
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic investigations of the Fe(II) mediated synthesis of squaraines†
Yu
Liu‡
 a,
Nathan T.
Coles‡
*a,
Nathalia
Cajiao
bc,
Laurence J.
Taylor
a,
E. Stephen
Davies
a,
Alistair
Barbour
a,
Patrick J.
Morgan
a,
Kevin
Butler
a,
Ben
Pointer-Gleadhill
a,
Stephen P.
Argent
a,
Jonathan
McMaster
a,
Michael L.
Neidig
b,
David
Robinson
d and
Deborah L.
Kays
*a
a,
Nathan T.
Coles‡
*a,
Nathalia
Cajiao
bc,
Laurence J.
Taylor
a,
E. Stephen
Davies
a,
Alistair
Barbour
a,
Patrick J.
Morgan
a,
Kevin
Butler
a,
Ben
Pointer-Gleadhill
a,
Stephen P.
Argent
a,
Jonathan
McMaster
a,
Michael L.
Neidig
b,
David
Robinson
d and
Deborah L.
Kays
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Deborah.Kays@nottingham.ac.uk; Nathan.Coles@nottingham.ac.uk
bInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK
cDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
dDepartment of Chemistry and Forensics, School of Science and Technology, Nottingham Trent University, Nottingham, NG11 8NS, UK
First published on 7th May 2024
Abstract
The scission and homologation of CO is a fundamental process in the Fischer–Tropsch reaction. However, given the heterogeneous nature of the catalyst and forcing reaction conditions, it is difficult to determine the intermediates of this reaction. Here we report detailed mechanistic insight into the scission/homologation of CO by two-coordinate iron terphenyl complexes. Mechanistic investigations, conducted using in situ monitoring and reaction sampling techniques (IR, NMR, EPR and Mössbauer spectroscopy) and structural characterisation of isolable species, identify a number of proposed intermediates. Crystallographic and IR spectroscopic data reveal a series of migratory insertion reactions from 1Mes to 4Mes. Further studies past the formation of 4Mes suggest that ketene complexes are formed en route to squaraine 2Mes and iron carboxylate 3Mes, with a number of ketene containing structures being isolated, in addition to the formation of unbound, protonated ketene (8). The synthetic and mechanistic studies are supported by DFT calculations.
1. Introduction
The reduction and homologation of CO to obtain high-value organic compounds is a long-standing area of research interest. In industry, the Fischer–Tropsch process utilises heterogeneous catalysts and forcing conditions to convert CO, in the presence of H2, to hydrocarbon fuels.1,2 However, such reactions are unselective, affording products with a range of chain lengths.3–5 As such, the reaction of CO with homogeneous species to selectively generate specific organic products remains a tantalising prospect. Over the years, many examples of such reactions have been published from across the periodic table, utilising s-block,6–11 p-block,12–16 d-block,17–25 and f-block elements.26–31 Not only do these reactions provide interesting organic products, but the mechanisms of these reactions are also of significant interest. Often, a complex series of steps facilitate this reduction and homologation process.32,33 Understanding these reactions allows us greater insight into not only the chemistry of the elements involved, but also the utilisation of CO on an industrial scale. The Fischer–Tropsch process, being a heterogeneous reaction, is challenging to study mechanistically. The investigation of CO reduction by soluble transition metal complexes can, therefore, provide valuable information about the reactions that may occur in such a system.The first example of insertion of CO into a low-coordinate Fe(II) m-terphenyl complex was reported by Ni and Power.34 Upon exposure of the complex Fe(C6H3-2,6-Dipp2)2 (Dipp = 2,6-iPr2C6H3) to an atmosphere of CO, the complex Fe(CO)2[C(O)C6H3-2,6-Dipp2]2 (Dipp = 2,6-iPr2C6H3) was obtained. Subsequently, we reported the selective reduction and homologation of CO by the less bulky Fe(II) m-terphenyl complexes (2,6-Ar2C6H3)2Fe (Ar = Mes (2,4,6-Me3C6H2), 1Mes; Ar = Xyl (2,6-Me2C6H3), 1Xyl).35 The reaction proceeded at room temperature and 1 atm CO in toluene over 10 days. The final isolated products were a highly unusual squaraine (2Mes, 2Xyl, Scheme 1) featuring broken conjugation between the C4 and aryl rings, as well as Fe(CO)5 and an iron carboxylate (3Mes, 3Xyl, Scheme 1). This reaction was particularly noteworthy for the complete scission of the strong C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond (1072 kJ mol−1), which is unusual for a process occurring under such mild conditions. Reactions with the 1-naphthyl (Naph) substituted analogue (2,6-Naph2C6H3)2Fe (1Naph) afforded an isolable iron carbene complex (CO)3Fe[C(2,6-Ar2C6H3)OC(O)(2,6-Ar2C6H3)] (Ar = 1-Naph 4Naph), but this species did not show further reactivity to 2 and 3. Regardless, analogous iron carbene complexes (4Mes, 4Xyl) were proposed as intermediates in the reaction between 1Mes and 1Xyl with CO.
O bond (1072 kJ mol−1), which is unusual for a process occurring under such mild conditions. Reactions with the 1-naphthyl (Naph) substituted analogue (2,6-Naph2C6H3)2Fe (1Naph) afforded an isolable iron carbene complex (CO)3Fe[C(2,6-Ar2C6H3)OC(O)(2,6-Ar2C6H3)] (Ar = 1-Naph 4Naph), but this species did not show further reactivity to 2 and 3. Regardless, analogous iron carbene complexes (4Mes, 4Xyl) were proposed as intermediates in the reaction between 1Mes and 1Xyl with CO.
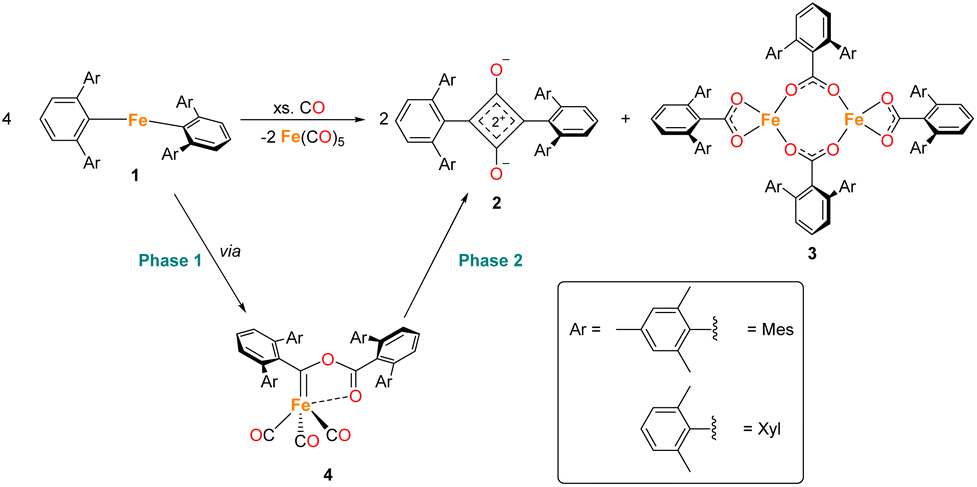 | ||
| Scheme 1 General reaction scheme for the reaction between 1 in the presence of CO, where Ar = Mes or Xyl. The two phases of the reaction are shown, phase 1 covers the reaction of 1 to 4, phase 2 covers the reaction from 4 to 2, 3 and Fe(CO)5. | ||
Herein, we report detailed mechanistic investigations of the homologation and scission of CO by a two-coordinate Fe(II) complex using in situ monitoring techniques (IR, NMR and EPR spectroscopy), frozen-solution Mössbauer (MB) spectroscopy and structural characterisation of intermediates via single crystal X-ray crystallography (scXRD). These experimental results are supported by DFT studies. From this, we propose a plausible mechanistic pathway, which highlights the unusual reactivity that can be facilitated using sterically demanding ligands which enforce low coordination environments at a metal centre.
2. Results and discussion
2.1 Overview
During the course of reaction monitoring (see discussion below) it became clear that the transformation of 1Mes–3Mes, and Fe(CO)5 proceeds in two distinct stages (Scheme 1). In phase 1, 1Mes reacts with CO, via several reactive intermediates, to afford an iron–carbene complex 4Mes (Scheme 1), which becomes the dominant species in solution. Phase 2 of the reaction involves 4Mes reacting with further equivalents of CO resulting in the formation of a number of species, and eventually the final products 2Mes, 3Mes and Fe(CO)5. For simplicity, we will discuss these two phases of the reaction separately. Sections 2.2 and 2.3 will focus on phase 1, covering all intermediates and observations up to the formation of 4Mes. Sections 2.4–2.7 will cover phase 2, looking at potential intermediates and pathways between 4Mes and the final products.2.2 Spectroscopic analysis of phase 1
In situ IR spectroscopy was conducted to monitor the transformation of the iron complex Fe(C6H3-2,6-Mes2)2 (1Mes) in the presence of CO over the course of the reaction. We postulate that the first step is the coordination of four CO molecules to 1Mes affording Fe(CO)4(C6H3-2,6-Mes2)2 (5, Scheme 2). IR spectra of the reaction between 1Mes and ca. 1 atm of CO in toluene were recorded at two-minute intervals. Within the first hour of the reaction, several carbonyl-containing species form which are subsequently consumed (Fig. 1). Initial intense signals at ν(CO) = 2014 cm−1, 2004 cm−1 and 1979 cm−1 were accompanied by less intense signals at ν(CO) = 2076 cm−1, 1942 cm−1 and 1935 cm−1. Following this, less intense peaks are observed at ν(CO) = 2063 cm−1 and 2035 cm−1 after approximately 30 minutes. The signals at ν(CO) = 2076 cm−1, 2014 cm−1 and 1979 cm−1 can be attributed to 6 which has been isolated and characterised independently in the solid state (See Section 2.3). After ca. 40 minutes of in situ monitoring the strongest IR bands occur at ν(CO) = 2049 cm−1, 1978 cm−1 and 1965 cm−1, which are assigned to 4Mes. These are similar to the previously reported 4Naph [ν(CO) = 2043 cm−1, 1972 cm−1 and 1954 cm−1], a structural analogue of 4Mes.35 It has not been possible to assign the remaining peaks to specific species, but they are postulated to be metal carbonyl complexes.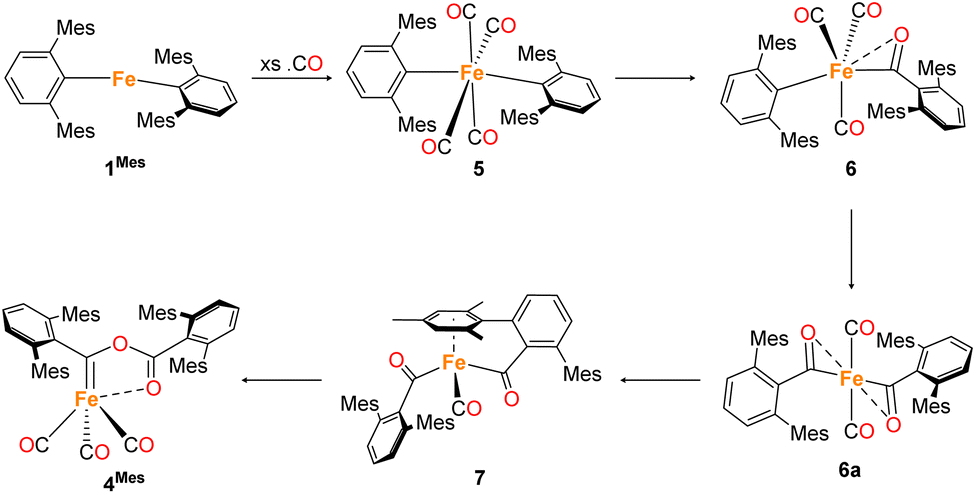 | ||
| Scheme 2 Proposed reaction pathway for formation of 4Mes from 1Mes. Note that 6a has only been observed spectroscopically in solution, all other species have been isolated and characterised by single crystal X-ray diffraction (See Section 2.3). | ||
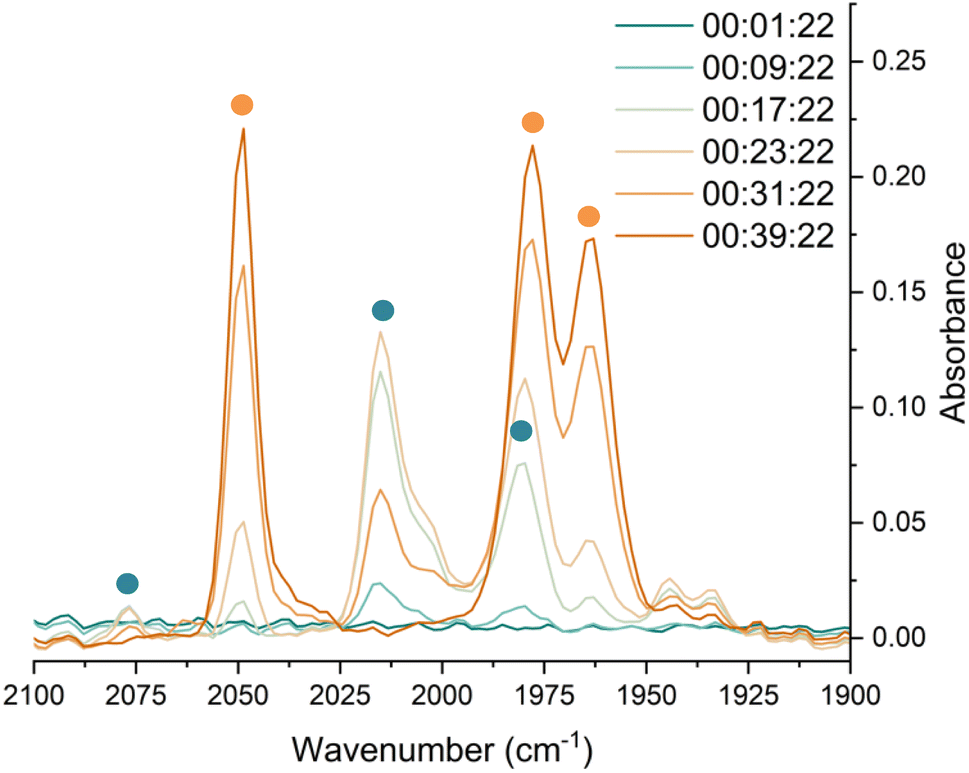 | ||
| Fig. 1 IR spectra for the first 40 minutes of the reaction between 1Mes and CO in toluene. CO added to the reaction after six minutes. The colour gradient goes from dark turquoise (earliest) to dark orange (latest). Orange circles highlight the signals for 4Mes, turquoise circles highlight the signals attributed to 6. | ||
The initial stages of the reaction were also monitored via1H NMR spectroscopy. 1Mes is a paramagnetic complex, and displays resonances over the range +80 to −180 ppm. However, upon introduction of CO, several new 1H NMR signals appear in the range 0–8 ppm. This is attributed to the formation of diamagnetic 18e− iron complexes. Forty minutes after addition of CO, several species form, resulting in a set of overlapping signals that cannot be resolved from one another (Fig. 2). As the reaction proceeds four signals become dominant at δH = 2.31 ppm, 2.23 ppm, 1.83 ppm and 1.65 ppm, consistent with a species featuring terphenyl moieties in two separate environments. The IR and 1H NMR spectroscopic data indicate one major species is formed, 4Mes (see Section 2.3). Full consumption of 1Mes takes ca. 36 h, with concomitant formation of a paramagnetic species (See Fig. S45–S47, ESI‡).
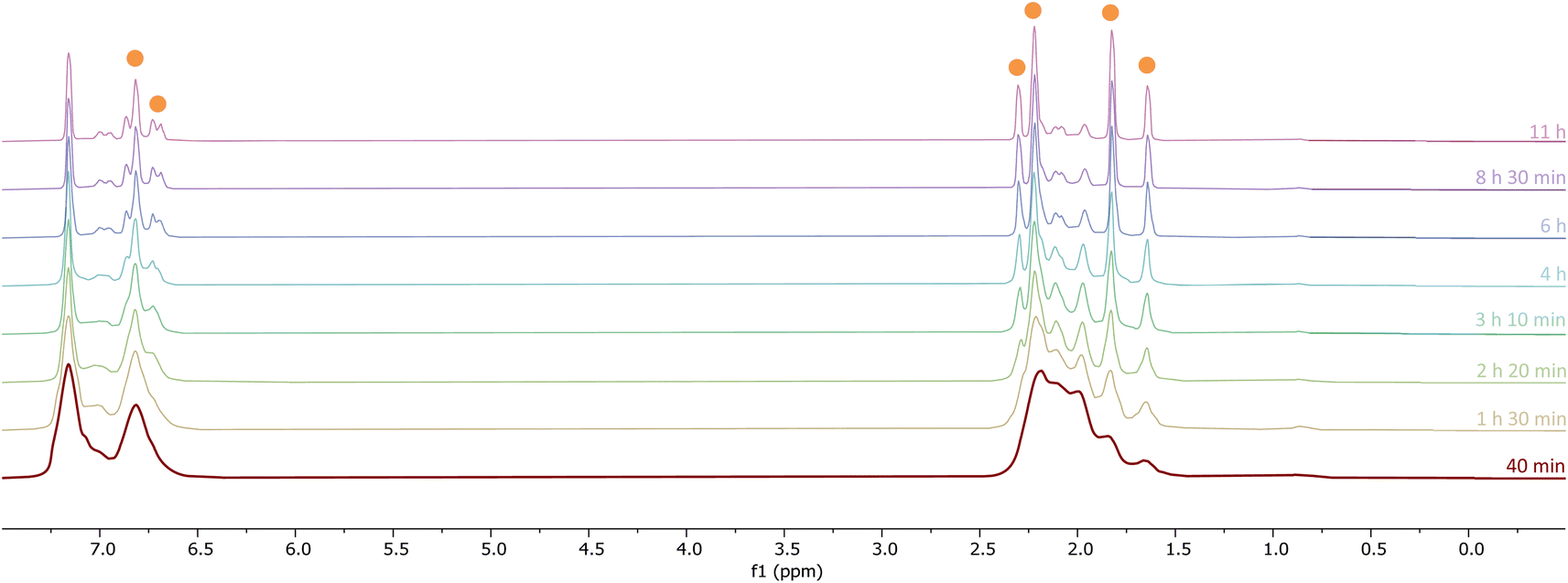 | ||
| Fig. 2 1H NMR spectra (−1 to 8 ppm) of the reaction between CO and 1Mes (C6D6, 1 atm CO, room temperature) recorded between 40 minutes and 11 hours of reaction. Orange circles highlight the signals for 4Mes. | ||
The use of 13CO allows the observation of intermediates by 13C{1H} NMR spectroscopy, showing the simultaneous formation of several compounds which contain CO in the form of acyl/carbene functional groups (250–270 ppm), metal-bound CO (220–200 ppm) and esters (176 ppm). Ni and Power have previously reported Fe(CO)2[C(O)C6H3-2,6-Dipp2]2 (Dipp = 2,6-iPr2C6H3) which showed carbonyl and acyl signals at δC = 214.8 ppm and 258.5 ppm, respectively. In our work, integration of the 13C{1H} NMR of spectra after ∼30 minutes of being placed under an atmosphere of 13CO shows that the signals at δC = 259.5 ppm and 206.0 ppm occur in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, while the signals at δC = 257.6 ppm and 214.7 ppm occur in a 1:1 ratio (Fig. S48, ESI‡). This suggests the formation of structures of the type 6 and 6a, respectively, en route to 4Mes.
:3 ratio, while the signals at δC = 257.6 ppm and 214.7 ppm occur in a 1:1 ratio (Fig. S48, ESI‡). This suggests the formation of structures of the type 6 and 6a, respectively, en route to 4Mes.
We note that, when monitoring the reaction using a ReactIR spectrometer, conversion to the carbene (4Mes) is complete within 40 minutes, but this takes ca. 36 hours when monitoring by in situ1H NMR spectroscopy. This is presumably due to a smaller headspace of CO, smaller interfacial surface area and, less efficient mixing. Paramagnetic species are also formed after 4Mes, which hinders further in situ monitoring by NMR spectroscopy.
2.3 Synthesis and structural characterisation of iron complexes observed during phase 1
Through careful control of the reaction conditions via solvent choice and reaction monitoring, it is possible to isolate the iron–carbene (CO)3Fe[C(2,6-Mes2C6H3)OC(O)(2,6-Mes2C6H3)] 4Mes, (Scheme 2), in preparative quantities. The reaction between 1Mes and an atmosphere of CO in hexane affords an orange solution from which 6 precipitates (Scheme 2, see discussion below), after ca. 10 minutes. The reaction mixture is then stirred until complete redissolution of 6 occurs, followed by an additional 10 minutes of stirring. Filtration of the dark orange-red solution followed by removal of the volatiles allows the isolation of 4Mes in 95% yield. 4Mes has been characterised by 1H and 13C{1H} NMR spectroscopy (Fig. S1 and S2, ESI‡), IR spectroscopy, mass spectrometry and elemental analysis. The NMR spectra confirm that the signals observed at δH = 2.31 ppm, 2.23 ppm, 1.83 ppm and 1.65 ppm in the 1H NMR spectroscopic monitoring of the reaction between CO and 1Mes (Fig. 2) are due to 4Mes. The ATR-FTIR spectrum of 4Mes (Fig. S18 and S19, ESI‡) displays three strong stretches which are observed at ν(CO) = 2046 cm−1, 1974 cm−1 and 1959 cm−1 corresponding to the metal-bound CO groups, with a less intense stretch at ν(CO) = 1612 cm−1 for the carboxyl group bound to the carbene. Again, this is consistent with 4Mes being the major species present after 40 minutes during in situ IR reaction monitoring (Fig. 1, Section 2.2).
4Mes has also been characterised by single crystal X-ray diffraction. Two solvatomorphs have been isolated; 4Mes (Fig. 3) and 4Mes·Et2O (see ESI, Fig. S36 and Table S2‡), both of which have been grown from concentrated Et2O solutions at low temperature. The Fe1–C1 bond length 4Mes is near identical to the analogous distance for the previously published 4Naph [1.8395(14) Å and 1.840(3) Å, respectively] suggesting the presence of an Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.35
C bond.35
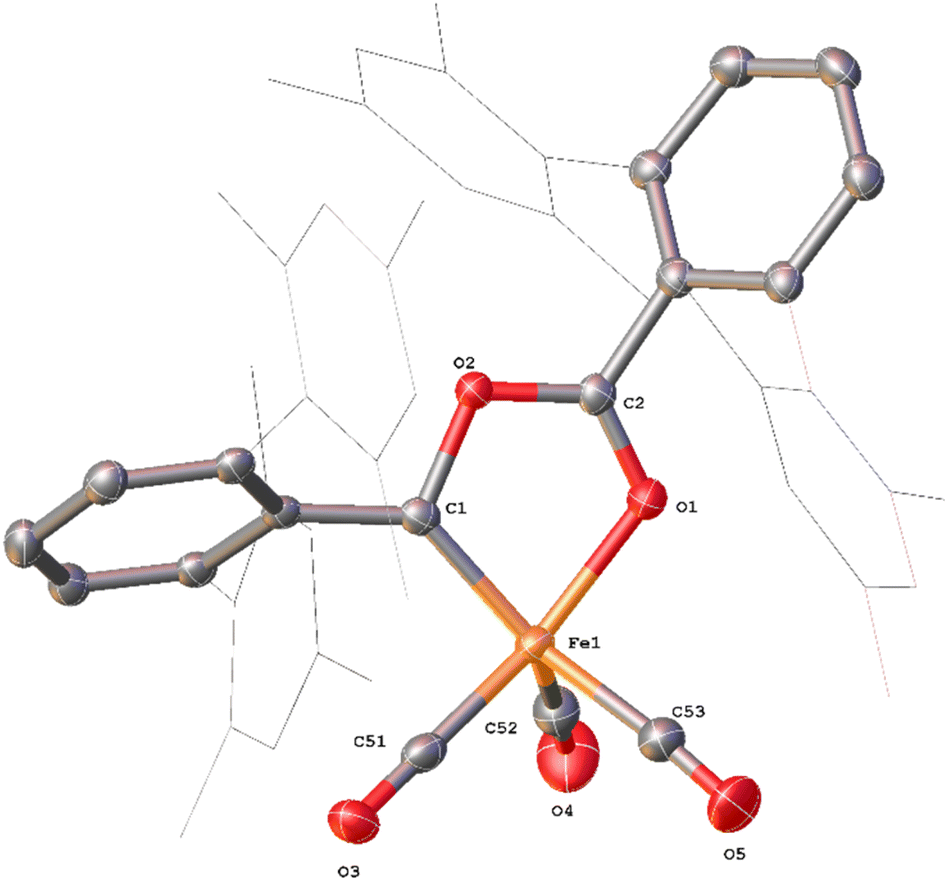 | ||
| Fig. 3 View of the metal complex from the crystal structure of 4Mes with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframe, hydrogen atoms, two co-crystallised diethyl ether and a second equivalent of 4Mes have been omitted for clarity. Selected bond distances (Å) and angles (°) for 4Mes shown: Fe1–C1 1.8395(14), Fe1–O1 1.9470(11), Fe1–C51 1.7549(16), Fe1–C52 1.828(2), Fe1–C53 1.8382(19), Fe1–C1–O1 80.81(5), O1–C2–O2 118.77(13). | ||
Whilst intermediate species between 1Mes and 4Mes are highly reactive, it has been possible to crystallise proposed intermediates of this transformation (Scheme 2). The reaction between 1Mes and 1 atm of CO in benzene resulted in a colour change from yellow to orange. Five minutes after the change in colour, the reaction mixture was flash frozen in liquid N2. The solvent was then sublimed off under vacuum, resulting in the formation of a red solid. This solid was then extracted three times with iso-hexane, keeping the extractions separate and these solutions were cooled to 8 °C for 48 hours. From these solutions, crystals of Fe(CO)4(C6H3-2,6-Mes2)2 (5), Fe(CO)3[C(O)C6H3-2,6-Mes2](C6H3-2,6-Mes2) (6) and Fe(CO)[C(O)C6H3-2,6-Mes2]2 (7) suitable for X-ray crystallography were obtained (Fig. 4–6). These represent the coordination of four CO molecules to 1Mes, followed by sequential migratory insertion reactions. Crystals of these complexes were isolated from mixtures containing several metal-containing species, therefore, it has not been possible to isolate 5, 6 or 7 in sufficient purity or quantities for full analysis.
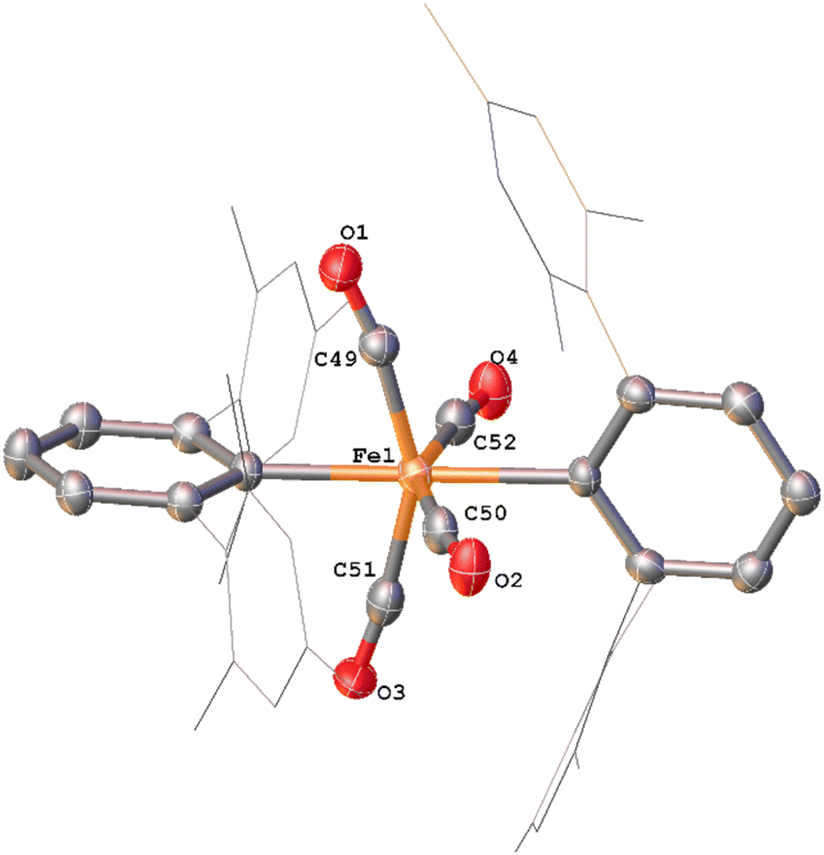 | ||
| Fig. 4 View of the metal complex from the crystal structure of 5 with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframe and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 5 shown: Fe1–C1 2.156(3), Fe1–C25 2.171(3), Fe1–C49 1.808(3), Fe1–C50 1.821(3), Fe1–C51 1.812(4), Fe1–C52 1.817(4), C1–Fe1–C25 174.58(11), C49–Fe1–C51 152.64(15), C50–Fe1–C52 153.63(15). | ||
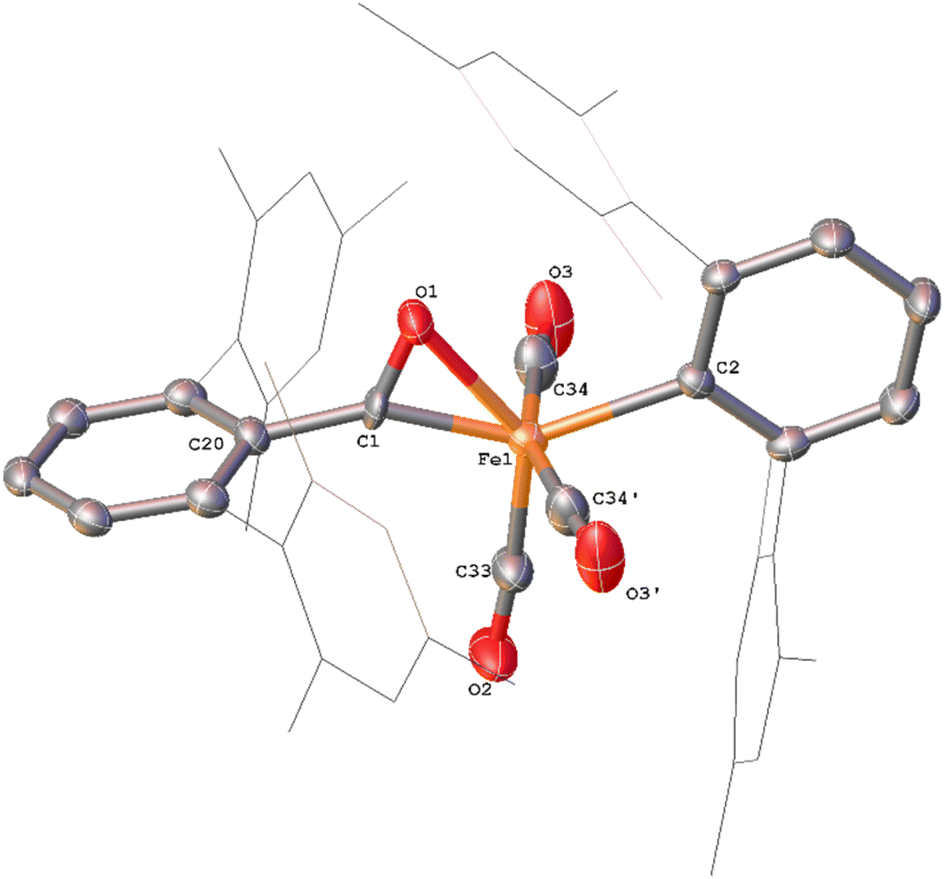 | ||
| Fig. 5 View of the metal complex from the crystal structure of 6 with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframe and hydrogen atoms have been omitted for clarity. The following symmetry operations were used to generate the marked atoms: +x,3/2 −y,+z Selected bond distances (Å) and angles (°) for 6 shown: C1–O1 1.221(4), C33–O2 1.148(5), C34–O3 1.130(4), Fe1–C1 1.872(3), Fe1–C2 2.104(4), Fe1–O1 2.110(3) C20–C1–O1 124.6(3) Fe1–C1–O1 83.2(2). | ||
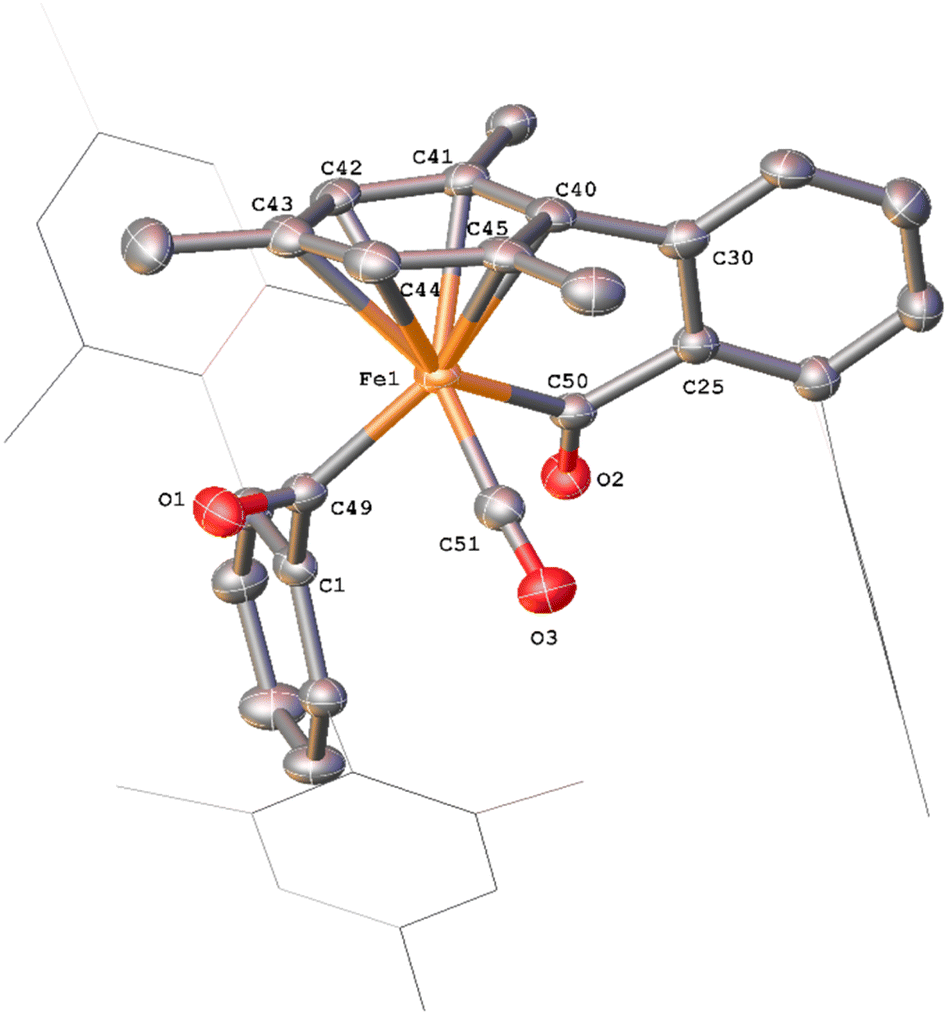 | ||
| Fig. 6 View of the metal complex from the crystal structure of 7 with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframe and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 7 shown: C49–O1 1.221(3), C50–O2 1.204(3), Fe1–C49 1.997(2), Fe1–C500 1.986(2), C1–C49–O1 116.24(19), C25–C50–O2 120.7(2), Fe1–C49–C1 129.13(15), Fe1–C50–C25 111.44(15), Fe1–C49–O1 114.59(17), Fe1–C500–O2 127.87(18). | ||
The solid-state structure of 5 (Fig. 4), features a rare trans-arrangement of the terphenyl ligands,36–40 which is closer to linearity than 1Mes [C1–Fe1–C25 angle of 174.57(10)°].41 Complex 5 shows a near-octahedral geometry at the iron(II) centre with bent CO ligands due to the steric hindrance of the terphenyl groups.
Complex 6 (Fig. 5) features one m-terphenyl ligand, three carbonyls and an acyl ligand formed through the migratory insertion of one CO ligand into the Fe–C bond of the second m-terphenyl ligand. The acyl ligand coordinates in an η2-bonding mode, affording an 18e− complex. These bond lengths and angles are similar to those observed for the diacyl complex Fe(CO)2[C(O)C6H3-2,6-Dipp2]2 which features a longer Fe–C bond than in 6 (1.8964(18) Å vs. 1.872(3) Å) but a decreased Fe–O distance (2.0229(14) Å vs. 2.110(3) Å) for the acyl group.34 Reaction of 1Mes (200 mg) in iso-hexane (20 mL) under an atmosphere of CO yielded an orange precipitate (15% yield) corresponding to intermediate 6. ATR-FTIR analysis of 6 revealed CO stretches at 2077 cm−1, 2013 cm−1 and 1975 cm−1 (Fig. S22 and 23, ESI‡) which correspond with those observed during the in situ measurements in toluene (Fig. 1; ν(CO) = 2076 cm−1, 2014 cm−1 and 1979 cm−1). The carbonyl stretch corresponding to the acyl ligand in this complex is observed at 1615 cm−1. Mass spectrometric analysis of 6 using MALDI-TOF allowed the observation of the [M−2(CO)]+ ion (see ESI‡). When dissolving 6 to obtain an NMR spectrum, 4Mes was detected within 10 minutes, even though there was no 4Mes present in the ATR-IR spectrum of the solid, demonstrating their intrinsic high reactivity. In situ13C{1H} NMR studies of the reaction between 1Mes and 13CO signals give signals at δC = 259.5 ppm and 206.0 ppm (integral ratio of 1:3, Fig. S48, ESI‡), which have been tentatively assigned to 6.
The solid state structure of 7 (Fig. 6) features one CO and two η1-acyl ligands, one of which also binds to the Fe via an η6-mesityl group, affording an 18e− metal centre. This differs substantially to the Dipp-substituted species Fe(CO)2[C(O)C6H3-2,6-Dipp2]2, in which the complex bears two η2-acyl moieties and two CO ligands.34 This difference is attributed to the lower steric demands of the mesityl substituents in 7 relative to the bulky 2,6-diisopropylphenyl moieties. As a result, the acyl ligands of 7 show significantly different bond lengths and angles relative to 6, consistent with η1-coordination, most notably increased Fe–C(acyl) bond lengths [1.997(2) Å (Fe1–C49) and 1.986(2) Å (Fe1–C50)]. We propose that 7 is likely only observed in the solid state as, unlike 6, there are no signals in the 13CO-labelled 13C{1H} NMR spectrum that can be assigned to 7. In a solution saturated with CO we propose 7 exists as 6a (Scheme 2) with two bound CO ligands, akin to Fe(CO)2(C(O)C6H3-2,6-Dipp2)2. This is based on similarities within the 13CO-labelled 13C{1H} NMR spectra where signals were observed at δC = 257.6 ppm and 214.7 ppm (1:1 ratio) for 6a, cf. acyl: δC = 258.5 ppm, carbonyl: 214.8 ppm for Fe(CO)2(C(O)C6H3-2,6-Dipp2)2.34
To further probe these structural insights, DFT calculations were employed. The calculations show a low barrier to the formation of 6 from 5 (21.9 kJ mol−1), while a further migratory insertion reaction occurs to form 6a (Fig. S86, ESI‡) before rapid formation of 7, with a barrier height relative to 5 of only 24.1 kJ mol−1. The reaction then proceeds further to, followed by rapid reaction to further intermediates. DFT calculations suggest the barrier to formation of 6a from 6 is ca. 70 kJ mol−1 without solvent, confirming the relative stability of 6.
2.4 Spectroscopic analysis of phase 2
Reaction monitoring of phase 2 towards the final products (Scheme 3) squaraine (2Mes), Fe(CO)5 and Fe2[O2C(2,6-MesC6H3)]4 (3), which takes up to an additional 8 days post formation of 4Mes, has proven more challenging. Through ReactIR, it was impossible to avoid ingress of small amounts of water and/or oxygen into the flask, even when a continuous positive atmosphere of CO was employed, leading to side reactions of the highly reactive intermediates. Additionally, 3 is an insoluble, paramagnetic solid that precipitates during the course of the reaction, which interferes with in situ NMR spectroscopic monitoring.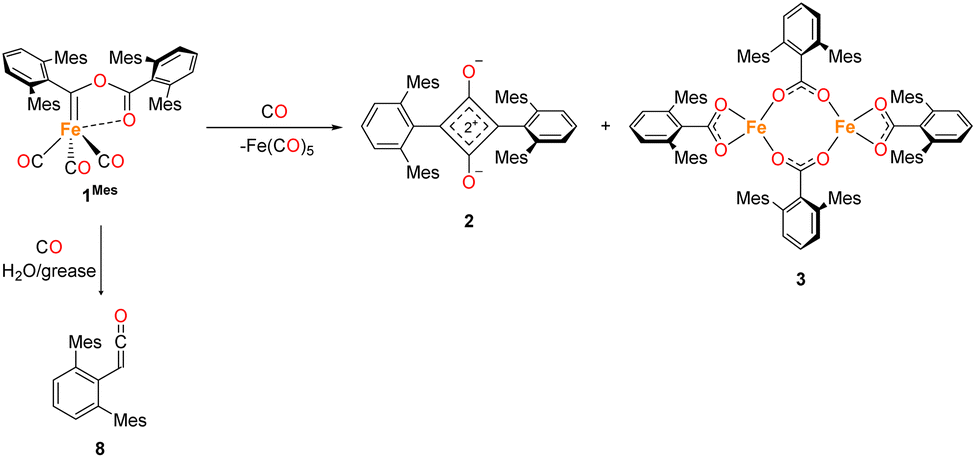 | ||
| Scheme 3 General reaction scheme post the formation of 4Mes and the observed degradation product upon reaction with moisture/grease. | ||
To obtain an endpoint IR spectrum upon completion of the reaction, 1Mes was reacted in toluene under ca. 1 atm of CO for 7 days. The reaction was filtered to remove insoluble 3 and an IR spectrum of the resulting solution was obtained (Fig. S45, ESI‡). The observed peaks were assigned as 2 (1674 cm−1), Fe(CO)5 (1996 cm−1, 2024 cm−1) and, at 2097 cm−1, a ketene (OCC(H)C6H3-2,6-Mes2, 8) which we propose results from reaction with moisture and/or silicone grease. Ketene 8 has been characterised in reaction mixtures by 1H NMR, 13C{1H} NMR and IR spectroscopy, and mass spectrometry (see ESI, Fig. S6, S7, S31, S32 and S50‡), although it has not been possible to isolate this as the sole product.
To monitor phase 2, by IR spectroscopy, a toluene solution of 4Mes was reacted with CO in a J. Young reaction flask. Periodically, an aliquot of the reaction was filtered to remove insoluble 3 and transferred to a sealable IR cell. Over the course of 8 days, signals corresponding to 4Mes (ν(CO) = 2049 cm−1, 1978 cm−1 and 1965 cm−1) are consumed (Fig. 7) and are replaced by signals for 2 (ν(CO) = 1674 cm−1) and ketene 8 (ν(CO) = 2097 cm−1). After 24 hours, an additional small signal is observed at 2107 cm−1 (Fig. S51, ESI‡), proposed to be a ketenyl–iron complex. This signal reaches a maximum intensity after ca. 48 hours, then decays. Signals relating to Fe(CO)5 and 2Mes are observed from the first spectra obtained after 24 hours.
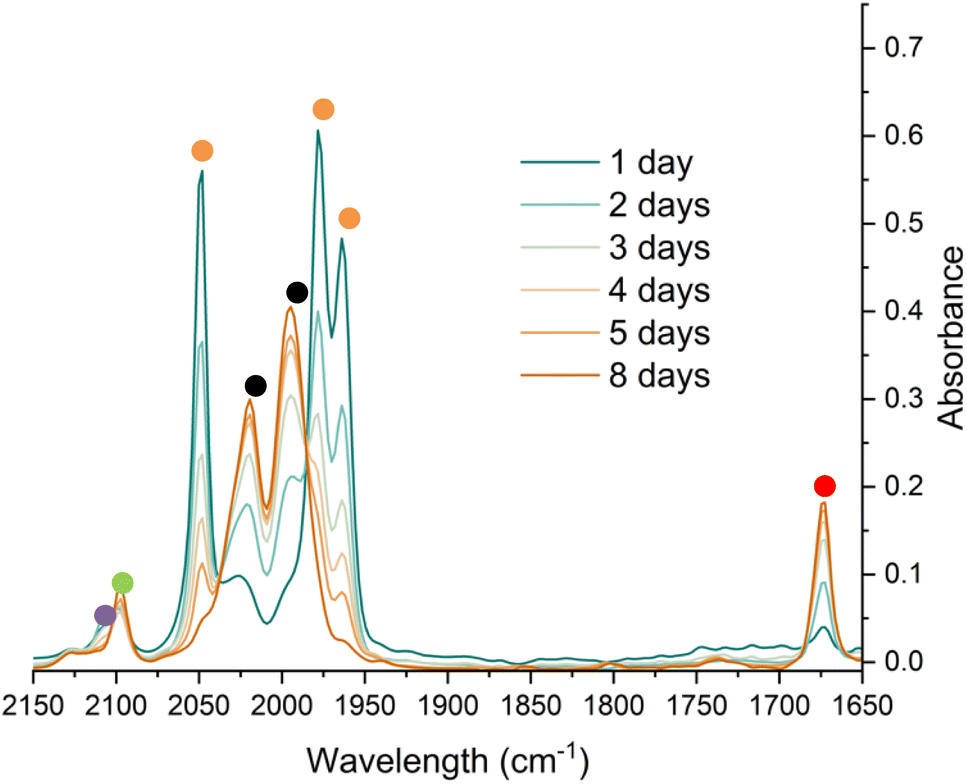 | ||
| Fig. 7 IR spectroscopic monitoring using a Harrick cell of the reaction of 4Mes under an atmosphere of CO in toluene over 8 days. Orange circles highlight the signals for 4Mes, red circles highlight the signals for 2Mes. Black circles highlight the signals for Fe(CO)5. The green circle highlights the signal for 8. The purple circle highlights a signal attributed to a proposed Fe–ketenyl complex, 9 (see Fig. 8 for proposed structure). See ESI‡ for a zoom in of the region between 2150–2075 cm−1. | ||
1H NMR spectroscopy was also used to investigate phase 2 of the reaction. As previously mentioned, 3Mes precipitates during the reaction, hindering NMR measurements. Additionally, in situ NMR spectroscopic monitoring in a J. Young NMR tube leads to a different distribution of products after 4Mes. This is attributed to the small headspace (see Section 2.2) leading to lower CO concentrations and side reactions. To circumvent this, aliquots of a larger reaction were filtered from a J. Young reaction flask into NMR tubes at different time points and the 1H NMR spectrum collected (Fig. 8). Signals for 2Mes are observed at δH = 1.98 ppm and 2.20 ppm within 24 hours of the start of the reaction. 8 was also observed (δH = 2.07 ppm, 2.22 ppm), which is consistent with IR spectroscopic measurements. Low intensity signals at δH = 2.36 ppm, 2.34 ppm and 2.14 ppm can be seen on day 2 and are consumed by day 6 (purple circles, Fig. 8). This is, again, in line with the IR spectroscopic monitoring where a ketene-type signal reaches a maximum concentration on day 3 and is subsequently consumed. Given the resolution of the peaks in the 1H NMR spectra, we conclude that this is likely an 18e−, diamagnetic complex. Complex 9 shown in Fig. 8 is the type of structure we propose that we are observing, however, this is only a tentative assignment based on the limited data and ketene containing complexes isolated (see Section 2.5). Additionally, a second diamagnetic complex, 10 (blue circles, Fig. 8) is consistently formed in small quantities. Full characterisation of 10 will be discussed further in Section 2.5.
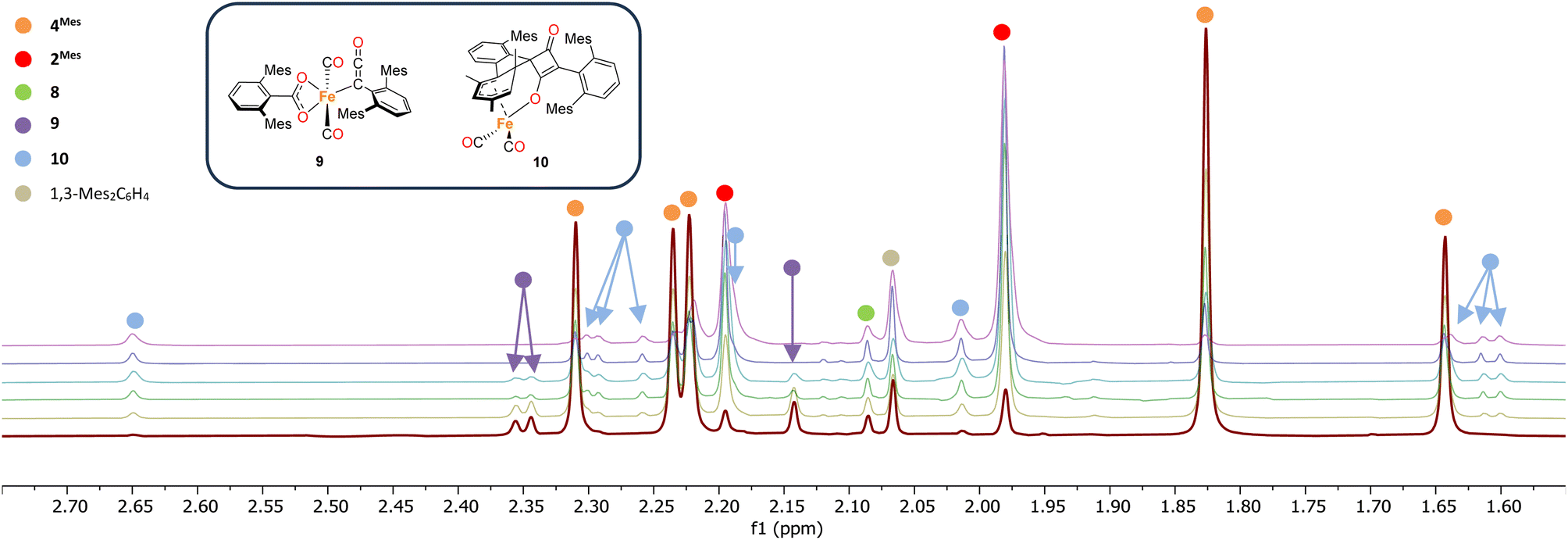 | ||
| Fig. 8 1H NMR spectra of the reaction of 4Mes with CO in C6H6. Spectra collected on after reacting for 1 (red), 2 (yellow), 3 (green), 4 (light blue), 5 (dark blue) and 8 (purple) days. Time increases from bottom to top. Corresponding para methyl signals for 8 and 1,3-Mes2C6H4 overlap with other signals and are not shown See the ESI for full spectrum (Fig. S56)‡ and further details on how the data was collected. Inset, structures of proposed Fe–ketenyl intermediate 9 and cyclobutenone byproduct 10. | ||
As Fe(CO)5 is a product, the reaction may proceed via disproportionation of an Fe(I) complex. To probe the two potential pathways, EPR and Mössbauer (MB) spectroscopies were employed to gain further insight into any intermediary complexes. During the course of the EPR monitoring we observe two signals at room temperature centred at ca. giso = 2.038 and giso = 2.003 (Fig. S57, ESI‡). The signal at giso = 2.003 is consistent with the previously synthesised radical anion [2]˙− (Fig. S58, ESI‡).35 The signal at giso = 2.038 appears as a singlet. Freezing the solution did not result in anisotropic splitting of this signal, indicating it is not due to an Fe(I) complex (Fig. S59, ESI‡).
MB spectroscopy required a higher concentration reaction solution relative to the IR and NMR spectroscopic experiments (8-fold increase, 0.23 mol L−1) to enable freeze-quenched solution monitoring with natural abundance iron. Freeze-quenched MB samples of the reaction of 4Mes with CO were collected over the course of the 5 days reaction. Within the first 24 h of reaction, the 80 K MB spectroscopy (Fig. 9) indicated the consumption of nearly 50% of 4Mes (δ = −0.10 mm s−1, |ΔEQ| = 1.56 mm s−1) together with the formation of the Fe(CO)5 product (δ = −0.08 mm s−1, |ΔEQ| = 2.54 mm s−1).42 Note that 3 is not observed in these spectra as it is filtered away prior to collection. Over the course of the next 4 days, further generation of Fe(CO)5 is observed as 4Mes is consumed (Fig. S59, ESI‡). Three additional iron species are also observed (10, A and B) that increase over the course of the reaction and are attributed to decomposition products, one of which is 10 that is observed by 1H NMR spectroscopy (Fig. 8). Signals for 10, A and B are also observed when reacting 4Mes in the absence of an atmosphere of CO (Fig. S85, ESI‡). While the identities of A and B remain unknown, the MB parameters of 10 are consistent with a diamagnetic, low-spin iron(II) species.43,44
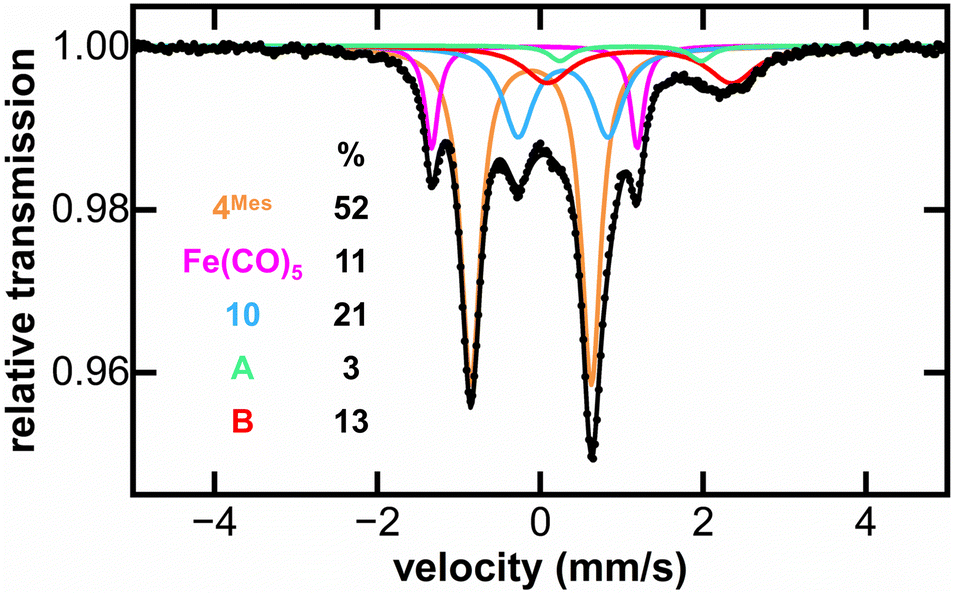 | ||
| Fig. 9 Freeze-trapped 80 K Mössbauer spectrum of the in situ formed iron species upon reaction of 4Mes with CO at 24 hours of reaction. The individual Mössbauer components are identified as 4Mes (orange. δ = −0.10 mm s−1, |ΔEQ| = 1.56 mm s−1) Fe(CO)5 (pink. δ = −0.08 mm s−1, |ΔEQ| = 2.54 mm s−1), 10 (light blue. δ = 0.30 mm s−1, |ΔEQ| = 1.10 mm s−1), A (light green. δ = 1.10 mm s−1, |ΔEQ| = 1.79 mm s−1) and B (red. δ = 1.21 mm s−1, |ΔEQ| = 2.33 mm s−1). Raw data are shown as black dots, total fit as a black line, and individual components as colored lines. Further reaction time points at 2, 3, 4 and 5 days are given in the ESI (Fig. S61).‡ | ||
2.5 Synthesis and structural characterisation of iron complexes in phase 2
Complex 10, which is commonly observed as a minor product, has been isolated as a mixture with 2Mes. Through in situ reaction monitoring, 10 was observed in larger quantities (relative to 2Mes) when the reaction was performed in an NMR tube. This was proposed to be due to less available CO in the reaction solution. Performing the reaction without an atmosphere of CO, greatly increases the proportion of 10. Both 10 and 2Mes precipitate readily from n-hexane, both forming intensely orange/red crystals. Therefore, 10 has not been isolated pure for IR and NMR spectroscopic analysis. However, structural characterisation of 10 was possible by scXRD (Fig. 10). 10 contains a dearomatized mesityl ring, which is bound to an [Fe(CO)2]2+ core as a cyclohexadienyl group with the anionic O of the cyclobutenone core also bound to the Fe. The dearomatization is the result of the remaining C of the formerly aromatic cyclohexadienyl ring forming a spirocycle with the cyclobutenone core. The cyclohexadienyl bond lengths are within error of other similar complexes,45–48 with the delocalised C–C bonds shorter than the single C–C bonds. The metal-bound cyclobutenone contains comparable bond lengths to a free cyclobutenone synthesised by Heimgartner et al.49 The structure fits well with the 1H NMR signals obtained during reaction monitoring. Nine signals are obtained for the methyl groups in 10 (δH = 0.70 ppm, 1.60 ppm (two overlapping signals), 1.62 ppm, 2.01 ppm, 2.18 ppm 2.25 ppm, 2.30 ppm and 2.65 ppm) due to steric hindrance preventing free rotation of one of the mesityl rings. The cyclohexadienyl group also has two distinct signals at δH = 3.54 ppm and 4.09 ppm for the protons of the cyclohexadienyl group, which is in agreement with other similar complexes.48,50,51 The 13C{1H} NMR spectrum for 10 also contains a number of indicative signals between δC = 50–115 ppm, clearly showing the signals for the cyclohexadienyl moiety, as well as two signals for the cyclobutenone at δC = 89.3 ppm and 123.2 ppm. Two M–CO signals (δC = 204.6 ppm and 207.4 ppm) and a further two distinct C–O signals for the cyclobutenone core (δC = 181.7 ppm and 191.1 ppm) are also observed. These signals compare well to those reported by Heimgartner et al.49 Further confirmation for 10 comes from 13CO-labelling (see Section 2.6) where the signals at δC = 204.6 ppm and 207.4 ppm are observed as a doublet (2JC–C = 15 Hz) and the four carbons of the cyclobutenone are observed as doublet-of-doublet-of-doublets (ddd, Fig. S12 and 13, ESI‡). ATR-IR spectroscopic analysis of a mixture of 2 and 10 in Fomblin® gave the characteristic signal for 2 at ν(CO) = 1674 cm−1 and three remaining strong stretches. The stretches at ν(CO) = 2027 cm−1 and 1980 cm−1 are attributed to the metal CO groups and the signal at ν(CO) = 1732 cm−1 to the ketone moiety of the cyclobutenone. The cyclobutenone CO stretch is again comparable to others in the literature.49,52,53
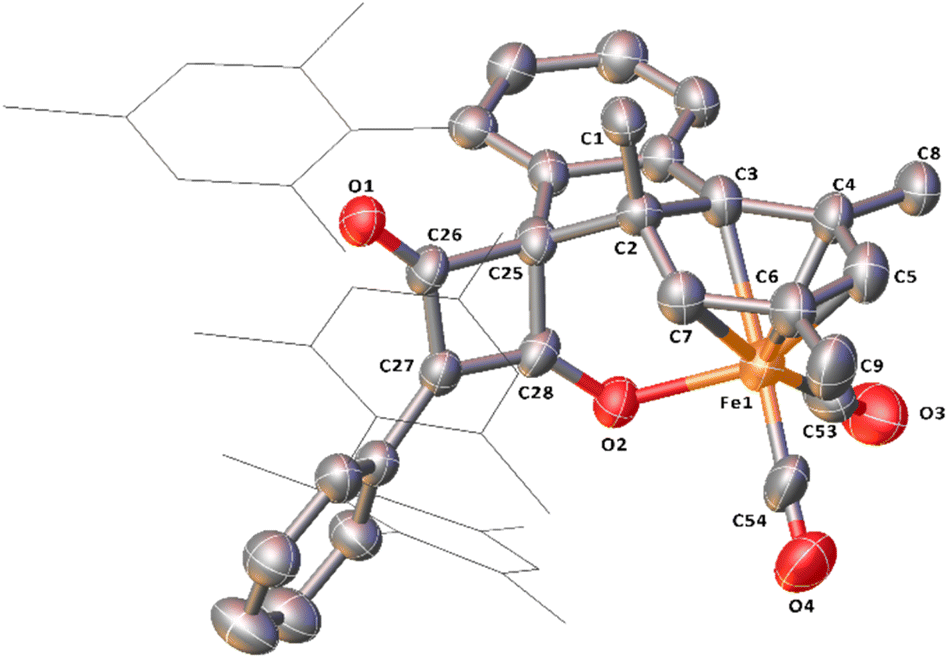 | ||
| Fig. 10 View of the metal complex from the crystal structure of 10 with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframes and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) for 10 shown: C2–C3 1.535(8), C2–C7 1.496(8), C3–C4 1.399(8), C4–C5 1.414(9), C5–C6 1.404(9), C6–C7 1.394(9), C25–C26 1.548(9), C25–C28 1.538(9), C26–C27 1.450(9), C27–C28 1.375(9), C26–O1 1.213(7), C28–O2 1.270(7), Fe1–C53 1.759(9), Fe1–C54 1.738(9), Fe1–O1 2.011(4). | ||
While in situ IR and NMR data from phase 2 of the reaction show some evidence for iron–ketene intermediates, this is further supported by the isolation and structural characterisation of three complexes containing ketenyl moieties (Fig. 11). Complex 11 (Fig. 12) was isolated as extremely air sensitive orange crystals from a concentrated Et2O solution of 4Mes in the absence of a CO atmosphere. Complex 11 features two ketenyl moieties bound to an iron centre, with one of the mesityl groups providing a stabilising η3 interaction, which shows a comparable η3-distance to [Zn(μ-Cl)(C6F5)(η3-C6Me6)]2 reported by Bochmann et al.54 The CC (C1–C2, 1.297(10) Å) and [C102–C103, 1.296(10) Å] and CO (C2–O1, 1.182(9) Å and C102–O8, 1.179(9) Å) bond distances in 11 are in accordance with the handful of iron–ketene complexes in the literature.14,55–72 So far, 11 has only been isolated once, and the crystallisation is challenging to replicate, so further characterisation of this species has not been possible.
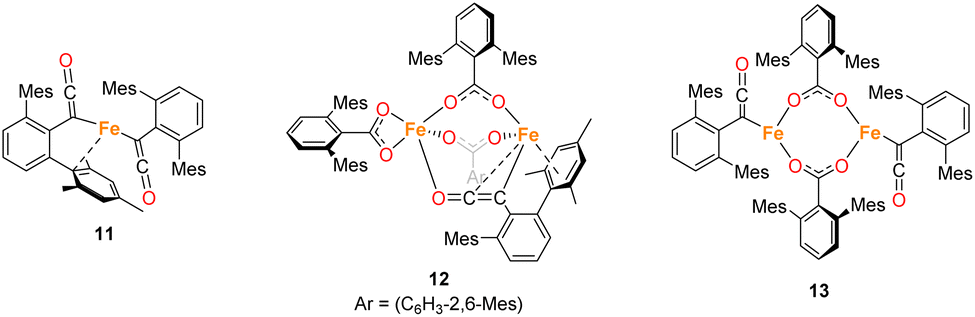 | ||
| Fig. 11 Ketene containing structures 11–13 isolated during the course of these studies. | ||
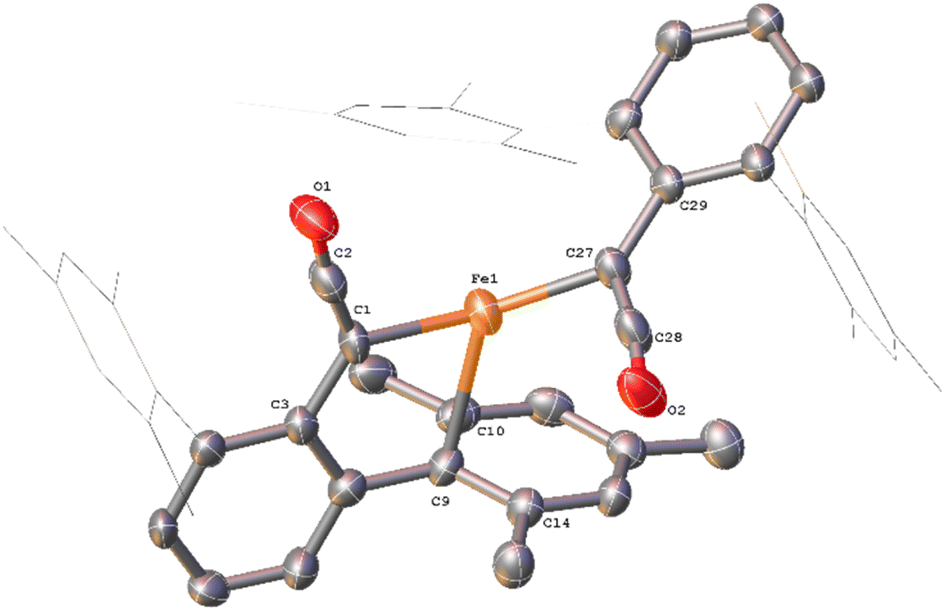 | ||
| Fig. 12 View of the metal complex from the crystal structure of 11 with anisotropic displacement ellipsoids set at 50% probability. Mesityl groups shown as wireframe, apart from the one bonding to the Fe, and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 11 shown: C1–C2 1.297(10), C2–O1 1.182(9), C27–C28 1.296(10), C28–O2 1.179(9), Fe1–C1 2.052(6), Fe1–C9 2.451(7), Fe1–C10 2.843(7), Fe1–C14 2.645(7), Fe1–C27 2.039(5), C1–C2–O1 171.6(7), C1–Fe1–C27 136.0(3), C27–C28–O2 172.2(6), Fe1–C1–C2 118.9(5), Fe1–C27–C29 127.9(5). | ||
Complexes 12 and 13 were isolated when performing the reaction in iso-hexane in the absence of an atmosphere of CO. 12 and 13 co-crystalised in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one equivalent of 12 and half an equivalent of 13 per asymmetric unit (Fig. 13a). 12 is composed of one ketenyl and three carboxylate groups, with two of the carboxylates and the ketenyl moiety bridging two iron centres. The ketenyl binds to Fe2 through an η2-interaction with the CC bond, and the remaining carboxylate group is bound in a terminal coordination. The ketenyl CC (C1–C2, 1.254(9) Å) and CO (C2–O1, 1.233(8) Å) bond lengths are similar to those in 11.
with one equivalent of 12 and half an equivalent of 13 per asymmetric unit (Fig. 13a). 12 is composed of one ketenyl and three carboxylate groups, with two of the carboxylates and the ketenyl moiety bridging two iron centres. The ketenyl binds to Fe2 through an η2-interaction with the CC bond, and the remaining carboxylate group is bound in a terminal coordination. The ketenyl CC (C1–C2, 1.254(9) Å) and CO (C2–O1, 1.233(8) Å) bond lengths are similar to those in 11.
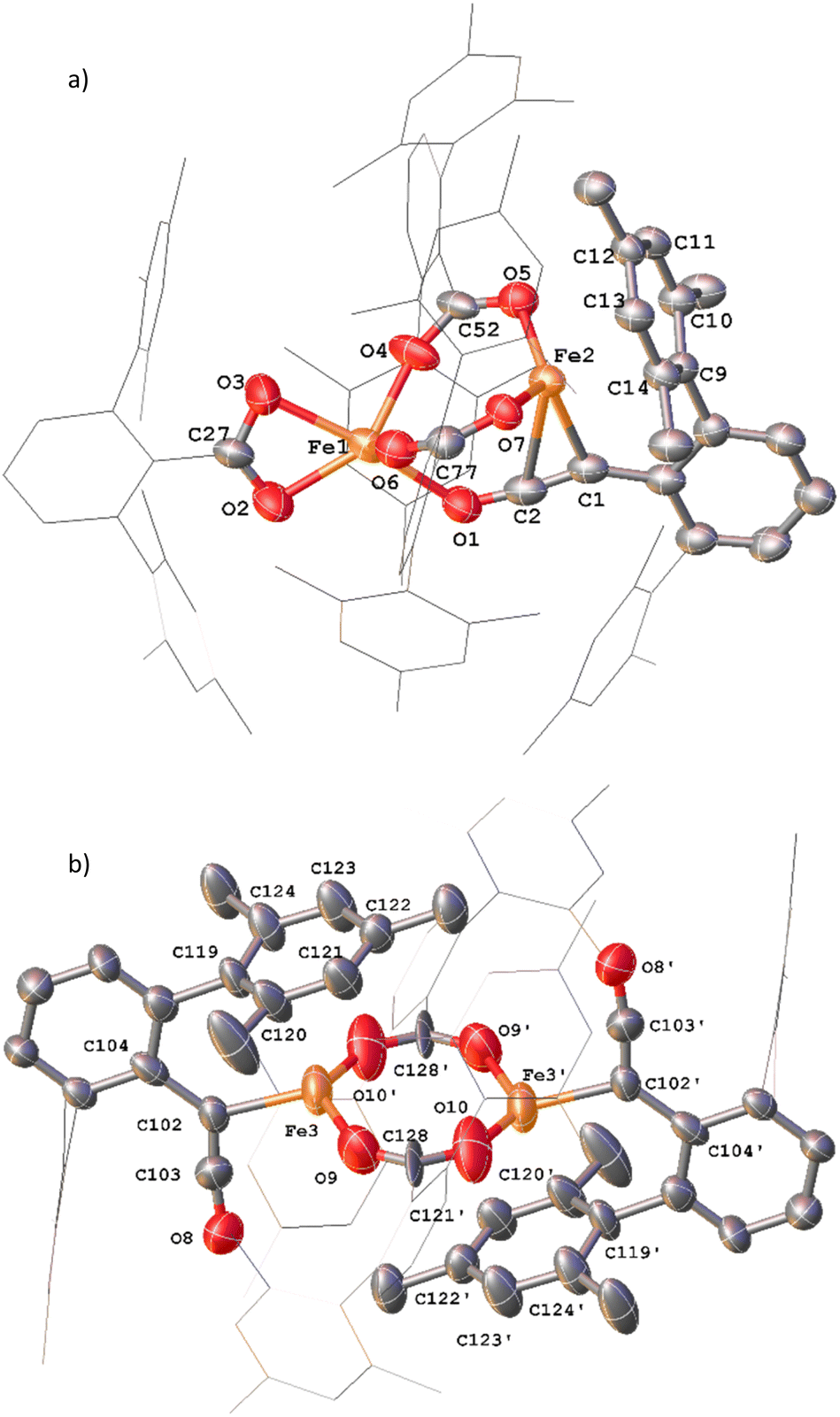 | ||
| Fig. 13 (a) View of the metal complex from the crystal structure of 12 with anisotropic displacement ellipsoids set at 50% probability. 2,6-Mes2C6H3 groups and non-bonding Mes groups shown as wireframe and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 12 shown: C1–C2 1.254(9), C2–O1 1.233(8), Fe2–C1 2.133(6), Fe2–C2 2.475(7), Fe1–O1 2.169(5), Fe1–O2 2.104(5), Fe1–O3 2.224(5), Fe1–O4 2.016(4), Fe1–O6 2.024(5), Fe2–O5 1.987(4), Fe2–O7 1.984(4), C1–C2–O1 173.4(7), O2–C27–O3 118.1(6), O4–C52–O5 121.8(6), O6–C77–O7 124.9(6). (b) View of the metal complex from the crystal structure of 13 with anisotropic displacement ellipsoids set at 30% probability. Atoms marked with ‘were generated using the following symmetry operator: 2 − X, −Y, 1 − Z. 2,6-Mes2C6H3 groups and non-bonding Mes groups shown as wireframe and hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 13 shown: C102–C103 1.213(15), C103–O8 1.250(14), Fe3–C102 2.026(8), Fe3–O9 2.09(1), Fe3–O10′ 2.018(11), C102–C103–O8 172.5(12), O9–C128–O10 115.6(11), Fe3–C102–C104 126.8(7). | ||
Complex 13 is situated across a special position in the asymmetric unit and contains two terminal ketenyl moieties and two bridging carboxylates (Fig. 13b). 13 shows comparable angles for the bridging carboxylates to 3 and 12, but a slight elongation of one of the Fe–O bonds [Fe3–O10, 2.09(1) Å]. For the ketene moiety in 13, the CC bonds are shorter than the CO, opposite to what is observed for 11 and 12. The Fe–C bond for the ketenyl group [2.026(5) Å] is also within error for a similar bond in 11.
Complex 14, which features a squaraine-like moiety bound to an Fe centre alongside a carboxylate ligand has also been identified by X-ray crystallography (Fig. 14). One of the mesityl groups of the squaraine-like moiety interacts in an η3 fashion to the Fe with bond lengths comparable to 11. The C4 moiety in 14 shows significant asymmetry as demonstrated by the differing C–C and C–O distances and is disordered by inversion of the C4 group. The C4 cycle can be best described as a cyclobutenone and is comparable to both Heimgartner's free cyclobutenone and complex 10.49 The carboxylate moiety is comparable to that previously reported for the terminal carboxylate of 3.
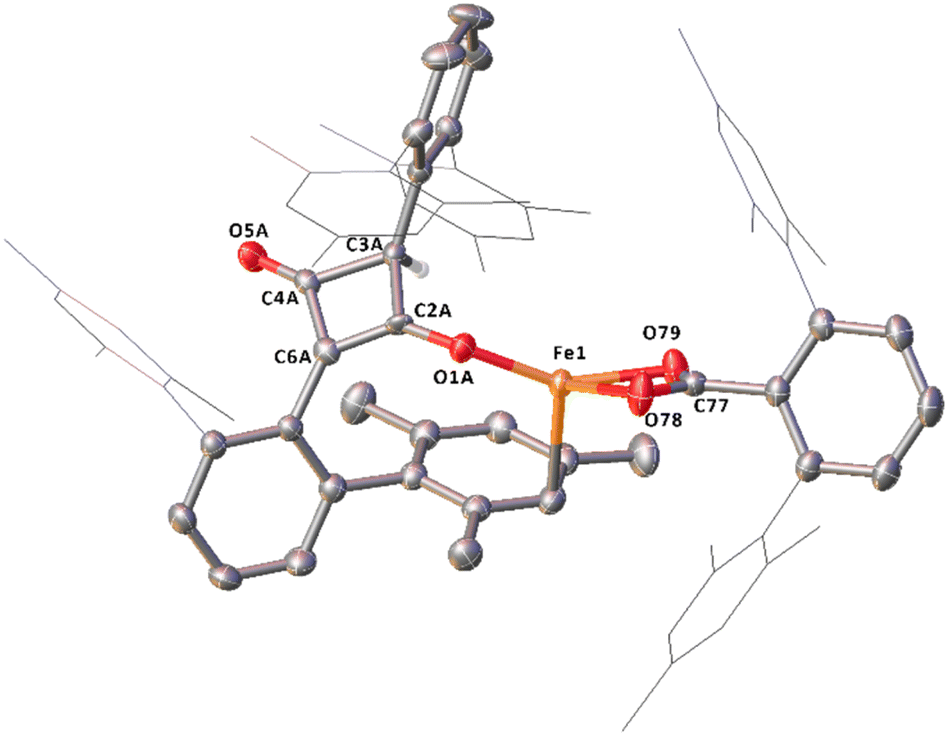 | ||
| Fig. 14 Molecular structure of 14 with anisotropic displacement ellipsoids set at 50% probability, major component of disordered C4 core shown. Mesityl groups shown as wireframe, except those interacting with the Fe and hydrogen atoms, except H4, have been omitted for clarity. Selected bond distances (Å) and angles (°) for 13 shown: Fe1–O1A 1.8702(9), Fe1–O78 2.1076(14), Fe1–O79 2.0442(10), C2A–O1A 1.306(17), C2A–C3A 1.560(17), C2A–C6A 1.362(18), C3A–C4A 1.576(13), C4A–C6A 1.456(17), C4A–O5A 1.20(2), O78–C77–O79 118.42(14). | ||
2.6 13C labelling experiments for phase 2
To further probe the mechanism, experiments with 13CO were conducted. 13CO labelled 4Mes (4Mes-13C) was obtained from the reaction between 1Mes and 13CO via the methodology described in Section 2.3. This was then reacted further under an atmosphere of either natural abundance CO (henceforth referred to as CO) or 13CO. When reacting 4Mes-13C with 13CO in C6H6 (see Section 4.2.10 of the ESI for details), to allow simultaneous IR and NMR spectroscopic analysis, the IR signals are red-shifted relative to the peaks for 4Mes. When 4Mes-13C was reacted under an atmosphere of CO, the IR spectrum recorded after 48 hours showed no evidence of CO exchange for 4Mes-13C as the signals for this species matched those observed for 4Mes-13C under an atmosphere of 13CO (Fig. 15). However, the signals for 8 (from degradation processes) and Fe(CO)5 are blue-shifted relative to the spectra obtained using 4Mes-13C and 13CO, suggesting de-enrichment. The signal for 8 is marginally red-shifted for 4Mes-13C + CO relative to 4Mes + CO (ν(CO) = 2091 cm−1vs. 2097 cm−1, 8. Cf.8-13Cν(CO) = 2037 cm−1), indicative of an Ar–13C = 12CO (Ar = 2,6-MesC6H3) isotope pattern in 8 formed from 4Mes-13C + CO. Three IR signals are present for 2Mes when reacting 4Mes-13CO under an atmosphere of natural abundance CO. This arises as the squaraine moiety has two C–O groups which can contain either 13C or 12C, and the three signals relate to squaraine C–O moieties containing 13C/13C, 13C/12C or 12C/12C. This suggests that uptake of CO is essential for the transformation of 4Mes into a ketenyl-containing species, as well as for the release of Fe(CO)5.
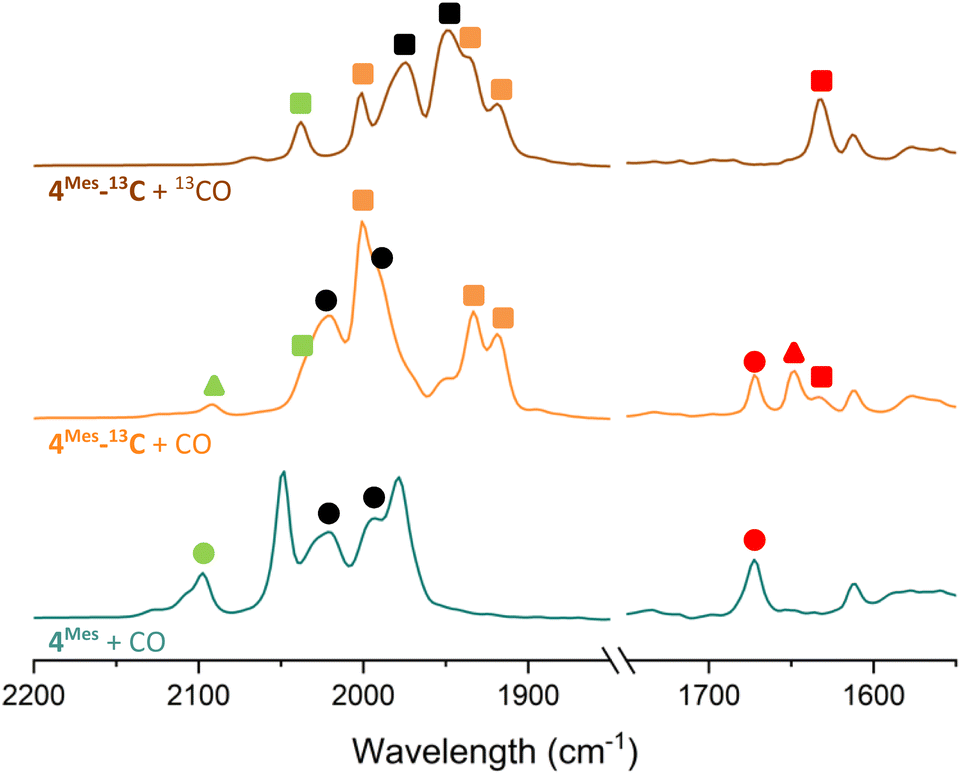 | ||
| Fig. 15 Comparison of the IR spectra of reactions after 2 days of 4Mes and 4Mes-13C under an atmosphere of CO or 13CO. All reactions performed in C6H6. Left, metal carbonyl and ketene region. Right, region for squaraine. Circles signify 12C products, triangles signify mixed 12C/13C and square signifies 13C products. Signals in green are for ketene (8). Signals in red are squaraine (2Mes). Signals in black are Fe(CO)5. | ||
The 13C{1H} NMR spectra of reactions between 4Mes-13C and CO gives additional insight into the reaction pathway. Both compounds 8, from decomposition, and 2Mes show uptake of carbon from the atmosphere, decreasing the degree of enrichment of the products (Fig. 16). For 8, the signal at 193.1 ppm is observed solely as a doublet with an integral of 0.62 while the signal at 24.8 ppm splits into a doublet and a singlet with, a total integral of 1. Taking both the coupling pattern and integrations into account, this means that the CO of the ketene (δC = 193.1 ppm) is partially incorporated from the atmosphere. Conversely, the CCO (δC = 24.8 ppm) is exclusively retained, presumably the carbene carbon in 4Mes-13C. This highlights the importance of an atmosphere of CO in the formation of the ketenyl complex. Furthermore, performing the same analysis for 2Mes indicates that there is a 56% depletion for the CO (δC = 177.2 ppm, integral = 0.44) relative to the (2,6-Mes2C6H3)–C (δC = 269.8 ppm, integral = 1). This is confirmed through analysis of the coupling patterns where the (2,6-Mes2C6H3)–C shows a ratio of 20 (t):48 (d):32 (s), consistent with 44% of the neighbouring carbons being 13C enriched. For the CO of 2Mes a ratio of 98 (t):2 (d), consistent with 98% carbons at the neighbouring positions being enriched, which is expected as the 13CO used is 99% enriched. This again shows that an atmosphere of CO is essential for the reaction to progress cleanly. It also suggests that there may be more than one step where the CO is incorporated due to the difference between the enrichment of 8 and 2Mes.
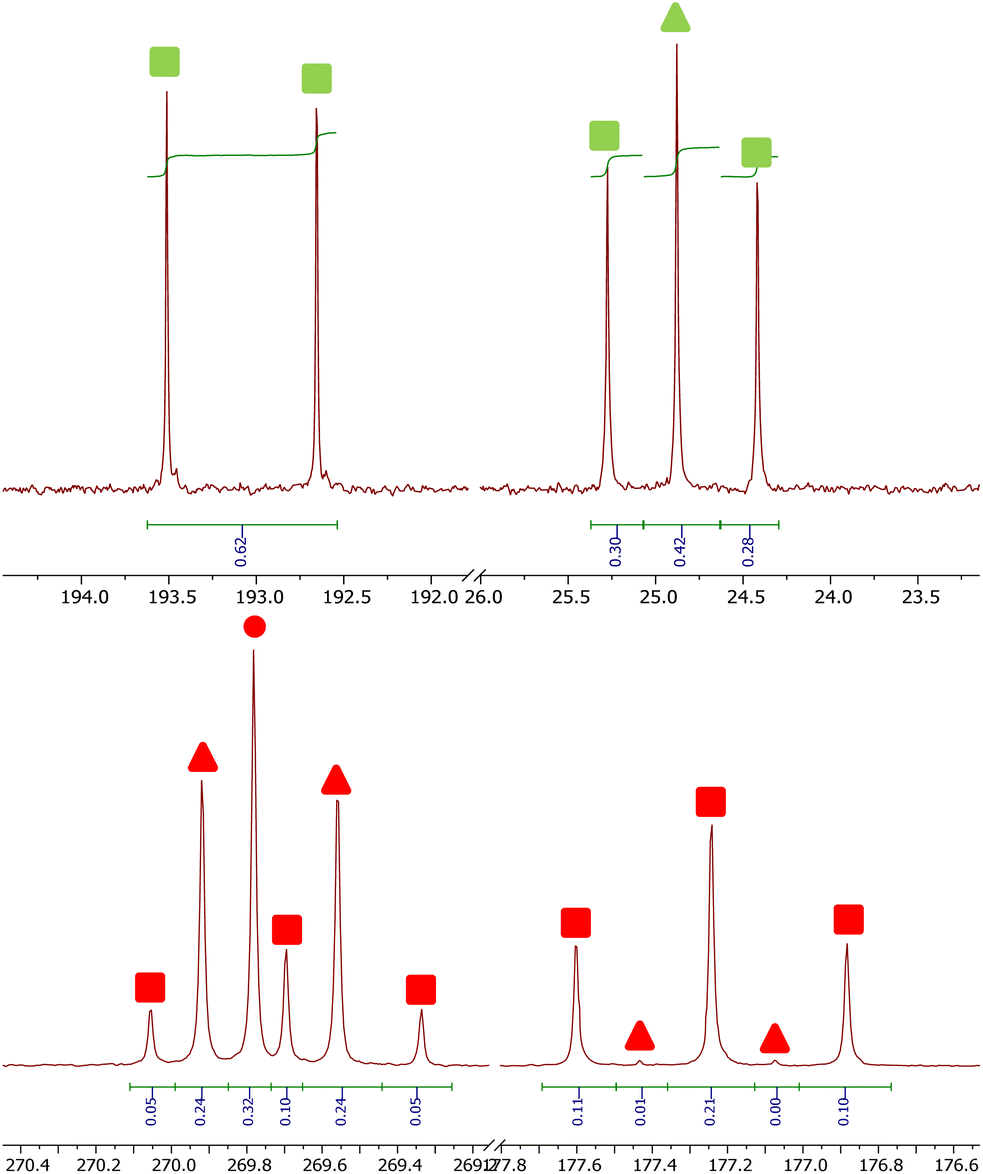 | ||
| Fig. 16 13C containing products from the reaction between 4Mes-13C and CO. Top, signals for 8. Bottom, signals for 2Mes. Circles signify 12C products, triangles signify mixed 12C/13C and square signifies 13C products. Markers in green are for ketene (8) and markers in red are squaraine (2Mes). | ||
The EPR studies (Section 2.4) indicate that radicals are present in the reactions. While one of the signals observed correlates to [2]˙−, the signal at giso = 2.038 has still not been assigned. When enriching the sample with 13CO, simulations of the experimental spectrum as a13C4-core using two pairs of 13C couplings, as is the case for [2]˙−, were unconvincing. A better simulation was obtained by considering lower symmetry in a 13C4-core with one larger coupling on a single 13C environment and a smaller coupling across three 13C atoms (Fig. S68, 69 and Table S3, ESI‡). This splitting is consistent with that expected for a radical centered on a species similar to a squaraine where the C4 core bears both a CO and C–O group with delocalisation of the radical over three carbon atoms. For reactions between CO and 4Mes-13C, the EPR signal obtained for [2]˙− shows depletion of the 13C for the CO, and the signal was modelled with a 43% 13C enrichment. This is consistent that observed in the 13C{1H} NMR spectra. The EPR studies show that the environment in which the reaction proceeds is highly reducing, with [2]˙− forming in the absence of an external reductant. Thus, single electron reactions are highly plausible.
2.7 Proposed mechanism for phase 2
The mechanism for phase 2 of the reaction (4Mes to 2Mes, 3Mes, Fe(CO)5) is more tentative than phase 1, but we propose a plausible route based on the data available. Some key observations are that ketene 8 and an iron–ketene complex (9) are observed spectroscopically (in situ IR and NMR) and that three iron–ketene containing byproducts (11–13) were isolated from the reaction. Thus, an iron–ketene complex is likely a key intermediate. We must also account for the formation of cyclobutenones 10 and 14 (Fig. 10 and 14), which are structurally related to squaraine 2Mes. We therefore propose that 4Mes initially rearranges to an iron ketene/carboxylate 9 (Scheme 4). While 9 has not been definitively characterised, its formation is consistent with the observed data, and we tentatively assign the iron–ketene signals (Fig. 7 and 8) to 9.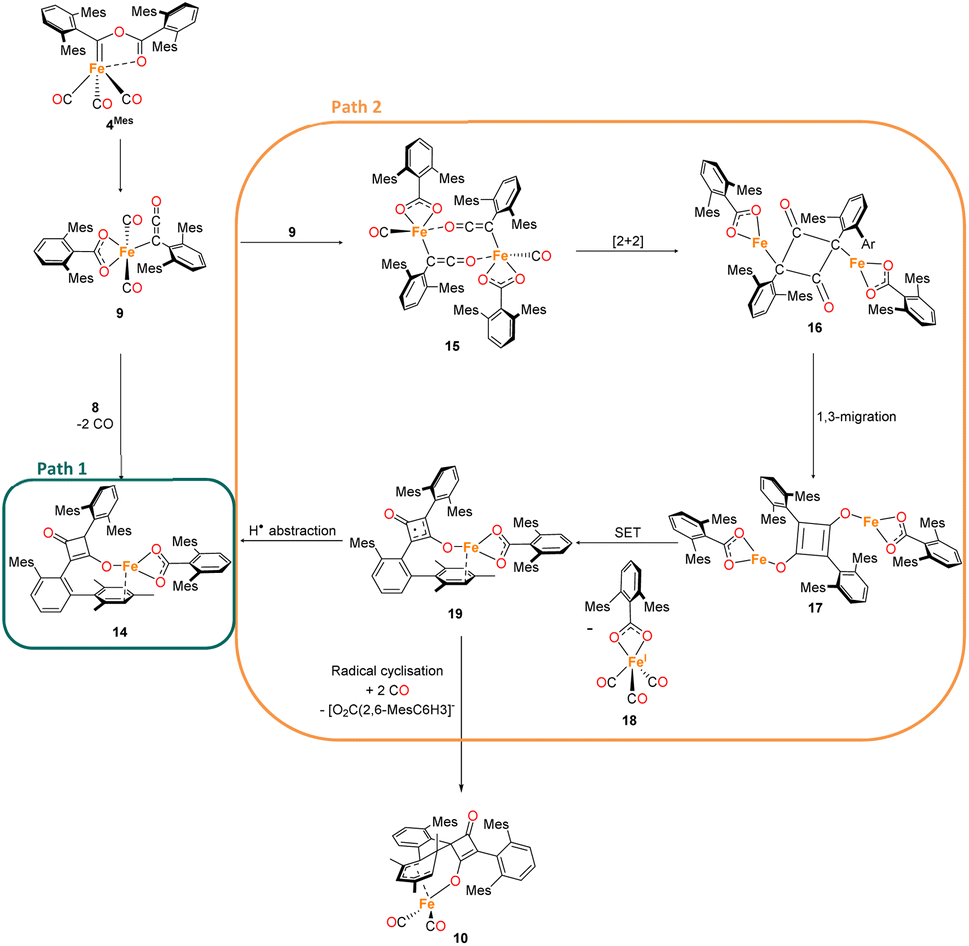 | ||
| Scheme 4 Two potential routes to 14via proposed complex 9. Path 1 forms 14via a [2 + 2] cycloaddition of 8 and 9. Path 2 forms 14 through dimerization of 9 then a [2 + 2] cyclisation, followed by 1,3 migration and a single electron transfer, giving 18 and 19. Path 2 also has the potential to form decomposition complex 10via radical cyclisation of 19. | ||
From 9, two possible paths to the isolated cyclobutanone complex 14 can be envisaged (Scheme 4). Path 1 involves a [2 + 2] cycloaddition between 9 and 8, followed by a 1,3-migration of the Fe centre, leading directly to 14. However, this route would not explain the presence of radical species detected by EPR spectroscopy. We therefore suggest that Path 2, which involves Single Electron Transfer (SET) processes, is more plausible (Scheme 4). In path 2, two molecules of 9 dimerise to form 15. 15 then undergoes a intramolecular [2 + 2] cycloaddition to form 16 followed by two 1,3-migrations to form 17, reducing steric crowding. From 17, an Fe(I) complex (18) and an Fe complex with a ligand centred radical (19) are formed by SET. Cyclobutenone complexes 10 and 14 can both be formed from the proposed 19, either by radical cyclisation (10) or H-atom abstraction (14).
From this, we propose the overall mechanism shown in Scheme 5. Here, the reaction proceeds as in Scheme 4 up to the formation of the ligand-centred radical 19. This undergoes an additional SET to form the squaraine 2Mes, along with another equivalent of Fe(I) species 18. We then propose that 18 undergoes disproportionation to form the other major products, iron carboxylate 3Mes and Fe(CO)5. Since it was not possible to observe Fe(I) signals by EPR or MB spectroscopy, we suggest the disproportionation of 18 is rapid. This is consistent with the formation of Fe(CO)5 in solution, and precipitation of 3Mes within 24 hours. Excess CO is required for clean formation of Fe(CO)5, otherwise the reactive intermediates undergo alternative reactions, resulting in cyclobutenone 10 and the unknown Fe complexes detected by MB spectroscopy (A and B, Fig. 9). It is worth noting that, A and B are always observed by MB spectroscopy due to the high concentrations required for this technique, meaning that it is not possible for excess CO in solution to react for 4Mes to react cleanly.
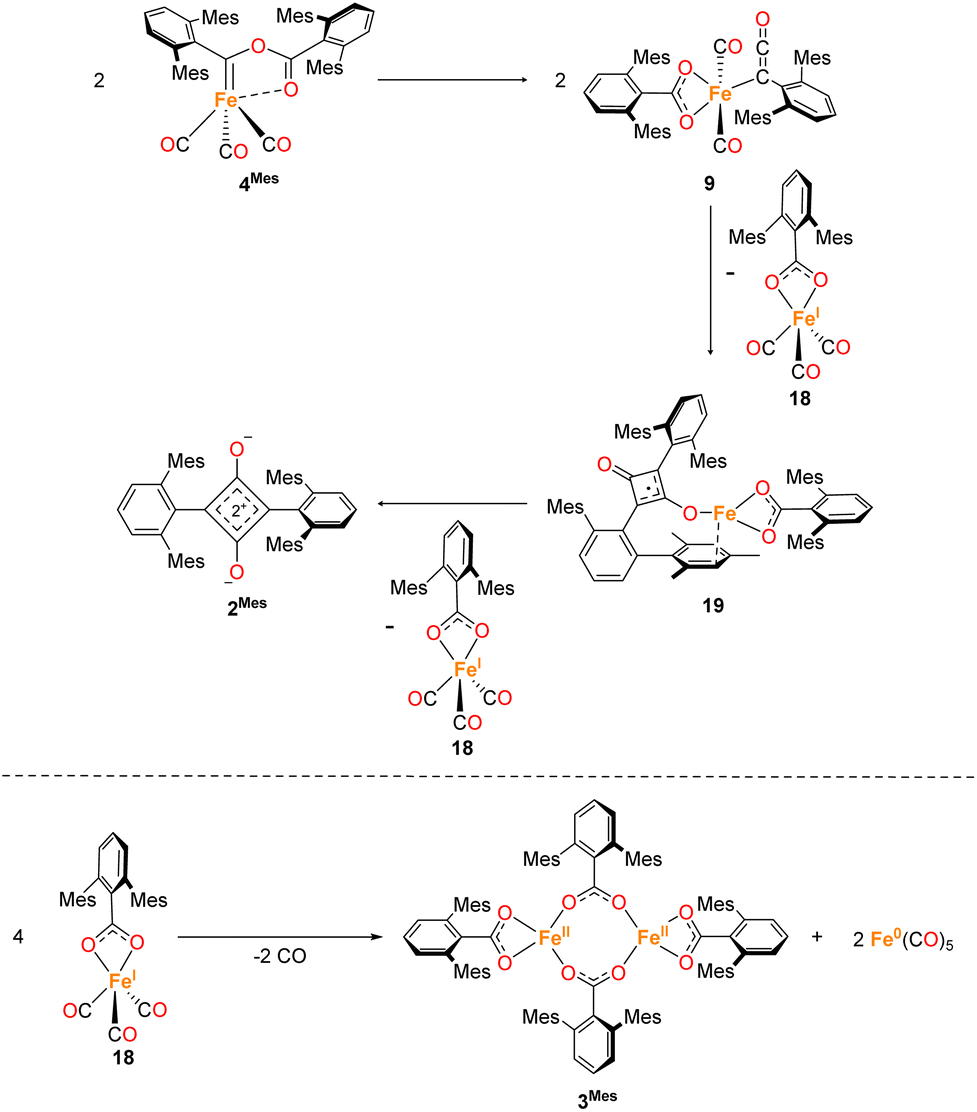 | ||
| Scheme 5 Proposed formation of 2Mes and 13via a ketenyl complex (9) by sequential single electron transfers, forming Fe(I) complex 18. 18 then undergoes disproportionation resulting in 3Mes and Fe(CO)5. | ||
Further DFT studies for phase 2 of the reaction proved difficult due to the size of the dimer like molecules. After the formation of 4Mes, high-spin Fe is strongly preferred according to the calculations. This is consistent with the observed formation of paramagnetic species in the 1H NMR spectra (see Fig. S61‡).
3. Conclusions
Through combined spectroscopic and structural studies we have developed a plausible mechanism for the formation of a squaraine (2Mes) from two-coordinate 1Mes, via reaction with CO. The first part of reaction clearly shows the formation of diamagnetic complexes by 1H NMR spectroscopy and a number of carbonyl containing complexes with complexes 5–7 isolated en route to the stable complex 4Mes, which are envisaged to form though sequential migratory insertion reactions. The reactivity post formation of 4Mes is significantly more difficult to follow due to the high sensitivity and reactivity of the intermediate species. Our initial studies proposed that Fe–ketenyl complexes were responsible for the formation of 2 due to a characteristic signal at 2097 cm−1 in the IR spectrum, however, this signal was found to relate to a protonated ketene (8) which forms in the presence of silicon grease and/or moisture. Other signals which are proposed to relate to Fe–ketenyl complexes are fleetingly observed by IR spectroscopy, but support them being key intermediates to the formation of 2. Pleasingly, other ketene complexes were isolated over the course of these studies (11–13) which gives strong evidence that ketenyl complexes are present en route to 2. However, these are proposed to be decomposition products and not active in the formation of 2. Complexes 10 and 14 both contain ligands bearing cyclobutenone moieties, providing further clues on potential intermediates in this reaction, all of which are derived from ketenes/ketenyl groups. Further spectroscopic studies using labelled materials and performing the reaction in the absence of an atmosphere of CO highlight the importance of excess CO in the reaction. Reactions of labelled 4Mes-13C under natural abundance showed selective loss of 13CO from the C–O of the squaraine product indicating that key steps post 4Mes forming ketenes requires uptake of CO from solution. The enriched carbene C in 4Mes-13C is retained adjacent to the terphenyl moiety in 2Mes. EPR spectra at room temperature showed evidence for the formation of radicals which are consumed over the course of the reaction, however, they could not be unequivocally identified. Mössbauer spectroscopy showed that the iron intermediates post 4Mes react rapidly, forming Fe(CO)5 within 24 hours. While there is no direct evidence for an Fe(I) complex by EPR and MB spectroscopy, it is possible to propose a series of SET reactions which form a fleeting Fe(I) complex (18) that undergoes rapid disproportionation to Fe(CO)5 and 3Mes. This work demonstrates how a multi-spectroscopic and structural approach is needed to truly understand highly complex reactions, showcasing the benefits of both in situ measurements and reaction sampling methodology.Data availability
A data repository containing spectroscopic data in their raw (IR and NMR) and processed forms (mass spectrometry, CHN, EPR and Mössbauer spectroscopy) can be found via the following https://doi.org/10.17639/nott.7407.Author contributions
NTC: drafted the manuscript. DLK, JM, NTC and LJT: conceptualization and supervision of the project. NTC, LJT, NC, YL, DR, MLN and DLK: writing, reviewing and editing of the manuscript. NTC, DR, NC: compiled the ESI. DR, AB, LJT: performed DFT analysis. YL, NTC, SPA: collected XRD data in house, solved and refined the crystal structures. NTC and SPA: performed XRD data collections at Diamond Light Source and validated the refined XRD data. ESD: collection and processing of EPR data. PJM: assistance with ReactIR methodology and setup. KB: implemented bespoke and high resolution NMR data collection. BP-G: performed MALDI mass spectrometry. MLN, NC: performed Mössbauer measurements and analysis. NTC, YL, NC: contributed to the experimental work, formal analysis and data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Emma Richards, Cardiff University, is thanked for valuable discussions on EPR experiments. DLK, JM, DR and NTC thank the Leverhulme Trust (RPG-2021-183) for funding. YL and DLK thank the University of Nottingham for a Vice-Chancellor's Scholarship for Research Excellence. DR thanks Nottingham Trent University for provision of the Hamilton and Avicenna high-performance computing clusters, on which the calculations were performed. The authors would like to thank Diamond Light Source for beamtime (proposal CY28766), and the staff of beamline I19 for assistance with crystal testing and data collection.References
- M. E. Dry, Appl. Catal., A, 1996, 138, 319–344 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- G. V. Schulz, Z. Phys. Chem., 1935, 30B, 379–398 CrossRef.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- R. A. Friedel and R. B. Anderson, J. Am. Chem. Soc., 1950, 72, 2307 CrossRef CAS.
- L. Gmelin, Ann. Phys., 1825, 80, 31–62 CrossRef.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed.
- A. Paparo, K. Yuvaraj, A. J. R. Matthews, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 630–634 CrossRef CAS PubMed.
- W. Yang, A. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2024, 63, e202319626 CrossRef CAS PubMed.
- H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS PubMed.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897–4901 CrossRef CAS PubMed.
- M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 8746–8750 CrossRef CAS PubMed.
- X. Wang, Z. Zhu, Y. Peng, H. Lei, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 6912–6913 CrossRef CAS PubMed.
- M. Xu, A. R. Jupp and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 3548–3552 CrossRef CAS PubMed.
- T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2005, 127, 17198–17199 CrossRef CAS PubMed.
- S. Hu, T. Shima and Z. Hou, J. Am. Chem. Soc., 2020, 142, 19889–19894 CrossRef CAS PubMed.
- T. Watanabe, Y. Ishida, T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2009, 131, 3474–3475 CrossRef CAS PubMed.
- T. Kurogi, Y. Ishida, T. Hatanaka and H. Kawaguchi, Dalton Trans., 2013, 42, 7510–7513 RSC.
- J. A. Buss and T. Agapie, Nature, 2016, 529, 72–75 CrossRef CAS PubMed.
- J. A. Buss and T. Agapie, J. Am. Chem. Soc., 2016, 138, 16466–16477 CrossRef CAS PubMed.
- R. Yadav, T. Simler, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 5765–5768 RSC.
- D. H. Berry, J. E. Bercaw, A. J. Jircitano and K. B. Mertes, J. Am. Chem. Soc., 1982, 104, 4712–4715 CrossRef CAS.
- A. S. Batsanov, J. A. Cabeza, M. G. Crestani, M. R. Fructos, P. García-Álvarez, M. Gille, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2016, 55, 4707–4710 CrossRef CAS PubMed.
- T. Shima and Z. Hou, J. Am. Chem. Soc., 2006, 128, 8124–8125 CrossRef CAS PubMed.
- W. J. Evans, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Chem. Soc., Chem. Commun., 1981, 706–708 RSC.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed.
- P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2010, 2, 77–79 RSC.
- B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed.
- A. M. Geer, D. L. Kays and L. J. Taylor, in Homologation Reactions, John Wiley & Sons, Ltd, 2023, pp. 813–846 Search PubMed.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC.
- C. Ni and P. P. Power, Chem. Commun., 2009, 5543–5545 RSC.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- G. Doyle, K. A. Eriksen and D. Van Engen, J. Am. Chem. Soc., 1985, 107, 7914–7920 CrossRef CAS.
- L. S. Crocker, B. M. Mattson, D. M. Heinekey and G. K. Schulte, Inorg. Chem., 1988, 27, 3722–3725 CrossRef CAS.
- P. A. Agron, R. D. Ellison and H. A. Levy, Acta Crystallogr., Sect. C, 1991, 47, 913–916 CrossRef.
- X. Tao, M. Feng, Y.-Y. Zhang, Y.-Q. Li, N. Wang and Y.-Z. Shen, Chin. J. Struct. Chem., 2009, 28, 287–290 CAS.
- H. Braunschweig, R. D. Dewhurst and C. Schneider, Organometallics, 2016, 35, 1002–1007 CrossRef CAS.
- D. L. Kays (née Coombs) and A. R. Cowley, Chem. Commun., 2007, 1053–1055 RSC.
- L. M. Epstein, J. Chem. Phys., 1962, 36, 2731–2737 CrossRef CAS.
- D. J. Evans, M. Jimenez-Tenorio and G. J. Leigh, J. Chem. Soc., Dalton Trans., 1991, 1785–1787 RSC.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry, Springer, Berlin, Heidelberg, 2011 Search PubMed.
- T. S. Cameron, M. D. Clerk, A. Linden, K. C. Sturge and M. J. Zaworotko, Organometallics, 1988, 7, 2571–2573 CrossRef CAS.
- R. S. Hay-Motherwell, B. Hussain-Bates, M. B. Hursthouse, B. E. Mann and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1993, 3219–3228 RSC.
- W. B. Wulff, A. L. Rheingold, and M. B. Allen, CSD Communication, Structure code: HIGREU Search PubMed.
- E. A. Trifonova, D. S. Perekalin, N. L. Loskutova, Y. V. Nelyubina and A. R. Kudinov, J. Organomet. Chem., 2014, 770, 1–5 CrossRef CAS.
- L. S. Trifonov, A. S. Orahovats, R. Prewo, J. H. Bieri and H. Heimgartner, J. Chem. Soc., Chem. Commun., 1986, 708–709 RSC.
- P. T. DiMauro and P. T. Wolczanski, Organometallics, 1987, 6, 1947–1954 CrossRef CAS.
- R. U. Kirss, A. Henriksen, D. A. Forsyth and W. Feighery, Inorg. Chim. Acta, 2006, 359, 4393–4397 CrossRef CAS.
- L. D. Foland, J. O. Karlsson, S. T. Perri, R. Schwabe, S. L. Xu, S. Patil and H. W. Moore, J. Am. Chem. Soc., 1989, 111, 975–989 CrossRef CAS.
- G. Verniest and N. D. Kimpe, Synlett, 2005, 2005, 0947–0950 CrossRef.
- Y. Sarazin, J. A. Wright, D. A. J. Harding, E. Martin, T. J. Woodman, D. L. Hughes and M. Bochmann, J. Organomet. Chem., 2008, 693, 1494–1501 CrossRef CAS.
- J. Martin-Gil, J. A. K. Howard, R. Navarro and F. G. A. Stone, J. Chem. Soc., Chem. Commun., 1979, 1168–1169 RSC.
- J. W. Kolis, E. M. Holt and D. F. Shriver, J. Am. Chem. Soc., 1983, 105, 7307–7313 CrossRef CAS.
- S. Ching, E. M. Holt, J. W. Kolis and D. F. Shriver, Organometallics, 1988, 7, 892–898 CrossRef CAS.
- P. L. Bogdan, M. Sabat, S. A. Sunshine, C. Woodcock and D. F. Shriver, Inorg. Chem., 1988, 27, 1904–1910 CrossRef CAS.
- Y. Tang, J. Sun and J. Chen, J. Chem. Soc., Dalton Trans., 1998, 4003–4008 RSC.
- H. Wadepohl, U. Arnold, H. Pritzkow, M. J. Calhorda and L. F. Veiros, J. Organomet. Chem., 1999, 587, 233–243 CrossRef CAS.
- R. Bertani, M. Casarin, P. Ganis, C. Maccato, L. Pandolfo, A. Venzo, A. Vittadini and L. Zanotto, Organometallics, 2000, 19, 1373–1383 CrossRef CAS.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2006, 25, 1317–1320 CrossRef CAS.
- M. A. Alvarez, M. E. García, M. E. Martínez, S. Menéndez and M. A. Ruiz, Organometallics, 2010, 29, 710–713 CrossRef CAS.
- M. Landman, B. van der Westhuizen, D. I. Bezuidenhout and D. C. Liles, Acta Crystallogr., Sect. E, 2012, 68, m308–m309 CrossRef CAS PubMed.
- H.-J. Liu, M. S. Ziegler and T. D. Tilley, Angew. Chem., Int. Ed., 2015, 54, 6622–6626 CrossRef CAS PubMed.
- S. A. Gonsales, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2016, 45, 15783–15785 RSC.
- V. Jakhar, D. Pal, I. Ghiviriga, K. A. Abboud, D. W. Lester, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2021, 143, 1235–1246 CrossRef CAS PubMed.
- W. Feuerstein, P. Varava, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Chem. Commun., 2021, 57, 11509–11512 RSC.
- T. Simler, K. N. McCabe, L. Maron and G. Nocton, Chem. Sci., 2022, 13, 7449–7461 RSC.
- R. J. Ward, I. del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Chem. Sci., 2023, 14, 2024–2032 RSC.
- R. Wei, X.-F. Wang, D. A. Ruiz and L. L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219211 CrossRef CAS PubMed.
- K. Yuvaraj, J. C. Mullins, T. Rajeshkumar, I. Douair, L. Maron and C. Jones, Chem. Sci., 2023, 14, 5188–5195 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2334282–2334296. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01286k |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |