
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring a new short-wavelength nonlinear optical fluoride material featuring unprecedented polar cis-[Zr6F34]10− clusters†
Mei
Yan
,
Ru-Ling
Tang
 *,
Wen-Dong
Yao
,
Wenlong
Liu
and
Sheng-Ping
Guo
*
*,
Wen-Dong
Yao
,
Wenlong
Liu
and
Sheng-Ping
Guo
*
School of Chemistry and Chemical Engineering, Yangzhou University, 180 Siwangting Road, Yangzhou 225002, P. R. China. E-mail: rltang@yzu.edu.cn; spguo@yzu.edu.cn
First published on 15th January 2024
Abstract
Traditional fluorides are rarely reported as candidates for nonlinear optical (NLO) materials featuring a deep-ultraviolet cutoff edge. Theoretical investigations suggest that the ZrF8 dodecahedron shows large polarizability anisotropy and benefits for large birefringence. Herein, a new fluorine-rich fluoride, K3Ba2Zr6F31, was synthesized by coupling the ZrF8 group, featuring acentric cis-[Zr6F34]10− clusters with a 63-screw axis. Significantly, K3Ba2Zr6F31 exhibits a short UV cutoff edge (below 200 nm) and moderate second-harmonic generation (SHG) response (0.5 × KH2PO4). It also possesses a relatively large birefringence (0.08@1064 nm), together with a broad transparency window (2.5–21.1 μm). First-principles calculations suggest that the cis-[Zr6F34]10− cluster built by ZrF8 dodecahedra are the dominant contributors to the large band gap (7.89 eV, cal.) and SHG response simultaneously. Such systematic work highlights that Zr-based fluorides afford a new paradigm for the development of efficient NLO materials with a short UV cutoff edge.
Introduction
Deep-ultraviolet (DUV) nonlinear optical (NLO) materials can produce coherent light below 200 nm through a second harmonic generation (SHG) process, which is useful for diverse applications in high-tech equipment and represents a leading edge in materials and civil science.1–6 Traditionally, research on such materials has been mainly limited to π-conjugated systems, expanding from borate, carbonates like AEB8O15H4 (AE = Sr, Ca),7 β-Rb2Al2B2O7,8 LiZn(OH)CO3,9 and K5Mg2La3(BO3)6 (ref. 10) to borate fluorides, fluorooxoborates, fluoride carbonates as RbAlB3O6F,11 Cs0.5Rb0.5AlB3O6F,11 CsB4O6F,12 M2B10O14F6 (M = Ca, Sr),13 BaBe2BO3F3,14 KMgCO3F.15 The latest, non-π-conjugated systems have provided a source for DUV transparent materials, and can be divided into phosphates, sulfates, silicates, and their derivatives. The discovery of LiRb2PO4,16 CsNaMgP2O7,17 Ba3P3O10X (X = Cl, Br),18 Li2BaSiO4,6 Cs2B4SiO9,19 Li3AlSiO5,20 and K2Zn3(SO4)(HSO4)2F4,21 have attracted more scientists to consider these systems as promising candidates for DUV NLO materials and to discover more compounds.The prerequisite of eminent NLO crystals is the ability to crystallize into noncentrosymmetric structures.22–26 Polar materials, one branch of noncentrosymmetric (NCS) symmetries, have been increasingly recognized as an indispensable part, along with various polar functional building blocks (FBBs). Wu’s group proposed a polar iodate K2Zn(IO3)3(I2O5(OH))(IO2(OH))(H2O) composed of “tumbler-like” [Zn(IO3)(I2O5(OH))] clusters.27 Luo’s group reported one DUV crystal Ba(SO3CH3)2, with a new polar non-π-conjugated NLO building unit, SO3CH3−.28 Wu’s group synthesized Zn(NH3)CO3 which displays a “three-in-one” NLO-active motif which includes a polar NH3 molecule, polar displaced Zn2+, and π-conjugated CO32− ions.29 The polar tetrahedral S2O3 group constructed of one UV NLO crystal (NH4)S2O3 was provided by Luo’s group.30 Additionally, to obtain a short UV cutoff edge, fluorine with large polarizability anisotropy and hyperpolarizability is frequently used in designing new NLO materials with a DUV cutoff edge.31–35 The borate fluorides, fluorooxoborates, fluoride carbonates, fluoride sulfates, and fluoride phosphates mentioned above with oxyfluoride moieties, have been reported with excellent overall NLO properties. The success of synthesizing these crystals draws our attention to pure fluorides. To date, the reported three fluorides (BaMgF4, BaZnF4, and SrAlF5) all feature short UV transparency (∼126, 155, and 145 nm).36–38 Nevertheless, severe drawbacks of small birefringence hinder the realization of phase-matchable (PM) behaviors.
To break the limits of non-PM behavior and impart a sense of polarity into a material, our work utilizes polar metal fluoride polyhedra. We herein comprise ZrF8 dodecahedra with large polarizability anisotropy and present a polar fluoride, K3Ba2Zr6F31, through assembling the alignment of acentric cis-[Zr6F34]10− clusters. Additionally, the K+ and Ba2+ cations without d–d/f–f transition are incorporated, and we survey the K–Ba–Zr–F system in detail. Since the band gap and optical properties are mainly decided by the Zr and F atoms, and the F-2p nonbonding states are the dominant contributors for the top of the valence band, speculation about the higher ratio of F atoms in the material may lead to a shorter UV cutoff edge, leading to an increase in the quantity of HF and altered ratio of reagents. Fortunately, our endeavor results in a new polar fluoride, K3Ba2Zr6F31. The cis-[Zr6F34]10− clusters, formed by vertex- and edge-sharing ZrF8 dodecahedra arranged in the same orientation, makes K3Ba2Zr6F31 exhibit a moderate SHG response and a DUV cutoff edge below 200 nm. It also features PM behavior, a large calculated band gap (7.89 eV), and large birefringence (0.08@1064 nm), which declares it as a potential NLO crystal and verifies the cis-[Zr6F34]10− cluster as a promising functional motif for constructing NLO materials.
Results and discussion
Colorless and transparent rod-like crystals of K3Ba2Zr6F31 were synthesized via a hydrothermal thermal reaction from a mixture of KF, ZrF4, BaCl2·2H2O, and HF solution at 200 °C (detailed description in the ESI†). The experimental powder X-ray diffraction (PXRD) pattern is characterized and shown in Fig. S1a,† matching well with the simulated one and confirming the purity of the powder sample. The result is also confirmed with EDS elemental analysis, as shown in Fig. S1b.†With a polar structure, K3Ba2Zr6F31 adopts the hexagonal space group P63mc (No. 186) (Table S1†). The asymmetric unit is made up of one unique K, one Ba, two Zr, and eight F atoms (Table S2†). The Zr(1) atom is eight-coordinated with F atoms to form a distorted ZrF8 bicapped triangular prism with wide Zr–F distances ranging from 1.991(3) to 2.348(3) Å (Table S3†). The Zr(2) atom also exhibits an eight-coordinated mode, being bonded to F atoms with bond lengths of 2.008(3)–2.169(3) Å and forming a ZrF8 bicapped triangular prism. Three Zr(1)F8 units share edges to form the [Zr(1)3F19]7− cluster and three Zr(2)F8 units share vertexes to form the [Zr(2)3F21]9− cluster (Fig. S2a and b†). The [Zr(1)3F19]7− and [Zr(2)3F21]9− clusters further share vertexes and edges to form the cis-[Zr6F34]10− cluster (Fig. 1a and b). The polar cis-[Zr6F34]10− clusters then share F(5) atoms to generate the {[Zr6F31]7−}∞ chain along the c-axis, which arrange parallel to the ab plane with the 63-screw axis (Fig. 1c). The K and Ba atoms are all bonded to twelve F atoms with bond distance ranges of d(K–F) = 2.647(5)–3.270(5) Å and d(Ba–F) = 2.795(3)–2.912(3) Å (Fig. S3†), situating at the space between the chains and serving as the counter ion to maintain charge balance (Fig. 1d). The BVS results of K, Ba, Zr, and F atoms are 0.92, 2.11, 4.06–4.09, 0.79–1.11, respectively, consistent with the normal valence states (Table S2†).39
 | ||
| Fig. 1 (a and b) Cis-[Zr6F34]10− cluster at the ac and ab plane; (c) the {[Zr6F31]7−}∞ chain along the c-axis; (d) the aligned arrangement of the {[Zr6F31]7−}∞ anionic chains and whole structure of K3Ba2Zr6F31 viewed along the c-axis. | ||
The polymeric Zr–F clusters, linkage association modes, and dimensions of Zr-based fluorides are concluded in Table S4.† It can be seen that the hexametric clusters are common in Zr-based fluorides, and small amounts are trimeric, tetrameric, or higher octal, decameric clusters. Nevertheless, the [Zr6F34]10− clusters are first reported here in zirconium fluorides, distinct from the [Zr6F36]12−, and [Zr6F38]14− clusters in other compounds.40,41 The {[Zr6F31]7−}∞ chain is also observed for the first time in K3Ba2Zr6F31, unlike the common {[Zr2F12]4−}∞ chain in BaZrF6,42 K2ZrF6,43 Li2ZrF6.44 As summarized in Tables S5 and S6,† K3Ba2Zr6F31 is not only the first with an asymmetric structure among the fluorides with 31 F elements in the formula, but also the first reported to be SHG-active among the fluorine-rich fluorides (F > 30), although (XeF)2(Ti9F38) and LiK10Zr6F35·2H2O also crystallizes in the NCS space groups.45,46
The thermal behavior of K3Ba2Zr6F31 was investigated using differential thermal analysis (DTA), thermogravimetric analysis (TGA), and PXRD characterization. As seen in Fig. S4a,† no distinct weight loss was apparent from the TGA curve until 650 °C, while an endothermic peak could be observed around 490 °C on the DTA curve. In order to find out the attribution of the peak at 490 °C, the powder sample was heated to 500 °C followed by PXRD (Fig. S4b†). The XRD peaks show obvious differences with the peaks in the unheated sample, and the residues correspond to BaF2 and K3Zr2F11. The XRD pattern of the sample heated to 630 °C is similar to that of the sample heated to 500 °C, which manifests the decomposition of K3Ba2Zr6F31. The weight loss covering 650–1000 °C could be assigned to the partial decomposition of the ZrF4, and the apparent weight loss (43.7%) detected is consistent with the calculated value (cal. 40.7%). The residues at 1000 °C are confirmed to be Ba4Zr2F16 and some unknown compounds, using the PXRD results.
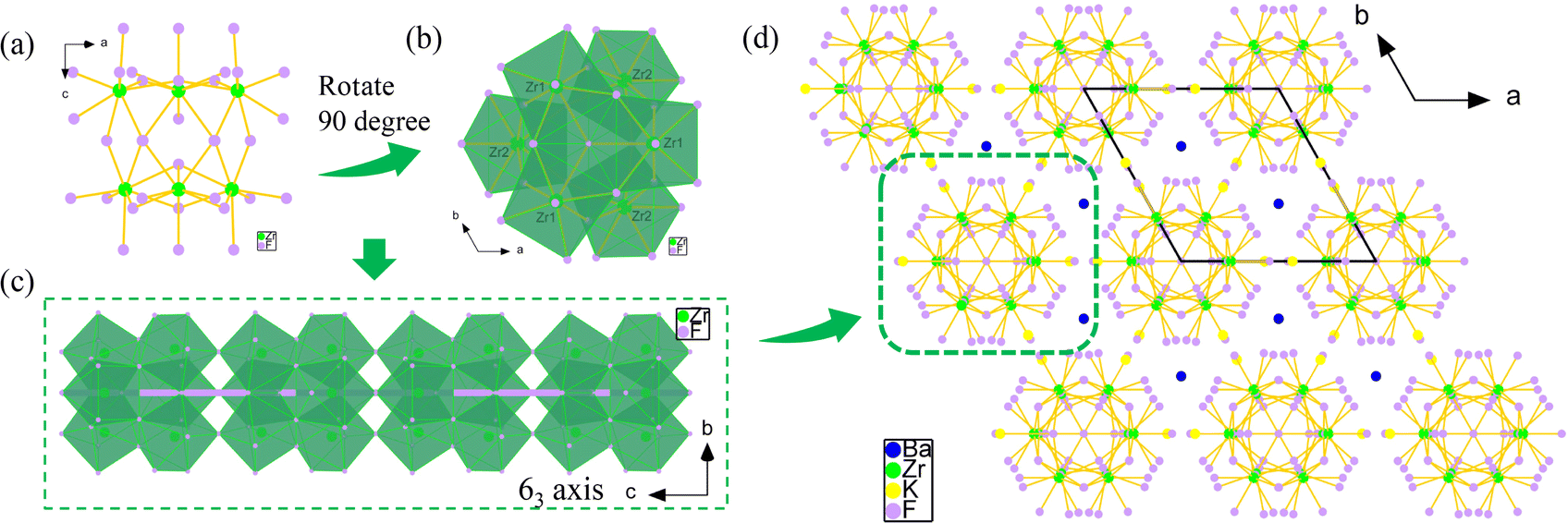
The UV-vis-NIR diffuse reflectance spectrum (Fig. 2a) was measured and showed that K3Ba2Zr6F31 is transparent down to 200 nm (corresponding to a band gap larger than 6.2 eV) with high reflectance exceeding 67% in the range of 200–1400 nm. The value is larger than some fluorides like KNa2ZrF7 (5.07 eV),37 Na2CeF6 (3.89 eV),47 and [Cd(C4H6N2)4]3[ZrF7]2 (C4H6N2 = 3methylpyrazole) (5.2 eV),48 and indicates its potential application in the UV region. The IR spectrum of K3Ba2Zr6F31 exhibits one obvious peak at 474 cm−1, which can be assigned to the stretching and bending vibrations of the Zr–F bonds in the ZrF8 units (Fig. 2b).49
 | ||
| Fig. 2 (a) UV-vis-NIR diffuse reflectance and (b) IR spectra of K3Ba2Zr6F31. | ||
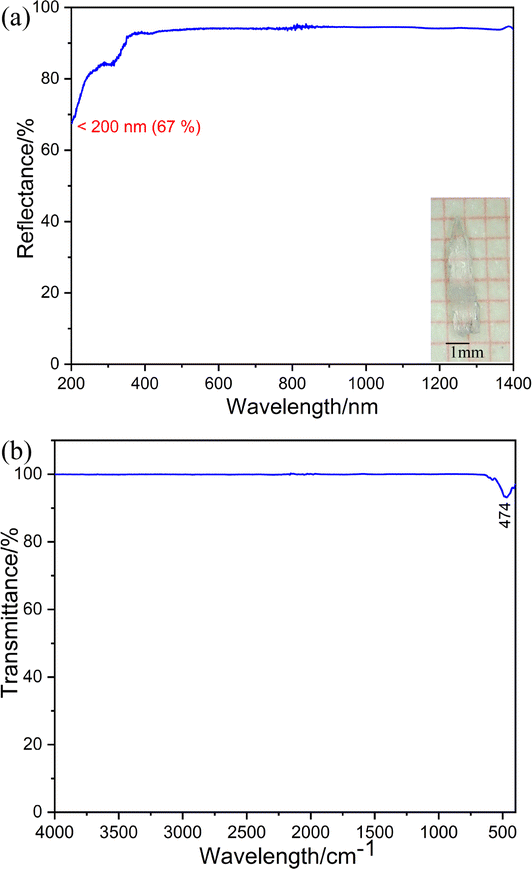
Powder SHG measurements under 1064 nm of K3Ba2Zr6F31 were performed using the Kurtz–Perry method to evaluate the NLO properties.50 The result indicates that PM behavior can be realized since the SHG intensities gradually increase with the increase of particle size and then reach a platform (Fig. 3a). Additionally, it showcases moderate SHG efficiency of about 0.5 times that of KH2PO4 (KDP) in the particle size range 200–250 μm (Fig. 3b), and is comparable with reported halides with a short UV cutoff edge, like SrAlF5 (0.65 × KDP).38 In fact, the SHG intensity of K3Ba2Zr6F31 (0.5 × KDP) is relatively weak when compared with those previously reported. To further improve the SHG efficiency of halides, Cl or Br atoms may be introduced for the favorable chemical covalence and flexibility, and relatively large polarizability. Halides with mixed halogen anions tend to show enhanced overall NLO performance, as demonstrated in previous studies involving Pb18O8Cl15I5,51 Pb7F12Cl2,52 and CsZnBO3X2 (X2 = F2, Cl2, and FCl).53 Additionally, the K site occupied by Rb/Cs may be helpful for larger SHG intensities for the increased polarizability of larger congener cations along with stronger interatomic interactions with adjacent F atoms, which is supported by the cases of LiA2PO4 (A = Rb, Cs),54–56 and A3VO(O2)2CO3 (A = K, Rb, and Cs).57–59
 | ||
| Fig. 3 (a) PM curves of K3Ba2Zr6F31 and KDP with 1064 nm laser radiation. (b) SHG intensities of K3Ba2Zr6F31 and KDP in the particle size range 200–250 μm. | ||
In order to elucidate the origin of the SHG response, the dipole moments of ZrF8 units have been calculated. Although the x and y components of the dipole moments in the unit cell of K3Ba2Zr6F31 did not completely offset, they only afford the values of 2.4205 and 0.0823 D in one unit cell. The z component provides much larger value of −19.8108 D that mainly determine the macroscopic polarization. The total dipole moment calculation of K3Ba2Zr6F31 affords a value of 19.9583 D for the net dipole moment in one unit cell, which corresponds with a dipole moment per unit cell of 0.034 D Å−3 (Table S7†). This value fits with the distorted coordination caused by the difference of the bond lengths and electronegativities, as mentioned above.
To clarify the microscopic mechanism of the properties of K3Ba2Zr6F31, theoretical calculations on the electronic structure and optical properties were performed in the CASTEP package based on density functional theory (DFT).60,61 The calculated band structure curve shows that K3Ba2Zr6F31 has an indirect band gap of 5.79 eV with the PBE function (Fig. S5a†). The HSE06 function was also performed and the calculated value is 7.89 eV for K3Ba2Zr6F31,62 corresponding to the very short cutoff edge of 157 nm (Fig. 4a). The underestimation of the calculated value is normal for the discontinuity of the local-density-approximation exchange–correlation function,63–66 and the scissor of 2.1 eV was adopted when analysing the optical properties of K3Ba2Zr6F31. The total and partial density of states (TDOS and PDOS) were also calculated to investigate the interrelation of atom orbitals. The top of the valence band near the Fermi level (Ef) is mainly occupied by F-2p nonbonding states, and the bottom of the conductive band is mainly dominated by Zr-4d and F-2p orbitals (Fig. S5b†), which illustrate that the band gap of K3Ba2Zr6F31 is determined by Zr and F atoms. Considering the Kleiman symmetry restriction and the space group, K3Ba2Zr6F31 has three nonzero independent SHG coefficients, d15, d31, and d33 (Fig. 4b). The largest SHG tensor of K3Ba2Zr6F31 is calculated to be χ15 = 0.17 pm V−1, which is 0.22 times that of KDP (d36 = 0.39 pm V−1). The calculated value is smaller than the experimental one (0.5 × KDP) and such underestimation has also been observed in other reports.34,67 The birefringence (Δn) is calculated to be 0.08@1064 nm (Fig. 4c), which is larger than some d0-TM based fluorides and oxyfluorides like CsNaTaF7 (0.01@1064 nm),68 K3ZrF4(SbF4)(SbF5) (0.038@1064 nm)49 and K8(ZrF6)(Sb2Zr2F20) (0.039@1064 nm).49 The birefringence (Δn) is large enough for PM ability and manifests the shortest PM wavelength (400 nm) calculated in Fig. S6.† The polarizability anisotropies of ZrF8, MgF6, AlF6, and ZnF6 species were calculated, and the value of ZrF8 (29.5) is larger than those of the other three species (10.2, 1.0, and 5.3), illustrating the greatly enhanced birefringence and PM behaviour of K3Ba2Zr6F31 (Fig. 4d). The birefringence is large enough to realize the PM behaviour and can be attributed to the strong local polarity generated by the ordered arrangement of the {[Zr6F31]7−}∞ chain along the c-axis.
 | ||
| Fig. 4 Calculation results for K3Ba2Zr6F31. (a) Band structure, (b) NLO coefficients, (c) refractive indexes, and (d) polarizability anisotropy of ZrF8, MgF6, AlF6, and ZnF6 species. The Fermi level is set at 0 eV. | ||
Conclusions
In summary, a new fluorine-rich NCS fluoride with polar structure K3Ba2Zr6F31, has been studied. It features cis-[Zr6F34]10− clusters formed of {[Zr6F31]7−}∞ anionic chains that formed by assembling ZrF8 dodecahedra with large polarizability anisotropy. It is a promising NLO material with moderate SHG response, DUV cutoff edge, and wide IR transition range. According to the theoretical calculations, it displays promising linear optical properties such as a large band gap (7.89 eV) and sufficient birefringence (0.08@1064 nm) for PM behaviour. This work opens the door to develop fluorides with polar structures composed of ZrF8 groups with large polarizability anisotropy, beneficial for large birefringence and accelerating the development of new NLO materials with DUV cutoff edge.Data availability
Data available in the ESI† includes experimental section, and additional tables and figures.Author contributions
This work was conceptualised by M. Yan and R. L. Tang. Experimentation was performed by M. Yan. Software was used by W. D. Yao. W. L. Liu supervised everything. R. L. Tang and S. P. Guo contributed funding acquisition and supervision. The first draft of the manuscript was prepared by M. Yan and the final draft was edited by all the authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22071212 and 22101248), the Lvyangjinfeng Talent Program of Yangzhou (YZLYJFJH2021YXBS083), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX22_3465).Notes and references
- M. Mutailipu, K. R. Poeppelmeier and S. L. Pan, Chem. Rev., 2021, 121, 1130–1202 CrossRef CAS PubMed.
- W. Q. Huang, S. G. Zhao and J. H. Luo, Chem. Mater., 2022, 34, 5–28 CrossRef CAS.
- Y. C. Liu, Y. G. Shen, S. G. Zhao and J. H. Luo, Coord. Chem. Rev., 2020, 407, 213152 CrossRef CAS.
- X. Liu, Y. C. Yang, M. Y. Li, L. Chen and L. M. Wu, Chem. Soc. Rev., 2023, 52, 8699 RSC.
- F. J. Hou, D. J. Mei, M. J. Xia and Y. D. Wu, Coord. Chem. Rev., 2021, 444, 214038 CrossRef CAS.
- H. P. Wu, B. B. Zhang, H. W. Yu, Z. G. Hu, J. Y. Wang, Y. C. Wu and P. S. Halasyamani, Angew. Chem., Int. Ed., 2020, 59, 8922–8926 CrossRef CAS PubMed.
- P. F. Gong, L. Kang and Z. S. Lin, J. Am. Chem. Soc., 2020, 142, 15157–15163 CrossRef CAS PubMed.
- T. T. Tran, N. Z. Koocher, J. M. Rondinelli and P. S. Halasyamani, Angew. Chem., Int. Ed., 2017, 56, 2969–2973 CrossRef CAS PubMed.
- X. M. Liu, L. Kang, P. F. Gong and Z. S. Lin, Angew. Chem., Int. Ed., 2021, 60, 13574–13578 CrossRef CAS PubMed.
- R. Q. Liu, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Chem. Mater., 2021, 33, 4240–4246 CrossRef CAS.
- H. K. Liu, B. B. Zhang, L. Li and Y. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 30853–30860 CrossRef CAS PubMed.
- X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS PubMed.
- M. Luo, F. Liang, Y. X. Song, D. Zhao, F. Xu, N. Ye and Z. S. Lin, J. Am. Chem. Soc., 2018, 140, 3884–3887 CrossRef CAS PubMed.
- S. Guo, X. X. Jiang, L. J. Liu, M. J. Xia, Z. Fang, X. Y. Wang, Z. S. Lin and C. T. Chen, Chem. Mater., 2016, 28, 8871–8875 CrossRef CAS.
- T. T. Tran, J. Young, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2017, 139, 1285–1295 CrossRef CAS PubMed.
- L. Li, Y. Wang, B. H. Lei, S. J. Han, Z. H. Yang, H. Y. Li and S. L. Pan, J. Mater. Chem. C, 2017, 5, 269–274 RSC.
- S. G. Zhao, Y. Yang, Y. G. Shen, X. D. Wang, Q. R. Ding, X. F. Li, Y. Q. Li, L. Li, Z. S. Lin and J. H. Luo, J. Mater. Chem. C, 2018, 6, 3910–3916 RSC.
- P. Yu, L. M. Wu, L. J. Zhou and L. Chen, J. Am. Chem. Soc., 2014, 136, 480–487 CrossRef CAS PubMed.
- H. P. Wu, H. W. Yu, S. L. Pan, Z. J. Huang, Z. H. Yang, X. Su and K. R. Poeppelmeier, Angew. Chem., Int. Ed., 2013, 52, 3406–3410 CrossRef CAS PubMed.
- X. L. Chen, F. F. Zhang, L. L. Liu, B. H. Lei, X. Y. Dong, Z. H. Yang, H.Y. Li and S. L. Pan, Phys. Chem. Chem. Phys., 2016, 18, 4362–4369 RSC.
- Y. Zhou, X. Y. Zhang, Z. Y. Xiong, X. F. Long, Y. Q. Li, Y. X. Chen, X. Chen, S. G. Zhao, Z. S. Lin and J. H. Luo, J. Phys. Chem. Lett., 2021, 12, 8280–8284 CrossRef CAS PubMed.
- Y. X. Ma, P. F. Li, C. L. Hu, J. G. Mao and F. Kong, Adv. Sci., 2023, 10, 2304463 CrossRef CAS PubMed.
- Y. Huang, B. X. Li, C. L. Hu, B. P. Yang and J. G. Mao, Inorg. Chem., 2023, 62, 3343–3348 CrossRef CAS PubMed.
- S.-J. Oh, D. W. Lee and K. M. Ok, Inorg. Chem., 2012, 51, 5393–5399 CrossRef CAS PubMed.
- G. H. Zou, C. S. Lin, H. Jo, G. Nam, T.-S. You and K. M. Ok, Angew. Chem., Int. Ed., 2016, 55, 12078–12082 CrossRef CAS PubMed.
- G. H. Zou and K. M. Ok, Chem. Sci., 2020, 11, 5404–5409 RSC.
- J. L. Chen, P. C. Yang, H. W. Yu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. P. Wu, ACS Mater. Lett., 2023, 5, 1665–1671 CrossRef CAS.
- H. T. Tian, C. S. Lin, X. Zhao, F. Xu, C. Wang, N. Ye and M. Luo, CCS Chem., 2022, 1–22 CrossRef.
- H. X. Tang, Q. R. Shui, R. B. Fu, Z. Q. Zhou, W. X. Bao, Z. J. Ma and X. T. Wu, J. Mater. Chem. C, 2021, 9, 16477–16484 RSC.
- S. X. Ke, H. X. Fan, C. S. Lin, N. Ye and M. Luo, Inorg. Chem. Front., 2023, 10, 2811–2817 RSC.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Angew. Chem., Int. Ed., 2023, 62, e202301420 CrossRef CAS PubMed.
- B. Zhang, J. H. Wu, C. L. Hu, Y. F. Li, F. Kong and J. G. Mao, Inorg. Chem. Front., 2023, 10, 1328–1337 RSC.
- M. Yan, R. L. Tang, W. L. Liu and S. P. Guo, Inorg. Chem., 2022, 61, 20709–20715 CrossRef CAS PubMed.
- X. Lian, W. D. Yao, W. L. Liu, R. L. Tang and S. P. Guo, Inorg. Chem., 2021, 60, 19–23 CrossRef CAS PubMed.
- E. Ko, H. Jo and K. M. Ok, Inorg. Chem., 2021, 60, 7914–7921 CrossRef CAS PubMed.
- J. G. Bergman, G. R. Crane and H. Guggenheim, J. Appl. Phys., 1975, 46, 4645–4646 CrossRef CAS.
- Y. Z. Tong, X. Y. Meng, Z. Z. Wang, C. T. Chen and M. H. Lee, J. Appl. Phys., 2005, 98, 033504 CrossRef.
- E. G. Víllora, K. Shimamura, K. Muramatsu, S. Takekawa, K. Kitamura and N. Ichinose, J. Cryst. Growth, 2005, 280, 145–150 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- J. H. Burns, R. D. Ellison and H. A. Levy, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 230–237 CrossRef CAS.
- J. P. Laval, B. Frit and J. Lucas, J. Solid State Chem., 1988, 72, 181–192 CrossRef CAS.
- J. P. Laval, R. Papiernik and B. Frit, Acta Crystallogr. B, 1978, 34, 1070–1074 CrossRef.
- R. Hoppe and B. Mehlhorn, Z. Anorg. Allg. Chem., 1976, 425, 200–208 CrossRef CAS.
- A. Grzechnik, V. Dmitriev and H. P. Weber, J. Phys. Chem. Solids, 2005, 66, 1769–1774 CrossRef CAS.
- K. A. Gayvoronskaya, N. A. Didenko, A. B. Slobodyuk, A. V. Gerasimenko and V. Y. Kavun, J. Fluorine Chem., 2011, 132, 1159–1164 CrossRef CAS.
- K. Radan, E. Goreshnik and B. Žemva, Angew. Chem., Int. Ed., 2014, 53, 13715–13719 CrossRef CAS PubMed.
- R. L. Tang, W. Xu, X. Lian, Y. Q. Wei, Y. L. Lv, W. L. Liu and S. P. Guo, Small, 2023, 2308348 CrossRef PubMed.
- B. Ahmed and K. M. Ok, J. Solid State Chem., 2022, 309, 122957 CrossRef CAS.
- T. K. Jiang, S. N. Yan, C. L. Hu, Y. F. Li, F. Kong and J. G. Mao, Inorg. Chem., 2022, 61, 4801–4805 CrossRef CAS PubMed.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- X. L. Chen, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2020, 59, 20712 CrossRef CAS.
- Q. Wu, X. Liu, F. Liang, S. R. Xu, H. B. Pi, X. Han, Y. Liu, Z. S. Lin and Y. J. Li, Dalton Trans., 2019, 48, 13529–13535 RSC.
- J. J. Zhou, Y. Q. Liu, H. P. Wu, H. W. Yu, Z. S. Lin, Z. G. Hu, J. Y. Wang and Y. C. Wu, Angew. Chem., Int. Ed., 2020, 59, 19006–19010 CrossRef CAS PubMed.
- X. Y. Cheng, M. H. Whangbo, G. C. Guo, M. C. Hong and S. Q. Deng, Angew. Chem., Int. Ed., 2018, 57, 3933–3937 CrossRef CAS PubMed.
- L. Li, Y. Wang, B. H. Lei, S. J. Han, Z. H. Yang, K. R. Poeppelmeier and S. L. Pan, J. Am. Chem. Soc., 2016, 138, 9101–9104 CrossRef CAS PubMed.
- L. Li, Y. Wang, B. H. Lei, S. J. Han, Z. H. Yang, H. Y. Li and S. L. Pan, J. Mater. Chem. C, 2017, 5, 269–274 RSC.
- G. H. Zou, H. Jo, S. Lim, T. You and K. M. Ok, Angew. Chem., Int. Ed., 2018, 57, 8619–8622 CrossRef CAS PubMed.
- G. H. Zou, Z. Lin, H. M. Zeng, H. Jo, S. Lim, T. You and K. M. Ok, Chem. Sci., 2018, 9, 8957–8961 RSC.
- Y. X. Song, M. Luo, F. Liang, N. Ye and Z. S. Lin, Chem. Commun., 2018, 54, 1445–1448 RSC.
- J. C. Stewart, D. S. Matthew, J. P. Chris, J. H. Phil, I. J. P. Matt, R. Keith and C. P. Mike, Z. Kristallogr.–Cryst. Mater., 2005, 220, 567–570 CrossRef.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- B. W. Liu, X. M. Jiang, H. Y. Zeng and G. C. Guo, J. Am. Chem. Soc., 2020, 142, 10641–10645 CrossRef CAS PubMed.
- H. D. Zhou, L. Xiong, L. Chen and L. M. Wu, Angew. Chem., Int. Ed., 2019, 58, 9979–9983 CrossRef CAS PubMed.
- A. K. Iyer, S. H. Ha, M. J. Waters, T. S. Ie, S. H. Shin, J. M. Rondinelli, J. I. Jang and M. G. Kanatzidis, Chem. Mater., 2023, 35, 8706–8713 CrossRef CAS.
- Y. K. Lian, R. A. Li, X. Liu, L. M. Wu and L. Chen, Cryst. Growth Des., 2020, 20, 8084–8089 CrossRef CAS.
- W. H. Xing, C. L. Tang, N. Z. Wang, C. X. Li, E. Uykur, J. Y. Wu, Z. S. Lin, J. Y. Yao, W. L. Yin and B. Kang, Adv. Opt. Mater., 2021, 9, 2100563 CrossRef CAS.
- R. L. Tang, X. Lian, X. H. Li, L. Huai, W. L. Liu and S. P. Guo, Chem.–Eur. J., 2022, 28, e202201588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and additional tables and figures. CCDC 2305228. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06683e |
| This journal is © The Royal Society of Chemistry 2024 |