoS2 through top and edge channels. An electrochemical cell is used to perform intercalation of Li+, Na+, and K+ into MoS2 with sealed and open edges. (b) In situ optical microscopy images of Li intercalation into MoS2 through top surface and edges, respectively. Scale bars, 5 μm. (c and e) In situ optical microscopy images of Na and K intercalation into MoS2 through the top surface. (d and f) In situ optical microscopy images of Na and K intercalation into MoS2 through the edges. Scale bars in (c–f), 5 μm. Reproduced with permission from ref. 128. Copyright 2018, Springer Nature. | ||
The large charge density of multivalent ions leads to a high ion intercalation and diffusion impedance in TMDs. Introducing defects can enhance the electrochemical reaction kinetics of TMDs, thus improving their storage capacity for multivalent ions. Xu et al. activated MoS2 for Zn2+ storage by introducing sulfur vacancies.44 Compared to pristine MoS2, MoS2 with sulfur vacancies exhibits much greater capacity, better rate performance, as well as high capacity retention. Theoretical calculations reveal that the numerous defects act as the preferential intercalation sites for Zn2+ storage. MoS2 nanosheets with sulfur vacancies were also reported by Zhu et al. for Mg2+ storage.130 They showed that introducing defects into MoS2 nanosheets improves the ability of MoS2 to store Mg2+. Bai et al. used defective VSe2 for Zn2+ storage by introducing selenium vacancies and reported an enhanced electrochemical performance.45 By using theoretical calculations, they found that the presence of selenium vacancies adjusts the adsorption energy of Zn2+, which makes the electrochemical reaction more reversible.
In short, introducing defects into TMDs can enhance their conductivity, provide more and stronger binding sites, create new ion transfer pathways, and shorten the diffusion paths of metal ions, which are beneficial for improving the ion storage capacity of TMDs. However, in some TMDs, the defects disappear after the first cycle and cannot be regenerated, yet electrochemical performance improvements can still be observed in the subsequent cycles. This phenomenon is interesting and has not been explained clearly yet. In addition, the clear relationship between the reaction types and defect retention also needs further studies for a better understanding.
3.3 Interlayer spacing expansion
A low ion diffusion barrier is essential for rapid ion transport in TMDs. Although the presence of defects has been demonstrated to accelerate ion diffusion, their positive effects only exist locally.124 Therefore, a strategy that can comprehensively reduce the ion diffusion barriers of TMDs is needed. It has been reported that although the interlayer spacing of ReS2 and MoS2 is very similar (6.14 Å for ReS2 and 6.15 Å for MoS2), the diffusion barriers of Li+ in ReS2 nanosheets are significantly lower than that in MoS2 nanosheets,131 and the main reason is attributed to the much weaker vdW interactions between the adjacent layers in ReS2 than that in MoS2.131,132 The weak vdW interactions between TMD interlayers are beneficial for rapid transport of ions with low impendence. As a result, weakening the vdW interactions between TMD interlayers could be an effective strategy to comprehensively reduce the ion diffusion barriers of TMDs. In addition to selecting some TMDs that exhibit weak vdW interactions on their own, such as ReS2, a more effective and broadly applicable strategy is to expand the interlayer spacing of TMDs to reduce their vdW interactions.131,133,134The relationship between the interlayer spacing and the diffusion of alkali metal ions in MoS2 was explored by theoretical calculations.135,136 With the diffusion pathway of Oh (stable site) → Th (metastable site) → Oh, the diffusion barriers of Na+ decrease from 1.14 eV to 0.2 eV with increasing the interlayer spacing from 6.5 Å to 9 Å. In the case of Li+, with the same diffusion pathway, the diffusion barriers decrease continuously from 0.54 eV with increasing the interlayer spacing from 6.5 Å to 7.5 Å. When the interlayer spacing is larger than 7.5 Å, the Th site becomes more stable than the Oh site, making the diffusion from Oh to Th endothermic.137,138 Therefore, the diffusion barriers of Li+ increase slightly to 0.31 eV with increasing the interlayer spacing from 7.5 Å to 9 Å. The relationship between the interlayer spacing and the diffusion of K+ in MoS2 was also explored by Zheng et al. with the diffusion path shown in Fig. 5a (the diffusion path of K+ is indicated by the blue dotted line).136 The results show that the diffusion barriers of K+ decrease from 3.94 eV to 0.045 eV with increasing the interlayer spacing from 6.2 Å to 9.2 Å (Fig. 5b). In general, taking MoS2 as an example, the diffusion barriers of alkali metal ions decrease with increasing the interlayer spacing of TMDs, and the increased interlayer spacing is beneficial for the rapid ion transport kinetics of TMDs.139,140
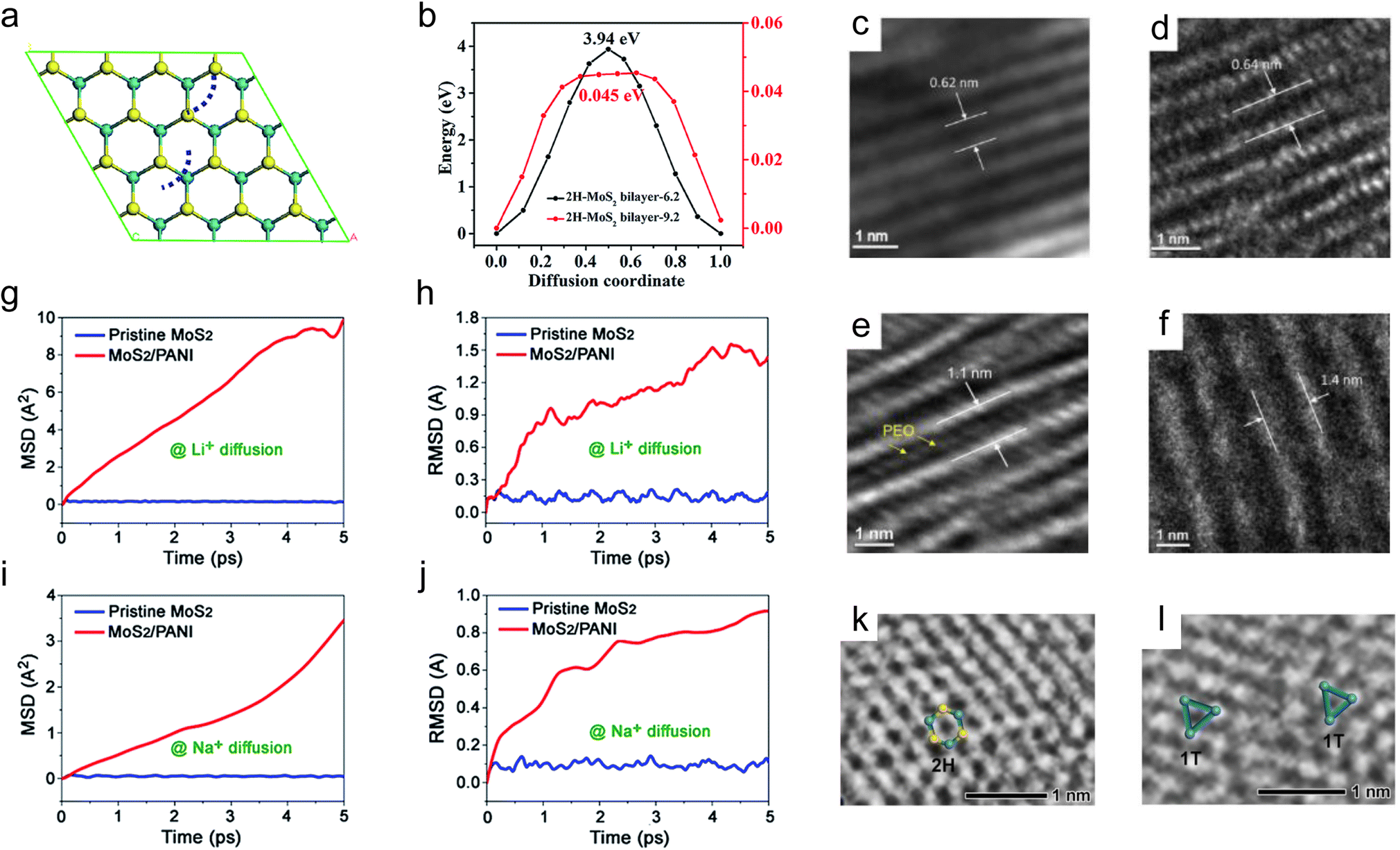 | ||
| Fig. 5 Interlayer spacing and diffusion of alkali metal-ions. (a) Top view of the optimized MoS2/MoS2 interface (the blue dotted line indicates the K+ diffusion path). (b) Energy profiles along the diffusion path in the selected interlayer spacing of MoS2/MoS2. (c and e) The mean square displacement and (d and f) the root mean square displacement of Li+/Na+ as a function of time of Li+/Na+ diffusion on the pristine MoS2 nanosheets and the MoS2/PANI hybrid nanosheets. (g–j) High-resolution TEM images show the cross-sectional view of com-MoS2, re-MoS2, PEO1L–MoS2, and PEO2L–MoS2. High-resolution TEM images and atomic models for (k) 2H-MoS2 and (l) 1T-MoS2, respectively. Reproduced with permission from (a and b) ref. 136. Copyright 2019, Royal Society of Chemistry; (c–f) ref. 46. Copyright 2017, Royal Society of Chemistry; (g–j) ref. 151. Copyright 2015, Elsevier; (k and l) ref. 149. Copyright 2020, Wiley-VCH. | ||
Xu et al. summarized the strategies for synthesizing interlayer-expanded TMDs as ‘top-down’ and ‘bottom-up’ approaches.133 Specifically, the methods for expanding interlayer spacing can be divided into two categories: one is the restacking of exfoliated TMD monolayer nanosheets, and the other is to introduce guests or guest precursors into TMD interlayers.133,141 Du et al. restacked MoS2 by an exfoliation and restacking process with enlarged interlayer spacing. The enlarged interlayer spacing of MoS2 provides a larger space for Li+ intercalation and reduces the diffusion barriers of Li+. Consequently, the restacked MoS2 exhibits improved electrochemical performance compared to raw MoS2.142 In the case of introducing guests or guest precursors into TMD interlayers, they include many foreign species, such as ions, organics, carbon, etc.133,143–145 The intercalated ions are often metal ions and NH4+.137,146,147 Zak et al. intercalated alkali metals into MS2 (M = W and Mo) nanoparticles by exposing MS2 to alkali metal (Na and K) vapor, and observed a substantial increase (≈3–5 Å) in the interlayer spacing of the intercalated phase.146 Ma et al. reported a multivalent ion intercalation method to construct 3D Co–MoS2 nanoflowers with expanded interlayer spacing. The intercalated Co2+ forms S–Co–S covalent bonds and enlarges the maximum interlayer spacing to 1.1 nm, which dramatically reduces the diffusion barriers of Na+.141 Hu et al. expanded the interlayer spacing of MoS2 by intercalating Na+ and NH4+. The expanded interlayers reduce the charge transfer resistance and provide more active sites for Na+ storage.147 As for the organic guests, a large number of organics has been reported that can be intercalated into TMD interlayers,14,145 such as poly(ethylene oxide) (PEO), polyaniline (PANI), oleylamine, methylamine, polypyrrole, etc.46,47,148–150 Yao and co-workers synthesized PEO-intercalated MoS2 composites (PEO–MoS2) using an exfoliation-restacking method.151 They prepared 4 samples, commercial MoS2 (com-MoS2), restacked MoS2 without PEO (re-MoS2), and monolayer and bilayer PEO-intercalated MoS2 composites (PEO1L–MoS2 and PEO2L–MoS2) for comparison. The com-MoS2 shows a sharp diffraction peak located at 14.4°, corresponding to an interlayer spacing of 0.62 nm (Fig. 5c). For re-MoS2, PEO1L–MoS2, and PEO2L–MoS2, the peaks become broadened and shift to lower angles, indicating the less ordered structures and the larger interlayer spacing. Compared to that of com-MoS2, the interlayer spacing of re-MoS2, PEO1L–MoS2, and PEO2L–MoS2 is increased to 0.64 nm, 1.1 nm, and 1.4 nm, respectively (Fig. 5d–f). As a result, PEO2L–MoS2, with a 160% increase in the interlayer spacing, exhibits the best electrochemical performance due to its greatest improvement in Na+ diffusivity. It is worth pointing out that the addition of excessive PEO will not further increase the interlayer spacing of MoS2. The incorporation of a conducting polymer can not only increase the interlayer spacing but also improve the conductivity of TMDs. Wang et al. prepared MoS2/polyaniline (PANI) hetero-structures with enlarged interlayer spacing for Li+ and Na+ storage,46 and they performed an AIMD simulation of the diffusion of Li+ and Na+ on pristine MoS2 nanosheets and the MoS2/PANI hybrid nanosheets (Fig. 5g–j). The mean square displacement (MSD) and the root mean square deviation (RMSD) values of Li+ and Na+ in MoS2/PANI hybrid nanosheets are both much larger than that in pristine MoS2 nanosheets, suggesting the faster diffusion mobility of Li+ and Na+, which is attributed to the improved conductivity and the enlarged interlayer spacing of MoS2/PANI composites. In the case of the carbon guests, they are usually formed via a subsequent carbonization process of MoS2 with pre-intercalated organics.152–154 For example, Jiang et al. prepared a 2D MoS2/mesoporous carbon hybrid nanostructure by annealing the MoS2/polydopamine (PDA) composites.155 Feng et al. synthesized interlayer-expanded MoS2/carbon hetero-aerogels by directly intercalating different polymers (polyethyleneimine and polyethylene glycol) into the MoS2 interlayers before the carbonization process,156 and the effects of intercalated carbon layers are similar to that of intercalated conducting polymers, such as PANI. In addition, the introduction of foreign species can also induce the phase transformation from the semiconducting phase to the metallic phase, which can enhance the electrical conductivity of Mo and W-based dichalcogenides. Ye et al. introduced methylamine into the MoS2 interlayers.149 The introduction of methylamine can not only expand the interlayer spacing, but also force the transformation of MoS2 from the 2H phase to the 1T phase, resulting in the formation of a metal-semiconducting mixed MoS2 phase (Fig. 5k and l).
The interlayer spacing expansion strategy is also widely used for multivalent ions to enhance their diffusion behaviors in TMDs.157–160 Similar to the theoretical research of the diffusion behaviors of alkali metal ions in MoS2, the diffusion behaviors of Mg2+ in MoS2 with varying interlayer spacing were also explored by Yao and co-workers in the diffusion pathway of Oh → Th → Oh (Fig. 6a).161 As shown in Fig. 6b and c, as the interlayer spacing increases from 0.65 nm to 0.9 nm, the diffusion barriers continuously decrease from 1.12 eV to 0.22 eV. The reason behind the continuous decrease in the diffusion barriers is the gradually weaker bonding between Mg and one of the two layers of MoS2 (Fig. 6d–f). It is worth pointing out that traditional interlayer engineering uses the TMDs (whose interlayer spacing has already been expanded) as electrode materials, which needs a pre-processing step. Yao and co-workers further developed an in situ interlayer expansion strategy for TiS2 for fast MgCl+ storage via electrolyte engineering.162 As shown in Fig. 6g, they added PY14Cl into the electrolyte, and Py14+ expanded the interlayer of TiS2in situ first during discharging in stage 1. From stage 1 to stage 3, a large amount of MgCl+ is intercalated continuously with a small amount of THF molecules, resulting in greatly improved electrochemical performance. The theoretical calculations show that for the same interlayer spacing, the diffusion barriers of MgCl+ are always lower than that of Mg2+, indicating the advantage of MgCl+ storage (Fig. 6h). For Zn2+ storage, Huang et al. intercalated PANI into MoS2 interlayers to expand the interlayer spacing.49 The intercalated PANI expands the diffusion channels of Zn2+ and reduces the electrostatic interaction between Zn2+ and MoS2, thus enhancing the electrochemical performance. Cui et al. introduced anionic organic layers (sodium dodecyl benzene sulfonate) into WSe2 to form superlattice-type WSe2 (S-WSe2) for Al3+ storage. The interlayer spacing expansion strategy makes S-WSe2 more active as a cathode in rechargeable Al batteries.163
 | ||
| Fig. 6 Mg diffusion in MoS2 and TiS2. (a) Calculated Mg diffusion path in MoS2. (b) The energy barrier for Mg diffusion continuously decreases as the interlayer distance of MoS2 increases. (c) Potential energy diagram for Mg migration at interlayer spacings of d = 0.65, 0.80, and 0.90 nm. As the interlayer distance increases from 0.65 nm (d) to 0.80 nm (e) and then 0.90 nm (f), the bond between Mg and MoS2 lengthens and is finally broken on one side. (g) A schematic of structural evolution of TiS2 at different stages of intercalation. (h) First-principles calculations for the diffusion of Mg-ions in TiS2. The energy barrier for the migration of Mg2+ and MgCl+ as a function of the interlayer distance of TiS2 at the dilute limit. The diffusion path from a Ti top site to another Ti top site via the adjacent hollow top site is shown in the inset. Energy diagrams along the diffusion path for the three representative cases of Mg2+ at c = 5.7 Å (green), Mg2+ at c = 10.9 Å (orange), and MgCl+ at c = 10.9 Å (cyan). Reproduced with permission from (a–f) ref. 161. Copyright 2015, American Chemical Society; (g and h) ref. 162. Copyright 2017, Springer Nature. | ||
In short, the interlayer spacing expansion strategy can comprehensively reduce the diffusion barriers of metal-ions in TMDs. The enlarged specific surface, the transformation of TMDs from the semiconducting phase to the metallic phase, the reduced lattice stress, and the improved electrical conductivity of TMDs are also observed.
3.4 Heteroatom doping
Similar to introducing defects into TMDs, heteroatom doping is also an effective strategy to modulate the local properties of TMDs, which has been reported to improve the conductivity, expand the interlayer spacing, and enhance the atom adsorption capacity of TMDs.164–166 The doped atoms can be divided into two categories: metal heteroatoms, such as Mn, Zn, Fe, Co, Ni, Cu, Re, Pd, Sn, and Au,50,167–174 and non-metal heteroatoms, such as N, O, As, P, F, Cl, Br, I, H, and B.51,170,174–176 The dopant type is an important factor that affects the electrochemical performance of TMDs.Sun et al. theoretically investigated the properties of heteroatom-doped monolayer MoS2 and the adsorption and diffusion of Li on heteroatom-doped MoS2 by substituting S with some nonmetallic elements (N, P, As, F, Cl, and I) and Mo with some metallic elements (Fe, Co, Ni, Cu, and Zn) (Fig. 7a–f).177 The DOS of monolayer MoS2 with different doping elements is shown in Fig. 7a and b. Although the doped monolayer MoS2 still exhibits semiconducting characteristics, the conductivity is improved to varying degrees. For metallic heteroatom doping, substituting Mo with Co, Ni, Cu, and Zn makes monolayer MoS2 exhibit p-type conducting behavior. For non-metallic heteroatom doping, substituting S with N, P, and As also makes monolayer MoS2 exhibit p-type conducting behavior due to their one less valence electron than S. In contrast, substituting S with F, Cl, and I makes monolayer MoS2 exhibit n-type conducting behavior. In the case of studying the adsorption of Li on heteroatom doped monolayer MoS2, two types of adsorption sites, the hollow site (H) and the top site above Mo (T), and the position of the adsorption sites as a function of distance from the doped site are considered. The adsorption energies of Li on pristine monolayer MoS2 are −0.95 and −1.24 eV for H and T sites. For all doped elements studied in this work,177 higher adsorption energies are observed near the doped sites, indicating the stronger bonding between Li and doped monolayer MoS2, and the adsorption energies decrease with increasing the distance from the adsorption sites to the doped sites. In addition, for non-metallic heteroatom doping, the adsorption energies of Li on N, P, and As doped monolayer MoS2 are higher than that on the other three elements (F, Cl, and I) doped monolayer MoS2, indicating that p-type doping is more beneficial for improving the adsorption of Li. This phenomenon can also be found in the case of metallic heteroatom doping. The diffusion energy barriers of Li on doped monolayer MoS2 were explored with the diffusion pathways shown in Fig. 7c and e. Similar to the effects of defects, when Li diffuses towards the doped sites, the diffusion energy barriers decrease, especially near the doped sites (Fig. 7d and f). In contrast, the diffusion energy barriers increase when Li diffuses away from the doped sites, suggesting the strong adsorption ability of the doped sites. For diffusion pathways far from the doped sites, the diffusion energy barriers will not be affected by the doped heteroatoms. Similarly, the adsorption and diffusion of alkali metals on heteroatom-doped monolayer TiS2 were then explored by Tian et al. via theoretical calculations (Fig. 7g–j).178 For the adsorption properties of alkali metals, monolayer TiS2 doped with C, N, and P (corresponding to p-type doping) exhibits higher adsorption energies (Fig. 7g), which is the same as the results obtained by Sun et al.177 And the adsorption energies of alkali metals on TiS2 with and without doped heteroatoms follow the order of Li > K > Na, which is thought to be affected by both the atomic radius and electronegativity. With the diffusion pathway shown in Fig. 7h and i, the diffusion energy barriers of alkali metals on heteroatom doped monolayer TiS2 increase to varying degrees compared to that on pristine monolayer TiS2 (Fig. 7j). But all of them are less than 0.6 eV, which assures the moderate diffusion kinetics of alkali metals on heteroatom doped monolayer TiS2. The calculation results also show that the diffusion energy barriers follow the order of K < Na < Li, and this tendency has also been observed in other materials.179–181
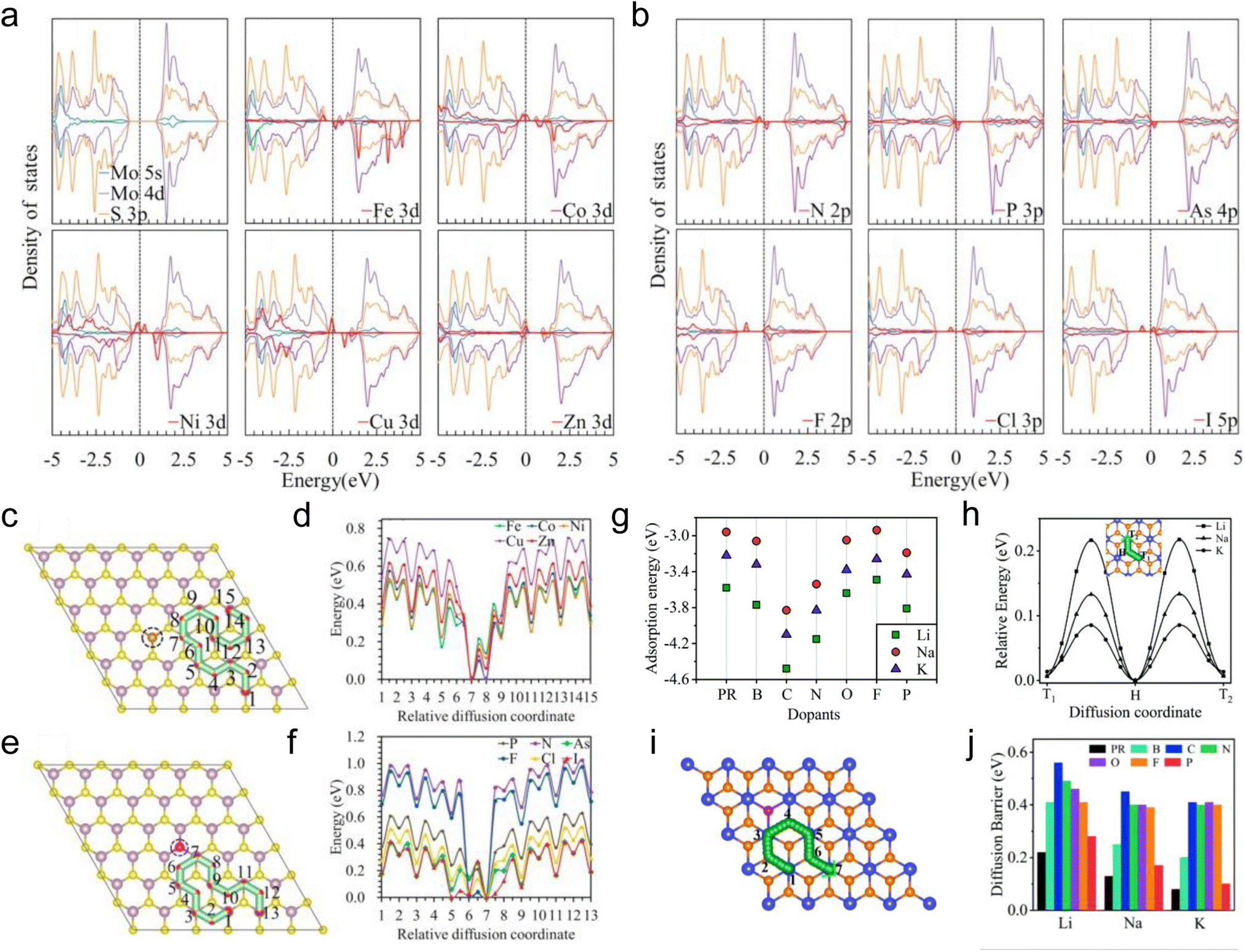 | ||
| Fig. 7 Density of states, dopant absorption energies, and diffusion pathways. (a) The projected density of states for pristine and monolayer MoS2 with substitution of Mo with Fe, Co, Ni, Cu, and Zn. (b) The projected density of states for monolayer MoS2 with substitution of S with N, P, As, F, Cl, and I in monolayer MoS2. Diffusion path and corresponding diffusion energy profiles for Li on (c and d) metal (e and f) nonmetal atom-doped monolayer MoS2. (g) The lowest adsorption energies for Li, Na, and K adsorbed on monolayer TiS2 without and with B, C, N, O, F, and P heteroatom doping. (h) Diffusion pathway and energy profiles for alkali metals on pristine monolayer TiS2. (i) Diffusion pathway of alkali metals on heteroatom doped monolayer TiS2. (j) The overall diffusion energy barriers for alkali metals on pristine and heteroatom-doped TiS2. Reproduced with permission from (a–f) ref. 177. Copyright 2018, American Chemical Society; (g–j) ref. 178. Copyright 2021, Royal Society of Chemistry. | ||
From the standpoint of theoretical calculations, it can be concluded that heteroatom doping enhances the conductivity and improves the adsorption capacity of TMDs for alkali metals. Although the diffusion energy barriers of alkali metals on TMDs increase, the moderate values can still assure moderate diffusion kinetics. For example, Qin et al. developed N-doped MoS2 nanosheets for Li+ storage. They found that the N-doped MoS2 nanosheets exhibit an improved conductivity of ca. 1.6 × 10−4 S m−1, while it is only ca. 1.1 × 10−5 S m−1 for pristine MoS2 nanosheets.182 Xie et al. prepared Co-doped MoS2 for Na+ storage.183 The Co-doping improves the conductivity of MoS2 and enhances the adsorption capacity of MoS2 for Na+. After being combined with N-doped graphene, Co-doped MoS2 exhibits higher specific capacity and better rate performance. The doping concentration is an important factor in maximizing the advantages of doping. Zhao et al. explored the optimal concentration by injecting different amounts of Mo into VS2,184 and 5% Mo-doping concentration exhibits the best electrochemical performance (Fig. 8a and b). Upon further increasing the doping concentrations, the electrochemical performance worsens and is almost the same as that of pristine VS2. This phenomenon is attributed to the distortion of atomic structures as a result of excessive doping.185 Apart from the single heteroatom doping, Zhang et al. explored NbS2 nanosheets with varying concentrations of M (M = Fe, Co, Ni) and Se codopants to store Li+ and Na+ (Fig. 8c).186 After optimizing the doping ratios, Fe0.3Nb0.7S1.6Se0.4 exhibits the best electrochemical performance. In addition to being able to manipulate the energy band structures of TMDs, the doping of heteroatoms can also lead to the transformation of Mo and W-based dichalcogenides from the semiconducting phase to the metallic phase, which can further enhance the conductivity. Wang et al. prepared N-doped MoS2 for Li storage.187 And N-doping induces the transformation of MoS2 from the 2H to the 1T phase (Fig. 8d), thus enhancing the conductivity of MoS2. He et al. developed a plasma-assisted method to introduce highly concentrated P-doping to trigger the phase transformation of MoSe2.109 The Se vacancies (VSe) induced by plasma treatment lead to the low crystallinity and structural instability of 2H-MoSe2, which decreases the energy barriers of phase transformation and initializes the phase transformation process. Then, the high concentration of P-doping induced by VSe prefers to donate electrons to Mo than Se, thereby triggering and stabilizing the formation of 1T phase MoSe2 with a high transition efficiency of 91%.
 | ||
| Fig. 8 Electrochemical performance and role of dopants. (a) Rate capability for bare VS2, 2.5% Mo–VS2, 5.0% Mo–VS2, and 7.5% Mo–VS2 cathodes at 0.1–1.0 A g−1. (b) Cycling performance for bare VS2, 2.5% Mo–VS2, 5.0% Mo–VS2, and 7.5% Mo–VS2 cathodes at 0.2 A g−1. (c) Mapping of Fe0.3Nb0.7S1.6Se0.4 nanosheets. (d) High-resolution TEM images of the synthetic composite and simulated image of (up) 1T and (down) 2H-MoS2 (S, blue; Mo, red). High-resolution TEM images of N-doped 1T-MoS2 (e), pure 1T-MoS2 (f), and pure 2H-MoS2 (g). Reproduced with permission from (a and b) ref. 184. Copyright 2023, Wiley-VCH; (c) ref. 186. Copyright 2017, American Chemical Society; (d) ref. 187. Copyright 2017, Elsevier; (e–g) ref. 188. Copyright 2021, American Chemical Society. | ||
For multivalent ion storage, Sheng et al. prepared N-doped 1T phase MoS2 for aqueous Zn2+ batteries.188 Although 1T-MoS2 exhibits a larger interlayer spacing than 2H-MoS2 (Fig. 8f and g), they found that N-doping could further increase the interlayer spacing of 1T-MoS2 (Fig. 8e), thus effectively decreasing the diffusion barriers of Zn2+ in MoS2. Li et al. introduced O-doping and defects into MoS2 for Zn2+ storage.52 O-doping increases the interlayer spacing and cooperates with structural defects to induce the transformation of MoS2 from the 2H phase to the 1T phase,51 resulting in improved Zn2+ diffusion kinetics, hydrophilicity, and conductivity of MoS2.
In short, heteroatom doping can enhance the conductivity, improve the metal adsorption ability, induce phase transformation, and expand the interlayer spacing of TMDs. Different types of doping exhibit different effects.
3.5 Alloy engineering
TMD alloys have been the subject of active research in energy conversion and storage. Unlike the broad selection of heterogeneous elements in doping engineering, the introduced and replaced elements are congeneric in alloy engineering. With a general formula of or
or 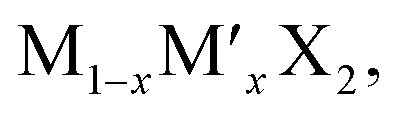 TMD alloys exhibit intriguing properties when the ratios of the components are changed.
TMD alloys exhibit intriguing properties when the ratios of the components are changed.
Among 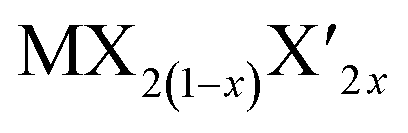 alloys, MoS2(1−x)Se2x is the representative one and has been widely studied. Ersan et al. studied the adsorption and diffusion of Li on MoS2(1−x)Se2x monolayers, where x varied from 0 to 1 (Fig. 9a and b).189 With the diffusion pathway shown in Fig. 9a, MoS2 and MoSe2 systems exhibit two symmetrical absolute energy maxima, an absolute energy minimum and a local energy minimum. The diffusion energy barriers of Li on MoS2 and MoSe2 are 0.194 and 0.237 eV. When x changes from 0.33 to 0.66, due to the introduction of congeneric elements, two asymmetrical energy maxima are observed, including an absolute maximum and a local energy maximum, and there are still one local and one absolute minimum in the case of x = 0.33 and 0.66. For x = 0.5, two local minima and one absolute minimum are observed. The diffusion energy barriers are 0.319, 0.541, and 0.391 eV for MoS2(1−x)Se2x alloys with x = 0.33, 0.5, and 0.66. From the calculations, it can be observed that Li prefers to bind with S. Although the diffusion energy barriers of these alloys increase, they are not very high values that are still acceptable for assuring moderate Li diffusion kinetics. The penetration barriers were also explored with varying x values (Fig. 9b). The results show that the penetration energy barriers decrease with increasing x from 0 to 1, indicating that the increasing concentration of Se is beneficial for the penetration of Li. However, a barrier energy of 1.283 eV is still large. One of the reasons may be attributed to the increased crystal constant with increasing Se concentration.
alloys, MoS2(1−x)Se2x is the representative one and has been widely studied. Ersan et al. studied the adsorption and diffusion of Li on MoS2(1−x)Se2x monolayers, where x varied from 0 to 1 (Fig. 9a and b).189 With the diffusion pathway shown in Fig. 9a, MoS2 and MoSe2 systems exhibit two symmetrical absolute energy maxima, an absolute energy minimum and a local energy minimum. The diffusion energy barriers of Li on MoS2 and MoSe2 are 0.194 and 0.237 eV. When x changes from 0.33 to 0.66, due to the introduction of congeneric elements, two asymmetrical energy maxima are observed, including an absolute maximum and a local energy maximum, and there are still one local and one absolute minimum in the case of x = 0.33 and 0.66. For x = 0.5, two local minima and one absolute minimum are observed. The diffusion energy barriers are 0.319, 0.541, and 0.391 eV for MoS2(1−x)Se2x alloys with x = 0.33, 0.5, and 0.66. From the calculations, it can be observed that Li prefers to bind with S. Although the diffusion energy barriers of these alloys increase, they are not very high values that are still acceptable for assuring moderate Li diffusion kinetics. The penetration barriers were also explored with varying x values (Fig. 9b). The results show that the penetration energy barriers decrease with increasing x from 0 to 1, indicating that the increasing concentration of Se is beneficial for the penetration of Li. However, a barrier energy of 1.283 eV is still large. One of the reasons may be attributed to the increased crystal constant with increasing Se concentration.
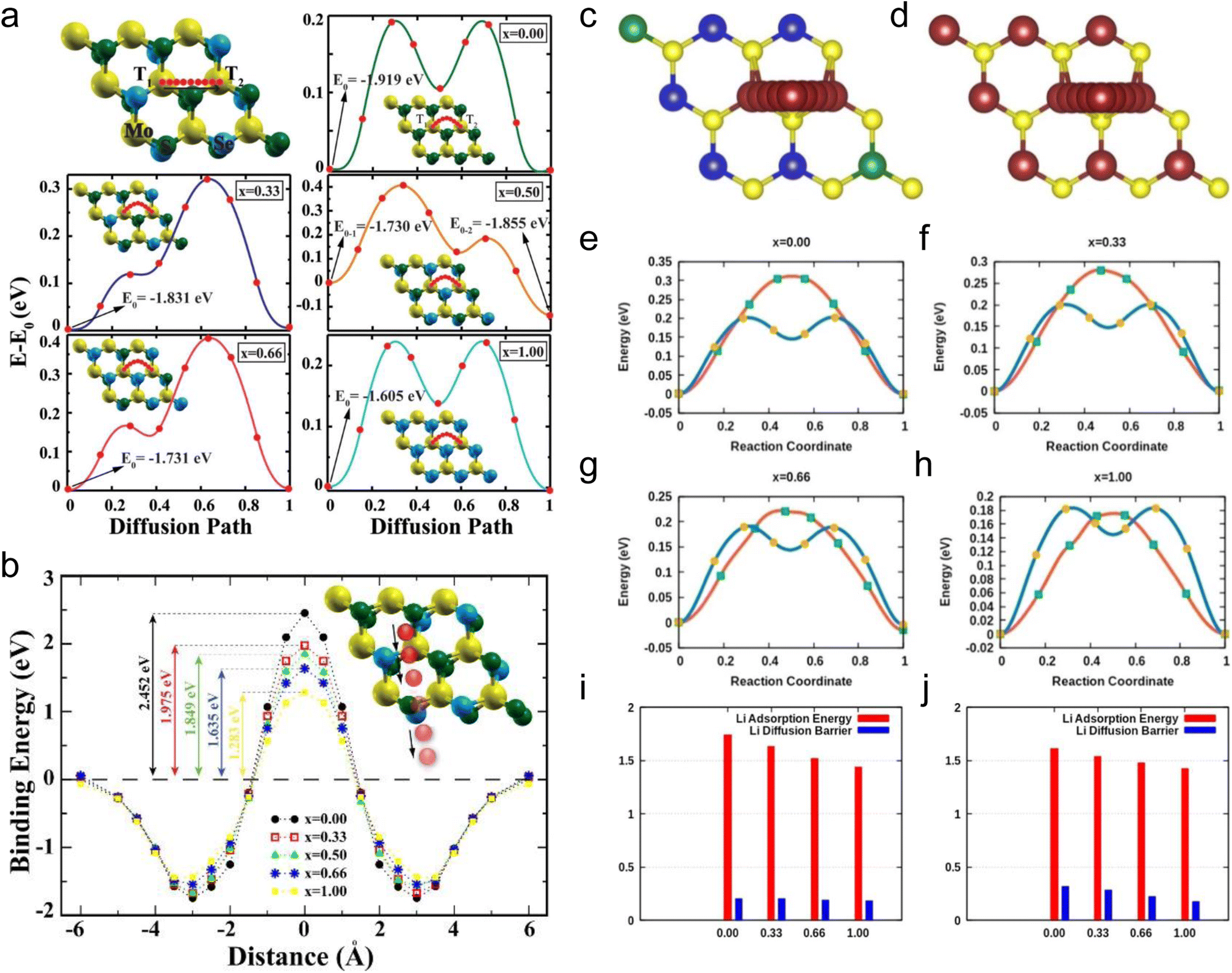 | ||
| Fig. 9 Diffusion path and binding energies of Li in monolayer TMDs. (a) Lowest energy diffusion paths of a single lithium atom and calculated energy profiles along the paths for one Li atom-adsorbed on the TMD monolayer. (b) Binding energies of a single lithium atom when it approaches and penetrates bare MoS2(1−x)Se2x monolayers from infinity. Schematic representation of the Li diffusion path in the Mo1−xWxS2 alloy of x = 0.33 from one top position to another through the hollow site. (c) Bare surface and (d) lithiated surface. The blue, green, yellow, and maroon balls denote Mo, W, S, and Li atoms, respectively. (e–h) Energy profiles for lithium diffusion on monolayer Mo1−xWxS2. The blue line indicates a bare surface, and the red line indicates a lithiated surface. Li adsorption energies and diffusion barriers on monolayer Mo1−xWxS2 compared to different compositions of x. (i) Migration of Li on the bare surface and (j) migration of Li on the lithiated surface. Reproduced with permission from (a and b) ref. 189. Copyright 2015, American Chemical Society; (c–j) ref. 190. Copyright 2018, American Chemical Society. | ||
Among  the Mo1−xWxS2 alloys have received much attention. Barik et al. explored monolayer Mo1−xWxS2 as anode materials for Li storage via theoretical calculations (Fig. 9c–j).190 For pristine monolayer Mo1−xWxS2, the DOS and the band gaps do not change much with different ratios of Mo and W. After lithiation, the conduction band minima of monolayer Mo1−xWxS2 move towards the Fermi level, suggesting the formation of the metallic phase, which is consistent with the fact that the intercalation of alkali metal ions into Mo and W-based dichalcogenides can induce the formation of the metallic phase.62,108 With the diffusion pathway shown in Fig. 9c and d, they studied the diffusion energy barriers of Li on pristine and lithiated Mo1−xWxS2 (Fig. 9e–h).190 The pristine MoS2 and WS2 systems exhibit two symmetrical absolute maxima. However, the two maxima are not symmetrical in the case of alloys. The maximum at the TMo site is always slightly higher than that at the TW site, indicating the stronger bonding interaction between Mo and Li. The lithiated Mo1−xWxS2 systems exhibit similar characteristics to pristine Mo1−xWxS2 systems except for the disappearance of the local energy minima due to the repulsion between Li atoms. The Li adsorption and its diffusion energy barriers in pristine and lithiated Mo1−xWxS2 indicate that they all decrease with increasing W ratios (Fig. 9i and j).
the Mo1−xWxS2 alloys have received much attention. Barik et al. explored monolayer Mo1−xWxS2 as anode materials for Li storage via theoretical calculations (Fig. 9c–j).190 For pristine monolayer Mo1−xWxS2, the DOS and the band gaps do not change much with different ratios of Mo and W. After lithiation, the conduction band minima of monolayer Mo1−xWxS2 move towards the Fermi level, suggesting the formation of the metallic phase, which is consistent with the fact that the intercalation of alkali metal ions into Mo and W-based dichalcogenides can induce the formation of the metallic phase.62,108 With the diffusion pathway shown in Fig. 9c and d, they studied the diffusion energy barriers of Li on pristine and lithiated Mo1−xWxS2 (Fig. 9e–h).190 The pristine MoS2 and WS2 systems exhibit two symmetrical absolute maxima. However, the two maxima are not symmetrical in the case of alloys. The maximum at the TMo site is always slightly higher than that at the TW site, indicating the stronger bonding interaction between Mo and Li. The lithiated Mo1−xWxS2 systems exhibit similar characteristics to pristine Mo1−xWxS2 systems except for the disappearance of the local energy minima due to the repulsion between Li atoms. The Li adsorption and its diffusion energy barriers in pristine and lithiated Mo1−xWxS2 indicate that they all decrease with increasing W ratios (Fig. 9i and j).
Different from the studies performed by Ersan and Barik et al.,189,190 Chaney and coworkers performed a comprehensive study of the adsorption and diffusion of Li on Janus Mo/WXY (X, Y = S, Se, Te), where the ratio of X and Y was fixed at 1:1.191 According to the adsorption energy results of both regular and Janus TMDs (Fig. 10a and b), it can be concluded that the binding interaction between chalcogen and Li follows the order of S > Se > Te, and for Mo and W, the binding interaction follows the order of Mo > W. The diffusion of Li on regular and Janus TMDs indicates that the diffusion barriers of Li on the X side of Janus MXYs are comparable to that on MX2 (Fig. 10c and d), while the diffusion barriers of Li on the Y side of Janus MXYs are always lower than that on the X side due to the weaker interaction between Li and Y.
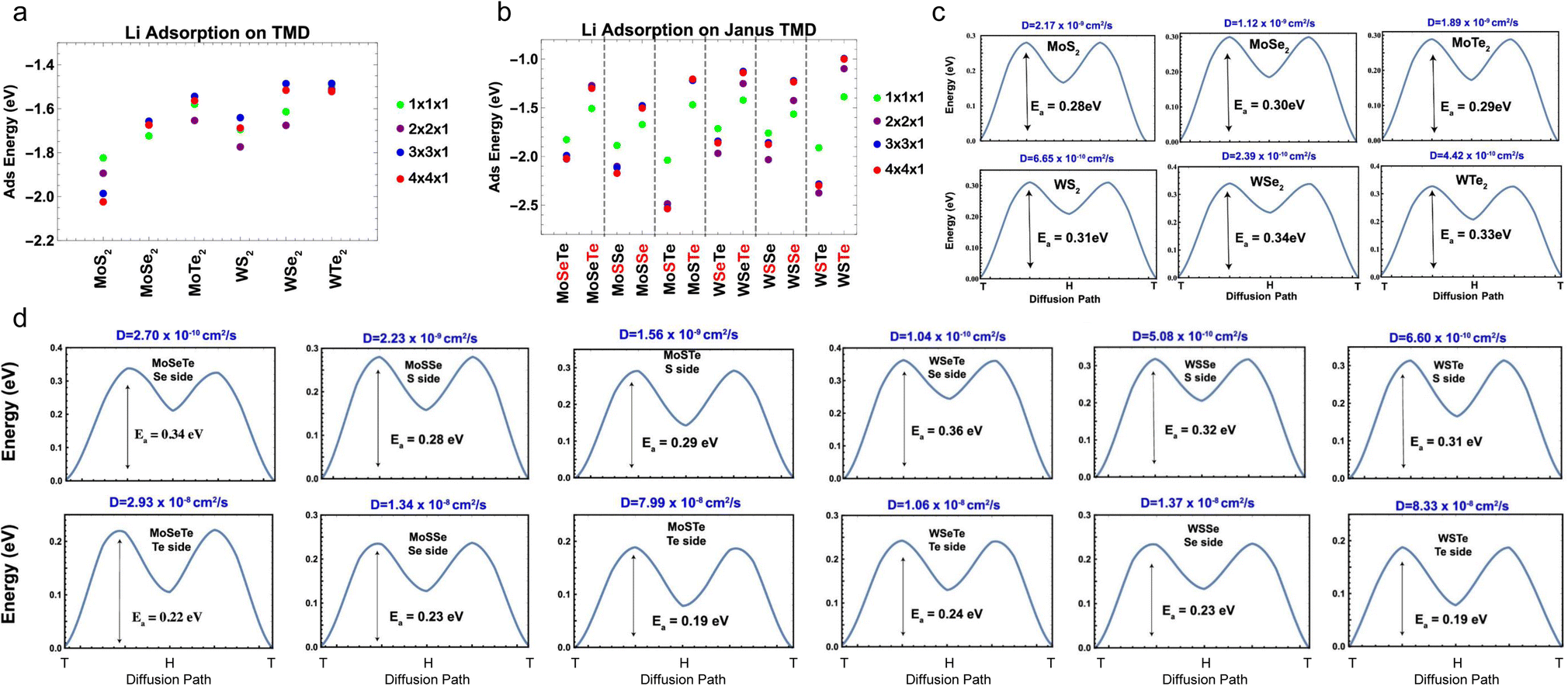 | ||
| Fig. 10 Adsorption energies for a Li atom over metal-top sites of all supercell sizes. (a) Mo- and W-based TMDs and (b) top and bottom sides of Janus structures. The red color on the chalcogen atom of Janus structures on the x-axis labeling indicates the side on which the Li atom is adsorbed. (c) Activation energy barriers for Li atom diffusion for regular TMD-MX2 (M = Mo, W; X = S, Se, Te), calculated with the nudged elastic band (NEB) method simulations. (d) Activation energy barriers for Li atom diffusion for Janus TMD MXY (M = Mo, W; X/Y = S, Se, Te), calculated with NEB simulations. Reproduced with permission from (a–d) ref. 191. Copyright 2021, American Chemical Society. | ||
According to the calculations, alloying is an effective strategy to balance the adsorption and diffusion of ions on TMDs. This also applies to some other properties for 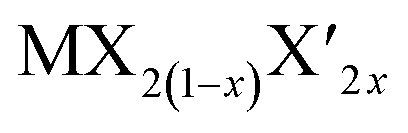 under similar conditions of the same ion storage capacity. The alloys will balance the theoretical specific capacity of TMD electrodes. For example, the relative atomic mass of S is lower than that of Se. Therefore, increasing the content of S in MSe2(1−x)S2x will reduce the relative molecular mass of MSe2(1−x)S2x. However, the ion storage capacity of MSe2(1−x)S2x will not change with the variation of the ratios of Se and S due to the same ion storage mechanism in MoS2 and MoSe2. As a result, the theoretical specific capacity of MSe2(1−x)S2x will increase with increasing the content of S.192,193 Although increasing the Se content would reduce the theoretical specific capacity of MSe2(1−x)S2x, Se has a larger atomic radius than S, which can enlarge the interlayer spacing of MSe2(1−x)S2x, resulting in reduced ion diffusion barriers (Fig. 11a and b).54,194,195 In addition to the increased specific capacity and the enlarged interlayer spacing, the incorporation of S or Se can also induce the structural rearrangement and the electron redistribution of MSe2(1−x)S2x, leading to the formation of abundant anion vacancies and the metallic phase, which can increase the active adsorption sites and enhance the conductivity of MSe2(1−x)S2x.56,196–198 He et al. fabricated MoS2(1−x)Se2x alloys through partial substitution of S for Se in MoSe2. They found that the alloying process can generate anion vacancies, and the vacancy concentration can be adjusted by tuning the ratios of S and Se.55 With a S:Se ratio close to 1:1, the largest vacancy concentration and the best electrochemical performance are achieved (Fig. 11c–e). Huang et al. synthesized MoSSe@rGo for Na+ storage,198 and in their Raman spectra, the 1T-MoSe2 and 1T-MoS2 modes were observed in both MoSSe and MoSSe@rGO, indicating the formation of a metallic phase by the alloying process (Fig. 11f). For
under similar conditions of the same ion storage capacity. The alloys will balance the theoretical specific capacity of TMD electrodes. For example, the relative atomic mass of S is lower than that of Se. Therefore, increasing the content of S in MSe2(1−x)S2x will reduce the relative molecular mass of MSe2(1−x)S2x. However, the ion storage capacity of MSe2(1−x)S2x will not change with the variation of the ratios of Se and S due to the same ion storage mechanism in MoS2 and MoSe2. As a result, the theoretical specific capacity of MSe2(1−x)S2x will increase with increasing the content of S.192,193 Although increasing the Se content would reduce the theoretical specific capacity of MSe2(1−x)S2x, Se has a larger atomic radius than S, which can enlarge the interlayer spacing of MSe2(1−x)S2x, resulting in reduced ion diffusion barriers (Fig. 11a and b).54,194,195 In addition to the increased specific capacity and the enlarged interlayer spacing, the incorporation of S or Se can also induce the structural rearrangement and the electron redistribution of MSe2(1−x)S2x, leading to the formation of abundant anion vacancies and the metallic phase, which can increase the active adsorption sites and enhance the conductivity of MSe2(1−x)S2x.56,196–198 He et al. fabricated MoS2(1−x)Se2x alloys through partial substitution of S for Se in MoSe2. They found that the alloying process can generate anion vacancies, and the vacancy concentration can be adjusted by tuning the ratios of S and Se.55 With a S:Se ratio close to 1:1, the largest vacancy concentration and the best electrochemical performance are achieved (Fig. 11c–e). Huang et al. synthesized MoSSe@rGo for Na+ storage,198 and in their Raman spectra, the 1T-MoSe2 and 1T-MoS2 modes were observed in both MoSSe and MoSSe@rGO, indicating the formation of a metallic phase by the alloying process (Fig. 11f). For 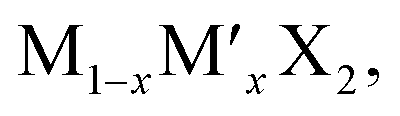 a similar phenomenon was also observed.199,200 Besides, there is an additional possibility that introducing the M elements from metallic phase MX2 into semiconducting phase M′X2 may endow
a similar phenomenon was also observed.199,200 Besides, there is an additional possibility that introducing the M elements from metallic phase MX2 into semiconducting phase M′X2 may endow  with interesting properties. Our group explored this possibility by introducing V into MoS2 to form Mo1−xVxS2 for Li storage.201 The introduction of V into MoS2 makes Mo1−xVxS2 exhibit semi-metallic conductivity, making Mo1−xVxS2 exhibit enhanced conductivity and better electrochemical performance than pristine 2H-MoS2.
with interesting properties. Our group explored this possibility by introducing V into MoS2 to form Mo1−xVxS2 for Li storage.201 The introduction of V into MoS2 makes Mo1−xVxS2 exhibit semi-metallic conductivity, making Mo1−xVxS2 exhibit enhanced conductivity and better electrochemical performance than pristine 2H-MoS2.
 | ||
| Fig. 11 Structural, electrochemical, and Raman properties of MoS2(1−x)Se2x alloys. (a) XRD patterns of MoSSeNSs@NC/hC-NC, MoSSe@C and MoS2@C. (b) HRTEM image of MoSSeNSs@NC/hC-NC. (c) EPR results of MoSe2, MoSSe, and MoS2. (d) Cycling performance at 100 mA g−1 for all the as-prepared samples. (e) Rate capability of MoSe2, MoSSe, and MoS2. (f) Raman spectra of as-prepared MoSSe@rGO, MoSSe, MoS2, and MoSe2 samples. MoSSeNSs@NC/hC-NC: MoSSe nanosheets supported on hollow cubic N-doped carbon. Reproduced with permission from (a and b) ref. 195. Copyright 2021, Wiley-VCH; (c–e) ref. 55. Copyright 2019, American Chemical Society; (f) ref. 198. Copyright 2020, Wiley-VCH. | ||
As discussed above, the different elements in 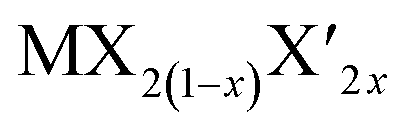 or
or 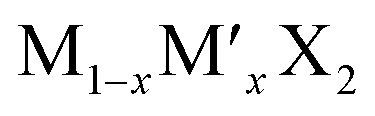 alloys have different influences. Therefore, it is important to manipulate the x values to optimize the electrochemical performance of TMD alloys. However, the optimal x values reported by different studies vary. For example, He and Yu et al. found that the electrochemical performance of MoS2(1−x)Se2x becomes the best when x = 0.5.55,197 However, Cai and coworkers believed that x = 0.25 is the optimal value for the MoS2(1−x)Se2x alloys.202 For Mo1−xWxS2, a similar issue also exists.53,203 The optimal values of x may be due to the other factors that can also affect the electrochemical performance of TMD alloys, such as the morphology and the types of stored ions, which need comprehensive and detailed studies.
alloys have different influences. Therefore, it is important to manipulate the x values to optimize the electrochemical performance of TMD alloys. However, the optimal x values reported by different studies vary. For example, He and Yu et al. found that the electrochemical performance of MoS2(1−x)Se2x becomes the best when x = 0.5.55,197 However, Cai and coworkers believed that x = 0.25 is the optimal value for the MoS2(1−x)Se2x alloys.202 For Mo1−xWxS2, a similar issue also exists.53,203 The optimal values of x may be due to the other factors that can also affect the electrochemical performance of TMD alloys, such as the morphology and the types of stored ions, which need comprehensive and detailed studies.
In short, alloy engineering is expected to balance different properties of TMDs, such as the metallic phase formation and the interlayer spacing enlargement. Defect introduction can also be achieved by alloying; however, the issue that needs to be clarified is determining the optimal x value and its influencing factors in TMD alloy systems.
3.6 Bond modulation
Different from the several mainstream molecular modulation strategies discussed above, the bond modulation strategy focuses on the impact of chemical bonds on the properties of TMDs, including bond length, bond angle, bond energy, and additionally created chemical bonds.170,204 For example, by manipulating the bond length, the interlayer spacing can be modulated,164,205 and the enhanced bond energy can stabilize the intercalation reaction, although it will make it more difficult for the conversion reaction to occur.168,169 The additional chemical bond formation in TMDs shows more possibilities in comparison and has therefore been studied more extensively. For instance, covalent interactions usually exist between the introduced guests and the transition metals or chalcogens in TMDs so they can be stably bonded at room temperature and modulate the TMDs' properties.145,154 Our group has demonstrated that the introduced Se atoms between the interlayers of MoSe2 form covalent Se–Se bonds with the Se in MoSe2,58 and the covalent Se–Se bonds not only enlarge the interlayer spacing of MoSe2 but also prevent the Se shuttle effects. The S–Co–S covalent bonds formed by intercalating Co into MoS2 interlayers can induce the formation of the 1T phase, expand the interlayer spacing of MoS2, and serve as electrical pathways to accelerate interlayer charge transfer, thus greatly improving the electrochemical performance of MoS2.141 In addition, introducing additional chemical bonds by incorporating additional atoms into TMDs can also lead to interesting phenomena. For example, Zhang et al. prepared unusual red MoSe2 nanosheets by introducing an oxygen gradient on the surface of MoSe2 nanosheets and forming Mo–O bonds.57 The formation of Mo–O bonds induces the formation of high valence Mo, thereby regulating the band gap of red MoSe2 and enhancing its electrical conductivity. A similar enhanced electrical conductivity was also observed when incorporating P and N atoms into TMDs due to the formation of M–P and M–N bonds.109,182,206In short, the bond modulation strategy modulates the properties of TMDs from a chemical bonding viewpoint. The bond length, bond angle, bond energy, and additional chemical bonding all impact the electrochemical performance of TMDs. However, they have not received widespread attention yet, and some systematic studies are lacking, thus needing further exploration in the future.
4. Summary and perspectives
In summary, this review outlines the basic properties of TMDs and discusses the molecular modulation strategies of TMDs by combining computational and experimental studies towards their application in metal ion batteries. In brief, due to the different coordination and stacking sequences of transition metals, TMDs show polymorphs and stacking polytypes. Among them, 1T and 2H phase TMDs are widely studied in metal ion batteries. The coordination environment of transition metals and the filling states of their d orbitals determine the electrical properties of TMDs. When the orbitals are fully occupied, semiconducting behaviour is exhibited, such as in 2H-MoS2 and 2H-MoSe2. When the orbitals are partially filled, metallic conductivity is exhibited, such as in 2H-NbS2 and 1T-VS2. In addition, the chalcogen atoms can also affect the electronic properties of TMDs, although their influence is not significant. When TMDs are used as alkali metal-ion battery electrodes, reversible intercalation and extraction reactions occur at a relatively high cutoff voltage (∼1 V). As the intercalation process continues, the subsequent reaction mechanism depends on the M–X bond strength of TMDs. If the M–X bonding interactions are stronger than the A–MX2 interactions, reversible intercalation and extraction reactions, such as in TiX2 and NbX2, will continue. In contrast, conversion reactions will occur if the M–X bonding interactions are weaker than the A–MX2 interactions, and the reversibility of the conversion reactions depends on the dynamic properties of TMDs. Excellent dynamic properties of TMDs contribute to reversible conversion reactions, such as in VX2, while poor dynamic properties of TMDs lead to irreversible conversion reactions, such as in MoX2, WX2, and ReX2. Only intercalation and extraction reactions have been reported when TMD electrodes are used in multivalent-ion batteries.Several molecular modulation strategies of TMDs, including phase engineering, defect engineering, interlayer spacing expansion, heteroatom doping, alloy engineering, and bond modulation, are also discussed. By combining theoretical and experimental studies, the basic mechanisms of different molecular modulation strategies and their specific effects on the properties of TMDs for storing metal ions are summarized, (1) phase engineering aims at improving the intrinsic electrical conductivity of semiconducting TMDs; (2) defect engineering can provide more active adsorption sites and decrease the length of the ion diffusion pathway; (3) the interlayer spacing expansion strategy is beneficial for reducing the diffusion energy barriers for metal ions in TMDs; (4) heteroatom doping is expected to improve the electrical conductivity and the atom adsorption capacity of TMDs; (5) alloy engineering is used to obtain comprehensive properties of TMDs; (6) bond modulation can enhance the electrical conductivity and the ion diffusion dynamics of TMDs. In addition to the main effects mentioned above, some extra advantages of molecular modulation strategies toward specific applications are also summarized (Fig. 12).
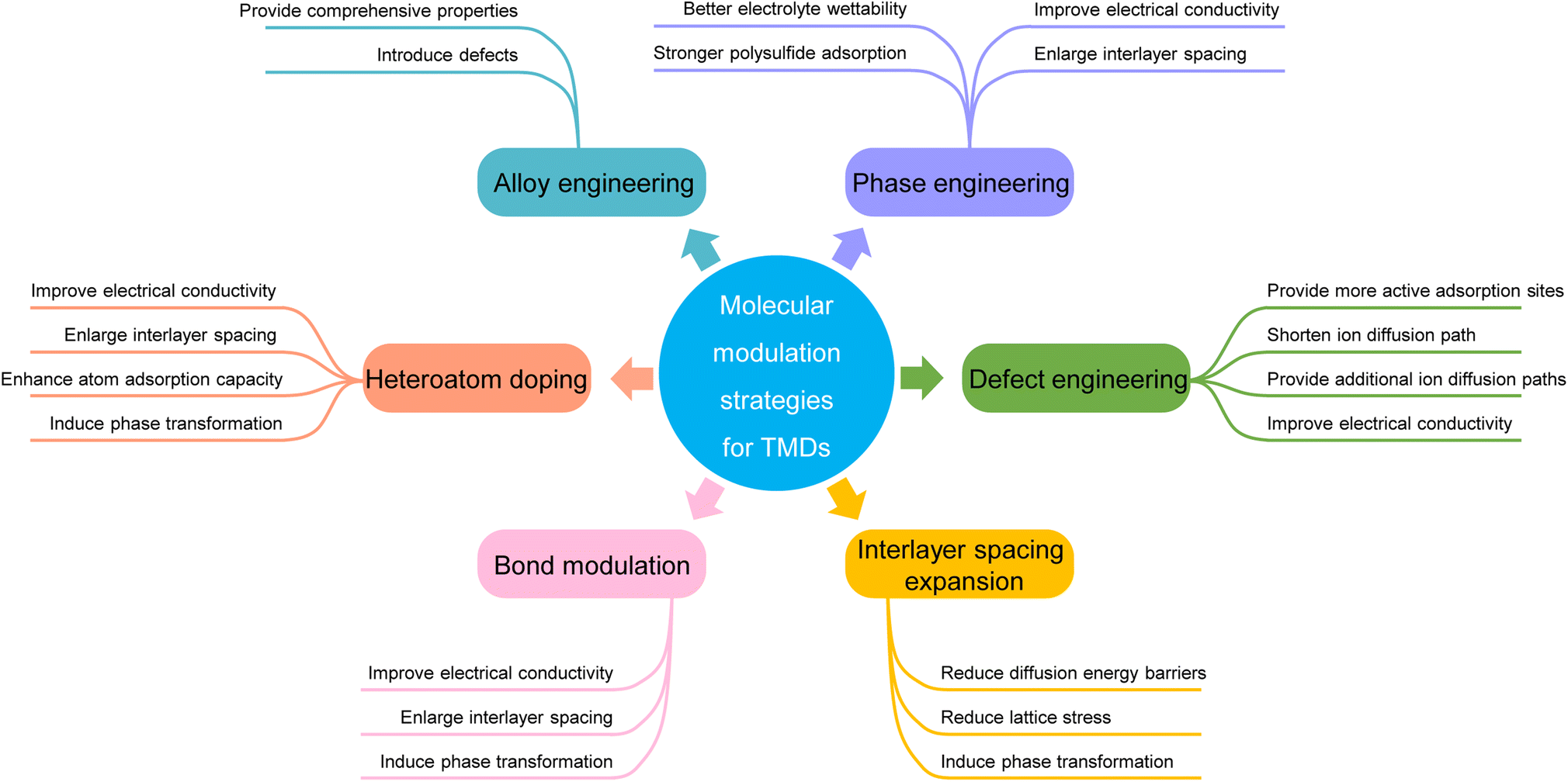 | ||
| Fig. 12 A schematic summarizing the influences of different molecular modulation strategies on the properties of TMDs. | ||
Although many advances have been achieved in developing TMD electrodes, much effort is still needed to further promote the development of TMDs as electrode materials from a molecular modulation viewpoint. (1) The conversion reaction provides high discharge capacity for some TMD electrodes, while the reversibility of the conversion reaction is not satisfactory. Although it has been reported that the dynamic properties of TMDs influence the reversibility of the conversion reaction, VX2 compounds with excellent dynamic properties only exhibit a partially reversible conversion reaction. Therefore, extensive theoretical and experimental research on the dynamic properties of the conversion reaction at the molecular and atomic level is needed to make it fully reversible, which is crucial for employing TMDs in metal-ion batteries. (2) The precise modulation methods for TMD molecules are necessary to be developed, which only exist in the theoretical models currently. By manipulating TMD molecules precisely, the specific mechanisms and effects of various molecular modulation strategies can be better understood. In addition, the properties of TMDs can be flexibly adjusted to meet different needs, which is beneficial for providing more potential opportunities and leveraging the maximum advantages of TMDs. (3) Deeper theoretical calculations need to be performed to help understand the reaction mechanisms of TMDs and direct the experimental designs. Currently, most theoretical calculations focus on the level of single-layer or double-layer TMDs, which is very different from the actual situation. Therefore, many conclusions drawn from calculations are inconsistent with the experimental data, and the real mechanisms cannot be verified. (4) In the future, various in situ characterization techniques which can reveal the basic reaction mechanisms of TMDs before and after molecular modulation need to be developed. Despite significant progress in research on TMDs in recent years, there are still a large number of problems which need to be solved. By developing advanced in situ characterization techniques, many problems can be understood and solved from the source, which is helpful for developing practice and high-performance TMD electrodes with balanced and optimized properties.
Author contributions
B. L. and J. Z. designed the scope of the paper. M. G. surveyed the relevant literature, and all authors wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. L. acknowledges financial support from the National Natural Science Foundation of China no. U20A20247 and 51922038. M. G. acknowledges financial support from the Postgraduate Scientific Research Innovation Project of Hunan Province no. CX20230407. A. M. R. acknowledges financial support through Clemson University's R. A. Bowen Fellowship funds.References
- M. Armand and J. M. Tarascon, Building better batteries, Nature, 2008, 451, 652–657 CrossRef CAS.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Rechargeable Batteries for Grid Scale Energy Storage, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS PubMed.
- K. Chen, X. Shen, L. Luo, H. Chen, R. Cao, X. Feng, W. Chen, Y. Fang and Y. Cao, Correlating the Solvating Power of Solvents with the Strength of Ion-Dipole Interaction in Electrolytes of Lithium-ion Batteries, Angew. Chem., Int. Ed., 2023, 62, e202312373 CrossRef CAS PubMed.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Ether-based electrolytes for sodium ion batteries, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Fundamentals, status and promise of sodium-based batteries, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, From Li-Ion Batteries toward Na-Ion Chemistries: Challenges and Opportunities, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- Y. Feng, Y. Lv, H. Fu, M. Parekh, A. M. Rao, H. Wang, X. Tai, X. Yi, Y. Lin, J. Zhou and B. Lu, Co-activation for enhanced K-ion storage in battery anodes, Natl. Sci. Rev., 2023, 10, nwad118 CrossRef PubMed.
- M. Gu, A. M. Rao, J. Zhou and B. Lu, In situ formed uniform and elastic SEI for high-performance batteries, Energy Environ. Sci., 2023, 16, 1166–1175 RSC.
- H. Ding, J. Wang, J. Zhou, C. Wang and B. Lu, Building electrode skins for ultra-stable potassium metal batteries, Nat. Commun., 2023, 14, 2305 CrossRef CAS PubMed.
- Y. Liu, Y. Shi, C. Gao, Z. Shi, H. Ding, Y. Feng, Y. He, J. Sha, J. Zhou and B. Lu, Low-Temperature Potassium Batteries Enabled by Electric and Thermal Field Regulation, Angew. Chem., Int. Ed., 2023, 62, e202300016 CrossRef CAS PubMed.
- G. Fang, J. Zhou, A. Pan and S. Liang, Recent Advances in Aqueous Zinc-Ion Batteries, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, Electrolyte Strategies toward Better Zinc-Ion Batteries, ACS Energy Lett., 2021, 6, 1015–1033 CrossRef CAS.
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Materials chemistry for rechargeable zinc-ion batteries, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- B. Chen, D. Chao, E. Liu, M. Jaroniec, N. Zhao and S.-Z. Qiao, Transition metal dichalcogenides for alkali metal ion batteries: engineering strategies at the atomic level, Energy Environ. Sci., 2020, 13, 1096–1131 RSC.
- H. Shuai, R. Liu, W. Li, X. Yang, H. Lu, Y. Gao, J. Xu and K. Huang, Recent Advances of Transition Metal Sulfides/Selenides Cathodes for Aqueous Zinc-Ion Batteries, Adv. Energy Mater., 2023, 13, 2202992 CrossRef CAS.
- L. Wang, Q. Zhang, J. Zhu, X. Duan, Z. Xu, Y. Liu, H. Yang and B. Lu, Nature of extra capacity in MoS2 electrodes: molybdenum atoms accommodate with lithium, Energy Storage Mater., 2019, 16, 37–45 CrossRef.
- M. S. Whittingham, Electrical Energy Storage and Intercalation Chemistry, Science, 1976, 192, 1126–1127 CrossRef CAS PubMed.
- T. Wang, S. Chen, H. Pang, H. Xue and Y. Yu, MoS2-Based Nanocomposites for Electrochemical Energy Storage, Adv. Sci., 2017, 4, 1600289 CrossRef.
- X. Ren, Q. Zhao, W. D. McCulloch and Y. Wu, MoS2 as a long-life host material for potassium ion intercalation, Nano Res., 2017, 10, 1313–1321 CrossRef CAS.
- W. S. V. Lee, T. Xiong, X. Wang and J. Xue, Unraveling MoS2 and Transition Metal Dichalcogenides as Functional Zinc-Ion Battery Cathode: A Perspective, Small Methods, 2021, 5, 2000815 CrossRef CAS.
- Y. Li, M. Wang and J. Sun, Molecular Engineering Strategies toward Molybdenum Diselenide Design for Energy Storage and Conversion, Adv. Energy Mater., 2022, 12, 2202600 CrossRef CAS.
- Q. Yun, L. Li, Z. Hu, Q. Lu, B. Chen and H. Zhang, Layered Transition Metal Dichalcogenide-Based Nanomaterials for Electrochemical Energy Storage, Adv. Mater., 2020, 32, 1903826 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Hierarchical MoS2 microboxes constructed by nanosheets with enhanced electrochemical properties for lithium storage and water splitting, Energy Environ. Sci., 2014, 7, 3302–3306 RSC.
- Y. Wang, W. Kang, X. Pu, Y. Liang, B. Xu, X. Lu, D. Sun and Y. Cao, Template-directed synthesis of Co2P/MoSe2 in a N-doped carbon hollow structure for efficient and stable sodium/potassium ion storage, Nano Energy, 2022, 93, 106897 CrossRef CAS.
- B. Jia, Q. Yu, Y. Zhao, M. Qin, W. Wang, Z. Liu, C.-Y. Lao, Y. Liu, H. Wu, Z. Zhang and X. Qu, Bamboo-Like Hollow Tubes with MoS2/N-Doped-C Interfaces Boost Potassium-Ion Storage, Adv. Funct. Mater., 2018, 28, 1803409 CrossRef.
- S. Wang, Y. Si, P. Wan, S. Zhu, W. Chu and Z. Yu, MoSe2 nanoflowers grown on 3D carbon network as an advanced anode for lithium ion batteries, Mater. Lett., 2022, 310, 131487 CrossRef CAS.
- M. Hong, J. Li, W. Zhang, S. Liu and H. Chang, Semimetallic 1T′ WTe2 Nanorods as Anode Material for the Sodium Ion Battery, Energy Fuels, 2018, 32, 6371–6377 CrossRef CAS.
- J. Ge, L. Fan, J. Wang, Q. Zhang, Z. Liu, E. Zhang, Q. Liu, X. Yu and B. Lu, MoSe2/N-Doped Carbon as Anodes for Potassium-Ion Batteries, Adv. Energy Mater., 2018, 8, 1801477 CrossRef.
- D. Sun, D. Ye, P. Liu, Y. Tang, J. Guo, L. Wang and H. Wang, MoS2/Graphene Nanosheets from Commercial Bulky MoS2 and Graphite as Anode Materials for High Rate Sodium-Ion Batteries, Adv. Energy Mater., 2018, 8, 1702383 CrossRef.
- S. H. Choi, Y. N. Ko, J.-K. Lee and Y. C. Kang, 3D MoS2–Graphene Microspheres Consisting of Multiple Nanospheres with Superior Sodium Ion Storage Properties, Adv. Funct. Mater., 2015, 25, 1780–1788 CrossRef CAS.
- J. Bai, B. Zhao, J. Zhou, J. Si, Z. Fang, K. Li, H. Ma, J. Dai, X. Zhu and Y. Sun, Glucose-Induced Synthesis of 1T-MoS2/C Hybrid for High-Rate Lithium-Ion Batteries, Small, 2019, 15, 1805420 CrossRef PubMed.
- Y. Zhou, Y. Liu, M. Zhang, Q. Han, Y. Wang, X. Sun, X. Zhang, C. Dong, J. Sun, Z. Tang and F. Jiang, Rationally designed hierarchical N, P co-doped carbon connected 1T/2H-MoS2 heterostructures with cooperative effect as ultrafast and durable anode materials for efficient sodium storage, Chem. Eng. J., 2022, 433, 133778 CrossRef CAS.
- W. Ding, L. Hu, J. Dai, X. Tang, R. Wei, Z. Sheng, C. Liang, D. Shao, W. Song, Q. Liu, M. Chen, X. Zhu, S. Chou, X. Zhu, Q. Chen, Y. Sun and S. X. Dou, Highly Ambient-Stable 1T-MoS2 and 1T-WS2 by Hydrothermal Synthesis under High Magnetic Fields, ACS Nano, 2019, 13, 1694–1702 CAS.
- D. Sun, D. Huang, H. Wang, G.-L. Xu, X. Zhang, R. Zhang, Y. Tang, D. Abd Ei-Hady, W. Alshitari, A. Saad Al-Bogami, K. Amine and M. Shao, 1T MoS2 nanosheets with extraordinary sodium storage properties via thermal-driven ion intercalation assisted exfoliation of bulky MoS2, Nano Energy, 2019, 61, 361–369 CrossRef CAS.
- H. He, X. Li, D. Huang, J. Luan, S. Liu, W. K. Pang, D. Sun, Y. Tang, W. Zhou, L. He, C. Zhang, H. Wang and Z. Guo, Electron-Injection-Engineering Induced Phase Transition toward Stabilized 1T-MoS2 with Extraordinary Sodium Storage Performance, ACS Nano, 2021, 15, 8896–8906 CrossRef CAS.
- Y. Li, X. Dong, Z. Xu, M. Wang, R. Wang, J. Xie, Y. Ding, P. Su, C. Jiang, X. Zhang, L. Wei, J.-F. Li, Z. Chu, J. Sun and C. Huang, Piezoelectric 1T Phase MoSe2 Nanoflowers and Crystallographically Textured Electrodes for Enhanced Low-Temperature Zinc-Ion Storage, Adv. Mater., 2023, 35, 2208615 CrossRef CAS.
- H. Fan, P. Mao, G. Lan, C. Liu, J. Chen, Z. Wang, Z. Li, R. Zheng, Y. Liu and H. Sun, Ultrathin Metallic-Phase Molybdenum Disulfide Nanosheets Stabilized on Functionalized Carbon Nanotubes Via Covalent Interface Interaction for Sodium- and Lithium-Ion Storage, ACS Appl. Energy Mater., 2021, 4, 9440–9449 CrossRef CAS.
- B. Chen, H. Lu, N. Zhao, C. Shi, E. Liu, C. He and L. Ma, Facile synthesis and electrochemical properties of continuous porous spheres assembled from defect-rich, interlayer-expanded, and few-layered MoS2/C nanosheets for reversible lithium storage, J. Power Sources, 2018, 387, 16–23 CrossRef CAS.
- Z. Lei, J. Zheng, X. He, Y. Wang, X. Yang, F. Xiao, H. Xue, P. Xiong, M. Wei, Q. Chen, Q. Qian and L. Zeng, Defect-rich WS2–SPAN nanofibers for sodium/potassium-ion batteries: ultralong lifespans and wide-temperature workability, Inorg. Chem. Front., 2023, 10, 1187–1196 RSC.
- K. Yao, Z. Xu, J. Huang, M. Ma, L. Fu, X. Shen, J. Li and M. Fu, Bundled Defect-Rich MoS2 for a High-Rate and Long-Life Sodium-Ion Battery: Achieving 3D Diffusion of Sodium Ion by Vacancies to Improve Kinetics, Small, 2019, 15, 1805405 CrossRef PubMed.
- J. Liang, Z. Wei, C. Wang and J. Ma, Vacancy-induced sodium-ion storage in N-doped carbon Nanofiber@MoS2 nanosheet arrays, Electrochim. Acta, 2018, 285, 301–308 CrossRef CAS.
- D. Sha, Y. You, R. Hu, X. Cao, Y. Wei, H. Zhang, L. Pan and Z. Sun, Comprehensively Understanding the Role of Anion Vacancies on K-Ion Storage: A Case Study of Se-Vacancy-Engineered VSe2, Adv. Mater., 2023, 35, 2211311 CrossRef CAS PubMed.
- Q. Zhu, W. Li, J. Wu, N. Tian, Y. Li, J. Yang, B. Liu and J. Jiang, Vacancy engineering in WS2 nanosheets for enhanced potassium-ion storage, J. Power Sources, 2022, 542, 231791 CrossRef CAS.
- W. Xu, C. Sun, K. Zhao, X. Cheng, S. Rawal, Y. Xu and Y. Wang, Defect engineering activating (boosting) zinc storage capacity of MoS2, Energy Storage Mater., 2019, 16, 527–534 CrossRef.
- Y. Bai, H. Zhang, B. Xiang, X. Liang, J. Hao, C. Zhu and L. Yan, Selenium Defect Boosted Electrochemical Performance of Binder-Free VSe2 Nanosheets for Aqueous Zinc-Ion Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 23230–23238 CrossRef CAS.
- H. Wang, H. Jiang, Y. Hu, N. Li, X. Zhao and C. Li, 2D MoS2/polyaniline heterostructures with enlarged interlayer spacing for superior lithium and sodium storage, J. Mater. Chem. A, 2017, 5, 5383–5389 RSC.
- L. Xing, K. Han, Q. Liu, Z. Liu, J. Chu, L. Zhang, X. Ma, Y. Bao, P. Li and W. Wang, Hierarchical two-atom-layered WSe2/C ultrathin crumpled nanosheets assemblies: engineering the interlayer spacing boosts potassium-ion storage, Energy Storage Mater., 2021, 36, 309–317 CrossRef.
- S. Di, P. Ding, Y. Wang, Y. Wu, J. Deng, L. Jia and Y. Li, Interlayer-expanded MoS2 assemblies for enhanced electrochemical storage of potassium ions, Nano Res., 2020, 13, 225–230 CrossRef CAS.
- M. Huang, Y. Mai, L. Zhao, X. Liang, Z. Fang and X. Jie, Tuning the kinetics of zinc ion in MoS2 by polyaniline intercalation, Electrochim. Acta, 2021, 388, 138624 CrossRef CAS.
- Y.-R. Liu, Z.-W. Lei, R.-P. Liu, X.-Y. Li, P.-X. Xiong, Y.-J. Luo, Q.-H. Chen, M.-D. Wei, L.-X. Zeng and Q.-R. Qian, Sn-doped induced stable 1T-WSe2 nanosheets entrenched on N-doped carbon with extraordinary half/full sodium/potassium storage performance, Rare Met., 2023, 42, 1557–1569 CrossRef CAS.
- Y. Zhang, H. Tao, T. Li, S. Du, J. Li, Y. Zhang and X. Yang, Vertically Oxygen-Incorporated MoS2 Nanosheets Coated on Carbon Fibers for Sodium-Ion Batteries, ACS Appl. Mater. Interfaces, 2018, 10, 35206–35215 CrossRef CAS.
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Molecular Engineering on MoS2 Enables Large Interlayers and Unlocked Basal Planes for High-Performance Aqueous Zn-Ion Storage, Angew. Chem., Int. Ed., 2021, 60, 20286–20293 CrossRef CAS PubMed.
- J. Li, H. Yan, W. Wei and L. Meng, Microwave-assisted mass synthesis of Mo1−xWxS2 alloy composites with a tunable lithium storage property, Dalton Trans., 2018, 47, 15148–15154 RSC.
- H.-N. Fan, X.-Y. Wang, H.-B. Yu, Q.-F. Gu, S.-L. Chen, Z. Liu, X.-H. Chen, W.-B. Luo and H.-K. Liu, Enhanced Potassium Ion Battery by Inducing Interlayer Anionic Ligands in MoS1.5Se0.5 Nanosheets with Exploration of the Mechanism, Adv. Energy Mater., 2020, 10, 1904162 CrossRef CAS.
- H. He, D. Huang, Q. Gan, J. Hao, S. Liu, Z. Wu, W. K. Pang, B. Johannessen, Y. Tang, J.-L. Luo, H. Wang and Z. Guo, Anion Vacancies Regulating Endows MoSSe with Fast and Stable Potassium Ion Storage, ACS Nano, 2019, 13, 11843–11852 CrossRef CAS.
- H. Li, B. Chen, R. Gao, F. Xu, X. Wen, X. Zhong, C. Li, Z. Piao, N. Hu, X. Xiao, F. Shao, G. Zhou and J. Yang, Integrating molybdenum sulfide selenide-based cathode with C-O-Mo heterointerface design and atomic engineering for superior aqueous Zn-ion batteries, Nano Res., 2023, 16, 4933–4940 CrossRef CAS.
- S. Zhang, G. Wang, J. Jin, L. Zhang, Z. Wen and J. Yang, Robust and Conductive Red MoSe2 for Stable and Fast Lithium Storage, ACS Nano, 2018, 12, 4010–4018 CrossRef CAS PubMed.
- T. Lei, M. Gu, H. Fu, J. Wang, L. Wang, J. Zhou, H. Liu and B. Lu, Bond modulation of MoSe2+x driving combined intercalation and conversion reactions for high-performance K cathodes, Chem. Sci., 2023, 14, 2528–2536 RSC.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- X. Qian, J. Liu, L. Fu and J. Li, Quantum spin Hall effect in two-dimensional transition metal dichalcogenides, Science, 2014, 346, 1344–1347 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Electronics and optoelectronics of two-dimensional transition metal dichalcogenides, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- D. Voiry, A. Mohite and M. Chhowalla, Phase engineering of transition metal dichalcogenides, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- I. Song, C. Park and H. C. Choi, Synthesis and properties of molybdenum disulphide: from bulk to atomic layers, RSC Adv., 2015, 5, 7495–7514 RSC.
- S. Jayabal, J. Wu, J. Chen, D. Geng and X. Meng, Metallic 1T-MoS2 nanosheets and their composite materials: Preparation, properties and emerging applications, Mater. Today Energy, 2018, 10, 264–279 CrossRef.
- J. Strachan, A. F. Masters and T. Maschmeyer, 3R-MoS2 in Review: History, Status, and Outlook, ACS Appl. Energy Mater., 2021, 4, 7405–7418 CrossRef CAS.
- A. Kumar and P. K. Ahluwalia, A first principle Comparative study of electronic and optical properties of 1H – MoS2 and 2H – MoS2, Mater. Chem. Phys., 2012, 135, 755–761 CrossRef CAS.
- Y. S. Huang, Preparation, Electrical and Modulation Optical Properties of 2H-MoSe2, Chin. J. Phys., 1984, 22, 43–53 Search PubMed.
- S. Zhao, T. Hotta, T. Koretsune, K. Watanabe, T. Taniguchi, K. Sugawara, T. Takahashi, H. Shinohara and R. Kitaura, Two-dimensional metallic NbS2: growth, optical identification and transport properties, 2D Mater., 2016, 3, 025027 CrossRef.
- J. Feng, X. Sun, C. Wu, L. Peng, C. Lin, S. Hu, J. Yang and Y. Xie, Metallic Few-Layered VS2 Ultrathin Nanosheets: High Two-Dimensional Conductivity for In-Plane Supercapacitors, J. Am. Chem. Soc., 2011, 133, 17832–17838 CrossRef CAS.
- W. Choi, N. Choudhary, G. H. Han, J. Park, D. Akinwande and Y. H. Lee, Recent development of two-dimensional transition metal dichalcogenides and their applications, Mater. Today, 2017, 20, 116–130 CrossRef CAS.
- J. Li, B. Zhao, P. Chen, R. Wu, B. Li, Q. Xia, G. Guo, J. Luo, K. Zang, Z. Zhang, H. Ma, G. Sun, X. Duan and X. Duan, Synthesis of Ultrathin Metallic MTe2 (M = V, Nb, Ta) Single-Crystalline Nanoplates, Adv. Mater., 2018, 30, 1801043 CrossRef.
- H. Liu, Y. Xue, J.-A. Shi, R. A. Guzman, P. Zhang, Z. Zhou, Y. He, C. Bian, L. Wu, R. Ma, J. Chen, J. Yan, H. Yang, C.-M. Shen, W. Zhou, L. Bao and H.-J. Gao, Observation of the Kondo Effect in Multilayer Single-Crystalline VTe2 Nanoplates, Nano Lett., 2019, 19, 8572–8580 CrossRef CAS.
- C. Zhu, D. Gao, J. Ding, D. Chao and J. Wang, TMD-based highly efficient electrocatalysts developed by combined computational and experimental approaches, Chem. Soc. Rev., 2018, 47, 4332–4356 RSC.
- E. Torun, H. Sahin, S. Cahangirov, A. Rubio and F. M. Peeters, Anisotropic electronic, mechanical, and optical properties of monolayer WTe2, J. Appl. Phys., 2016, 119, 074307 CrossRef.
- M. Rahman, K. Davey and S.-Z. Qiao, Advent of 2D Rhenium Disulfide (ReS2): Fundamentals to Applications, Adv. Funct. Mater., 2017, 27, 1606129 CrossRef.
- H.-X. Zhong, S. Gao, J.-J. Shi and L. Yang, Quasiparticle band gaps, excitonic effects, and anisotropic optical properties of the monolayer distorted 1T diamond-chain structures ReS2 and ReSe2, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 115438 CrossRef.
- J. A. Wilson and A. D. Yoffe, The transition metal dichalcogenides discussion and interpretation of the observed optical, electrical and structural properties, Adv. Phys., 1969, 18, 193–335 CrossRef CAS.
- L. Peng, Y. Zhu, D. Chen, R. S. Ruoff and G. Yu, Two-Dimensional Materials for Beyond-Lithium-Ion Batteries, Adv. Energy Mater., 2016, 6, 1600025 CrossRef.
- S. Hao, X. Shen, M. Tian, R. Yu, Z. Wang and L. Chen, Reversible conversion of MoS2 upon sodium extraction, Nano Energy, 2017, 41, 217–224 CrossRef CAS.
- X. Fang, C. Hua, X. Guo, Y. Hu, Z. Wang, X. Gao, F. Wu, J. Wang and L. Chen, Lithium storage in commercial MoS2 in different potential ranges, Electrochim. Acta, 2012, 81, 155–160 CrossRef CAS.
- T. Zhao, H. Shu, Z. Shen, H. Hu, J. Wang and X. Chen, Electrochemical Lithiation Mechanism of Two-Dimensional Transition-Metal Dichalcogenide Anode Materials: Intercalation versus Conversion Reactions, J. Phys. Chem. C, 2019, 123, 2139–2146 CrossRef CAS.
- L. Zhang, D. Sun, J. Kang, J. Feng, H. A. Bechtel, L.-W. Wang, E. J. Cairns and J. Guo, Electrochemical Reaction Mechanism of the MoS2 Electrode in a Lithium-Ion Cell Revealed by in Situ and Operando X-ray Absorption Spectroscopy, Nano Lett., 2018, 18, 1466–1475 CrossRef CAS.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao, Y. Li and Y. Li, Hierarchical VS2 Nanosheet Assemblies: A Universal Host Material for the Reversible Storage of Alkali Metal Ions, Adv. Mater., 2017, 29, 1702061 CrossRef.
- L. Zhang, D. Sun, Q. Wei, H. Ju, J. Feng, J. Zhu, L. Mai, E. J. Cairns and J. Guo, Understanding the electrochemical reaction mechanism of VS2 nanosheets in lithium-ion cells by multiple in situ and ex situ x-ray spectroscopy, J. Phys. D: Appl. Phys., 2018, 51, 494001 CrossRef.
- C. Yang, J. Feng, F. Lv, J. Zhou, C. Lin, K. Wang, Y. Zhang, Y. Yang, W. Wang, J. Li and S. Guo, Metallic Graphene-Like VSe2 Ultrathin Nanosheets: Superior Potassium-Ion Storage and Their Working Mechanism, Adv. Mater., 2018, 30, 1800036 CrossRef.
- G. Wang, Y. Zhang, H. S. Cho, X. Zhao, F. Kim and J. Zou, Revisiting the Structural Evolution of MoS2 During Alkali Metal (Li, Na, and K) Intercalation, ACS Appl. Energy Mater., 2021, 4, 14180–14190 CrossRef CAS.
- V. P. H. Huy, Y. N. Ahn and J. Hur, Recent Advances in Transition Metal Dichalcogenide Cathode Materials for Aqueous Rechargeable Multivalent Metal-Ion Batteries, Nanomaterials, 2021, 11, 1517 CrossRef CAS PubMed.
- K. Yao, Z. Xu, M. Ma, J. Li, F. Lu and J. Huang, Densified Metallic MoS2/Graphene Enabling Fast Potassium-Ion Storage with Superior Gravimetric and Volumetric Capacities, Adv. Funct. Mater., 2020, 30, 2001484 CrossRef CAS.
- S. Yan, W. Qiao, X. He, X. Guo, L. Xi, W. Zhong and Y. Du, Enhancement of magnetism by structural phase transition in MoS2, Appl. Phys. Lett., 2015, 106, 012408 CrossRef.
- T. Xiang, S. Tao, W. Xu, Q. Fang, C. Wu, D. Liu, Y. Zhou, A. Khalil, Z. Muhammad, W. Chu, Z. Wang, H. Xiang, Q. Liu and L. Song, Stable 1T-MoSe2 and Carbon Nanotube Hybridized Flexible Film: Binder-Free and High-Performance Li-Ion Anode, ACS Nano, 2017, 11, 6483–6491 CrossRef CAS.
- K. Wu, X. Cao, M. Li, B. Lei, J. Zhan and M. Wu, Bottom-Up Synthesis of MoS2/CNTs Hollow Polyhedron with 1T/2H Hybrid Phase for Superior Potassium-Ion Storage, Small, 2020, 16, 2004178 CrossRef CAS.
- M. Srinivaas, C.-Y. Wu, J.-G. Duh and J. M. Wu, Highly Rich 1T Metallic Phase of Few-Layered WS2 Nanoflowers for Enhanced Storage of Lithium-Ion Batteries, ACS Sustain. Chem. Eng., 2019, 7, 10363–10370 CrossRef CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- L. Liu, W. Yang, H. Chen, X. Chen, K. Zhang, Q. Zeng, S. Lei, J. Huang, S. Li and S. Peng, High zinc-ion intercalation reaction activity of MoS2 cathode based on regulation of thermodynamic metastability and interlayer water, Electrochim. Acta, 2022, 410, 140016 CrossRef CAS.
- Y. Liu and X. Wu, Recent Advances of Transition Metal Chalcogenides as Cathode Materials for Aqueous Zinc-Ion Batteries, Nanomaterials, 2022, 12, 3298 CrossRef CAS.
- J. He, G. Hartmann, M. Lee, G. S. Hwang, Y. Chen and A. Manthiram, Freestanding 1T MoS2/graphene heterostructures as a highly efficient electrocatalyst for lithium polysulfides in Li–S batteries, Energy Environ. Sci., 2019, 12, 344–350 RSC.
- J. Liu, N. Gong, W. Peng, Y. Li, F. Zhang and X. Fan, Vertically aligned 1 T phase MoS2 nanosheet array for high-performance rechargeable aqueous Zn-ion batteries, Chem. Eng. J., 2022, 428, 130981 CrossRef CAS.
- J. Liu, P. Xu, J. Liang, H. Liu, W. Peng, Y. Li, F. Zhang and X. Fan, Boosting aqueous zinc-ion storage in MoS2 via controllable phase, Chem. Eng. J., 2020, 389, 124405 CrossRef CAS.
- W.-H. Ryu, J.-W. Jung, K. Park, S.-J. Kim and I.-D. Kim, Vine-like MoS2 anode materials self-assembled from 1-D nanofibers for high capacity sodium rechargeable batteries, Nanoscale, 2014, 6, 10975–10981 RSC.
- B. Yu, Y. Chen, Z. Wang, D. Chen, X. Wang, W. Zhang, J. He and W. He, 1T-MoS2 nanotubes wrapped with N-doped graphene as highly-efficient absorbent and electrocatalyst for Li–S batteries, J. Power Sources, 2020, 447, 227364 CrossRef CAS.
- M. Wang, L. Fan, D. Tian, X. Wu, Y. Qiu, C. Zhao, B. Guan, Y. Wang, N. Zhang and K. Sun, Rational Design of Hierarchical SnO2/1T-MoS2 Nanoarray Electrode for Ultralong-Life Li–S Batteries, ACS Energy Lett., 2018, 3, 1627–1633 CrossRef CAS.
- H. Shu, F. Li, C. Hu, P. Liang, D. Cao and X. Chen, The capacity fading mechanism and improvement of cycling stability in MoS2-based anode materials for lithium-ion batteries, Nanoscale, 2016, 8, 2918–2926 RSC.
- Y. Zhang, Z. Mu, C. Yang, Z. Xu, S. Zhang, X. Zhang, Y. Li, J. Lai, Z. Sun, Y. Yang, Y. Chao, C. Li, X. Ge, W. Yang and S. Guo, Rational Design of MXene/1T-2H MoS2-C Nanohybrids for High-Performance Lithium–Sulfur Batteries, Adv. Funct. Mater., 2018, 28, 1707578 CrossRef.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Recent Development of Metallic (1T) Phase of Molybdenum Disulfide for Energy Conversion and Storage, Adv. Energy Mater., 2018, 8, 1703482 CrossRef.
- W.-j. Tang, X.-l. Wang, D. Xie, X.-h. Xia, C.-d. Gu and J.-p. Tu, Hollow metallic 1T MoS2 arrays grown on carbon cloth: a freestanding electrode for sodium ion batteries, J. Mater. Chem. A, 2018, 6, 18318–18324 RSC.
- J. Wu, J. Liu, J. Cui, S. Yao, M. Ihsan-Ul-Haq, N. Mubarak, E. Quattrocchi, F. Ciucci and J.-K. Kim, Dual-phase MoS2 as a high-performance sodium-ion battery anode, J. Mater. Chem. A, 2020, 8, 2114–2122 RSC.
- Z. Zhao, T. Xu and X. Yu, Unlock the Potassium Storage Behavior of Single-Phased Tungsten Selenide Nanorods via Large Cation Insertion, Adv. Mater., 2023, 35, 2208096 CrossRef CAS.
- F. Liu, Y. Zou, X. Tang, L. Mao, D. Du, H. Wang, M. Zhang, Z. Wang, N. Yao, W. Zhao, M. Bai, T. Zhao, Y. Liu and Y. Ma, Phase Engineering and Alkali Cation Stabilization for 1T Molybdenum Dichalcogenides Monolayers, Adv. Funct. Mater., 2022, 32, 2204601 CrossRef CAS.
- H. He, H. Zhang, D. Huang, W. Kuang, X. Li, J. Hao, Z. Guo and C. Zhang, Harnessing Plasma-Assisted Doping Engineering to Stabilize Metallic Phase MoSe2 for Fast and Durable Sodium-Ion Storage, Adv. Mater., 2022, 34, 2200397 CrossRef CAS PubMed.
- P. Li, Y. Yang, S. Gong, F. Lv, W. Wang, Y. Li, M. Luo, Y. Xing, Q. Wang and S. Guo, Co-doped 1T-MoS2 nanosheets embedded in N, S-doped carbon nanobowls for high-rate and ultra-stable sodium-ion batteries, Nano Res., 2019, 12, 2218–2223 CrossRef CAS.
- P. Xiong, R. Ma, N. Sakai, L. Nurdiwijayanto and T. Sasaki, Unilamellar Metallic MoS2/Graphene Superlattice for Efficient Sodium Storage and Hydrogen Evolution, ACS Energy Lett., 2018, 3, 997–1005 CrossRef CAS.
- Y. Fang, Y. Lv, F. Gong, A. A. Elzatahry, G. Zheng and D. Zhao, Synthesis of 2D-Mesoporous-Carbon/MoS2 Heterostructures with Well-Defined Interfaces for High-Performance Lithium-Ion Batteries, Adv. Mater., 2016, 28, 9385–9390 CrossRef CAS PubMed.
- M. Jiang, Y. Hu, B. Mao, Y. Wang, Z. Yang, T. Meng, X. Wang and M. Cao, Strain-regulated Gibbs free energy enables reversible redox chemistry of chalcogenides for sodium ion batteries, Nat. Commun., 2022, 13, 5588 CrossRef CAS PubMed.
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Electrochemistry of Nanostructured Layered Transition-Metal Dichalcogenides, Chem. Rev., 2015, 115, 11941–11966 CrossRef CAS PubMed.
- R. Zhou, H. Wang, J. Chang, C. Yu, H. Dai, Q. Chen, J. Zhou, H. Yu, G. Sun and W. Huang, Ammonium Intercalation Induced Expanded 1T-Rich Molybdenum Diselenides for Improved Lithium Ion Storage, ACS Appl. Mater. Interfaces, 2021, 13, 17459–17466 CrossRef CAS.
- J. Yang, J. Wang, L. Zhu, X. Wang, X. Dong, W. Zeng, J. Wang and F. Pan, Boosting magnesium storage in MoS2 via a 1T phase introduction and interlayer expansion strategy: theoretical prediction and experimental verification, Sustain. Energy Fuels, 2021, 5, 5471–5480 RSC.
- W. Zong, C. Yang, L. Mo, Y. Ouyang, H. Guo, L. Ge, Y.-E. Miao, D. Rao, J. Zhang, F. Lai and T. Liu, Elucidating dual-defect mechanism in rhenium disulfide nanosheets with multi-dimensional ion transport channels for ultrafast sodium storage, Nano Energy, 2020, 77, 105189 CrossRef CAS.
- J. Mei, Y. Zhang, T. Liao, Z. Sun and S. X. Dou, Strategies for improving the lithium-storage performance of 2D nanomaterials, Natl. Sci. Rev., 2018, 5, 389–416 CrossRef CAS.
- Y. Zhao, D. Yang, T. He, J. Li, L. Wei, D. Wang, Y. Wang, X. Wang, G. Chen and Y. Wei, Vacancy engineering in VS2 nanosheets for ultrafast pseudocapacitive sodium ion storage, Chem. Eng. J., 2021, 421, 129715 CrossRef CAS.
- T. Liu, N. Peng, X. Zhang, R. Zheng, M. Xia, H. Yu, M. Shui, Y. Xie and J. Shu, Controllable defect engineering enhanced bond strength for stable electrochemical energy storage, Nano Energy, 2021, 79, 105460 CrossRef CAS.
- F. Liu and Z. Fan, Defect engineering of two-dimensional materials for advanced energy conversion and storage, Chem. Soc. Rev., 2023, 52, 1723–1772 RSC.
- Y. Jia, K. Jiang, H. Wang and X. Yao, The Role of Defect Sites in Nanomaterials for Electrocatalytic Energy Conversion, Chem, 2019, 5, 1371–1397 CAS.
- P. Gao, Z. Chen, Y. Gong, R. Zhang, H. Liu, P. Tang, X. Chen, S. Passerini and J. Liu, The Role of Cation Vacancies in Electrode Materials for Enhanced Electrochemical Energy Storage: Synthesis, Advanced Characterization, and Fundamentals, Adv. Energy Mater., 2020, 10, 1903780 CrossRef CAS.
- G. Barik and S. Pal, Defect Induced Performance Enhancement of Monolayer MoS2 for Li- and Na-Ion Batteries, J. Phys. Chem. C, 2019, 123, 21852–21865 CrossRef CAS.
- J. Kunstmann, T. B. Wendumu and G. Seifert, Localized defect states in MoS2 monolayers: electronic and optical properties, Phys. Status Solidi B, 2017, 254, 1600645 CrossRef.
- Y. Li, R. Zhang, W. Zhou, X. Wu, H. Zhang and J. Zhang, Hierarchical MoS2 Hollow Architectures with Abundant Mo Vacancies for Efficient Sodium Storage, ACS Nano, 2019, 13, 5533–5540 CrossRef CAS PubMed.
- T. Liu, X. Zhang, M. Xia, H. Yu, N. Peng, C. Jiang, M. Shui, Y. Xie, T.-F. Yi and J. Shu, Functional cation defects engineering in TiS2 for high-stability anode, Nano Energy, 2020, 67, 104295 CrossRef CAS.
- J. Zhang, A. Yang, X. Wu, J. van de Groep, P. Tang, S. Li, B. Liu, F. Shi, J. Wan, Q. Li, Y. Sun, Z. Lu, X. Zheng, G. Zhou, C.-L. Wu, S.-C. Zhang, M. L. Brongersma, J. Li and Y. Cui, Reversible and selective ion intercalation through the top surface of few-layer MoS2, Nat. Commun., 2018, 9, 5289 CrossRef CAS PubMed.
- Y. Liu, B. V. Merinov and W. A. Goddard, Origin of low sodium capacity in graphite and generally weak substrate binding of Na and Mg among alkali and alkaline earth metals, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3735–3739 CrossRef CAS.
- F. Zhu, H. Zhang, Z. Lu, D. Kang and L. Han, Controlled defective engineering of MoS2 nanosheets for rechargeable Mg batteries, J. Energy Storage, 2021, 42, 103046 CrossRef.
- Q. Zhang, S. Tan, R. G. Mendes, Z. Sun, Y. Chen, X. Kong, Y. Xue, M. H. Rümmeli, X. Wu, S. Chen and L. Fu, Extremely Weak van der Waals Coupling in Vertical ReS2 Nanowalls for High-Current-Density Lithium-Ion Batteries, Adv. Mater., 2016, 28, 2616–2623 CrossRef CAS PubMed.
- S. Tongay, H. Sahin, C. Ko, A. Luce, W. Fan, K. Liu, J. Zhou, Y.-S. Huang, C.-H. Ho, J. Yan, D. F. Ogletree, S. Aloni, J. Ji, S. Li, J. Li, F. M. Peeters and J. Wu, Monolayer behaviour in bulk ReS2 due to electronic and vibrational decoupling, Nat. Commun., 2014, 5, 3252 CrossRef PubMed.
- J. Xu, J. Zhang, W. Zhang and C.-S. Lee, Interlayer Nanoarchitectonics of Two-Dimensional Transition-Metal Dichalcogenides Nanosheets for Energy Storage and Conversion Applications, Adv. Energy Mater., 2017, 7, 1700571 CrossRef.
- L. Wu, M. Gu, Y. Feng, S. Chen, L. Fan, X. Yu, K. Guo, J. Zhou and B. Lu, Layered Superconductor Cu0.11TiSe2 as a High-Stable K-Cathode, Adv. Funct. Mater., 2022, 32, 2109893 CrossRef CAS.
- J. Shuai, H. D. Yoo, Y. Liang, Y. Li, Y. Yao and L. C. Grabow, Density functional theory study of Li, Na, and Mg intercalation and diffusion in MoS2 with controlled interlayer spacing, Mater. Res. Express, 2016, 3, 064001 CrossRef.
- N. Zheng, G. Jiang, X. Chen, J. Mao, Y. Zhou and Y. Li, Rational design of a tubular, interlayer expanded MoS2–N/O doped carbon composite for excellent potassium-ion storage, J. Mater. Chem. A, 2019, 7, 9305–9315 RSC.
- Y. Li, D. Wu, Z. Zhou, C. R. Cabrera and Z. Chen, Enhanced Li Adsorption and Diffusion on MoS2 Zigzag Nanoribbons by Edge Effects: A Computational Study, J. Phys. Chem. Lett., 2012, 3, 2221–2227 CrossRef CAS PubMed.
- H. J. Chen, J. Huang, X. L. Lei, M. S. Wu, G. Liu, C. Y. Ouyang and B. Xu, Adsorption and Diffusion of Lithium on MoS2 Monolayer: The Role of Strain and Concentration, Int. J. Electrochem. Sci., 2013, 8, 2196–2203 CrossRef CAS.
- H. Hwang, H. Kim and J. Cho, MoS2 Nanoplates Consisting of Disordered Graphene-like Layers for High Rate Lithium Battery Anode Materials, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Highly Ordered Mesoporous MoS2 with Expanded Spacing of the (002) Crystal Plane for Ultrafast Lithium Ion Storage, Adv. Energy Mater., 2012, 2, 970–975 CrossRef CAS.
- K. Ma, Y. Liu, H. Jiang, Y. Hu, R. Si, H. Liu and C. Li, Multivalence-Ion Intercalation Enables Ultrahigh 1T Phase MoS2 Nanoflowers to Enhanced Sodium-Storage Performance, CCS Chem., 2020, 3, 1472–1482 CrossRef.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Superior stability and high capacity of restacked molybdenum disulfide as anode material for lithium ion batteries, Chem. Commun., 2010, 46, 1106–1108 RSC.
- E. Benavente, M. A. S. Ana, F. Mendizábal and G. González, Intercalation chemistry of molybdenum disulfide, Coord. Chem. Rev., 2002, 224, 87–109 CrossRef CAS.
- K. D. Rasamani, F. Alimohammadi and Y. Sun, Interlayer-expanded MoS2, Mater. Today, 2017, 20, 83–91 CrossRef CAS.
- W. M. R. Divigalpitiya, R. F. Frindt and S. R. Morrison, Inclusion Systems of Organic Molecules in Restacked Single-Layer Molybdenum Disulfide, Science, 1989, 246, 369–371 CrossRef CAS PubMed.
- A. Zak, Y. Feldman, V. Lyakhovitskaya, G. Leitus, R. Popovitz-Biro, E. Wachtel, H. Cohen, S. Reich and R. Tenne, Alkali Metal Intercalated Fullerene-Like MS2 (M = W, Mo) Nanoparticles and Their Properties, J. Am. Chem. Soc., 2002, 124, 4747–4758 CrossRef CAS.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, MoS2 Nanoflowers with Expanded Interlayers as High-Performance Anodes for Sodium-Ion Batteries, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- G. González, M. A. S. Ana, E. Benavente, J. P. Donoso, T. J. Bonagamba, N. C. Mello and H. Panepucci, Electrical conductivity and lithium diffusion in molybdenum disulfide intercalated with poly(ethylene oxide), Solid State Ionics, 1996, 85, 225–230 CrossRef.
- W. Ye, F. Wu, N. Shi, H. Zhou, Q. Chi, W. Chen, S. Du, P. Gao, H. Li and S. Xiong, Metal–Semiconductor Phase Twinned Hierarchical MoS2 Nanowires with Expanded Interlayers for Sodium-Ion Batteries with Ultralong Cycle Life, Small, 2020, 16, 1906607 CrossRef CAS PubMed.
- R. Bissessur and P. K. Y. Liu, Direct insertion of polypyrrole into molybdenum disulfide, Solid State Ionics, 2006, 177, 191–196 CrossRef CAS.
- Y. Li, Y. Liang, F. C. R. Hernandez, H. D. Yoo, Q. An and Y. Yao, Enhancing sodium-ion battery performance with interlayer-expanded MoS2–PEO nanocomposites, Nano Energy, 2015, 15, 453–461 CrossRef CAS.
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, Graphene-like MoS2/amorphous carbon composites with high capacity and excellent stability as anode materials for lithium ion batteries, J. Mater. Chem., 2011, 21, 6251–6257 RSC.
- Y. Liu, X. Wang, X. Song, Y. Dong, L. Yang, L. Wang, D. Jia, Z. Zhao and J. Qiu, Interlayer expanded MoS2 enabled by edge effect of graphene nanoribbons for high performance lithium and sodium ion batteries, Carbon, 2016, 109, 461–471 CrossRef CAS.
- Y. Liu, Y. Yuan, L. Peng, L. Cheng, B. An, Y. Wang, Q. Wei, X. Xia and H. Zhou, Study on the construction of interlayer adjustable C@MoS2 fiber anode by biomass confining and its lithium/sodium storage mechanism, ChemSusChem, 2023, e202300576 CrossRef CAS PubMed.
- H. Jiang, D. Ren, H. Wang, Y. Hu, S. Guo, H. Yuan, P. Hu, L. Zhang and C. Li, 2D Monolayer MoS2–Carbon Interoverlapped Superstructure: Engineering Ideal Atomic Interface for Lithium Ion Storage, Adv. Mater., 2015, 27, 3687–3695 CrossRef CAS PubMed.
- N. Feng, R. Meng, L. Zu, Y. Feng, C. Peng, J. Huang, G. Liu, B. Chen and J. Yang, A polymer-direct-intercalation strategy for MoS2/carbon-derived heteroaerogels with ultrahigh pseudocapacitance, Nat. Commun., 2019, 10, 1372 CrossRef PubMed.
- H. Liang, Z. Cao, F. Ming, W. Zhang, D. H. Anjum, Y. Cui, L. Cavallo and H. N. Alshareef, Aqueous Zinc-Ion Storage in MoS2 by Tuning the Intercalation Energy, Nano Lett., 2019, 19, 3199–3206 CrossRef CAS PubMed.
- P. Jing, H. Lu, W. Yang and Y. Cao, Interlayer-expanded and binder-free VS2 nanosheets assemblies for enhanced Mg2+ and Li+/Mg2+ hybrid ion storage, Electrochim. Acta, 2020, 330, 135263 CrossRef CAS.
- M. Wang, H. Ye, V. Vasudevan and N. V. Medhekar, Enhancing kinetic and electrochemical performance of layered MoS2 cathodes with interlayer expansion for Mg-ion batteries, J. Power Sources, 2022, 542, 231722 CrossRef CAS.
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao and X. Qu, Sandwich-Like Heterostructures of MoS2/Graphene with Enlarged Interlayer Spacing and Enhanced Hydrophilicity as High-Performance Cathodes for Aqueous Zinc-Ion Batteries, Adv. Mater., 2021, 33, 2007480 CrossRef CAS PubMed.
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. R. Hernandez, L. C. Grabow and Y. Yao, Interlayer-Expanded Molybdenum Disulfide Nanocomposites for Electrochemical Magnesium Storage, Nano Lett., 2015, 15, 2194–2202 CrossRef CAS PubMed.
- H. D. Yoo, Y. Liang, H. Dong, J. Lin, H. Wang, Y. Liu, L. Ma, T. Wu, Y. Li, Q. Ru, Y. Jing, Q. An, W. Zhou, J. Guo, J. Lu, S. T. Pantelides, X. Qian and Y. Yao, Fast kinetics of magnesium monochloride cations in interlayer-expanded titanium disulfide for magnesium rechargeable batteries, Nat. Commun., 2017, 8, 339 CrossRef PubMed.
- F. Cui, M. Han, W. Zhou, C. Lai, Y. Chen, J. Su, J. Wang, H. Li and Y. Hu, Superlattice-Stabilized WSe2 Cathode for Rechargeable Aluminum Batteries, Small Methods, 2022, 6, 2201281 CrossRef CAS PubMed.
- Q. Liu, X. Weijun, Z. Wu, J. Huo, D. Liu, Q. Wang and S. Wang, The origin of the enhanced performance of nitrogen-doped MoS2 in lithium ion batteries, Nanotechnology, 2016, 27, 175402 CrossRef PubMed.
- Y. Cai, H. Yang, J. Zhou, Z. Luo, G. Fang, S. Liu, A. Pan and S. Liang, Nitrogen doped hollow MoS2/C nanospheres as anode for long-life sodium-ion batteries, Chem. Eng. J., 2017, 327, 522–529 CrossRef CAS.
- A. A. Kotsun, V. A. Alekseev, S. G. Stolyarova, A. A. Makarova, M. A. Grebenkina, A. P. Zubareva, A. V. Okotrub and L. G. Bulusheva, Effect of molybdenum disulfide doping with substitutional nitrogen and sulfur vacancies on lithium intercalation, J. Alloys Compd., 2023, 947, 169689 CrossRef CAS.
- Y.-C. Lin, D. O. Dumcenco, H.-P. Komsa, Y. Niimi, A. V. Krasheninnikov, Y.-S. Huang and K. Suenaga, Properties of Individual Dopant Atoms in Single-Layer MoS2: Atomic Structure, Migration, and Enhanced Reactivity, Adv. Mater., 2014, 26, 2857–2861 CrossRef CAS PubMed.
- H. Shan, J. Qin, J. Wang, H. M. K. Sari, L. Lei, W. Xiao, W. Li, C. Xie, H. Yang, Y. Luo, G. Zhang and X. Li, Doping-Induced Electronic/Ionic Engineering to Optimize the Redox Kinetics for Potassium Storage: A Case Study of Ni-Doped CoSe2, Adv. Sci., 2022, 9, 2200341 CrossRef CAS PubMed.
- P. Wen, H. Wang, X. Wang, H. Wang, Y. Bai and Z. Yang, Exploring the physicochemical role of Pd dopant in promoting Li-ion diffusion dynamics and storage performance of NbS2 at the atomic scale, Phys. Chem. Chem. Phys., 2022, 24, 14877–14885 RSC.
- S. Sovizi and R. Szoszkiewicz, Single atom doping in 2D layered MoS2 from a periodic table perspective, Surf. Sci. Rep., 2022, 77, 100567 CrossRef CAS.
- X.-L. Fan, Y.-R. An and W.-J. Guo, Ferromagnetism in Transitional Metal-Doped MoS2 Monolayer, Nanoscale Res. Lett., 2016, 11, 154 CrossRef PubMed.
- Y. C. Cheng, Z. Y. Zhu, W. B. Mi, Z. B. Guo and U. Schwingenschlögl, Prediction of two-dimensional diluted magnetic semiconductors: Doped monolayer MoS2 systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 100401 CrossRef.
- Q. Ding, W. Zheng, A. Zhao, Y. Zhao, K. Chen, X. Zhou, H. Zhang, Q. Li, X. Ai, H. Yang, Y. Fang and Y. Cao, W-Doping Induced Efficient Tunnel-to-Layered Structure Transformation of Na0.44Mn1-xWxO2: Phase Evolution, Sodium-Storage Properties, and Moisture Stability, Adv. Energy Mater., 2023, 13, 2203802 CrossRef CAS.
- L. Xiao, F. Ji, J. Zhang, X. Chen and Y. Fang, Doping Regulation in Polyanionic Compounds for Advanced Sodium-Ion Batteries, Small, 2023, 19, 2205732 CrossRef CAS PubMed.
- X. Zhao, P. Chen, C. Xia, T. Wang and X. Dai, Electronic and magnetic properties of n-type and p-doped MoS2 monolayers, RSC Adv., 2016, 6, 16772–16778 RSC.
- S. Lu, C. Li, Y. F. Zhao, Y. Y. Gong, L. Y. Niu and X. J. Liu, Tunable redox potential of nonmetal doped monolayer MoS2: first principle calculations, Appl. Surf. Sci., 2016, 384, 360–367 CrossRef CAS.
- X. Sun and Z. Wang, Adsorption and diffusion of lithium on heteroatom-doped monolayer molybdenum disulfide, Appl. Surf. Sci., 2018, 455, 911–918 CrossRef CAS.
- R. Tian, A. Wu, G. Zhang, J. Liu, R. A. P. Camacho, W. Yu, S. Zhou, M. Yao and H. Huang, Adsorption and diffusion of alkali metals (Li, Na, and K) on heteroatom-doped monolayer titanium disulfide, Dalton Trans., 2021, 50, 7065–7077 RSC.
- D. Wang, Y. Liu, X. Meng, Y. Wei, Y. Zhao, Q. Pang and G. Chen, Two-dimensional VS2 monolayers as potential anode materials for lithium-ion batteries and beyond: first-principles calculations, J. Mater. Chem. A, 2017, 5, 21370–21377 RSC.
- F. Li, Y. Qu and M. Zhao, Germanium sulfide nanosheet: a universal anode material for alkali metal ion batteries, J. Mater. Chem. A, 2016, 4, 8905–8912 RSC.
- H. R. Jiang, W. Shyy, M. Liu, L. Wei, M. C. Wu and T. S. Zhao, Boron phosphide monolayer as a potential anode material for alkali metal-based batteries, J. Mater. Chem. A, 2017, 5, 672–679 RSC.
- S. Qin, W. Lei, D. Liu and Y. Chen, Advanced N-doped mesoporous molybdenum disulfide nanosheets and the enhanced lithium-ion storage performance, J. Mater. Chem. A, 2016, 4, 1440–1445 RSC.
- H. Xie, B. Chen, C. Liu, G. Wu, S. Sui, E.-Z. Liu, G. Zhou, C. He, W. Hu and N. Q. Zhao, Engineering the Interfacial Doping of 2D Heterostructures with Good Bidirectional Reaction Kinetics for Durably Reversible Sodium-Ion Batteries, Energy Storage Mater., 2023, 60, 102830 CrossRef.
- J. Zhao, D. Xiao, Q. Wan, X. Wei, G. Tao, Y. Liu, Y. Xiang, K. Davey, Z. Liu, Z. Guo and Y. Song, Molybdenum Atom Engineered Vanadium Disulfide for Boosted High-Capacity Li-Ion Storage, Small, 2023, 2301738 CrossRef CAS PubMed.
- P. Wang, Z. Shen, Y. Xia, H. Wang, L. Zheng, W. Xi and S. Zhan, Atomic Insights for Optimum and Excess Doping in Photocatalysis: A Case Study of Few-Layer Cu-ZnIn2S4, Adv. Funct. Mater., 2019, 29, 1807013 CrossRef.
- J. Zhang, C. Du, Z. Dai, W. Chen, Y. Zheng, B. Li, Y. Zong, X. Wang, J. Zhu and Q. Yan, NbS2 Nanosheets with M/Se (M = Fe, Co, Ni) Codopants for Li+ and Na+ Storage, ACS Nano, 2017, 11, 10599–10607 CrossRef CAS PubMed.
- T. Wang, C. Sun, M. Yang, G. Zhao, S. Wang, F. Ma, L. Zhang, Y. Shao, Y. Wu, B. Huang and X. Hao, Phase-transformation engineering in MoS2 on carbon cloth as flexible binder-free anode for enhancing lithium storage, J. Alloys Compd., 2017, 716, 112–118 CrossRef CAS.
- Z. Sheng, P. Qi, Y. Lu, G. Liu, M. Chen, X. Gan, Y. Qin, K. Hao and Y. Tang, Nitrogen-Doped Metallic MoS2 Derived from a Metal–Organic Framework for Aqueous Rechargeable Zinc-Ion Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 34495–34506 CrossRef CAS PubMed.
- F. Ersan, G. Gökoğlu and E. Aktürk, Adsorption and Diffusion of Lithium on Monolayer Transition Metal Dichalcogenides (MoS2(1–x)Se2x) Alloys, J. Phys. Chem. C, 2015, 119, 28648–28653 CrossRef CAS.
- G. Barik and S. Pal, Monolayer Transition-Metal Dichalcogenide Mo1–xWxS2 Alloys as Efficient Anode Materials for Lithium-Ion Batteries, J. Phys. Chem. C, 2018, 122, 25837–25848 CrossRef CAS.
- G. Chaney, A. Ibrahim, F. Ersan, D. Çakır and C. Ataca, Comprehensive Study of Lithium Adsorption and Diffusion on Janus Mo/WXY (X, Y = S, Se, Te) Using First-Principles and Machine Learning Approaches, ACS Appl. Mater. Interfaces, 2021, 13, 36388–36406 CrossRef CAS PubMed.
- J. Shi, H. Li, Y. Liu, Q. Lu and F. Gao, Molybdenum Sulfide Selenide Nanosheets Synergized with Nitrogen-Rich Carbon Frameworks toward High Performance and Stable Sodium Storage, Adv. Mater. Interfaces, 2022, 9, 2102408 CrossRef CAS.
- Y. Yang, S. Wang, J. Zhang, H. Li, Z. Tang and X. Wang, Nanosheet-assembled MoSe2 and S-doped MoSe2−x nanostructures for superior lithium storage properties and hydrogen evolution reactions, Inorg. Chem. Front., 2015, 2, 931–937 RSC.
- G. Jia, D. Chao, N. H. Tiep, Z. Zhang and H. J. Fan, Intercalation Na-ion storage in two-dimensional MoS2-xSex and capacity enhancement by selenium substitution, Energy Storage Mater., 2018, 14, 136–142 CrossRef.
- B. Liu, Y. Liu, X. Hu, G. Zhong, J. Li, J. Yuan and Z. Wen, N-Doped Carbon Modifying MoSSe Nanosheets on Hollow Cubic Carbon for High-Performance Anodes of Sodium-Based Dual-Ion Batteries, Adv. Funct. Mater., 2021, 31, 2101066 CrossRef CAS.
- K. Zhang, H. Zhang, C. Li and X. Ma, Vertically oriented MoSe0.4S1.6/N-doped C nanostructures directly grown on carbon nanotubes as high-performance anode for potassium-ion batteries, J. Electroanal. Chem., 2022, 923, 116846 CrossRef CAS.
- X. Yu, G. Zhao, C. Wu, H. Huang, C. Liu, X. Shen, M. Wang, X. Bai and N. Zhang, Constructing anion vacancy-rich MoSSe/G van der Waals heterostructures for high-performance Mg–Li hybrid-ion batteries, J. Mater. Chem. A, 2021, 9, 23276–23285 RSC.
- Y. Huang, Z. Wang, M. Guan, F. Wu and R. Chen, Toward Rapid-Charging Sodium-Ion Batteries using Hybrid-Phase Molybdenum Sulfide Selenide-Based Anodes, Adv. Mater., 2020, 32, 2003534 CrossRef CAS PubMed.
- A. Kutana, E. S. Penev and B. I. Yakobson, Engineering electronic properties of layered transition-metal dichalcogenide compounds through alloying, Nanoscale, 2014, 6, 5820–5825 RSC.
- C. Tan, W. Zhao, A. Chaturvedi, Z. Fei, Z. Zeng, J. Chen, Y. Huang, P. Ercius, Z. Luo, X. Qi, B. Chen, Z. Lai, B. Li, X. Zhang, J. Yang, Y. Zong, C. Jin, H. Zheng, C. Kloc and H. Zhang, Preparation of Single-Layer MoS2xSe2(1-x) and MoxW1-xS2 Nanosheets with High-Concentration Metallic 1T Phase, Small, 2016, 12, 1866–1874 CrossRef CAS PubMed.
- Q. Zhang, L. Wang, J. Wang, X. Yu, J. Ge, H. Zhang and B. Lu, Semimetallic vanadium molybdenum sulfide for high-performance battery electrodes, J. Mater. Chem. A, 2018, 6, 9411–9419 RSC.
- G. Cai, L. Peng, S. Ye, Y. Huang, G. Wang and X. Zhang, Defect-rich MoS2(1−x)Se2x few-layer nanocomposites: a superior anode material for high-performance lithium-ion batteries, J. Mater. Chem. A, 2019, 7, 9837–9843 RSC.
- J. Xu, L. Dong, H. Tang and C. Li, Facile synthesis of Mo0.91W0.09S2 ultrathin nanosheets/amorphous carbon composites for lithium-ion battery anode, Ceram. Int., 2016, 42, 7803–7809 CrossRef CAS.
- J. Dong, Y. Zhao, G. Ouyang and G. Yang, A perspective on optimizing photoelectric conversion process in 2D transition-metal dichalcogenides and related heterostructures, Appl. Phys. Lett., 2022, 120, 080501 CrossRef CAS.
- P. Tao, J. He, T. Shen, Y. Hao, J. Yan, Z. Huang, X. Xu, M. Li and Y. Chen, Nitrogen-Doped MoS2 Foam for Fast Sodium Ion Storage, Adv. Mater. Interfaces, 2019, 6, 1900460 CrossRef.
- T. Wang, C. Sun, M. Yang, L. Zhang, Y. Shao, Y. Wu and X. Hao, Enhanced reversible lithium ion storage in stable 1T@2H WS2 nanosheet arrays anchored on carbon fiber, Electrochim. Acta, 2018, 259, 1–8 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |