
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide macrocyclisation via intramolecular interception of visible-light-mediated desulfurisation†
Frances R.
Smith
a,
Declan
Meehan
a,
Rhys C.
Griffiths
a,
Harriet J.
Knowles
a,
Peiyu
Zhang
b,
Huw E. L.
Williams
 c,
Andrew J.
Wilson
bd and
Nicholas J.
Mitchell
*a
c,
Andrew J.
Wilson
bd and
Nicholas J.
Mitchell
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: nicholas.mitchell@nottingham.ac.uk
bSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
cBiodiscovery Institute, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
dSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 14th May 2024
Abstract
Synthetic methods that enable the macrocyclisation of peptides facilitate the development of effective therapeutic and diagnostic tools. Herein we report a peptide cyclisation strategy based on intramolecular interception of visible-light-mediated cysteine desulfurisation. This method allows cyclisation of unprotected peptides in an aqueous solution via the installation of a hydrocarbon linkage. We explore the limits of this chemistry using a range of model peptides of increasing length and complexity, including peptides of biological/therapeutic relevance. The method is applied to replace the native disulfide of the peptide hormone, oxytocin, with a proteolytically/redox-stable hydrocarbon, and internal macrocyclisation of an MCL-1-binding peptide.
Introduction
Despite increasing interest in peptide therapeutics within the pharmaceutical industry over the past three decades, relatively few peptides have made it through pre-clinical development to challenge the dominance of small-molecule pharmaceuticals and protein-based biologics.1 This is primarily due to the low membrane permeability and poor stability of linear peptides.2 Conversely, constrained macrocyclic peptides display enhanced stability relative to their linear counterparts and higher binding affinities due to a rigid conformation. Furthermore, macrocyclic peptides offer an effective tool to enable selective interference of the myriad of traditionally intractable protein–protein interactions (PPIs) that mediate cellular biochemistry.3,4 Thus, this modality of therapeutic bridges the gap between the advantageous physicochemical properties of small molecules and the exceptional activity, specificity, and bioavailability inherent to protein-based biologics. While numerous synthetic methods have been developed to enable effective peptide macrocyclisation, biocompatible chemistries that work with readily accessible building blocks in environmentally acceptable solvents are still required to enable the significant impact of macrocyclic peptides as a next-generation drug discovery tool to be realised.Many of the synthetic methods developed to cyclise peptides5–8 take inspiration from nature and utilise the canonical residues, cysteine (Cys) and lysine (Lys), or the native N- and C-terminal functionality to form disulfide,5 thioether,9,10 and amide bonds (Fig. 1). Cyclisation via amide bond formation has been applied using both synthetic methods (peptide ligation11–13) and biological approaches exploiting intein chemistry (e.g., SICLOPPS14,15), mRNA display,10 and ligase enzymes.16 Due to the nucleophilicity of the thiol sidechain, numerous Cys-selective reactions (beyond disulfide and thioether formation) have been developed/repurposed for peptide cyclisation. These include the formation of bicycles17via alkylating scaffolds,18–21 bridging Cys residues using perfluoroaryl braces/fluorine displacement22,23 (and alternative bridging groups24,25), thiol coordination to bismuth,26 thiol-addition chemistry,27,28 and desulfurative replacement of a disulfide bridge.29 Incorporation of non-standard amino acids enables the exploitation of bioorthogonal chemistry such as azide–alkyne cycloaddition30,31 (i.e., ‘Click Chemistry’), Staudinger ligation32 and azide-phosphonite chemistry.33 Simple imine34 and oxime35 bond formation has been utilized, as well as more complex transition metal (TM)-catalysed36–41 and multi-component chemistry.42 Radical reactions such as thiol–ene43–45 and, more recently, decarboxylative photoredox catalysis46 and C–H alkylation47 have also been successfully applied. However, among the broad range of available synthetic techniques, Grubbs' ruthenium (Ru)-catalysed ring-closing olefin metathesis (RCM)48 reaction has found global application as a method-of-choice for peptide cyclisation due to effective formation of a proteolytic, hydrolytic, and redox-stable hydrocarbon linkage.49–53 Whilst an undeniably powerful method, RCM is usually conducted on protected peptides in organic solvent, and access to the saturated hydrocarbon necessitates reduction under harsh conditions. New, operationally simple techniques that retain the benefits of RCM, but that work effectively in more sustainable solvents, would offer an impactful alternative to this universally popular method.
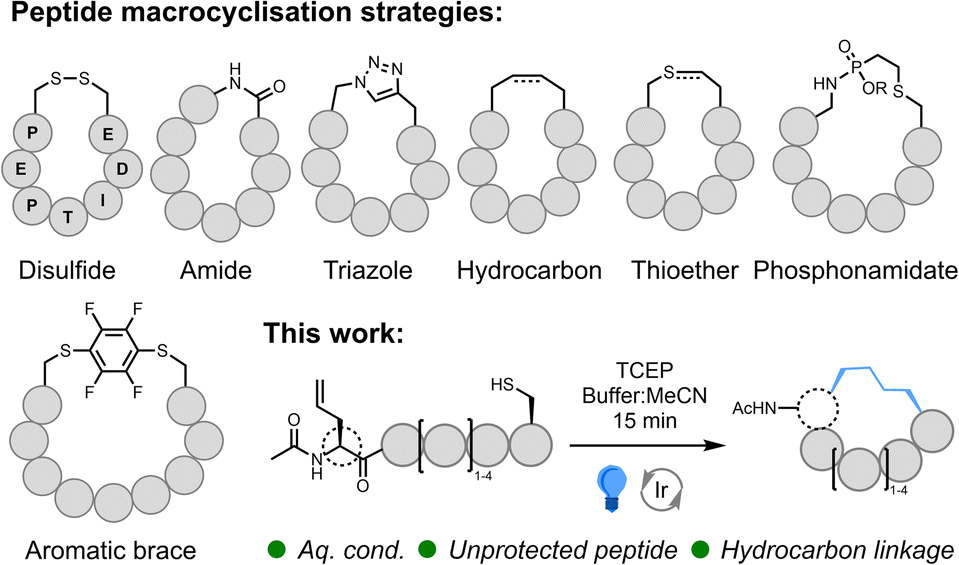 | ||
| Fig. 1 Commonly applied peptide macrocyclisation strategies; interception of desulfurisation as an approach to macrocyclisation of unprotected peptides. | ||
We recently reported a novel strategy for the site-selective modification of peptides and proteins via visible-light-mediated desulfurative C(sp3)–C(sp3) bond formation.54,55 Desulfurisation of Cys (and alternative non-proteinogenic thiol-containing amino acids56–58) can be applied post-peptide ligation as an elegant method to access a broad range of ligation junctions and facilitate chemical protein synthesis.56,59–67 A widely used free-radical-mediated Cys desulfurative protocol,59 developed by Danishefsky and co-workers, proceeds via a thiophosphoranyl radical species generated using a radical initiator (VA-044) to form a thiyl radical from the thiol sidechain of Cys in the presence of the water-soluble phosphine, tris(2-carboxyethyl)phosphine hydrochloride (TCEP). β-Scission of the thiophosphoranyl radical62 produces a peptide ‘alanyl’ radical which, in the presence of a suitable thiol additive (e.g. glutathione), will abstract an H-atom to yield the residue, alanine (Ala), at the ligation junction.59 In previous work, we demonstrated desulfurisation using an iridium(III) photocatalyst (PC) and employed alkenes to intercept the alanyl radical species, enabling installation of Lys sidechains carrying natural modifications as well as effective mimics of this modified sidechain.54,55 This reaction is initiated via excitation of the Ir(III) PC by a photon of visible light. The activated catalyst is then reduced by the thiol group producing a thiol radical cation which forms a thiyl radical on deprotonation. In the presence of TCEP, the thiophosphoranyl radical is formed; β-scission of this species produces the ‘alanyl’ radical60 which is trapped by the alkene. Due to the requirement to out-compete H-atom abstraction during this process, a large excess of the alkene is required (a minimum of 200 equivalents). By installing an appropriate alkene into a peptide containing a Cys residue, we postulated that intramolecular trapping of the radical produced upon desulfurisation may proceed preferentially to H-atom abstraction, essentially allowing us to use an equimolar equivalent of the alkene (Fig. S1†). The cyclic peptide radical produced during this reaction will be quenched to form the macrocyclic product; H-atom transfer (HAT) from the thiol group of remaining starting peptide is a likely pathway. The resulting thiyl radical can then continue the cycle or be reduced by the catalyst and protonated.68 Oxygen in the buffer has also been identified as an oxidant for the catalyst during Ru-mediated desulfurisation.60
The chemoselectivity of this chemistry should ensure that the reaction enables efficient cyclisation of unprotected peptides in aqueous solution. If realised, this technique would be a valuable addition to the toolkit available to researchers for the production of cyclic peptides.
Results and discussion
Optimisation of reaction conditions and radical trap exploration
Initial trials of this strategy focused on the utilisation of both 2-methylallyl and allyl moieties to facilitate peptide cyclisation in the presence of a Cys residue (Table 1). Addition of the alanyl radical to a 2-methylallyl group would generate a tertiary radical intermediate previously shown to improve the efficiency of this chemistry;55 however, diastereomers of the desired macrocycle will be produced due to the methyl branch of the linkage. Employing an allyl group as the radical trap would result in a single product, however, the reaction will be less effective and may not fully out-compete H-atom abstraction. Both of these strategies were initially explored. Amino acid building blocks 1 and 2 were synthesised via alkylation of Boc-Ser-OH with 1-bromo-3-methylbut-3-ene and allyl bromide, respectively (ESI†). These residues were incorporated into a simple peptide sequence using solid phase peptide synthesis (SPPS) to afford peptides 3 (H–S(O-2-methylallyl)AFAC-NH2) and 4 (H–S(OAllyl)AFAC-NH2). Both peptides (3/4, 0.5 mM) were subjected to desulfurisation conditions in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 conjugation buffer (6 M Gdn·HCl, 0.1 M Na2HPO4, pH 7.5–8.0):acetonitrile (MeCN) in the presence of a phosphine (TCEP; 5 mM) and an Ir(III) PC (5 mol%, (Ir[dF(CF3)ppy]2(dtbpy))PF6), under irradiation of blue light (450 nm) using inexpensive blue LED light strips (see ESI† for details). The reaction progress was monitored using analytical HPLC. Excess phosphine was employed to ensure that the thiyl radical did not interfere with the reaction via thiol–ene radical addition.45 Promisingly, we observed complete and clean conversion of the starting peptide 3 within 10 min. As anticipated, two products were observed, each with the same mass. Purification of the material via preparative HPLC was followed by NMR analysis which determined that the two products were diastereomers of the desired cyclised peptide, isolated in an excellent combined yield of 77% (5a/5b, 53% and 24%, respectively) (Table 1, entry 1). No resonances were observed corresponding to the allyl group in the 1H NMR spectrum, which precludes the misidentification of the desulfurised starting peptide as the product; a concern considering these structures share the same molecular weight.
:1 conjugation buffer (6 M Gdn·HCl, 0.1 M Na2HPO4, pH 7.5–8.0):acetonitrile (MeCN) in the presence of a phosphine (TCEP; 5 mM) and an Ir(III) PC (5 mol%, (Ir[dF(CF3)ppy]2(dtbpy))PF6), under irradiation of blue light (450 nm) using inexpensive blue LED light strips (see ESI† for details). The reaction progress was monitored using analytical HPLC. Excess phosphine was employed to ensure that the thiyl radical did not interfere with the reaction via thiol–ene radical addition.45 Promisingly, we observed complete and clean conversion of the starting peptide 3 within 10 min. As anticipated, two products were observed, each with the same mass. Purification of the material via preparative HPLC was followed by NMR analysis which determined that the two products were diastereomers of the desired cyclised peptide, isolated in an excellent combined yield of 77% (5a/5b, 53% and 24%, respectively) (Table 1, entry 1). No resonances were observed corresponding to the allyl group in the 1H NMR spectrum, which precludes the misidentification of the desulfurised starting peptide as the product; a concern considering these structures share the same molecular weight.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Peptide | [Peptide]a/mM | [TCEP]/mM | Ir(III) PC (mol%) | Reaction durationb (min) | Isolated yieldc [% conversiond] |
| a Reaction conducted in 10% acetonitrile (MeCN)/6 M Gdn·HCl, 0.1 M Na2HPO4, pH 7.5–8.0. b LED strips, photochem set up 1 (ESI†). c Peptide products isolated by preparative HPLC. d % Conversion calculated via analytical HPLC. e Combined yield from both diastereomers. f Diastereomeric ratio calculated via analytical HPLC. g No reaction observed in the absence of blue light. h Reaction completed in 15 min using a PhotoRedOx box (photochem set up 2 – ESI†). | ||||||
| 1 | 3 | 0.5 | 5 | 5 | 10 | 77%e (dr 69:31)f |
| 2 | 4 | 0.5 | 5 | 5 | 60 | [72%] |
| 3 | 4 | 0.5 | 2.5 | 5 | 60 | [38%] |
| 4 | 4 | 0.5 | 5 | 1 | 60 | [36%] |
| 5 | 4 | 0.5 | 25 | 5 | 60 | 78% |
| 6g | 7a | 0.5 | 25 | 1 | 45 [15]h | 79% |
| 7 | 10 | 0.5 | 25 | 1 | 60 | 32% |
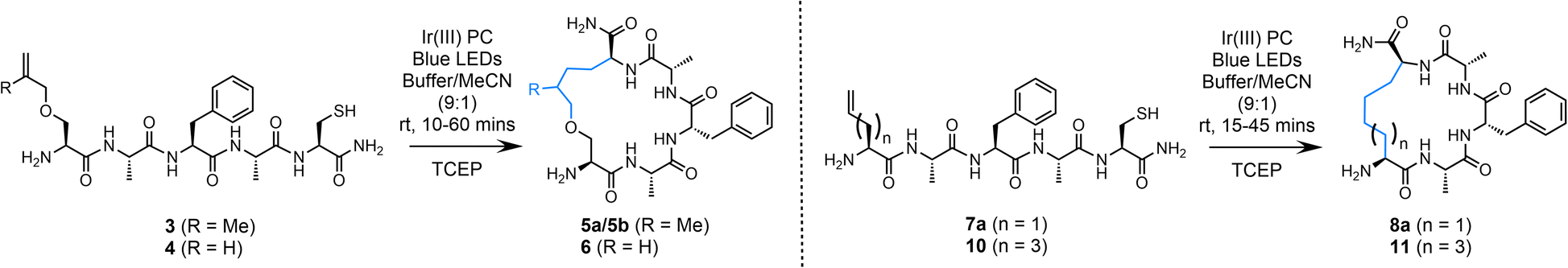
Quantitative conversion of the starting sequence to cyclised product, with negligible formation of the linear desulfurised by-product (formed via the alanyl radical abstracting an H-atom), is a gratifying initial result that allows high-yielding and rapid access to macrocyclic peptides. However, the formation of diastereomers is not ideal. Peptide 4 carries an allyl group as the radical trap; while this moiety would generate a less stable secondary radical upon addition of the alanyl radical, our previous results gave us confidence that intramolecular trapping should still out-compete H-atom abstraction. Under the conditions described, the starting peptide (4) was consumed within 60 min leading to the production of a major product (72% conversion to product by analytical HPLC, entry 2). It was noted that the reaction was equally effective without using degassed buffer; therefore, this step was omitted from the protocol. Prior to scaling the reaction up for isolation, a brief optimisation study was undertaken. It was observed that a reduction in the equivalents of TCEP led to a dramatic decrease in conversion to the product (Entry 3; 38% conversion to product 6), as did decreasing the mol% loading of the PC from 5 to 1 mol% (entry 4). When scaling up to an isolable yield it was observed that the conditions detailed in entry 2 were not optimal; an increase in the equivalents of TCEP to 50 (25 mM) was necessary to maintain high conversion to product. Using these adjusted conditions (entry 5) the desired product (6) was isolated in 78% yield and characterised via MS and NMR spectroscopy. The remaining mass balance for these reactions was the linear desulfurised by-product. No peptide degradation or epimerisation was observed over the course of the reaction.
Due to the need to synthesise the allyl-protected serine (Ser) building block, a more readily accessible option was sought. The commercially available amino acid, Fmoc-allyl-Gly-OH (alGly, alG), would afford a cyclic peptide with a butyl hydrocarbon linker. Therefore, Fmoc-protected alGly was incorporated into a model peptide (H-(alG)AFAC-NH2; 7a) and cyclised using the optimised conditions based on entry 5, Table 1 with a decrease in the PC loading to 1 mol% (entry 6), a change which which did not hinder conversion to the product. The reaction proceeded to completion as expected over 45 min and the desired product (8a) was isolated in an excellent yield of 79% by preparative HPLC (Fig. 2). The following conditions were therefore identified as optimal: 0.5 mM peptide, 1 mol% PC, 25 mM TCEP, in 10% MeCN/6 M Gdn·HCl, 0.1 M Na2HPO4, pH 7.5–8.0 (degassing step omitted). The reaction was then repeated using a PhotoRedOx Box equipped with a 34 mW cm−2, 450 nm LED (HeptatoChem). The ratio of desired product to by-product remained consistent with the reaction performed using blue LED light strips, however the rate of the reaction was enhanced, reaching completion in just 15 min (Fig. S49–S54†).
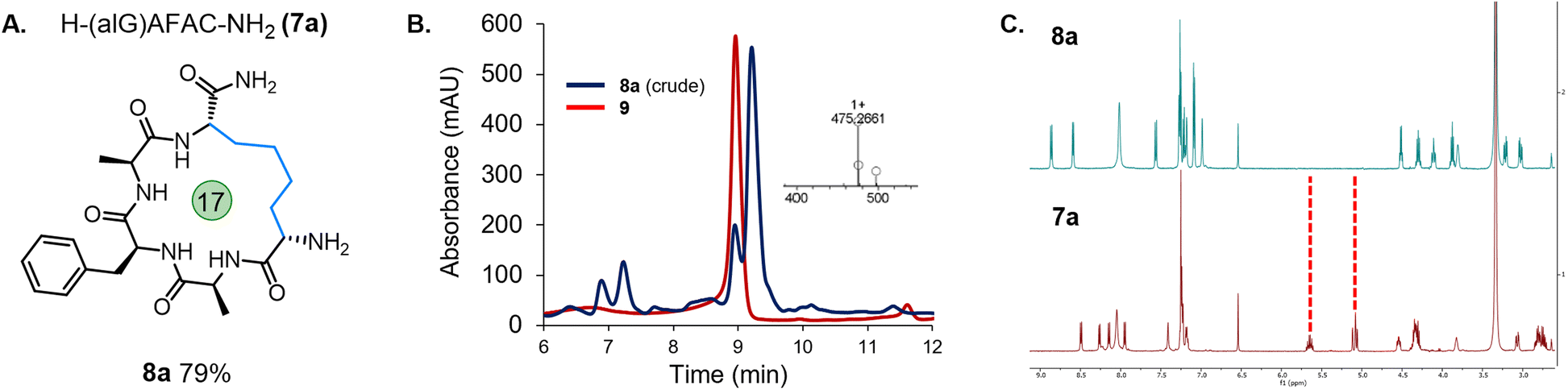 | ||
| Fig. 2 (A) Cyclised peptide 8a from H-(alG)AFAC-NH2 (7a); (B) analytical HPLC of the crude cyclisation of 7a (blue trace) overlaid with peptide H-(alG)AFAA-NH2 (9), inset – ESI MS showing the mass of the desired cyclised product (8a); (C) 1H NMR spectrum for linear peptide 7a (red trace) and peptide macrocycle 8a (blue trace) showing the absence of the allylic protons. | ||
While 1H NMR analysis of 8a confirmed that the allylic protons were not present (indicating successful cyclisation) (Fig. 2), further analysis was sought to fully characterise the macrocycle for peptide 8a. A TOCSY NMR experiment was carried out on this model and a complete assignment of the macrocycle was achieved (Fig. 3). Proton environments in the hydrocarbon linker were found to couple to Hα signals on residues at each end of the macrocycle indicating successful cyclisation. Moreover, the Hα signal from what was initially the Cys residue prior to cyclisation coupled to the Hα of the first Ala residue, which could only occur as a result of macrocycle formation. A significant chemical shift dispersion suggests an ordered structure. Furthermore, the phenylalanine (Phe) Hα resonances show an NOE interaction to the Hα of the allyl glycine position, suggesting that the macrocycle is strained.
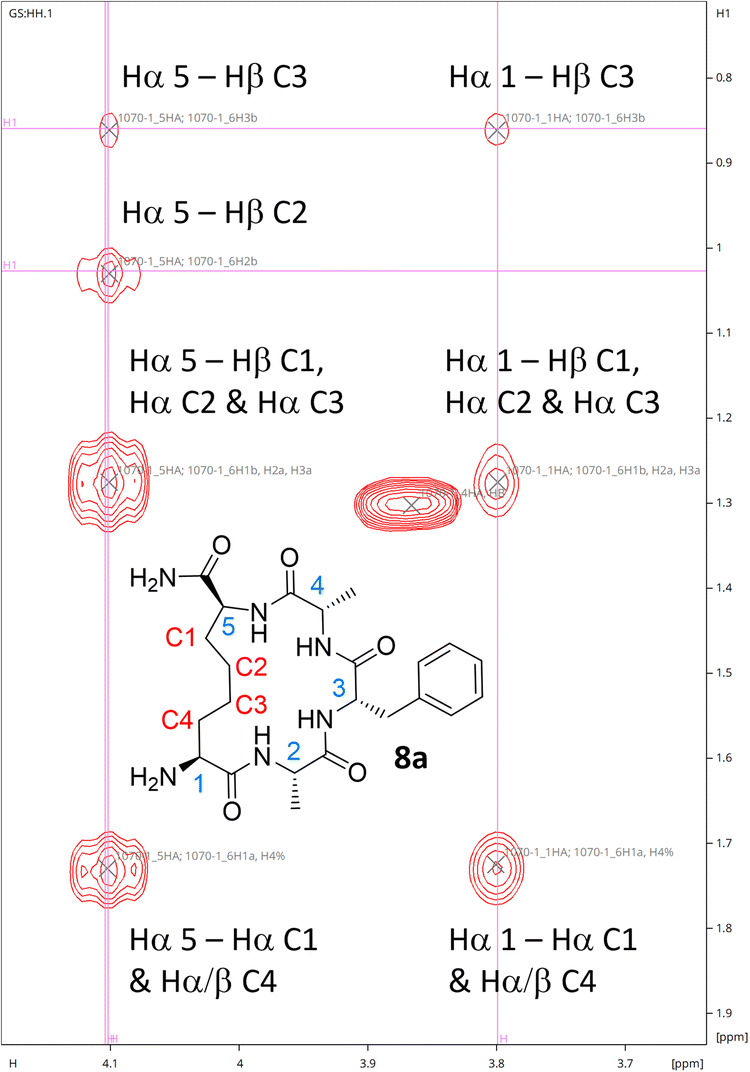 | ||
| Fig. 3 Hα region for the TOCSY NMR spectrum of 8a confirming successful formation of the macrocycle. | ||
To further explore the optimised reaction, we considered buffer composition, the phosphine additive, and alternative desulfurisation conditions. Cyclisation of model 7a in PBS did not proceed cleanly (Fig. S55†), while employing HEPES buffer did lead to clean conversion of the starting peptide, however, the undesired desulfurised linear by-product was the dominant product (Fig. S56†). Thus, we conclude that cyclisation is most effective in a buffer containing a high concentration of chaotropic salt. Replacing TCEP with the water soluble phosphine, 3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt (TPPTS) did not lead to effective conversion of the starting peptide 7a (Fig. S57†). Employing 1,3,5-triaza-7-phosphaadamantane (PTA) initially appeared to eliminate the production of the undesired linear desulfurised by-product (Fig. S58†). However, repetition of the reaction on an isolable scale and purification by preparative HPLC revealed the by-product and other impurities eluting under the broader product peak (Fig. S59–S62†) which ultimately limited the isolated yield. Finally, the peptide H-(alG)AFAC-OH (7b) was synthesised on 2-CTC resin to probe the cyclisation on peptides bearing a C-terminal carboxylic acid. While conversion to the desired product was observed (Fig. S68†), the ratio of by-product to macrocycle was less favourable when compared to C-terminal amide peptides. In addition, for this simple model, the retention times for the desired product and by-product were very similar making separation a significant challenge.
Several methods for the desulfurisation of Cys residues have been reported in the literature, these include; photo-induced desulfurisation69 using a ruthenium PC,60 photo-desulfurisation in flow,61 accelerated desulfurisation using tetraethylborate (NaBEt4),63 desulfurative borylation,64 and the exploitation of phosphite66 and phosphine-dependent pathways.65 We attempted to adapt and explore two appropriate examples.60,63 However, both the use of NaBEt463 and a ruthenium photocatalyst60 failed to improve on our reported conditions (Fig. S63 and S64–S67†).
To enable the synthesis of peptide macrocycles with longer hydrocarbon linkages, the amino acid pentenyl glycine (pGly, pG) was incorporated into the N-terminus of a simple model peptide (H-(pG)AFAC-NH2; 10) and this peptide subjected to the optimised cyclisation conditions. The desired product (11) was successfully formed but in low yield compared to the alGly example (Table 1 and Fig. 4). Furthermore, in addition to the use of non-proteinogenic amino acids carrying alkenes, we also explored the on-resin installation of similar radical traps. The N-terminus of H-AFAC-NH2 was functionalised on-resin using a solution of pentafluorophenyl acrylate46 (12) to afford the linear acrylamide peptide, 13. When subjected to the optimised cyclisation conditions the starting peptide was fully consumed within 60 min; LC-MS analysis of the crude reaction material suggested the formation of an interesting phosphonium salt by-product (Fig. S74†) but no cyclised material was observed.
 | ||
| Fig. 4 Linear starting peptides (10, 14–20) and the macrocyclic peptide products formed via desulfurative C–C bond formation (11, 21–27); macrocycle size indicated. | ||
Exploration of reaction scope and tolerance
To further explore the tolerance and scope of this cyclisation methodology we synthesised several pentapeptides carrying a range of proteinogenic amino acids (14–20; Fig. 4). When positioning the alGly residue at the N-terminus and the Cys at the C-terminus, effective N- to C-terminal macrocyclisation was realised for all sequences explored to yield 17-membered macrocycles (wrt the number of bonds within the macrocycle) in moderate to excellent yields (21–27, 48–74% isolated yield). The model peptides explored carried the majority of the 20 canonical amino acids, demonstrating the tolerance of this reaction to the diverse chemical functionality displayed across the proteome. Interestingly, switching the terminal residues eliminated or hindered conversion to the desired product for this size of macrocycle (Table S1†). To explore macrocyclisation of a peptide carrying the radical trap residue at an internal position, peptide 20 was synthesised with alGly as the penultimate N-terminal residue. For this model, cyclisation was successful in high yield (27, 82%). Moving to longer sequences we explored several 6, 7, & 8-residue peptides carrying a range of proteinogenic residues (28–38) to yield 20, 23, and 26-membered peptide macrocycles in moderate to excellent yield (39–48, 32–76%, Fig. 5). All macrocyclic products were characterised by analytical HPLC, MS and 1H NMR; CD spectroscopy was run for examples of each size of macrocycle (ESI†). Switching the N- and C-terminal residues for model 29 (H-CKISY(alG)-NH2) had no effect on yield for this size of macrocycle, unlike the smaller model peptides explored (Table S1†). In several cases of low or negligible yield (e.g., 40a) acetylation of the N-terminus reinstated a moderate yield (40b, 48%). To confirm the effect that buffer composition has on the isolated yield, cyclisation of model 28 was carried out in PBS. Conversion to the desired product was again not as effective in this buffer compared to cyclisation in conjugation buffer. However, for this model, the ratio of product to desulfurised linear by-product was comparable to that of the conjugation buffer example. Additionally, PTA was revisited for the low-yielding model 31. While this additive did not give an improvement in yield for model 7a, we found that it improved the conversion to the product for the more complex model 31, increasing the yield from 37% to 52% (41). This result indicates that, while TCEP gives more favourable results for the majority of sequences, alternative phosphines can be employed.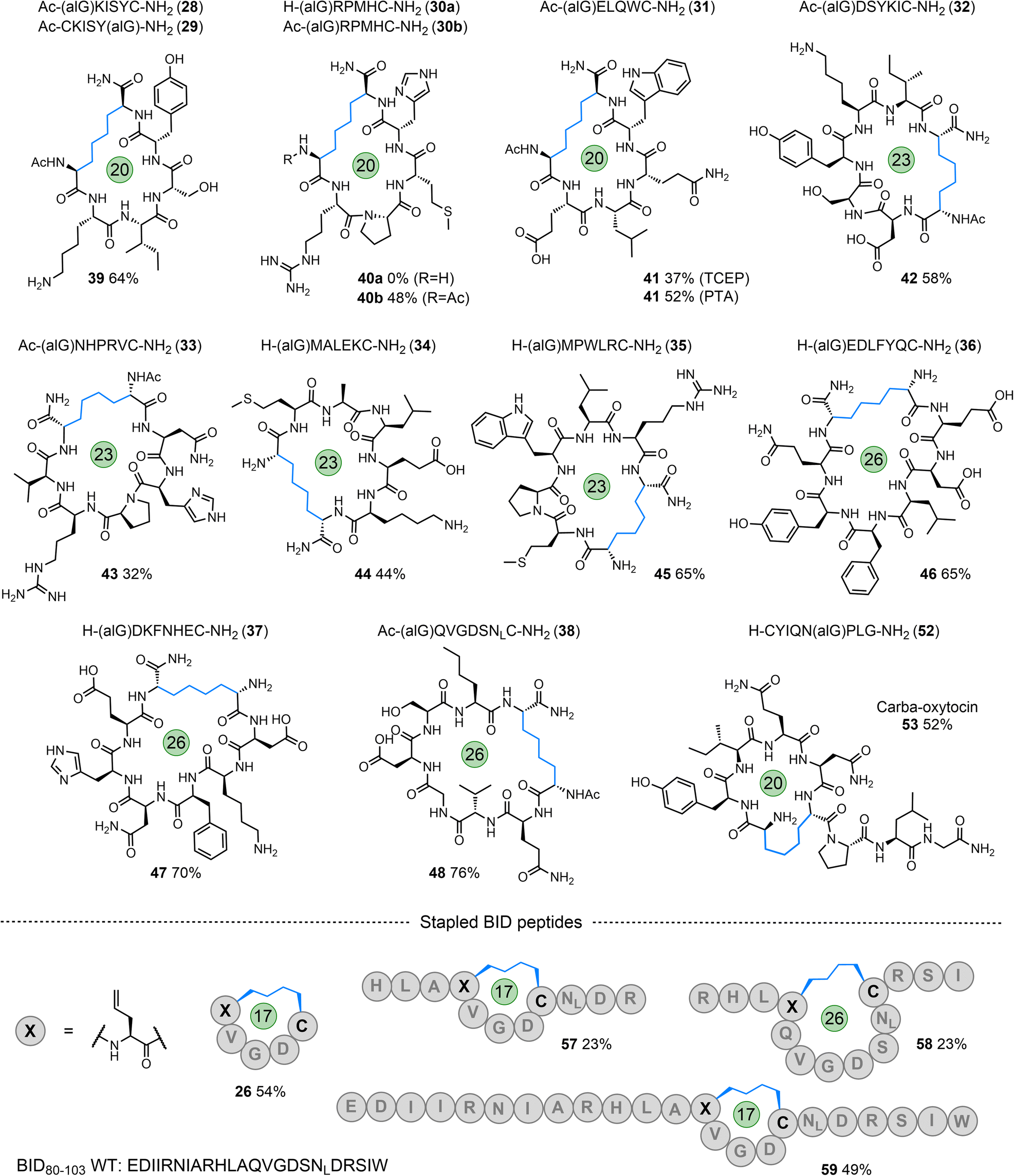 | ||
| Fig. 5 Starting peptides (28–38) and the macrocyclic peptide products formed via desulfurative C–C bond formation (39–48); carba-oxytocin (53); internally ‘stapled’ MCL-1 binding peptides (57–59), initial positions of alGly and Cys indicated within the sequence of these products; macrocycle size indicated. | ||
A final 14-residue model peptide (49) was prepared to explore the preparation of larger (44-membered) macrocycles. Under the optimised cyclisation conditions the starting peptide was fully consumed to afford an inseparable mixture of the desired cyclised product and the desulfurised linear by-product. Increasing the loading of the PC to 5 mol% resulted in formation of the postulated phosphonium by-product observed for acrylamide model 13 (Fig. S144–S146†). This by-product (50) was isolated in 58% yield; 31P NMR analysis gave a single phosphorus resonance at 36.6 ppm. Our strategy is, therefore, highly effective for the formation of peptide macrocycles up to a 26-membered ring, and tolerates the full range of proteinogenic chemical functionality found across the proteome. Access to larger macrocycles may be possible, but will be dependent on successful separation of the desired product from the linear desulfurised by-product.
Macrocyclisation of therapeutic peptides
To apply this technology to biomedically relevant peptides, the hormone oxytocin was synthesised with alGly replacing one of the Cys residues in the peptide to install a hydrocarbon ‘brace’ in place of the native disulfide (carba-oxytocin). This modification has been previously explored to enhance the proteolytic and hydrolytic stability of this peptide.70 Positioning alGly at the N-terminus and Cys at an internal position for this model (51, ESI†) failed to afford the desired product when applying the cyclisation conditions, instead producing desulfurised linear material only. Interestingly, Scanlan, Petracca and co-workers observed the same challenge when attempting to cyclise oxytocin via a thiol–ene reaction using an N-terminal alGly residue.71 We found that switching the positions of the Cys and alGly residues (sequence 52) re-instated the cyclisation, affording the desired macrocycle in moderate yield (53, 52% isolated yield, Fig. 5).Finally, to explore internal peptide ‘stapling’ using this strategy, we selected a region of a BH3 protein – BID80–102, that binds MCL-1 to regulate apoptosis.53,72 This PPI is known to play a significant role in cancer development and progression.53,72 Three sequences (54–56) were synthesised with alGly and Cys positioned to afford either an internal i, i + 4 staple (57, 59; Fig. 5 and ESI†) or i, i + 7 staple (58; Fig. 5 and ESI†), fixing the length of either one or two full helical turns of the peptide, respectively. In addition, peptide macrocycle 26 is a head-to-tail macrocycle representing the i, i + 4 binding region of these longer sequences. The staple for all three peptides was successfully formed under the standard conditions, albeit in lower yield than the N- to C-terminal cyclisations previously explored (57–59). The alpha-helical structure of peptide 59 was assessed via CD spectroscopy. While macrocyclisation did increase the helicity by a few percent compared to the native BH3 sequence (22% compared to 19%), the starting peptide carrying the allyl glycine residue had a relatively high helical content (38%, Fig. S159†). This can be rationalised by considering the strain on the macrocycle imposed by the linker, and the fact that the native residues glutamine (Gln) and Ser were switched for alGly and Cys, respectively. These original residues have higher helical propensities relative to their replacements. Competitive inhibition studies (measured via fluorescence anisotropy) using MCL-1 and the fluorophore-labelled WT BH3 sequence demonstrated slightly lower inhibitory potency (26 ± 4 mM) for 59 compared to BID-wt (7.4 ± 0.9 mM) (Fig. S160†).
Conclusions
Herein, we report a powerful method for the cyclisation of peptides via desulfurative C(sp3)–C(sp3) bond formation. Our approach is operationally simple, effective ‘on the bench’ under ambient conditions with irradiation of blue light, utilising readily available starting building blocks. The reaction is rapid, high yielding (for most cases studied) on unprotected peptides in aqueous solution and tolerant to all proteinogenic chemical functionality. The technique enables the preparation of a range of macrocycle sizes and extends to internal macrocyclisation (peptide stapling). This technology presents a more sustainable alternative to the widely employed RCM, offering an effective new method for peptide cyclisation.Data availability
The experimental procedures and compound/peptide characterisation data can be found in the ESI.†Author contributions
R. C. G. and N. J. M. conceived the project, F. R. S., R. C. G., D. M., H. J. K., P. Z., and H. E. L. W. carried out the experimental work. A. J. W. and N. J. M. supervised the work, N. J. M. wrote the manuscript. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the UKRI Engineering and Physical Sciences Research Council [EP/S028323/1, EP/S017739/1, and EP/N013573/1] and the Biotechnology and Biological Sciences Research Council [BB/V008412/1 and BB/V003577/1], the Leverhulme Trust [RPG-2023-022], and from the University of Nottingham (PhD studentship for RCG). We thank Dr Mattia Silvi (School of Chemistry, University of Nottingham) for helpful discussions.References
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- Y. H. Lau, P. De Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- H. C. Hayes, L. Y. Luk and Y.-H. Tsai, Org. Biomol. Chem., 2021, 19, 3983–4001 RSC.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554 RSC.
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 CrossRef CAS PubMed.
- F. Rohrbacher, G. Deniau, A. Luther and J. W. Bode, Chem. Sci., 2015, 6, 4889–4896 RSC.
- L. Zhang and J. P. Tam, J. Am. Chem. Soc., 1997, 119, 2363–2370 CrossRef CAS.
- N. Ollivier, T. Toupy, R. C. Hartkoorn, R. Desmet, J.-C. M. Monbaliu and O. Melnyk, Nat. Commun., 2018, 9, 2847 CrossRef PubMed.
- A. Tavassoli, Curr. Opin. Chem. Biol., 2017, 38, 30–35 CrossRef CAS PubMed.
- A. Tavassoli and S. J. Benkovic, Nat. Protoc., 2007, 2, 1126–1133 CrossRef CAS PubMed.
- R. Wills, V. Adebomi and M. Raj, ChemBioChem, 2021, 22, 52–62 CrossRef CAS PubMed.
- C. A. Rhodes and D. Pei, Chem.–Eur. J., 2017, 23, 12690–12703 CrossRef CAS PubMed.
- S. Chen, D. Bertoldo, A. Angelini, F. Pojer and C. Heinis, Angew. Chem., Int. Ed., 2014, 53, 1602–1606 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- X.-D. Kong, J. Moriya, V. Carle, F. Pojer, L. A. Abriata, K. Deyle and C. Heinis, Nat. Biomed. Eng., 2020, 4, 560–571 CrossRef CAS PubMed.
- J. R. Pace, B. J. Lampkin, C. Abakah, A. Moyer, J. Miao, K. Deprey, R. A. Cerulli, Y.-S. Lin, J. D. Baleja, D. Baker and J. A. Kritzer, J. Am. Chem. Soc., 2021, 143, 15039–15044 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- M. S. Islam, S. L. Junod, S. Zhang, Z. Y. Buuh, Y. Guan, M. Zhao, K. H. Kaneria, P. Kafley, C. Cohen, R. Maloney, Z. Lyu, V. A. Voelz, W. Yang and R. E. Wang, Nat. Commun., 2022, 13, 350 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhang, C. T. Wong and X. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef CAS PubMed.
- B. Li, Z. Wan, H. Zheng, S. Cai, H.-W. Tian, H. Tang, X. Chu, G. He, D.-S. Guo, X.-S. Xue and G. Chen, J. Am. Chem. Soc., 2022, 144, 10080–10090 CrossRef CAS PubMed.
- S. Voss, J. Rademann and C. Nitsche, Angew. Chem., Int. Ed., 2022, 61, e202113857 CrossRef CAS PubMed.
- A. J. Cameron, P. W. R. Harris and M. A. Brimble, Angew. Chem., Int. Ed., 2020, 59, 18054–18061 CrossRef CAS PubMed.
- X. Y. Liu, X. Ji, C. Heinis and J. Waser, Angew. Chem., Int. Ed., 2023, 62, e202306036 CrossRef CAS PubMed.
- S. Gary and S. Bloom, ACS Cent. Sci., 2022, 8, 1537–1547 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- Y. H. Lau, P. De Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Sköld, T. J. Sum, P. J. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- R. Kleineweischede and C. P. Hackenberger, Angew. Chem., Int. Ed., 2008, 47, 5984–5988 CrossRef CAS PubMed.
- M.-A. Kasper, M. Glanz, A. Oder, P. Schmieder, J. P. von Kries and C. P. Hackenberger, Chem. Sci., 2019, 10, 6322–6329 RSC.
- L. R. Malins, J. N. deGruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233–5241 CrossRef CAS PubMed.
- L. J. Davies, L. M. Shuttleworth, X. Zhang, S. Peng and C. Nitsche, Org. Lett., 2023, 25, 2806–2809 CrossRef CAS PubMed.
- Z. Bai, C. Cai, W. Sheng, Y. Ren and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 14686–14692 CrossRef CAS PubMed.
- D. G. Rivera, G. M. Ojeda-Carralero, L. Reguera and E. V. Van der Eycken, Chem. Soc. Rev., 2020, 49, 2039–2059 RSC.
- P. Yang, X. Wang, B. Li, Y. Yang, J. Yue, Y. Suo, H. Tong, G. He, X. Lu and G. Chen, Chem. Sci., 2021, 12, 5804–5810 RSC.
- X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540–548 CrossRef CAS PubMed.
- B. Li, X. Li, B. Han, Z. Chen, X. Zhang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 9401–9407 CrossRef CAS PubMed.
- P. Yang, M. J. Širvinskas, B. Li, N. W. Heller, H. Rong, G. He, A. K. Yudin and G. Chen, J. Am. Chem. Soc., 2023, 145, 13968–13978 CrossRef CAS PubMed.
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS PubMed.
- Y. Wang and D. H. C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- L. Raynal, N. C. Rose, J. R. Donald and C. D. Spicer, Chem.–Eur. J., 2021, 27, 69–88 CrossRef CAS PubMed.
- M. Ahangarpour, I. Kavianinia, P. W. Harris and M. A. Brimble, Chem. Soc. Rev., 2021, 50, 898–944 RSC.
- S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed.
- X. Hu, X. Chen, B. Li, G. He and G. Chen, Org. Lett., 2021, 23, 716–721 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- E. C. Gleeson, W. R. Jackson and A. J. Robinson, Tetrahedron Lett., 2016, 57, 4325–4333 CrossRef CAS.
- Y.-W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed.
- J. A. Miles, D. J. Yeo, P. Rowell, S. Rodriguez-Marin, C. M. Pask, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Sci., 2016, 7, 3694–3702 RSC.
- R. C. Griffiths, F. R. Smith, J. E. Long, D. Scott, H. E. Williams, N. J. Oldham, R. Layfield and N. J. Mitchell, Angew. Chem., Int. Ed., 2022, 61, e202110223 CrossRef CAS PubMed.
- R. C. Griffiths, F. R. Smith, D. Li, J. Wyatt, D. M. Rogers, J. E. Long, L. M. Cusin, P. J. Tighe, R. Layfield, J. D. Hirst, M. M. Muller and N. J. Mitchell, Chem. - Eur. J., 2023, 29, e202202503 CrossRef CAS PubMed.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem, 2018, 2, 0122 CrossRef CAS.
- L. R. Malins, N. J. Mitchell and R. J. Payne, J. Pept. Sci., 2014, 20, 64–77 CrossRef CAS PubMed.
- R. C. Griffiths, F. R. Smith, J. E. Long, H. E. Williams, R. Layfield and N. J. Mitchell, Angew. Chem., Int. Ed., 2020, 59, 23659–23667 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- X.-F. Gao, J.-J. Du, Z. Liu and J. Guo, Org. Lett., 2016, 18, 1166–1169 CrossRef CAS PubMed.
- T. S. Chisholm, D. Clayton, L. J. Dowman, J. Sayers and R. J. Payne, J. Am. Chem. Soc., 2018, 140, 9020–9024 CrossRef CAS PubMed.
- J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, ACS Catal., 2020, 10, 7250–7261 CrossRef CAS PubMed.
- Z. Sun, W. Ma, Y. Cao, T. Wei, X. Mo, H. Y. Chow, Y. Tan, C. H. Cheung, J. Liu, H. K. Lee, E. C. Tse, H. Liu and X. Li, Chem, 2022, 8, 2542–2557 CAS.
- R. Jing, W. C. Powell, K. J. Fisch and M. A. Walczak, J. Am. Chem. Soc., 2023, 145, 22354–22360 CrossRef CAS PubMed.
- N. M. Venneti, G. Samala, R. M. Morsy, L. G. Mendoza, A. Isidro-Llobet, J. K. Tom, S. Mukherjee, M. E. Kopach and J. L. Stockdill, J. Am. Chem. Soc., 2023, 145, 1053–1061 CrossRef CAS PubMed.
- R. M. Morsy, G. Samala, A. Jalan, M. E. Kopach, N. M. Venneti and J. L. Stockdill, Chem. Sci., 2023, 14, 9016–9023 RSC.
- J. Zhang, H. Liu, S. Teng, Z. Liao, L. Meng, Q. Wan and S. Dong, Chem. Commun., 2023, 59, 6513–6516 RSC.
- X. Bao, W. Yu and G. Wang, Adv. Synth. Catal., 2023, 365, 2299–2309 CrossRef CAS.
- L. Geniller, M. Taillefer, F. Jaroschik and A. Prieto, ChemCatChem, 2023, 15, e202300808 CrossRef CAS.
- J. L. Stymiest, B. F. Mitchell, S. Wong and J. C. Vederas, Org. Lett., 2003, 5, 47–49 CrossRef CAS PubMed.
- M. D. Nolan, C. Shine, E. M. Scanlan and R. Petracca, Org. Biomol. Chem., 2022, 20, 8192–8196 RSC.
- P. E. Czabotar and A. J. Garcia-Saez, Nat. Rev. Mol. Cell Biol., 2023, 24, 732–748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05865d |
| This journal is © The Royal Society of Chemistry 2024 |