
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[Br4F21]− – a unique molecular tetrahedral interhalogen ion containing a μ4-bridging fluorine atom surrounded by BrF5 molecules†
Martin
Möbs
a,
Tim
Graubner
a,
Antti J.
Karttunen
b and
Florian
Kraus
 *a
*a
aAnorganische Chemie, Fluorchemie, Philipps-Universität Marburg, Hans-Meerwein-Str. 4, 35032 Marburg, Germany. E-mail: f.kraus@uni-marburg.de
bDepartment of Chemistry and Materials Science, Aalto University, 00076 Espoo, Finland
First published on 20th January 2024
Abstract
The reaction of [NMe4][BrF6] with an excess of BrF5 leads to the compound [NMe4][Br4F21]·BrF5. It features molecular [(μ4-F)(BrF5)4]− anions of tetrahedron-like shape containing central μ4-bridging F atoms coordinated by four BrF5 molecules. It is the most BrF5-rich fluoridobromate anion by mass. Quantum-chemical calculations showed that the μ4-F–Br bonds within the anion are essentially ionic in nature. The compound is the first example where F atoms bridge μ4-like neither to metal nor to hydrogen atoms. It was characterized by Raman spectroscopy and by single-crystal X-ray diffraction. The latter showed surprisingly that its crystal structure is related to the intermetallic half-Heusler compound and structure type MgAgAs.
Introduction
The halogen fluorides form a class of chemically most reactive compounds, some of them even exceeding the reactivity of F2 itself. BrF3, BrF5, and ClF3 are hypergolic oxidizers that may react vigorously, sometimes explosively, with many organic and inorganic substrates and materials if conditions are not controlled very carefully.1,2 If non-oxidizable starting materials are used, such violent reactions are not to be expected, the respective halogen fluoride is not decomposed and may act as a ligand. Towards Lewis acids, such as BF3, AsF5 or SbF5, the halogen fluorides can act as fluoride ion donors, forming the corresponding fluoridohalonium cations,3,4 whereas towards Lewis basic compounds, such as the alkali metal fluorides, many halogen fluorides can act as a fluoride ion acceptors, forming fluoridohalogenate anions.5,6 Currently known poly halogen and interhalogen anions are summarized in Table S1.† Currently known poly (inter-) halogen cations are listed in Table S2.† Besides numerous “mononuclear” halogen fluoride ions, only a few examples have been discovered, where halogen fluoride molecules and their derived ions are linked by bridging F atoms forming “oligonuclear” halogen fluoride ions. To the best of our knowledge, only two cations, namely [Br2F8]+ and [Br3F8]+,7 and seven anions of this type are currently known. These include [(μ3-F)(ClF)3]−,8 [(μ3-F)(ClF3)3]−,9 [(μ-F)(BrF3)2]−,10–12 [(μ3-F)(BrF3)3],11 two different isomers of the [Br4F13]−,13 [(μ3-F)(BrF5)3]−,14 and [(μ3-F)(IF5)3]−.15,16 All those “oligonuclear” anions consist of a central μ- or μ3-bridging fluoride ion acting as a linker between two or three halogen fluoride moieties. Depending on the isomer, the [Br4F13]− anion formally consists of a [(μ3-F)(BrF3)3]− anion with a further BrF3 unit attached to one of the terminal F atoms or of a [(μ-F)(BrF3)2]− anion which is linked to two further BrF3 units via two different terminal F atoms.13Tetracoordinated F atoms are present in ionic compounds such as CaF2. In contrast, only a few complexes with μ4-F atoms are known in Y, V, Mo, and Al compounds.17–21 Molecular ions containing fluorine atoms that are tetracoordinated by non-metal atoms are generally very scarce, with the tetrahydrogen pentafluoride ion, [(μ4-F)(HF)4]−,22 likely being the most prominent example. We recently reported the compound Cs[Br3F16],14 which contained the first fluoride-bridged oligonuclear Br(V) anion, [(μ3-F)(BrF5)3]−. It was obtained in the reaction of CsF with an excess of BrF5. By variation of the counter ion using KF or RbF instead of CsF as a starting material, we did not succeed in the isolation of other compounds containing fluoride-bridged fluoridobromate(V) anions, as of yet, but obtained the corresponding [BrF6]− salts.14,23
By introducing the sterically more demanding [NMe4]+ ion as a counterion, the reaction favors the formation of the bulkier [(μ4-F)(BrF5)4]− ion instead of the smaller [(μ3-F)(BrF5)3]− anion that is formed in the Cs compound. Here we present the first interhalogen compound, [NMe4][Br4F21]·BrF5, in which a central fluorine atom is coordinated simultaneously by four halogen fluoride molecules forming a molecular [(μ4-F)(BrF5)4]− anion. The compound was characterized by single-crystal X-ray diffraction and Raman spectroscopy and was investigated by quantum-chemical methods.
Results and discussion
Synthesis
The compound tetramethylammonium μ4-fluorido-tetrakis(pentafluoridobromate(V))—bromine pentafluoride(1/1), [NMe4][Br4F21]·BrF5, was obtained from the reaction of [NMe4][BrF6] with an excess of BrF5. Therefore, [NMe4][BrF6] was cooled to −196 °C, BrF5 was distilled onto the solid, and the reaction mixture was allowed to warm up to room temperature giving a colorless solution. The solution was stored at −36 °C. After several hours, colorless cube-shaped single crystals of [NMe4][Br4F21]·BrF5 had formed. The reaction can be described with (Eq. 1).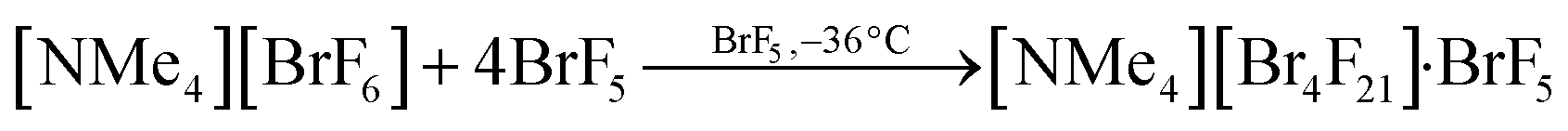 | (Eq. 1) |
Crystals of the title compound are not stable in BrF5 solution at room temperature and decompose to the starting materials. If crystals are warmed slightly above room temperature in the absence of excess BrF5, then they decompose under evolution of brown vapors of Br2. The left-overs were not characterized further.
When reacting [NMe4]F directly with an excess of neat BrF5, a very powerful oxidant, the utmost care must be taken. The reaction is hardly controllable and often results in explosions even at temperatures as low as −196 °C (see Fig. S1†). The metathesis reaction between Cs[BrF6] and [NMe4]F, as reported by Christe and coworkers,24 however, offers an elegant and dependable method for obtaining [NMe4][BrF6], see (Eq. 2).
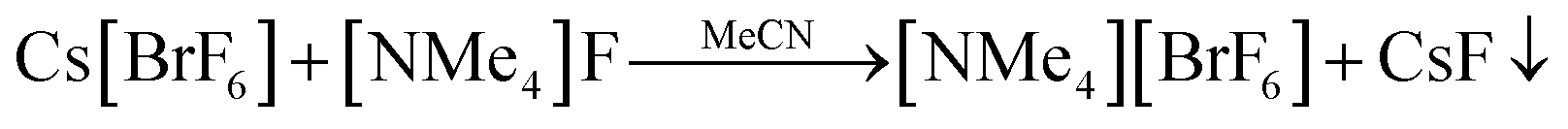 | (Eq. 2) |
[NMe4][BrF6] is stable at room temperature, not shock sensitive,24 and did not exhibit any violent reaction tendencies when exposed to BrF5. Further details on the synthesis are given in the ESI.†
Crystal structure of [NMe4][Br4F21]·BrF5
Single-crystal X-ray structure determination showed [NMe4][Br4F21]·BrF5 to crystallize in the cubic space group P213 (no. 198, b27a5, cP192) with the lattice parameter a = 13.389(4) Å, V = 2400(2) Å3, Z = 4 at T = 100 K. The structure consists of [(μ4-F)(BrF5)4]− fluoridobromate(V) anions (Fig. 1), [NMe4]+ cations and a BrF5 molecule of crystallization per formula unit.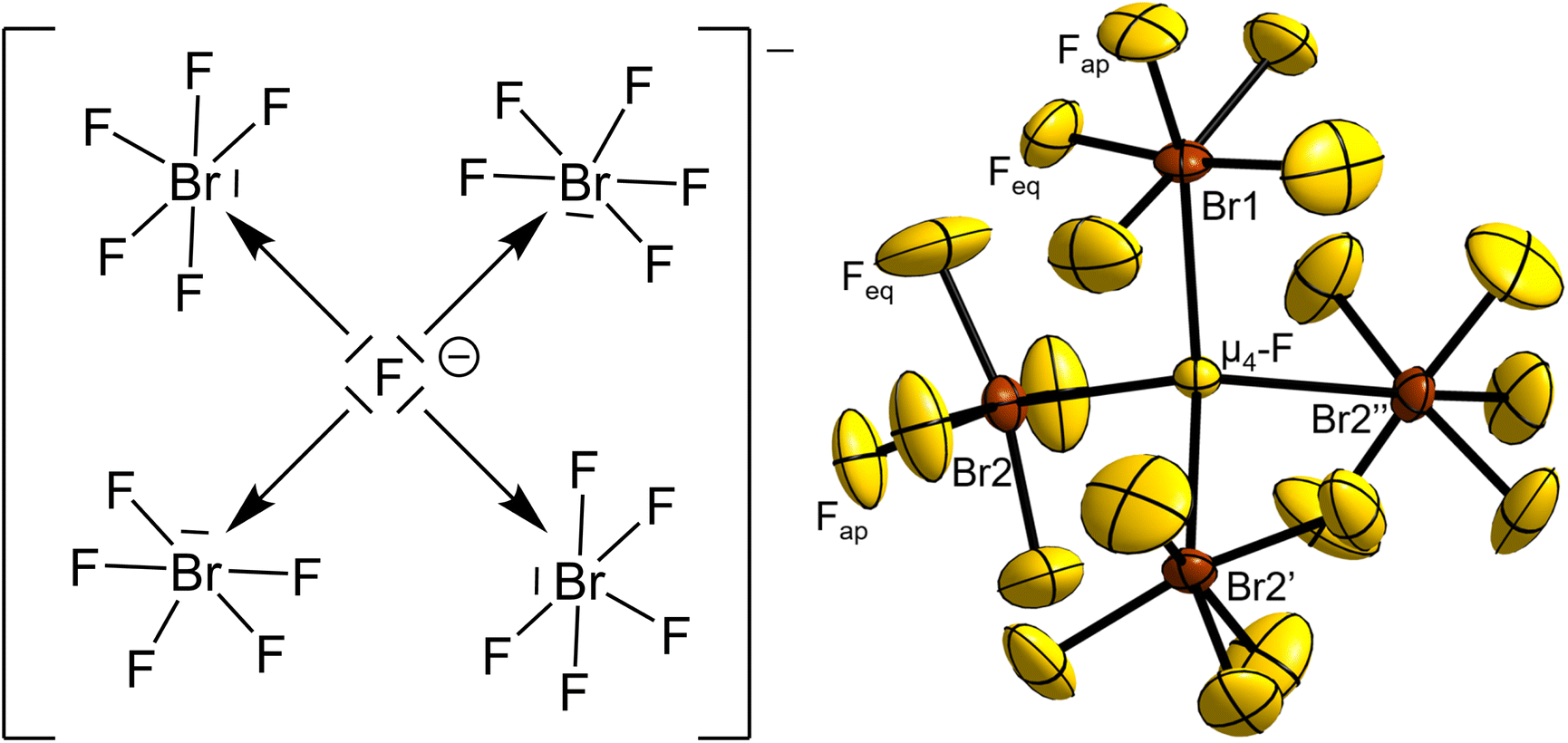 | ||
| Fig. 1 Valence structural formula of the [(μ4-F)(BrF5)4]− anion and its molecular structure in the salt [NMe4][Br4F21]·BrF5. Displacement ellipsoids are shown at 50% probability level at 100 K. Symmetry-equivalent, disordered F atoms bound to Br1 are omitted for clarity. Selected bond lengths (Å) and angles (°): μ4-F–Br1 2.474(3), μ4-F–Br2 2.5447(14), Br1–Fap 1.668(8), Br2–Fap 1.696(3), Br1–Feq 1.685(13) to 1.864(16), Br2–Feq 1.743(5) to 1.783(4), Br1–μ4-F–Br2 111.01(7), Br2–μ4-F–Br2 107.89(7), μ4-F–Br1–Fap 166.6(5), μ4-F–Br2–Fap 160.26(18). Symmetry codes: ′y, z, x, ′′z, x, y. | ||
The crystal structure presents a borderline scenario between twinning and/or disorder affecting the cation, a BrF5 unit of the anion, and the BrF5 molecule of crystallization. These three reside on the threefold rotation axis of the space group and are therefore disordered by symmetry. A corresponding reduction of symmetry by eliminating the threefold rotation axis leads to the orthorhombic space group P212121 and permits a crystallographic description of the structure as a three-component twin. However, the cubic unit cell metric results in too few symmetry-independent reflections to refine the structure properly in the orthorhombic crystal system and to construct a satisfying structural model. Therefore, the choice of the cubic space group P213 with a description of the disorder lead to the clearly superior structure model. Further crystallographic details are given in the ESI.†
The central μ4-F atom (4a, .3.) of the anion is surrounded tetrahedron-like by BrF5 moieties. One BrF5 moiety is 2.474(3) Å distant from the μ4-F atom with its Br atom residing on a 4a position with 3. symmetry, and therefore is affected by disorder over three positions. The remaining three symmetry-equivalent BrF5 units reside on the 12a position (site symmetry 1), with a μ4-F–Br distance of 2.5447(14) Å.
For comparison, the observed distances for the two μ3-bridged fluoridohalogenate(V) anions [Br3F16]−14 and [I3F16]−16 are almost equal with those present here, with X–μ3-F distances of 2.462(2) Å for X = Br and 2.477 to 2.514 Å for X = I, respectively. The Br–μ4-F–Br angles of the [Br4F21]− anion are 3 × 111.01(7)° and 3 × 107.89(7)°, showing a small distortion from the ideal tetrahedral angle with 109.5°.
The atomic distances of the Br to the terminally bound F atoms within the BrF5 units closely agree with those determined for pure BrF5 at 100 K.23 The bond lengths between Br2 and the equatorial F atoms, Br2–Feq, range from 1.743(5) to 1.783(4) Å and are therefore essentially similar within their tripled standard uncertainties with those in pure BrF5 that range from 1.744(3) to 1.779(3) Å. The bond length of the apical Br–F bond, Br2–Fap, is slightly longer with 1.696(3) Å compared to 1.686(2) Å for the one in pure BrF5, however, there is an overlap of the two bond lengths considering their tripled standard uncertainties. For the disordered BrF5 unit, the Br1–Fap bond length is 1.668(8) Å and the Br1–Feq bond lengths range from 1.685(13) to 1.864(16) Å. This broader range and the larger standard uncertainties are caused by the disorder mentioned above. From a chemical point of view, the intramolecular atomic distances and angles of the BrF5 units should differ only insignificantly among themselves, which is also confirmed by our quantum-chemical calculations (see below).
The Fap–Br–Feq angles of the ordered BrF5 units of the anion lie in the range from 83.1(3)° to 84.7(3)°, while for the disordered BrF5 unit and BrF5 molecule of crystallization they are observed around 83° to 85°. As observed for pure BrF5, the Br atoms are located slightly below a virtual plane spanned by the four surrounding Feq atoms by 0.126(7) Å in the case of Br1 and 0.190(3) Å for Br2, respectively. The μ4-F–Br–Fap angles are 166.6(5)° for Br1 and 160.26(18)° for Br2 and can be interpreted as an indication for the extent of the stereochemical activity of the free electron pair located on the Br atoms. The more the μ4-F–Br–Fap angle deviates from 180°, the stronger the stereochemical influence of the lone pair. In the octahedral [BrF6]− anion, no stereochemical effect of the lone pair is observed,16,23,25–27 while it is also present in the [(μ3-F)(BrF5)3]− anion, as evidenced in the μ3-F–Br–Fap bond angle of 165.0(3)°, also deviating from 180°.14 The stereochemical effect of the free electron pair is even more apparent in the analogous [(μ3-F)(IF5)3]− anion, whose μ3-F–I–Fap angles are in the range from 141.4° to 149.0°.16
Except for Coulomb interaction between the [NMe4]+ cations and the [Br4F21]− anions we observe, due to the disorder, no additional intermolecular interactions such as hydrogen bonds in the crystal structure of [NMe4][Br4F21]·BrF5. C–H⋯F hydrogen bonds are however likely present as H⋯F distances are overall in the proper range.28 Each [Br4F21]− anion is octahedrally surrounded by [NMe4]+ cations, and vice versa. Thus, cations and anions are arranged in a cubic close packing, respectively, in which all octahedral voids are occupied by counterions. The ionic part of the crystal structure is therefore related to the NaCl structure type. The BrF5 molecules of crystallization occupy half of its tetrahedral voids and are arranged tetrahedron-like. So, the overall arrangement of these building blocks corresponds to the intermetallic half-Heusler compound and structure type MgAgAs. Fig. 2 shows the pseudo-face centered cubic unit cell in blue and the arrangement of the N atoms of the cations, the μ4-F atoms of the anions, and the Br atoms of the BrF5 molecules of crystallization corresponding to the MgAgAs structure type.
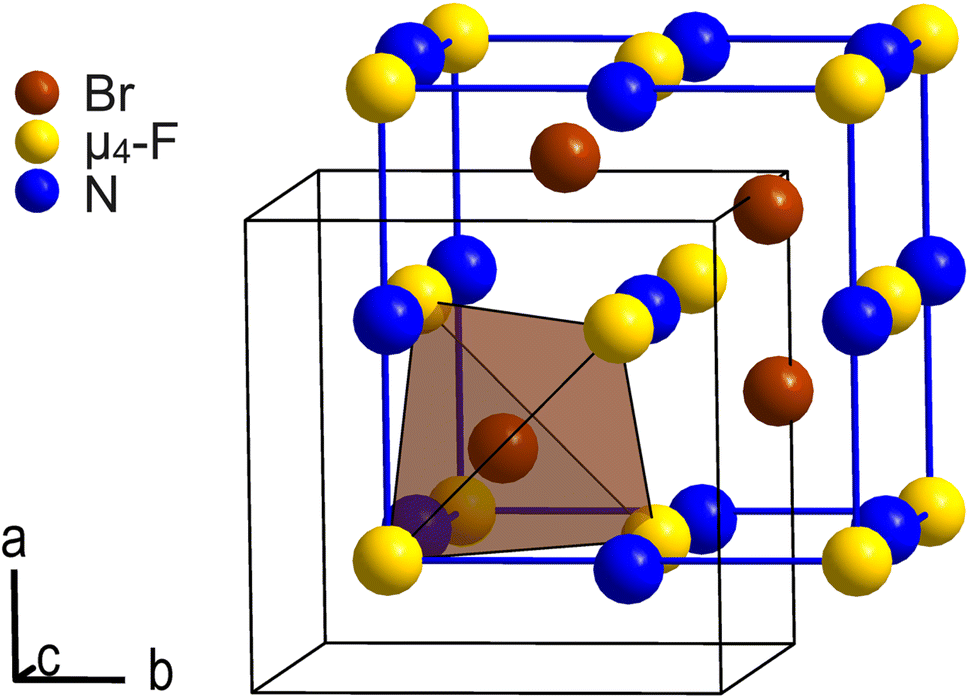 | ||
| Fig. 2 Section of the crystal structure of [NMe4][Br4F21]·BrF5 showing the arrangement of the μ4-F atoms of the anions (yellow), the N atoms of the [NMe4]+ cations (blue), and the Br atoms of the BrF5 molecules of crystallization (brown) according to the MgAgAs structure type. The pseudo-face centered cubic unit cell is shown in blue. It is shifted by +(1/4, 1/4, 1/4) with respect to the origin of the original unit cell shown in black. Atoms are shown as spheres with arbitrary radii. | ||
Quantum-chemical calculations on the crystal structure of [NMe4][Br4F21]·BrF5
The lattice parameters and atomic positions of the crystal structure were optimized by using hybrid density functional methods (DFT-PBE0/TZVP level of theory, see ESI† for the computational details). Since the structure model in space group P213 derived from the X-ray structure determination includes disordered atoms, the structure was optimized in the orthorhombic subgroup P212121 so that a description of the structure without disorder was possible. A harmonic frequency calculation for the optimized structure confirmed it as a true local minimum on the potential energy surface. The optimized lattice parameters deviate less than 2.3% from the lattice parameter determined by X-ray diffraction and bond lengths and angles are in accordance with the experiment. The calculated four μ4-F–Br distances of the [Br4F21]− anion are in the range of 2.456 to 2.577 Å compared to the experimentally determined distances of 2.474(3) and 2.5446(14) Å. The calculated Br–Fap bond lengths range from 1.704 to 1.708 Å, while the calculated Br–Feq bond lengths are in the range from 1.777 to 1.798 Å. Compared to the experimentally determined bond lengths, the agreement is significantly better for the BrF5 units that are not affected by disorder, as expected. For these, the Br–Fap distance was determined to be 1.696(3) Å and the Br–Feq distances are 1.742(5) to 1.783(4) Å. However, the experimentally determined Br–F distances for the disordered parts, which are 1.668(8) Å for the Br–Fap bond and 1.685(13)–1.864(16) Å for the Br–Feq bonds, are of lower precision and therefore spread around the calculated values. A comparison of other calculated and experimentally determined atomic distances and angles is available in Tables S6 and S7.†Vibrational spectroscopy
A Raman spectrum of the compound was recorded at 213 K and is shown in Fig. 3 in black in comparison to the calculated spectrum at the DFT-PBE0/TZVP level of theory for the solid-state structure in red.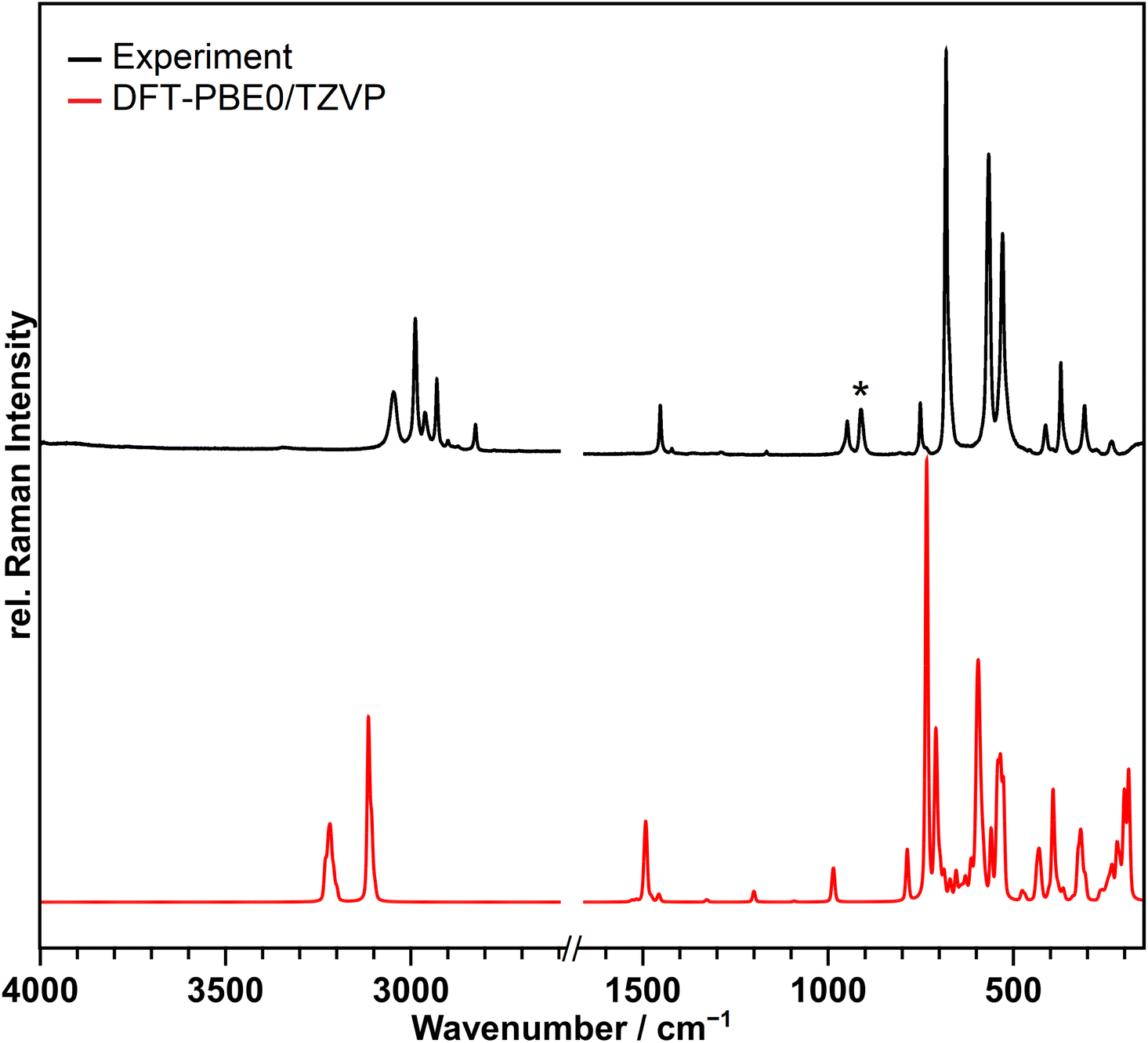 | ||
| Fig. 3 Observed Raman spectrum of [NMe4][Br4F21]·BrF5 suspended in perfluorinated oil at 213 K in black and the calculated spectrum at the DFT-PBE0/TZVP level of theory in red. No bands were observed or calculated in the range from 1600–2700 cm−1, so it was omitted in the Figure. The band marked with an asterisk originates form BrO2F, which is a hydrolysis product formed during the Raman measurement.29 The calculated spectrum in red shows many bands not observed in the recorded one. This is because of the lower resolution of the recorded spectrum. | ||
The bands above 700 cm−1 belong to the [NMe4]+ cation, and those below 700 cm−1 to the anion and the BrF5 molecule of crystallization, however, the latter two could not be clearly separated. The bands from 529 to 682 cm−1 are due to the Br–F stretching vibrations, those from 235 to 413 cm−1 are assigned to wagging, scissoring and umbrella modes. Table S8† contains the observed and calculated band positions and their detailed assignments.
With the exception of two points the recorded spectrum agrees with the calculated one in which the frequencies are slightly overestimated as anharmonic effects are not considered.30 Additional bands not reproduced by the frequency calculations are observed in the range of 2800–2970 cm−1. However, these are assignable to second-order bands of the [NMe4]+ ion.31,32 The band marked with an asterisk at 911 cm−1, does not originate from the title compound and also increased in intensity during the measurement. It can be assigned to the symmetric ν(Br–O) stretching vibration of BrO2F, which is obtained as a hydrolysis product of the title compound.29 The other bands expected for BrO2F are significantly weaker in intensity and overlap with the bands of the title compound, so that they cannot be observed. Contact of the sample with the atmosphere causing hydrolysis could not be completely excluded during the transfer of the cold sample into the spectrometer. Additional details of the Raman setup and sample preparation are given in the ESI.†
Intrinsic bonding orbital analysis
Quantum-chemical calculations on the isolated [(μ4-F)(BrF5)4]− anion and its hypothetical homologues [(μ4-F)(ClF5)4]− and [(μ4-F)(IF5)4]− were conducted at the DFT-PBE0/def2-TZVP level of theory with a consecutive analysis of the bonding situation using intrinsic bonding orbitals (IBOs).33 The structures of the molecular anions [(μ4-F)(XF5)4]−, with X = Cl–I, were all optimized for the gas phase at 0 K in point group S4 and were confirmed to be true local minima on the potential energy surfaces by means of harmonic frequency calculations. The calculated μ4-F–Br bond length in the [(μ4-F)(BrF5)4]− anion is 2.576 Å and agrees with the experimentally determined ones of 3 × 2.5447(14) and 2.474(3) Å. The IBO analysis showed that none of the Br atoms contribute to the μ4-F–Br bond with more than 1%, which indicates a purely ionic bonding situation, and suggests the description of the μ4-F atom as a F− anion. For comparison: In a gas-phase NaF molecule, in which the bond should be highly ionic, the contribution of the F atom is 96%. In the H2 molecule, which has a purely covalent bond, the contribution of each H atom is 50%. The partial charge of the μ4-F atom is −0.77 e, which is significantly more negative than that of the F atoms in a BrF5 unit with partial charges between −0.41 and −0.51 e. The μ4-F atom is even more ionic than the μ3-F atom in the [(μ3-F)(BrF5)3]− anion with −0.73 e.14 The partial charges of the μ4-F atoms in the hypothetical anions [(μ4-F)(ClF5)4]− and [(μ4-F)(IF5)4]−, calculated to be −0.76 and −0.77 e, respectively, are similar to the one observed for [(μ4-F)(BrF5)4]−. The partial charges of the Br atoms of the [(μ4-F)(BrF5)4]− anion and the previously reported [(μ3-F)(BrF5)3]− anion are almost the same with +2.38 e and +2.37 e, respectively.14The high negative partial charge of the μ4-F atom as well as the high positive partial charge of the Br atoms of the [(μ4-F)(BrF5)4]− anion strengthen the suggestion of describing the μ4-F atom as an F− anion and the μ4F–Br bonds as essentially ionic (see Fig. 4).
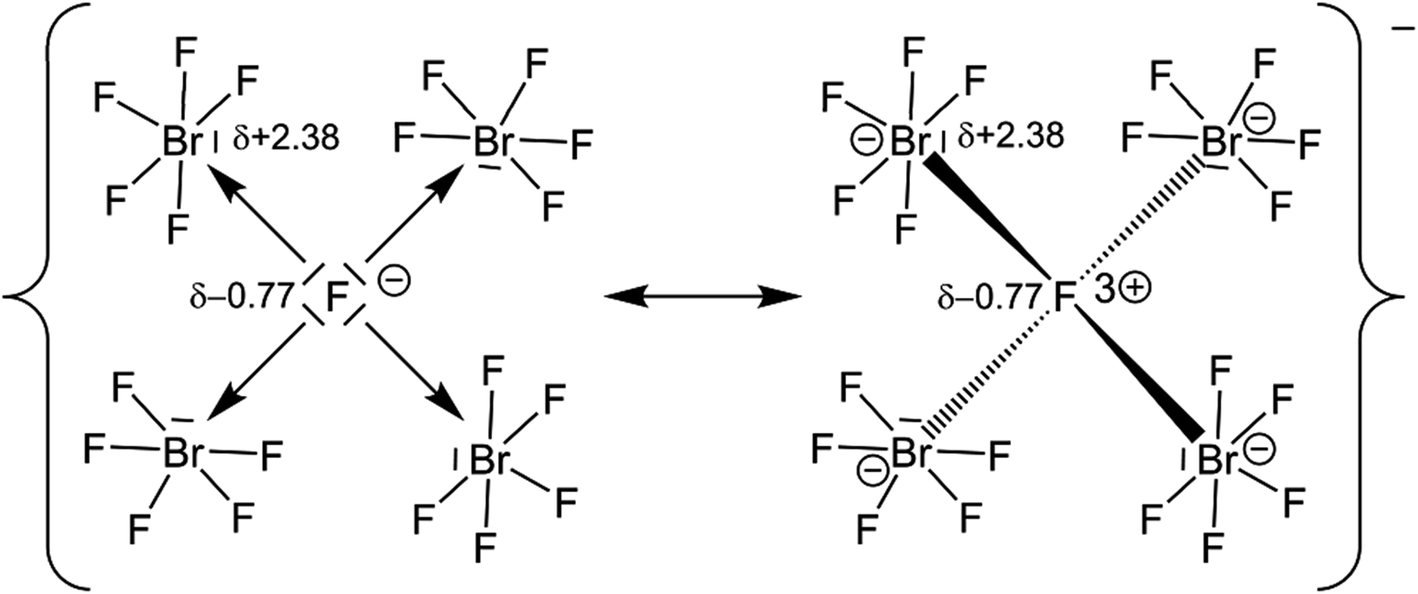 | ||
| Fig. 4 Selected resonance valence structural formulae of the [(μ4-F)(BrF5)4]− anion. Numbers next to the atoms indicate their calculated partial charges. The result of our IBO analysis shows that the one on the left with an essentially ionic Br–F bond is to be preferred. | ||
Conclusions
The compound [NMe4][Br4F21]·BrF5 was obtained by reacting [NMe4][BrF6] with an excess of BrF5. Surprisingly, its crystal structure is closely related to the intermetallic half-Heusler compound and structure type MgAgAs, as the cations, anions and BrF5 molecules of crystallization are ordered as in MgAgAs. In the [(μ4-F)(BrF5)4]− anion, the μ4-F atom is surrounded by four BrF5 molecules in a tetrahedron-like shape. It is the first example of a F atom μ4-bridging to neither metal nor H atoms. IBO analyses of the anion showed that the covalence of the μ4-F–Br interaction is negligibly small and therefore the bond is best described as ionic. The recorded Raman spectrum of [NMe4][Br4F21]·BrF5 agrees with the quantum-chemically calculated one for the solid state. The tetrahedral [(μ4-F)(BrF5)4]− and the propeller-shaped [(μ3-F)(BrF5)3]− anions are so far the only known polynuclear anions of BrF5. We have currently no experimental evidence for the existence of the putative dinuclear [(μ-F)(BrF5)2]− anion.Data availability
Upon request from the authors.Author contributions
Martin Möbs: planning and conducting the experiments, main data acquisition and interpretation, quantum-chemical calculations and interpretation of the results, manuscript preparation. Tim Graubner: quantum-chemical calculations and interpretation of the results, manuscript preparation. Antti J. Karttunen: quantum-chemical calculations, CRYSTAL basis set development, manuscript preparation. Florian Kraus: project supervision, data interpretation, manuscript preparation.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Dr Sergei Ivlev and Dr Matthias Conrad for helpful discussions. We thank Solvay for the kind donations of fluorine and we thank the Deutsche Forschungsgemeinschaft for funding via a Koselleck project, KR3595/10-1. A. J. K. thanks CSC – The Finnish IT Center for Science for computational resources.References
- E. W. Lawless and I. C. Smith, Inorganic High-Energy Oxidizers - Synthesis, Structure, and Properties, Marcel Dekker, Inc., New York, 1968 Search PubMed.
- W. I. Bailey and A. J. Woytek, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000 Search PubMed.
- F. Seel and O. Detmer, Z. Anorg. Allg. Chem., 1959, 301, 113–136 CrossRef CAS.
- R. J. Gillespie and M. J. Morton, Q. Rev., Chem. Soc., 1971, 25, 553–570 RSC.
- K. O. Christe, in XXIVth International Congress of Pure and Applied Chemistry, Butterworth, London, 1974, pp. 115–141 Search PubMed.
- D. Naumann, Fluor und Fluorverbindungen, Dr. Dietrich Steinkopf Verlag, Darmstadt, 1980 Search PubMed.
- S. I. Ivlev, A. J. Karttunen, M. R. Buchner, M. Conrad and F. Kraus, Angew. Chem., Int. Ed., 2018, 57, 14640–14644 CrossRef CAS PubMed.
- P. Pröhm, N. Schwarze, C. Müller, S. Steinhauer, H. Beckers, S. M. Rupf and S. Riedel, Chem. Commun., 2021, 57, 4843–4846 RSC.
- B. Scheibe, A. J. Karttunen, U. Müller and F. Kraus, Angew. Chem., Int. Ed., 2020, 59, 18116–18119 CrossRef CAS PubMed.
- V. F. Sukhoverkhov, N. D. Takanova and A. A. Uskova, Zh. Neorg. Khim., 1976, 21, 2245–2249 CAS.
- S. I. Ivlev, A. J. Karttunen, R. V. Ostvald and F. Kraus, Chem. Commun., 2016, 52, 12040–12043 RSC.
- S. Ivlev, P. Woidy, V. Sobolev, I. Gerin, R. Ostvald and F. Kraus, Z. Anorg. Allg. Chem., 2013, 639, 2846–2850 CrossRef CAS.
- J. Bandemehr, S. I. Ivlev, A. J. Karttunen and F. Kraus, Eur. J. Inorg. Chem., 2020, 48, 4568–4576 CrossRef.
- M. Möbs, T. Graubner, A. J. Karttunen and F. Kraus, Chem.–Euro. J., 2023, e202301876 CrossRef PubMed.
- K. O. Christe, Inorg. Chem., 1972, 11, 1215–1219 CrossRef CAS.
- A. R. Mahjoub, A. Hoser, J. Fuchs and K. Seppelt, Angew. Chem., Int. Ed., 1989, 28, 1526–1527 CrossRef.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef CAS.
- Q. Chen and J. Zubieta, J. Chem. Soc., Chem. Commun., 1994, 2663–2664 RSC.
- D. L. Thorn, R. L. Harlow and N. Herron, Inorg. Chem., 1995, 34, 2629–2638 CrossRef CAS.
- N. Buchholz and R. Mattes, Angew. Chem., Int. Ed., 1986, 25, 1104–1105 CrossRef.
- Y. Yang, J. Pinkas, M. Schäfer and H. W. Roesky, Angew. Chem., Int. Ed., 1998, 37, 2650–2653 CrossRef CAS PubMed.
- B. A. Coyle, L. W. Schroeder and J. A. Ibers, J. Solid State Chem., 1970, 1, 386–393 CrossRef.
- M. Möbs, T. Graubner, K. Eklund, A. J. Karttunen and F. Kraus, Chem.–Euro. J., 2022, 28, e202202466 CrossRef PubMed.
- W. W. Wilson and K. O. Christe, Inorg. Chem., 1989, 28, 4172–4175 CrossRef CAS.
- K. O. Christe, Inorg. Chem., 1972, 11, 1215–1219 CrossRef CAS.
- K. O. Christe and H. Oberhammer, Inorg. Chem., 1981, 20, 296–297 CrossRef CAS.
- K. O. Christe and W. W. Wilson, Inorg. Chem., 1989, 28, 3275–3277 CrossRef CAS.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423–436 RSC.
- R. J. Gillespie and P. H. Spekkens, J. Chem. Soc., Dalton Trans., 1977, 1539–1546 RSC.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- G. Kabisch and M. Klose, J. Raman Spectrosc., 1978, 7, 311–315 CrossRef.
- G. Kabisch, J. Raman Spectrosc., 1980, 9, 279–285 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2311055. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06688f |
| This journal is © The Royal Society of Chemistry 2024 |