
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilized four-electron aqueous zinc–iodine batteries by quaternary ammonium complexation†
Pengjie
Jiang‡
,
Qijun
Du‡
,
Chengjun
Lei
 ,
Chen
Xu
,
Tingting
Liu
,
Xin
He
and
Xiao
Liang
*
,
Chen
Xu
,
Tingting
Liu
,
Xin
He
and
Xiao
Liang
*
State Key Laboratory of Chem/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: xliang@hnu.edu.cn
First published on 31st January 2024
Abstract
Four-electron aqueous zinc–iodine batteries (4eZIBs) leveraging the I−/I0/I+ redox couple have garnered attention for their potential high voltage, capacity, and energy density. However, the electrophilic I+ species is highly susceptible to hydrolysis due to the nucleophilic attack by water. Previous endeavors to develop 4eZIBs primarily relied on highly concentrated aqueous electrolytes to mitigate the hydrolysis issue, nonetheless, it introduced challenges associated with dissolution, high electrolyte viscosity, and sluggish electrode kinetics. In this work, we present a novel complexation strategy that capitalizes on quaternary ammonium salts to form solidified compounds with I+ species, rendering them impervious to solubilization and hydrolysis in aqueous environments. The robust interaction in this complexation chemistry facilitates a highly reversible I−/I0/I+ redox process, significantly improving reaction kinetics within a conventional ZnSO4 aqueous electrolyte. The proposed 4eZIB exhibits a superior rate capability and an extended lifespan of up to 2000 cycles. This complexation chemistry offers a promising pathway for the development of advanced 4eZIBs.
Introduction
The development of four-electron aqueous zinc–iodine batteries (4eZIBs) with consecutive I−/I0/I+ redox couples has sparked considerable interest in the realm of energy storage, driven by their impressive theoretical specific capacity, abundant resources, and environmental sustainability.1–3 However, the widespread adoption of these batteries faces significant challenges that impact their stability and overall performance.3–5 Among these challenges, the hydrolysis of positive valence I+ species (such as ICl or ICl2−) in aqueous electrolytes stands out as a primary concern.5–8 While the formation of ICl interhalogens during the electro-oxidation of iodine is thermodynamically favorable in the presence of nucleophiles like halides, it is highly susceptible to hydrolysis due to the nucleophilic attack by water's OH moieties.9–11 This hydrolysis process results in low coulombic efficiency, voltage degradation, and poor cycling performance.Previous attempts to address these issues in I−/I0/I+ redox couples involved the use of super-concentrated aqueous electrolytes or non-aqueous electrolytes.8,12–15 Nevertheless, the latter approach, while proficient at stabilizing the oxidized iodine species, has been plagued by complications related to the dissolution of active materials. This predicament arises from the high solubility of iodine and ICl interhalogens in organic solvents.16–18 In contrast, the super-concentrated aqueous electrolyte approach we previously established, known as the 19–19–8 formula (specific molality of ZnCl2, LiCl, and acetonitrile in water), has demonstrated notable success in mitigating the hydrolysis of I+ species in an aqueous media. Such an electrolyte provides the requisite free chloride ions for the stabilization of I+ species while simultaneously suppressing free water activity to participate in the hydrolysis. However, it introduced challenges associated with high electrolyte viscosity, which, in turn, hindered ion transport and led to sluggish electrode kinetics. To advance the development of 4eZIBs towards practicality, there is a compelling need to explore solutions that effectively suppress hydrolysis and dissolution, while simultaneously enhancing electrode redox kinetics in low-concentration aqueous electrolytes.
This study explores the application of quaternary ammonium salts to resolve these challenges in a dilute aqueous electrolyte composed of the commonly used ZnSO4. Quaternary ammonium salts, known for their ability to form stable complexes with iodine species, hold the potential to significantly improve the stability and overall performance of the 4eZIB. In this work, we delve into the electrochemical behavior, complexation chemistry, and mechanisms governing the I−/I0/I+ redox couple in zinc–iodine batteries, offering insights into the transformation of iodine states and the provision of a stable environment for I+ within a dilute aqueous electrolyte. These findings pave the way for advancements in the design and implementation of zinc–iodine batteries, with a focus on mitigating solubility and stability challenges, and thereby contributing to the development of more sustainable energy storage solutions.
Results and discussion
The complexation chemistry between ICl2− and quaternary ammonium cations
Previous research has established the possibility of quaternary ammonium cations forming complexes with polyhalides, including Br3−, ICl2−, and I3−.19–22 The complexation approaches have been validated in halogen batteries with the I−/I0 and Br−/Br0 redox couples.23–25 In this work, the complexation chemistry between ICl2− and quaternary ammonium cations was investigated to elucidate the stability of I+ in aqueous media. Various quaternary ammonium cations with differing chain lengths as the complexing agents were selected. The corresponding quaternary ammonium dichloroiodates (QICl2 compounds) were synthesized by simple redox reactions (see the Experimental section for details), with their molecular structures shown in Fig. S1.† Fig. S2a† visually illustrates the QICl2 compounds, which are in the powder form at room temperature.Raman spectroscopy was employed to characterize the as-prepared precipitates. Fig. 1a demonstrates that the symmetric stretching of the I–Cl band occurs around 270 cm−1, while the asymmetric stretching occurs around 250 cm−1.26–30 There is an observable blue-shift and subsequent red-shift of the specific Raman signal corresponding to the I–Cl bond with increasing chain lengths of the quaternary ammonium cations, indicating a varying intensity of the complexation effect. Among all QICl2 compounds, Pr4NICl2 exhibits the most pronounced complexation effect as revealed by the Raman spectrum (Fig. 1a), detailed Raman results for QICl2 compounds within a broader measuring range can be found in Fig. S3.† This is consistent with the binding energies among all QICl2 compounds in a vacuum environment, as determined by the first principles calculations based on density functional theory (DFT) (Fig. 1b). However, the complexation energy of quaternary ammonium cations with ICl2− would be influenced if water was considered in the simulation system (Fig. S4†). The results indicate that, in the presence of water, Hexy4NICl2 exhibits the highest binding energy, followed by Am4NICl2 and Pr4NICl2. We also assessed the complexation effect of QICl2 compounds through observing their status in distilled water visually. Me4NICl2 and Et4NICl2 are soluble in distilled water and exhibit the typical ICl2− Raman signal right after the preparation of the solution (Fig. 1c and S5†).
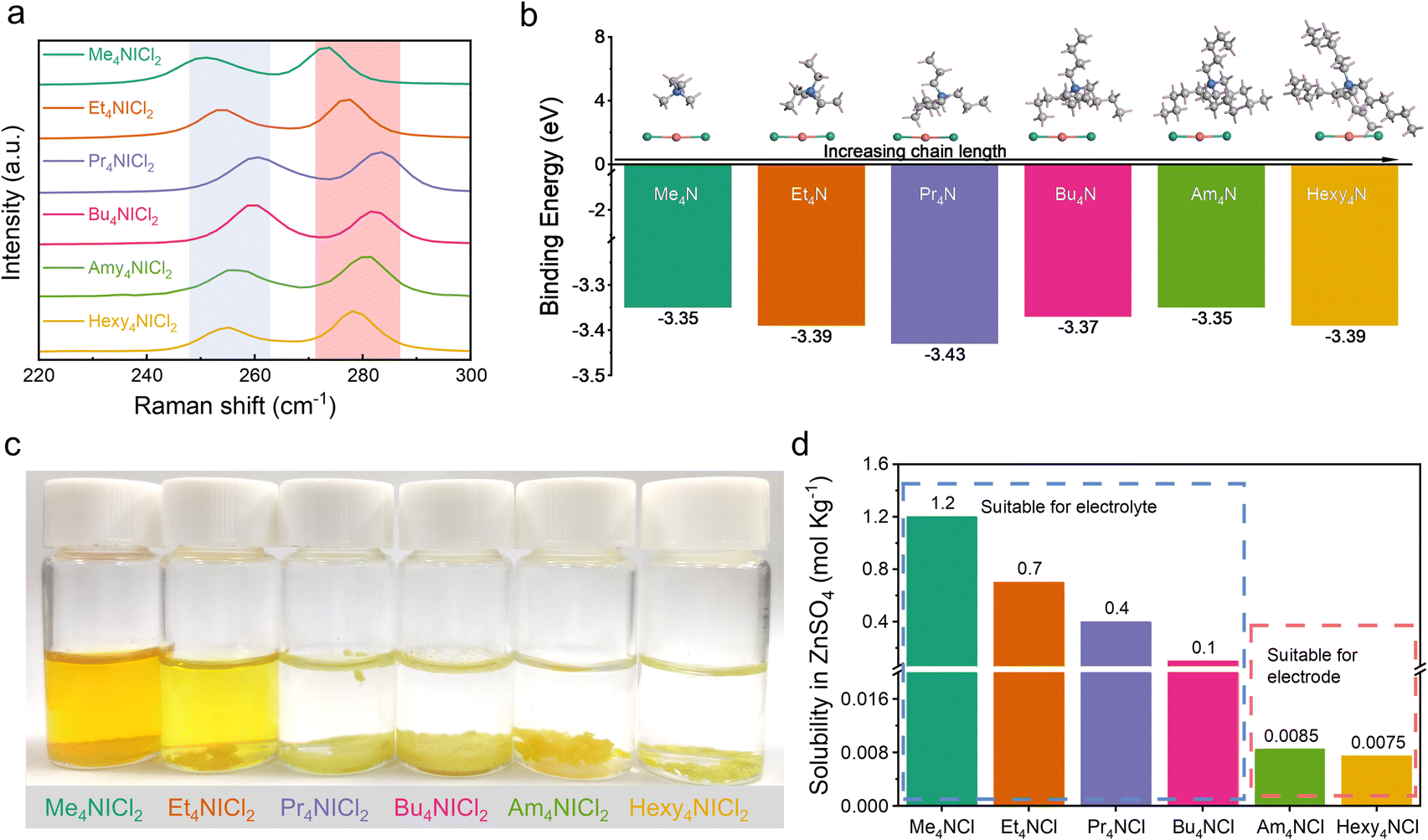 | ||
| Fig. 1 Complexation of quaternary ammonium cations with ICl2−. (a) Raman spectra of the QICl2 compounds. (b) The calculated binding energy of quaternary ammonium cations with ICl2− in a vacuum environment. (c) Digital photo of QICl2 compounds in H2O for a week. (d) The solubility of QICl2 compounds in ZnSO4 electrolyte. | ||
In contrast, the remaining QICl2 compounds were insoluble in distilled water, forming a separated phase. This observation can be elucidated by the similarity–intermiscibility theory, suggesting that the QICl2 compounds become progressively more lipophilic and hydrophobic with increasing chain length.31 Even after 6 months immersion, these initially insoluble QICl2 compounds remain as a separated phase in water (Fig. S2b†), albeit with some degree of aggregation in shape. This is consistent with the calculated binding energy results of these QICl2 compounds (Fig. S4†), which shows that the long-term stability of these materials in water mainly depends on the strength of their complexation interaction. On the other hand, the corresponding quaternary ammonium chloride (QCl) has distinct solubility in water and electrolytes (Fig. 1d and ESI Table 1†), which could be attributed to their alkyl chain-length dependent hydrophobicity.
The solubility tests of the QICl2 compounds were also conducted in commonly used ZnSO4 based aqueous electrolytes. Similar to their behavior in distilled water, all QICl2 compounds, except for Me4NICl2 and Et4NICl2, are insoluble in electrolytes of 1 m ZnSO4 + 1 m ZnCl2 (Fig. S2c†) and 3 m ZnSO4 + 1 m ZnCl2 (Fig. S2d†). Notably, for Me4NICl2 and Et4NICl2, their solubility in the electrolyte was lower compared to that in water, as the color of the electrolyte solution was much clearer. These results indicate that electrolyte solutions also offer a more stable environment for QICl2 compounds than distilled water.
Furthermore, the stability of QICl2 compounds was evaluated using thermogravimetry analysis (Fig. S6a and b†), verifying the strong complexation from a thermal decomposition perspective. The melting points of all QICl2 compounds are above 100 °C, which are higher than that of free ICl (27.2 °C in the α-form or 13.9 °C in the β-form),32 indicating their exceptional thermal stability. Moreover, QICl2 compounds remain in the solid state at room temperature, even after a long-term storage in air (Fig. S2a†), unlike ICl, which is a volatile liquid, and prone to hydrolysis.33
QICl2 compounds with the strong complexation exhibit superior chemical stability in mitigating hydrolysis compared to the free ICl without complexation (Fig. 2a). Equimolar amounts of these compounds were dispersed/dissolved in water (∼0.1 M). The solution/suspensions after relaxation were diluted with acetonitrile before the UV-vis spectroscopy test. Free ICl is highly susceptible to hydrolysis due to the nucleophilic attack by water.9–11 As depicted in Fig. S7,† the intensity of the UV absorption peak associated with free ICl (335 nm) quickly diminished when subjected to H2O attack, while the UV absorption peak ascribed to I2 (450 nm) increased. This shift indicates the hydrolysis of iodine monochloride, resulting in the formation of I2.34 QICl2 compounds exhibited varying degrees of attenuation in the intensity of their specific UV absorption peak at 335 nm in response to water attack (Fig. 2b and S7†), with Pr4NICl2 showing the slowest rate of hydrolysis by normalization of their UV adsorption intensities (Fig. 2c).
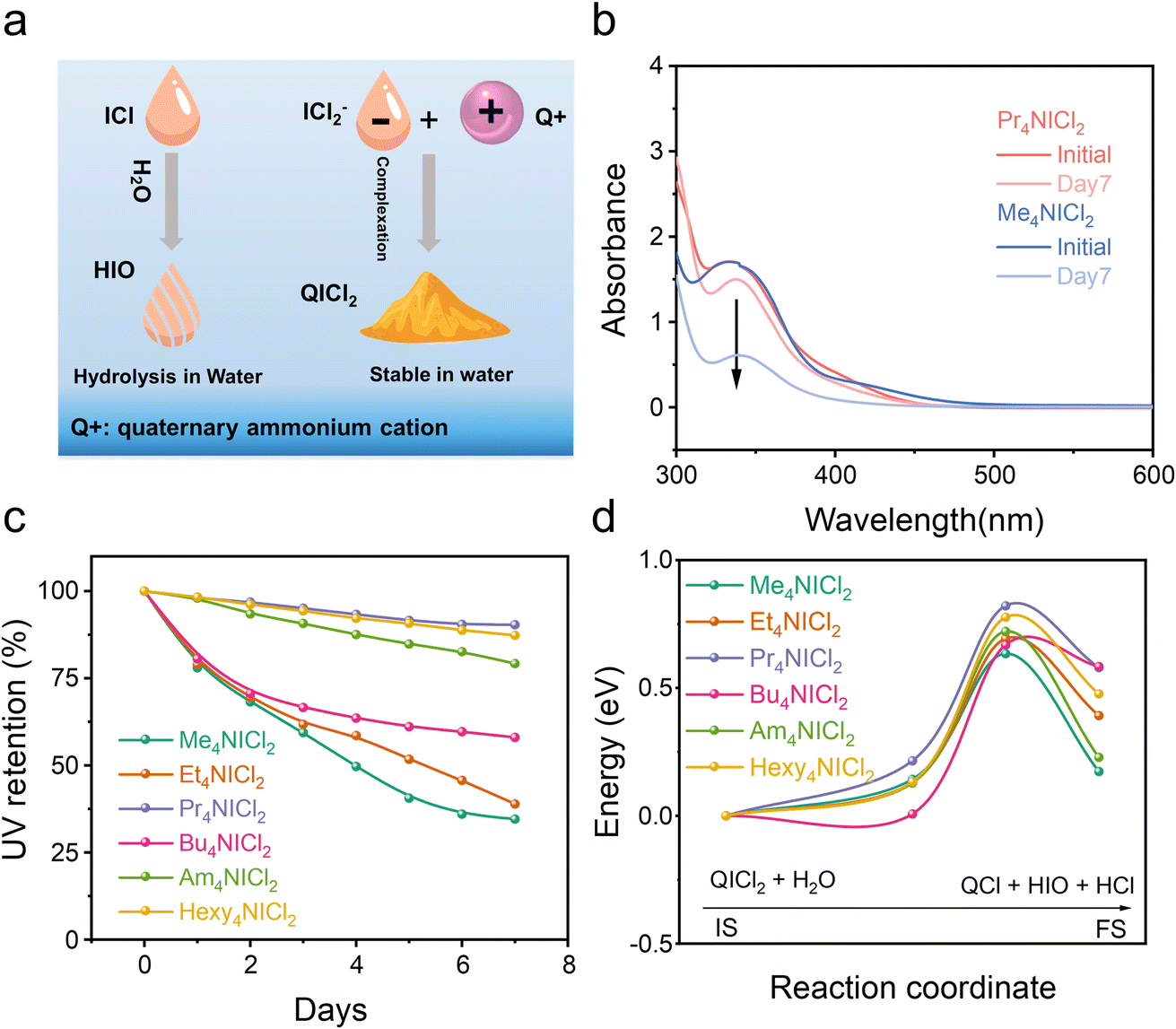 | ||
| Fig. 2 Stability of QICl2 compounds in aqueous media. (a) Schematic diagram of the complexation and stability of quaternary ammonium salts with ICl− in aqueous media. (b) Typical UV-vis spectra of QICl2 compounds before and after H2O attack. (c) Retention of QICl2 compounds by UV-vis spectral analysis. (d) Calculated energy barrier for the hydrolysis process of QICl2 compounds. | ||
The hydrolysis process was further calculated (Fig. 2d), confirming that Pr4NICl2 possesses the highest hydrolysis energy barrier among various QICl2 compounds. Detailed representations of the initial state, transition state, and final state of various QICl2 compounds during hydrolysis can be found in Fig. S9–S14.† These results collectively suggest that the dissolution and hydrolysis of I+ species in an aqueous electrolyte could be effectively mitigated by the complexation strategy.
Validating the complexation chemistry through electrolyte additives
To assess the stability of the I0/I+ redox couple employing a quaternary ammonium salt complexation approach, we introduced Me4NCl, Et4NCl, Pr4NCl and Bu4NCl into a conventional ZnSO4 electrolyte for evaluation. These cells were composed of a PAC/I2 cathode and a zinc foil anode. It is important to note that other quaternary ammonium salts with longer chain lengths cannot be used as electrolyte additives due to their limited solubility in the ZnSO4 electrolyte (Fig. 1d and ESI Table 1†). These will be studied as the cathode in the following sections.Focusing on the I0/I+ redox couple within the voltage range of 1.25–1.8 V versus Zn/Zn2+, we observed that batteries supplemented with KCl and Me4NCl additive exhibited an inclined charge/discharge profile with a short platform (Fig. S15a†). In contrast, the batteries incorporating Et4NCl, Pr4NCl, and Bu4NCl displayed a flat charge/discharge profile, indicative of a more reversible redox process. Furthermore, Pr4NCl exhibited the highest coulombic efficiency, as shown in Fig. S15b,† suggesting that the I+ hydrolysis during the charge/discharge process is effectively mitigated. The stability of I+ was quantified by measuring the retained capacity of the I0/I+ couple (discharged to 1.25 V) after extended storage using a fully charged Zn–I2 cell (charged to 1.8 V) at open-circuit voltage. As illustrated in Fig. S15c,† following a 24 hour resting period, the battery with Pr4NCl as an additive maintained a discharge plateau, while batteries with other additives displayed a declining discharge profile.
We also investigated the stability of the battery over an extended resting time for the I−/I0/I+ multiple electron transformation process within the voltage range of 0.6–1.8 V. As anticipated, the battery supplemented with Pr4NCl exhibited both the highest coulombic efficiency and the most distinct I+ discharge platform, as depicted in Fig. 3a and S16a.† These results strongly support the complexation strategy to improve I+ stability in a dilute aqueous electrolyte environment, especially when using Pr4NCl as an additive. In the context of the 4eZIB employing quaternary ammonium chloride electrolyte additives, all the batteries exhibit two pairs of charge/discharge plateaus (Fig. 3b). Specifically, Pr4NCl displayed the flattest I0/I+ discharge platform and the lowest capacity degradation rate (Fig. S16b†). The Pr4NCl concentration was further investigated (Fig. S16c†). The discharge plateaus at 1.6 V (I0/I+) are elongated rationally with the increase of the Pr4NCl additive in the electrolyte. However, when an excessive additive was introduced (0.2 m Pr4NCl in the electrolyte), an excess of Pr4NICl2 passivation layer on the electrode might be formed, resulting in significant polarization during charge/discharge. Galvanostatic intermittent titration technique (GITT) profiles were collected to investigate the kinetic mechanism (Fig. S17†). The calculated diffusion coefficient indicates a substantial drop by 2 orders of magnitude at the high plateau, which can be attributed to the formation of the Pr4NICl2 phase that is hydrophobic in aqueous media thus impeding ion migration.
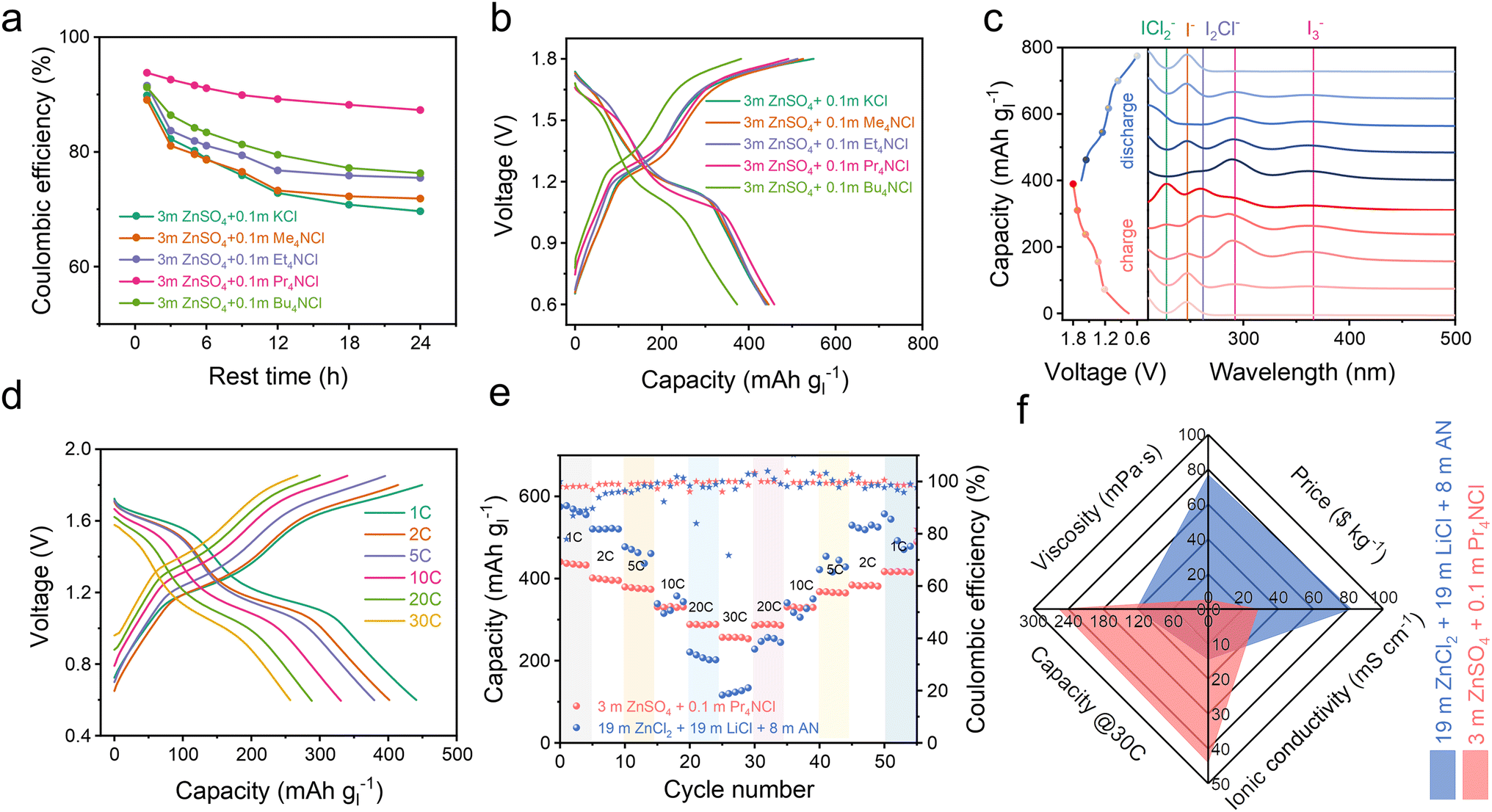 | ||
| Fig. 3 Quaternary ammonium chlorides as electrolyte additives. (a) Self-discharge of the 4eZIBs after various resting periods in different electrolytes. (b) Voltage profiles of 4eZIBs within the voltage range of 0.6–1.8 V. (c) Ex situ UV-vis absorption spectrum of the 4eZIBs with the 3 m ZnSO4 + 0.1 m Pr4NCl electrolyte. (d) Voltage profiles of 4eZIBs with different rates, the electrolyte is 3 m ZnSO4 + 0.1 m Pr4NCl. (e) The rate capability of the 4eZIBs. (f) The characteristics of different electrolytes. | ||
We conducted ex situ UV-vis absorption spectroscopy and in situ Raman spectroscopy focusing on the electrode to elucidate the electrode composition. Fig. 3c shows the ex situ UV-vis spectrum during the charge/discharge process in a 3 m ZnSO4 + 0.1 m Pr4NCl electrolyte. During the charging process, the detected iodine species undergo a transformation from I− (244 nm)35 into I3− (295 nm and 365 nm),36 I2Cl− (264 nm),37 ICl2− (230 nm).37,38 Notably, 230 nm (C-/D-absorption band) and 335 nm (A-/B-absorption band) are both the characteristic absorption bands of ICl2−, which are concentration sensitive due to the UV absorption efficiency.37,39 The characteristic Raman signal around 167 cm−1 exhibits changes during charge/discharge to 1.6 V/1.4 V (Fig. S18†), corresponding to the bending vibration for ICl2−.27 These results are similar to our previous report,8 indicating an I−/I0/I+ redox process, however, this is achieved in a dilute aqueous electrolyte enabled by the complexation chemistry.
After 1000 cycles, the battery still exhibits distinct charge/discharge plateaus (Fig. S19a†), revealing an unchanged primary electrochemical reaction process (I−/I0 and I0/I+). Impressively, the battery employing the 3 m ZnSO4 + 0.1 m Pr4NCl electrolyte achieves a capacity of 442 mA h g−1 at 1C, and 252 mA h g−1 at 30C based on iodine mass (Fig. 3d). As demonstrated in Fig. 3e, the zinc–iodine battery shows an excellent rate performance for the 3 m ZnSO4 + 0.1 m Pr4NCl than in a conventional high concentration 19–19–8 electrolyte (19 m ZnCl2 + 19 m LiCl + 8 m AN in water). These could be ascribed to the low viscosity (5.3 mPa s) and high ionic conductivity (44 mS cm−1) of the 3 m ZnSO4 + 0.1 m Pr4NCl electrolyte, as shown in Fig. 3f. Furthermore, the quite dilute electrolyte also proves to be more cost-effective with 28.4 $ per kg than the concentrated electrolyte (81.9 $ per kg). The zinc–iodine battery with the 3 m ZnSO4 + 0.1 m Pr4NCl electrolyte demonstrates exceptional longevity, sustaining over 1100 cycles at 5C with 74% capacity retention (Fig. S19b†). The reliability of the complexation strategy was also accessed at a high current density of 4420 mA g−1 (10C), where it underwent more than 5000 cycles with minor degradation (Fig. S19c†).
Application of the complexation chemistry in an electrode
While the use of quaternary ammonium salts as electrolytes has demonstrated success, the solubility of these salts in electrolytes (i.e. 0.4 m for Pr4NCl in 3 m ZnSO4 electrolyte), along with the required volume of the electrolytes for a high areal capacity, imposes constraints on the energy density of 4eZIBs. In response to this challenge, we propose employing QICl2 compounds directly as cathodes. In these cases, the insoluble but strong complexing Hexy- and Amy-quaternary ammonium salts can be utilized, while the electrolyte is composed of 3 m ZnSO4 + 1 m ZnCl2. Note that the specific capacity was based on the mass fraction of iodine, if not specified, so as to facilitate comparison.As shown in Fig. 4a and S20,† the Me4NICl2 and Et4NICl2 cathodes in the 3 m ZnSO4 + 1 m ZnCl2 electrolyte exhibited fast capacity decay due to their solubility in the electrolyte as discussed in Fig. 1c. Conversely, the Hexy4NICl2 cathode showed minimal capacity fade and a consistent charge/discharge profile for long-term cycling, which is correlated with their strong complexation ability to ICl2− anion. An intriguing observation is the shift in the equilibrium potential of quaternary ammonium dichloroiodates from ∼1.7 V for Me4NICl2 and Et4NICl2 to ∼1.6 V for others. This shift can be attributed to the change of ICl2− from a partially dissociated/hydrolyzed environment to a tightly associated hydrophobic environment in aqueous electrolytes. When using an electrolyte comprising 1 m ZnSO4 + 1 m ZnCl2, the Hexy4NICl2 cathode showed robust performance with negligible capacity degradation, while Amy4NICl2 exhibited a relatively fast decay (Fig. S21†). These indicate that, in addition to the complexation chemistry, the kosmotropic ZnSO4 electrolyte with its strong salting-out effect due to the intrinsic structured water formation ability, also plays a pivotal role in suppressing the dissolution/hydrolysis of the chaotropic quaternary ammonium dichloroiodates.40,41
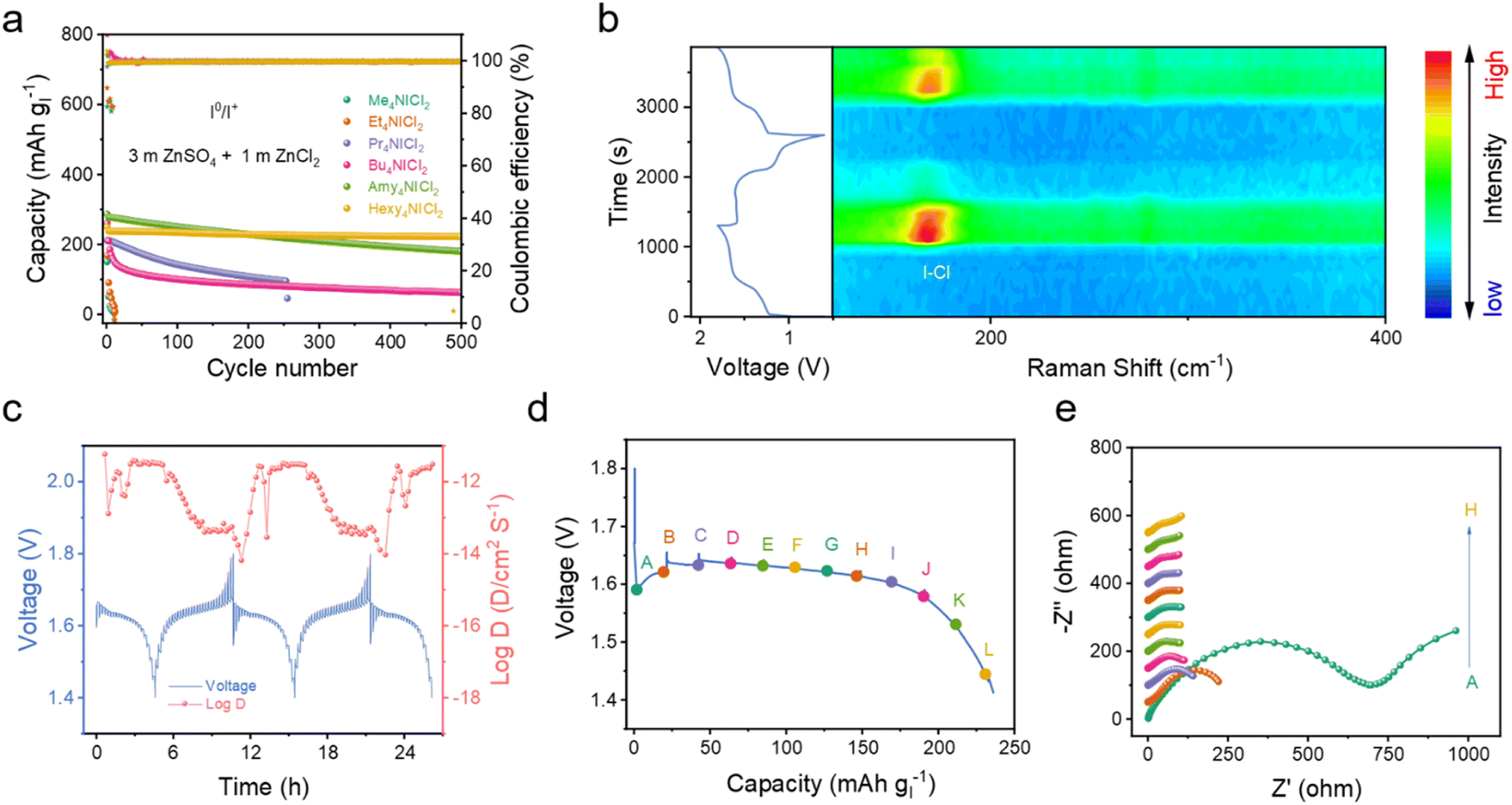 | ||
| Fig. 4 Electrochemical mechanism of the 4eZIBs using the QICl2 cathode. (a) Cycling performance of various QICl2 compounds. (b) In situ Raman spectrum of Hexy4NICl2 during charge/discharge. (c) The GITT profile and corresponding diffusion coefficient of the Hexy4NICl2 electrode. (d) The voltage profile of the Hexy4NICl2 electrode and (e), the corresponding EIS spectrum at different depths of discharge. The electrolytes were 3 m ZnSO4 + 1 m ZnCl2. | ||
Real-time monitoring of iodine species on the electrode by in situ Raman spectroscopy captured the typical Raman signal around 167 cm−1 (Fig. 4b), which either rose or disappeared during the charge/discharge process to 1.6 V/1.4 V, corresponding to the bending vibration (v2) of the I–Cl band.29,30 Besides, the weak signal of the symmetric stretching and asymmetric stretching of the I–Cl band was also found when charged to 1.8 V (Fig. S22†). These relatively weaker signals compared to those of the synthesized Hexy4NICl2 might be attributed to the decreased crystallinity of the material formed by the electrochemical process.26,42 These are in accordance with the redox process when using the quaternary ammonium salt as the electrolyte additive. The diffusion coefficient of the Hexy4NICl2 electrode reveals a reduction of nearly one order of magnitude at the beginning of the discharge process (Fig. 4c). This substantial decrease is likely attributed to the necessity of chemical bond deformation for the reduction/decomposition of Hexy4NICl2. The charge transfer resistance of the Hexy4NICl2 electrode at the beginning of the discharge process (point A) is the highest, further aligning with these observations (Fig. 4d and e). Additionally, Fig. S23† demonstrates that when the battery operates at 60 °C, the voltage drop during the initial discharge process is notably smaller compared to that at room temperature. This can be attributed to the improved reaction kinetics by the elevated temperature that facilitates the breaking of chemical interaction within Hexy4NICl2. However, it is important to note that the high temperature also increases the solubility of Hexy4NICl2, diminishing its stability in the electrolyte and resulting in a relatively low coulombic efficiency (about 88% at 60 °C).
By employing Hexy4NICl2 as the cathode, the Zn–I2 battery based on the I0/I+ couple showed nearly consistent charge and discharge curves throughout 500 cycles (Fig. 5a), and 250 mA h g−1 at 1C. It should be noted that Super P contributes a small fraction of capacity in the operational voltage window (Fig. S24†). This Zn–I2 battery based on the I0/I+ couple also showed a remarkable rate performance of 120 mA h g−1 at 100C (Fig. 5b) and sustained long-term cycle performance over 2000 cycles with an 88% capacity retention at 5C (Fig. 5c). For comprehensively understanding the role of the electrolyte in this battery system, we examined an electrolyte containing only ZnSO4 (Fig. S25†). An electrolyte composed solely of ZnSO4 resulted in rapid fading and pronounced polarization, highlighting that an extra Cl− carrier in the electrolyte is also essential to accomplish the multi-electron conversion.
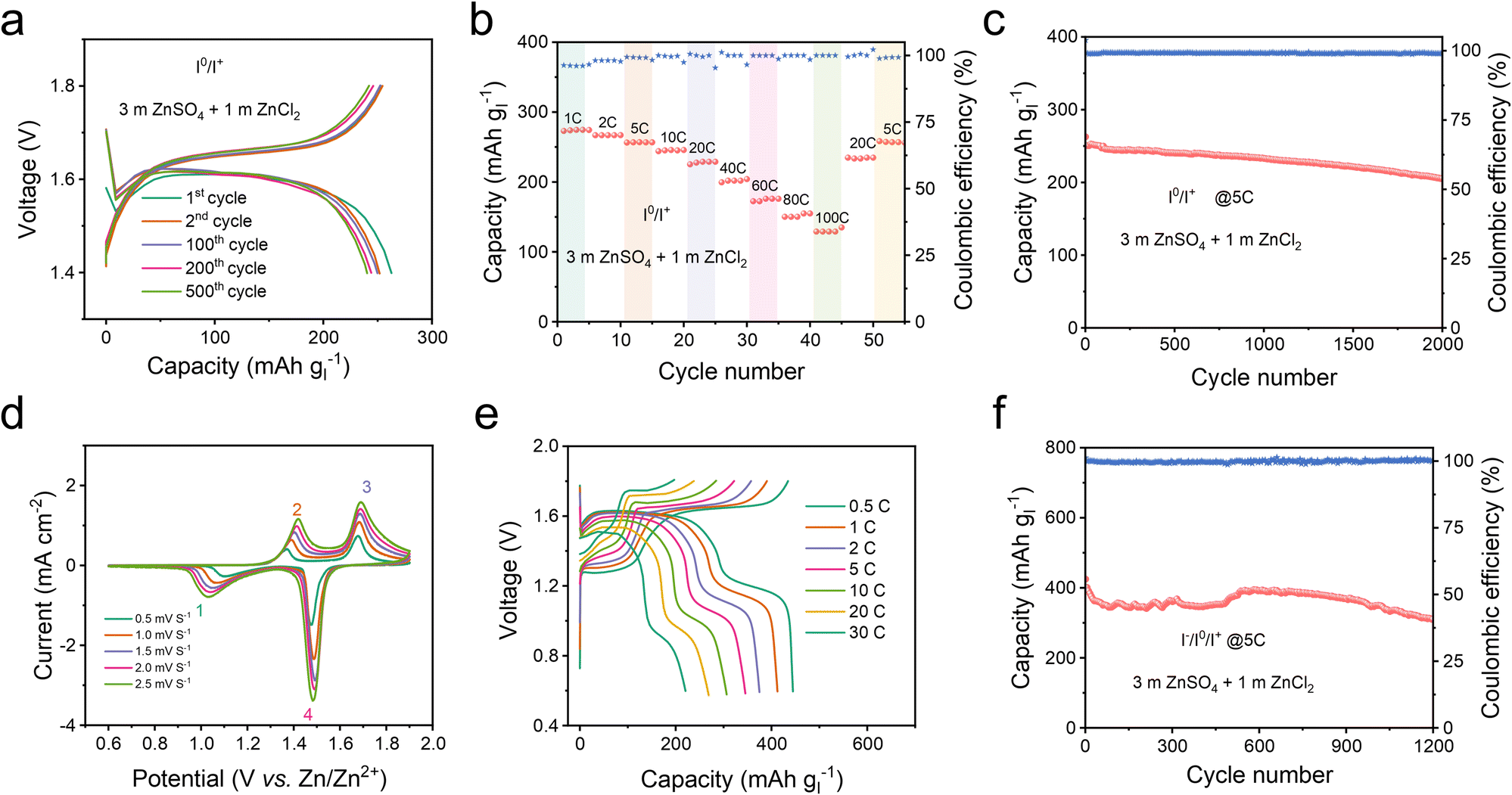 | ||
| Fig. 5 Cycling performance of the 4eZIBs with the Hexy4NICl2 cathode. (a) Voltage profile, (b) rate behavior and (c) long term battery performance of Hexy4NICl2 within the voltage range of 1.4–1.8 V. (d) CV curves of Hexy4NICl2 at sweep rates from 0.5 to 2.5 mV s−1. (e) Voltage profiles at various rates and (f) long term cycling performance of 4eZIBs within the voltage range of 0.6–1.8 V. The electrolyte was 3 m ZnSO4 + 1 m ZnCl2. | ||
In the context of the I−/I0/I+ redox couple within the Hexy4NICl2 cathode, the 4eZIB has two redox couples around 1.3 V and 1.6 V (Fig. 5d). The fitted b values of the four redox peaks are 0.664, 0.644, 0.519, and 0.517 respectively (Fig. S26†), indicating that the energy storage process is governed by a combination of both the diffusion process and the pseudocapacitive process. The proposed 4eZIB exhibits a self-discharging rate of only 2.5% after charging to 1.8 V with a 5 hour rest period (Fig. S27†). This 4eZIB demonstrates two well-defined charge/discharge plateaus, even at a high rate of 30C, with a discharge capacity of about 445 mA h g−1 (or 118 mA h g−1 based on Hexy4NI mass) at 0.5C and 200 mA h g−1 (or 52 mA h g−1 base on Hexy4NI mass) at 30C (Fig. 5e and S28†). Additionally, the 4eZIB with the Hexy4NICl2 cathode can sustain operation at 5C (2110 mA h g−1) for 1200 cycles while retaining 75% capacity (Fig. 5f and S29†).
We noticed that a trace of the quaternary ammonium salt in the electrolyte could also regulate the zinc anode electroplating process. In the blank electrolyte, the zinc anode exhibited an uneven deposition morphology (Fig. S30a†), which gave rise to a pronounced dendrite formation and a reduced lifespan of the battery. In contrast, the introduction of the Hexy4NCl additive (5 mmol kg−1, close to its saturation) led to a dense plating of zinc under the same conditions (Fig. S30b†). In Zn‖Ti asymmetric cells, the high surface area dendrites induced severe side reactions with the electrolyte, resulting in a low coulombic efficiency in the blank ZnSO4 electrolyte (Fig. S30c†). In comparison, the Zn‖Ti cells achieved an improved coulombic efficiency of 99.2% in the 5 mmol Hexy4NCl electrolyte. Moreover, repeated plating/stripping tests in Zn‖Zn symmetric cells demonstrated the robustness of the zinc anode in the presence of the Hexy4NCl additive (Fig. S30d†).
Conclusions
In summary, this study focused on enhancing the stability and performance of 4eZIBs in the commonly used ZnSO4 electrolyte through the utilization of symmetric quaternary ammonium salts with distinct chain lengths. These salts provide substantial complexation ability to I+ species, which mitigated its hydrolysis/dissolution thus the electrochemical performance was improved. This study not only delved into the complexation chemistry but also elucidated the underlying mechanisms governing the I−/I0/I+ redox couple, shedding light on the transformation of iodine states. Particularly, Hexy4NICl2 demonstrated excellent stability in aqueous media and served as an effective cathode, leading to superior rate capability and an extended lifespan of 4eZIB. These findings offer promising prospects for the practical application of 4eZIBs, addressing the solubility and stability concerns and providing a sustainable solution for energy storage systems. While we note that the symmetric quaternary ammonium cations take up a large amount of mass in comparison to iodine, further optimization of the ammonium salts with asymmetric structures or smaller molecular weights is necessary, which are currently being explored in our laboratory. Additionally, the formation of a stable hydrophobic QICl2 layer on the iodine electrode might also prevent the hydrolysis of internally oxidized I+ at a high iodine content.Data availability
The data supporting this article have been uploaded as part of the ESI.† Additional data is available from the authors on reasonable request.Author contributions
P. Jiang, Q. Du: electrochemical experiments and material synthesis; C. Lei: theoretical discussions; C. Xu, T. Liu: instrumental characterization; X. He: commentary or revision of the manuscript; X. Liang: proposed, designed and organized the work and wrote the manuscript; all the authors participated in the discussions and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program of China (2019YFA0210600 and 2022YFB3807700) and National Natural Science Foundation of China (51972107 and 22309048).Notes and references
- F. Zhang, Q. Wang and Y. Tang, Matter, 2021, 4, 2637–2639 CrossRef CAS.
- Y. Yang, S. Liang and J. Zhou, Curr. Opin. Electrochem., 2021, 30, 100761 CrossRef CAS.
- D. Lin and Y. Li, Adv. Mater., 2022, 2108856 CrossRef CAS PubMed.
- X. Li, M. Li, Z. Huang, G. Liang, Z. Chen, Q. Yang, Q. Huang and C. Zhi, Energy Environ. Sci., 2021, 14, 407–413 RSC.
- Z. Zhang, Y. Zhu, M. Yu, Y. Jiao and Y. Huang, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- L. Yan, S. Zhang, Q. Kang, X. Meng, Z. Li, T. Liu, T. Ma and Z. Lin, Energy Storage Mater., 2023, 54, 339–365 CrossRef.
- Y. Yang, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2022, 15, 1192–1200 RSC.
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170 CrossRef CAS PubMed.
- Q. Guo, K.-I. Kim, S. Li, A. M. Scida, P. Yu, S. K. Sandstrom, L. Zhang, S. Sun, H. Jiang, Q. Ni, D. Yu, M. M. Lerner, H. Xia and X. Ji, ACS Energy Lett., 2021, 459–467 CrossRef CAS.
- Z. Xue, Z. Gao and X. Zhao, Energy Environ. Mater., 2022, 5, 1155–1179 CrossRef CAS.
- F. A. Philbrick, J. Am. Chem. Soc., 1934, 56, 1257–1259 CrossRef CAS.
- S. Lv, T. Fang, Z. Ding, Y. Wang, H. Jiang, C. Wei, D. Zhou, X. Tang and X. Liu, ACS Nano, 2022, 16, 20389–20399 CrossRef CAS.
- J. Xu, T. P. Pollard, C. Yang, N. K. Dandu, S. Tan, J. Zhou, J. Wang, X. He, X. Zhang, A.-M. Li, E. Hu, X.-Q. Yang, A. Ngo, O. Borodin and C. Wang, Joule, 2023, 7, 83–94 CrossRef CAS.
- X. Li, S. Wang, D. Zhang, P. Li, Z. Chen, A. Chen, Z. Huang, G. Liang, A. L. Rogach and C. Zhi, Adv. Mater., 2023, 2304557 Search PubMed.
- X. Li, Y. Wang, Z. Chen, P. Li, G. Liang, Z. Huang, Q. Yang, A. Chen, H. Cui, B. Dong, H. He and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202113576 CrossRef CAS PubMed.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- J. J. Hong, L. Zhu, C. Chen, L. Tang, H. Jiang, B. Jin, T. C. Gallagher, Q. Guo, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 15910–15915 CrossRef CAS.
- F. Ascoli and F. M. Kahan, J. Biol. Chem., 1966, 241, 428–431 CrossRef CAS PubMed.
- S. Kajigaeshi, M. Moriwaki, S. Fujisaki, T. Kakinami and T. Okamoto, Bull. Chem. Soc. Jpn., 1990, 63, 3033–3035 CrossRef CAS.
- J. J. Novoa, F. Mota and S. Alvarez, J. Phys. Chem., 1988, 92, 6561–6566 CrossRef CAS.
- E. H. Wiebenga, E. E. Havinga and K. H. Boswijk, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeleus and A. G. Sharpe, Academic Press, 1961, vol. 3, pp. 133–169 Search PubMed.
- S. J. Yoo, B. Evanko, X. Wang, M. Romelczyk, A. Taylor, X. Ji, S. W. Boettcher and G. D. Stucky, J. Am. Chem. Soc., 2017, 139, 9985–9993 CrossRef CAS PubMed.
- L. Gao, Z. Li, Y. Zou, S. Yin, P. Peng, Y. Shao and X. Liang, iScience, 2020, 23, 101348 CrossRef CAS PubMed.
- M. Wang, T. Li, Y. Yin, J. Yan, H. Zhang and X. Li, Adv. Energy Mater., 2022, 12, 2200728 CrossRef CAS.
- P. Li, X. Li, Y. Guo, C. Li, Y. Hou, H. Cui, R. Zhang, Z. Huang, Y. Zhao, Q. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2022, 12, 2103648 CrossRef CAS.
- A. G. Maki and R. Forneris, Spectrochim. Acta, Part A, 1967, 23, 867–880 CrossRef CAS.
- W. B. Person, G. R. Anderson, J. N. Fordemwalt, H. Stammreich and R. Forneris, J. Chem. Phys., 1961, 35, 908–914 CrossRef CAS.
- T. J. Emge, H. H. Wang, P. C. W. Leung, P. R. Rust, J. D. Cook, P. L. Jackson, K. D. Carlson, J. M. Williams and M. H. Whangbo, et al. , J. Am. Chem. Soc., 1986, 108, 695–702 CrossRef CAS.
- M. S. Abdelbassit, O. J. Curnow, M. K. Dixon and M. R. Waterland, Chem.–Eur. J., 2019, 25, 11650–11658 CrossRef CAS PubMed.
- W. Gabes and H. Gerding, J. Mol. Struct., 1972, 14, 267–279 CrossRef CAS.
- K. J. Shah, M. K. Mishra, A. D. Shukla, T. Imae and D. O. Shah, J. Colloid Interface Sci., 2013, 407, 493–499 CrossRef CAS.
- D. P. Day, N. I. Alsenani and A. A. Alsimaree, Eur. J. Org Chem., 2021, 2021, 4299–4307 CrossRef CAS.
- D. Janas, A. P. Herman, S. Boncel and K. K. K. Koziol, Carbon, 2014, 73, 225–233 CrossRef CAS.
- Y. L. Wang, J. C. Nagy and D. W. Margerum, J. Am. Chem. Soc., 1989, 111, 7838–7844 CrossRef CAS.
- J. Jortner and A. Treinin, Trans. Faraday Soc., 1962, 58, 1503–1510 RSC.
- W. Gabes and D. J. Stufkens, Spectrochim. Acta, Part A, 1974, 30, 1835–1841 CrossRef.
- S. Schott, L. Ress, J. Hrušák, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2016, 18, 33287–33302 RSC.
- A. I. Popov and R. F. Swensen, J. Am. Chem. Soc., 1955, 77, 3724–3726 CrossRef CAS.
- W. Gabes and D. J. Stufkens, Spectrochim. Acta, Part A, 1974, 30, 1835–1841 CrossRef.
- N. J. Bridges, K. E. Gutowski and R. D. Rogers, Green Chem., 2007, 9, 177–183 RSC.
- W.-Y. Kim, H.-I. Kim, K. M. Lee, E. Shin, X. Liu, H. Moon, H. Adenusi, S. Passerini, S. K. Kwak and S.-Y. Lee, Energy Environ. Sci., 2022, 15, 5217–5228 RSC.
- R. S. Halford, J. Chem. Phys., 1946, 14, 8–15 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06155h |
| ‡ P. Jiang and Q. Du contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |