
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSinglet fission in TIPS-anthracene thin films†
Damon M.
de Clercq
 a,
Miles I.
Collins
b,
Nicholas P.
Sloane
b,
Jiale
Feng
a,
Dane R.
McCamey
b,
Murad J. Y.
Tayebjee
c,
Michael P.
Nielsen
c and
Timothy W.
Schmidt
*a
a,
Miles I.
Collins
b,
Nicholas P.
Sloane
b,
Jiale
Feng
a,
Dane R.
McCamey
b,
Murad J. Y.
Tayebjee
c,
Michael P.
Nielsen
c and
Timothy W.
Schmidt
*a
aSchool of Chemistry, ARC Centre of Excellence in Exciton Science, UNSW Sydney, NSW 2052, Australia. E-mail: timothy.schmidt@unsw.edu.au
bSchool of Physics, ARC Centre of Excellence in Exciton Science, UNSW Sydney, NSW 2052, Australia
cSchool of Photovoltaic and Renewable Energy Engineering, UNSW Sydney, NSW 2052, Australia
First published on 25th March 2024
Abstract
Singlet fission is an exciton multiplication process that allows for the conversion of one singlet exciton into two triplet excitons. Organic semiconductors, such as acenes and their soluble bis(triisopropylsilylethynyl) (TIPS) substituted counterparts, have played a major role in elucidating the understanding of the underlying mechanisms of singlet fission. Despite this, one prominent member of the acene family that has received little experimental attention to date is TIPS-anthracene, even with computational studies suggesting potential high singlet fission yields in the solid state. Here, time-resolved spectroscopic and magneto-photoluminescence measurements were performed on spin-cast films of TIPS-anthracene, showing evidence for singlet fission. A singlet fission yield of 19% (out of 200%) is estimated from transient absorption spectroscopy. Kinetic modeling of the magnetic field effect on photoluminescence suggests that fast rates of triplet dissociation lead to a low magnetic photoluminescence effect and that non-radiative decay of both the S1 and 1(TT) states is the cause for the low triplet yield.
1 Introduction
Singlet fission (SF) involves partitioning the energy of a singlet (S1) exciton into two triplet (T1) excitons on separate chromophores (Fig. 1). The fission of a high-energy excitation into two lower-energy T1 states is of interest to the photovoltaic community, due to its potential to overcome the detailed balance limit of single junction silicon photovoltaics.1–7 To overcome this limit, chromophores with high SF yields and triplet energies above the band gap of silicon are required.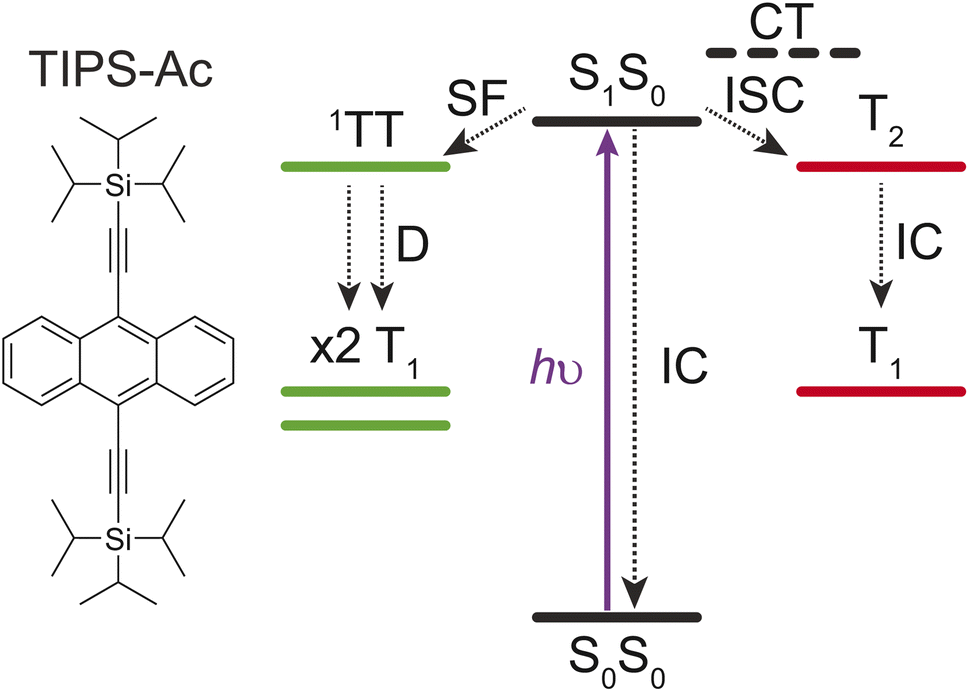 | ||
| Fig. 1 The structure and some possible fates for the singlet state (S1S0) in TIPS-anthracene. Upon excitation (hν), coupled chromophores (S1S0) can undergo singlet fission to a strongly-coupled triplet pair (1(TT)) before dissociating (D) into two free triplets (2 × T1). Possible loss mechanisms from S1S0 include (1) intersystem crossing (ISC) to the T2 state facilitated by a virtual charge transfer (CT) state, followed by internal conversion (IC) to a T1 state, and (2) IC to the ground-state (S0S0). | ||
Solution-processable triisopropylsilylethynyl (TIPS) substituted acenes, such a TIPS-tetracene and TIPS-pentacene, have served as the workhorse molecules having informed the mechanism, intermediate species, and loss pathways involved in SF.8–16 However, these molecules have triplet energies below the bandgap of Si (1.1 eV), and are thus not suitable for inclusion in Si-photovoltaic cells.17
The triplet energy levels of anthracene derivatives make them prime candidates for coupling with Si-photovoltaic cells. Derivatives such as 9,10-bis(phenylethynyl)anthracene (BPEA, T1 > 1.24 eV)18 and TIPS-anthracene (TIPS-Ac, T1 = 1.37 eV)19 have T1 energies above the Si bandgap. BPEA has previously been reported to undergo SF.18 Conversely TIPS-anthracene has predominantly been used as an annihilator for photochemical upconversion in solution.20,21 Recently, thermally evaporated thin films of TIPS-Ac were reported to undergo SF with free triplet yields of less than 40% (out of a possible 200%).22 These low yields contradict previous computational studies that estimated the SF yield to be 150%.23 This implies that there is a loss mechanism that competes with SF in TIPS-Ac. Possible loss mechanisms include internal conversion of the singlet (S1S0) to the ground state (S0S0) or rapid high-level level-intersystem crossing facilitated by a virtual charge transfer intermediate (Fig. 1).
High-level reverse intersystem crossing (RISC) has emerged as a promising aspect to increase the spin statistical factor (f) of triplet–triplet annihilation (TTA) – a process that operates in the reverse sense to SF.24–26 An increase in f can result in more efficient organic light-emitting diodes due to more dark T1 excitons being converted to bright S1 excitons.27–29 Although high-level RISC offers a potential boost to TTA efficiency, the opposite process of high-level intersystem crossing (ISC) to a low-lying T2 state could negatively impact SF if the ISC rate is competitive with SF (Fig. 1).
Here high-level ISC to the T2 state, triplet dissociation, and non-radiative decay are investigated for their roles as loss mechanisms in TIPS-Ac SF. Transient absorption (TA) spectroscopy and magnetic field effect photoluminescence (MPL) measurements are used to fully characterize the SF process in TIPS-Ac thin films. Kinetic modeling of the MPL indicates that ISC to the T2 state is not a major contributor to the low SF-yield of 19%. Instead non-radiative decay from both the S1 and 1(TT) states is determined as the main cause of low SF yield.
2 Experimental
2.1 Preparation of thin films
For film fabrication 9,10-bis(triisopropylsilylethynyl)anthracene (Sigma Aldrich) was used without further purification. The synthesis of 5,12-bis(triisopropylsiylethynyl)tetracene is reported elsewhere.10,30 All sample preparation was performed in a nitrogen-filled glovebox. A 15 mg mL−1 stock solution of TIPS-Ac in anhydrous toluene (Sigma Aldrich) was prepared for spin-coating. Spin-coating was performed by depositing 15 μL of the stock solution onto pre-treated quartz (1 cm × 1 cm). Quartz was pre-treated with a 15 min plasma clean followed by subsequent 10 min ultrasonic cleaning in Deacon 90, water, acetone, and ethanol before being placed in a vacuum oven (120 °C) overnight. The spin-coater was set to 1500 rpm for 50 s. After spin-coating, the samples were annealed at 60 °C to remove excess toluene. Annealed samples were encapsulated with a quartz coverslip by applying epoxy to the edges, and then cured by short-wave UV radiation. A shadow mask was placed over the active area to prevent degradation of the sample during the curing process. Sensitization experiments were conducted with Pd(II) octaethyl porphyrin (Frontier Scientific) in toluene at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio of PdOEP to TIPS-Ac.
:10 molar ratio of PdOEP to TIPS-Ac.
2.2 X-ray diffraction
X-Ray diffraction was measured with a X'Pert3 MRD (Malvern Panalytical) Cu Kα radiation (λ = 1.54056 Å), and an accelerating voltage of 45 kV with a current of 40 mA.2.3 Steady-state measurements
Absorption spectra were measured using a Shimadzu UV-Vis Spectrophotometer (UV2600). Fluorescence spectra were taken with an Edinburgh Instruments FS5 spectrofluorometer.2.4 Time correlated single photon counting
Time-correlated single photon counting (TCSPC) was measured with a benchtop microscope. The sample was excited with a 405 nm picosecond pulsed laser (EKSPLA, PT200, 10 ps pulse-width, 1 MHz). A 0.3NA (15×) reflective objective was used to collect emission from the sample. The excitation beam was filtered out with a 450 nm long pass filter and the emission was detected with a silicon photodiode detector (IQ Quantique, ID110). The signal was digitized using a TCSPC card (PicoQuant, TimeHarp 260).2.5 Transient absorption
TA measurements were performed on a commercial set-up (Ultrafast Systems, Helios) pumped by a Ti:Sapphire amplifier (Spectra Physics Solstice Ace) that generates an 800 nm pulse train at a repetition rate of 1 kHz with a pulse duration of nominally 100 fs. The 800 nm output was split with a beam splitter and one part of the pulse was sent to an optical parametric amplifier (TOPAS Prime, Light Conversion) to generate the pump pulse at 380 nm. A 450 nm short-pass filter was placed in the pump line to block residual 800 and 532 nm. Before the sample, the pump polarization was scrambled with a depolarizer. The second part of the 800 nm pulse was focused onto a CaF2 crystal to generate the white light continuum probe. The CaF2 crystal was kept in constant motion with a motorized stage to avoid damage. An 8 ns optical delay line with a minimum step size of 14 fs varied the pump-probe delay. The signal was detected with a silicon linear array detector for the UV-Vis region. At least three individual scans were taken, and each scan was averaged over 3 s per time delay. For the long-time TA, an Nd:YAG laser (Piccolo Innolas, 355 nm 800 ps pulse duration) set to external frequency (500 Hz) control was used as the pump beam.2.6 Magnetic-field photoluminescence
Samples were excited with a 405 nm wavelength continuous-wave laser. Photoluminescence was imaged by off-axis parabolic mirrors into an optical fiber and detected by an Ocean Optics Flame spectrometer. An electromagnet (Magnetech MFG-6-24) controlled by a DC power supply (Keithley 2230G-30-1) was used to provide the magnetic field. The field was calibrated with a Gaussmeter (Lakeshore 475).3 Results and discussion
The X-ray diffraction (XRD) pattern of the spin cast TIPS-Ac thin films shows that they are largely amorphous with some preferential packing along the 1,0,-1 crystal plane (Fig. S1†), with the powder XRD patterns exhibiting only one polymorph. Fig. 2 shows the steady-state absorbance of TIPS-Ac in a 1 mM solution in toluene and as a thin film. There is an evident 20 meV (4 nm) bathochromic shift of the thin film spectrum relative to the solution, which is accompanied by a low energy tail extending out to 600 nm. This long tail suggests strong chromophore coupling which is further evinced by the presence of excimer emission in the photoluminescence spectrum (Fig. S2†). The thin film spectrum has clear Davydov splitting (100 meV, 10 nm) of the 0–0 band. Davydov splitting is an indication that there are two translational nonequivalent molecules within the crystal lattice which are excitonically coupled.31 There are two known polymorphs of TIPS-Ac. One is highly fluorescent and thus unlikely to undergo SF.32 This polymorph is not expected to exhibit Davydov splitting. However, the presence of Davydov splitting in our thin film indicates that its structure is most similar to the TIPS-Ac polymorph that has been observed to undergo singlet fission.22 The arrangement of non-parallel chromophores undergoing singlet fission is observed in thin films of pentacene and tetracene, two of the most well-studied singlet fission chromophores.11,33–35 The photophysics of TIPS-Ac was further investigated with TA.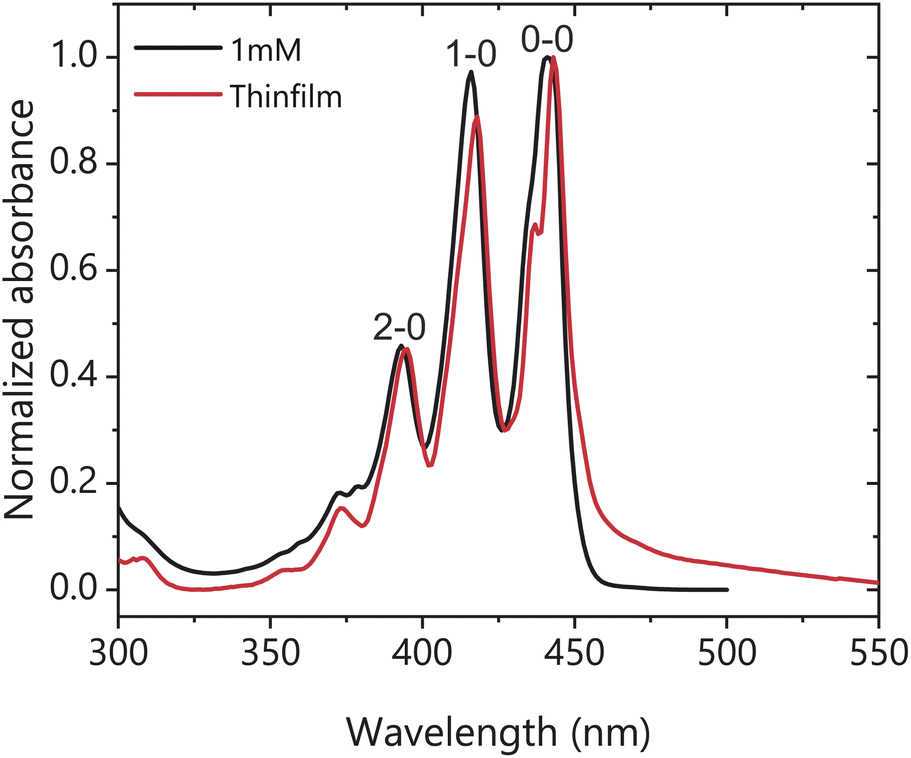 | ||
| Fig. 2 Normalised absorbance of a 1 mM toluene solution (black) and thin film (red) of TIPS-Ac. Thin film spectrum was corrected for scattering. | ||
3.1 Transient absorption spectroscopy
Fig. 3 shows the TA spectrum of a thin film of TIPS-Ac. The negative signal between 400 and 450 nm overlaps with the 0–0 and 1–0 absorption bands (Fig. 2) and is therefore assigned to the ground state bleach (GSB) (Fig. 3a). Stimulated emission (SE) from S1 → S0 is attributed to the negative transient at 475 nm. The 475 nm SE is accompanied by an excited state absorption (ESA) at 535 nm. A notable feature is the decay of the GSB at 450 nm into an ESA, which indicates the formation of a new species. This ESA rise is not present in a 1 mM solution of TIPS-Ac (Fig. S3†). Instead, the TA of the 1 mM sample appears to be consistent with the S1 state with no evidence of T1 formation.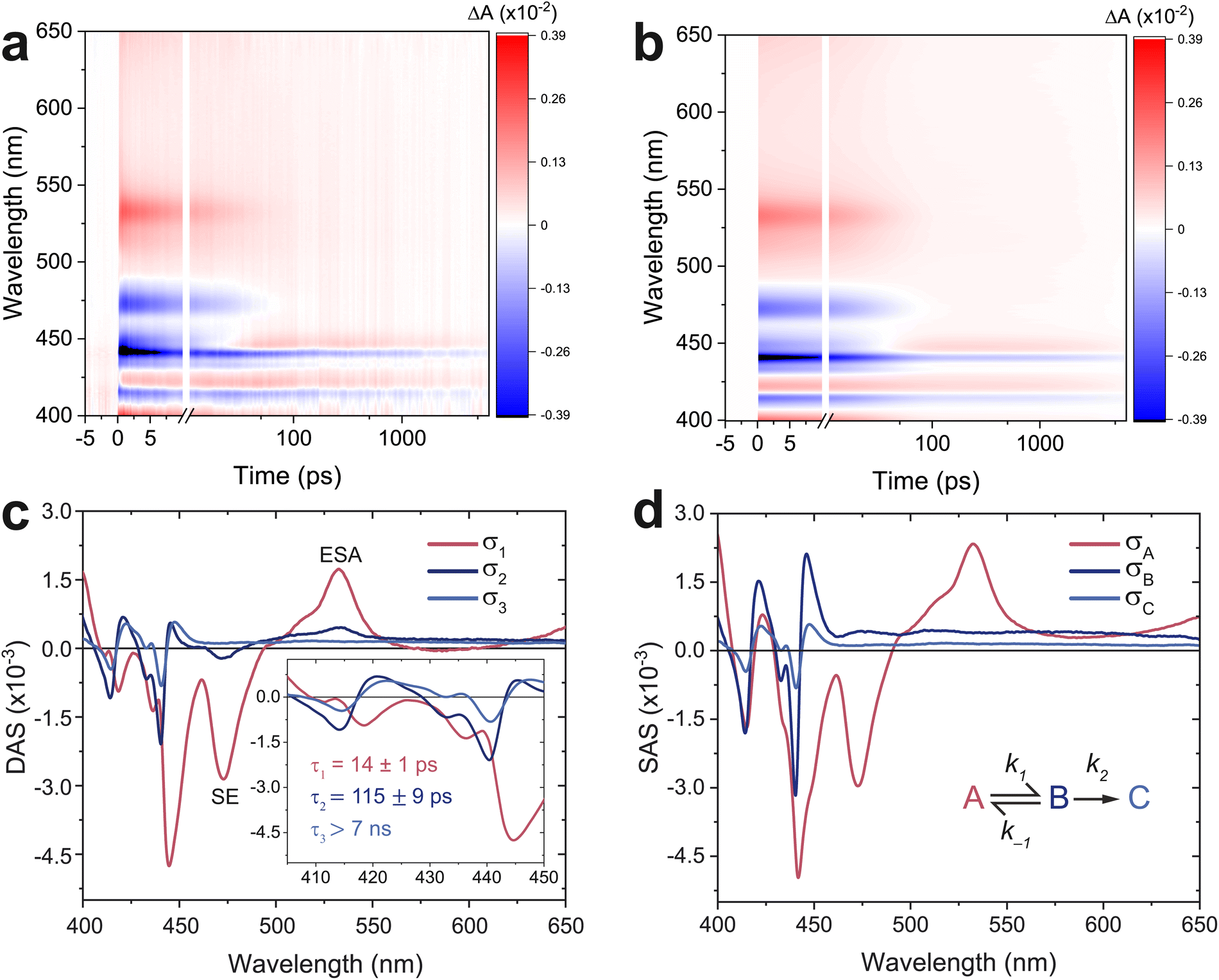 | ||
| Fig. 3 Transient absorption spectroscopy of TIPS-Ac thin films. (a) Experimental TA spectrum of thin films excited with a 380 nm pump (13 μJ cm−2). (b) A tri-exponential global fit of the experimental TA results. (c) Decay-associated spectra (DAS) along with their corresponding lifetimes (τ), the singlet excited state absorption (ESA), and stimulated emission (SE) are labeled. Inset depicts the region between 400 and 450 nm, highlighting the anti-correlation between σ1 and σ2. (d) The species-associated spectra derived from applying the displayed sequential rate model to the DAS. | ||
Notwithstanding early-time few-picosecond exciton thermalization, the TA spectrum of TIPS-Ac thin films is accurately reproduced with a tri-exponential global fit (Fig. 3b, S4a and b†), suggesting that there are three decays present. To confirm that the spectrum was not over parameterized a bi-exponential fit was also performed (Fig. S5a†). It is clear from the residuals of the bi-exponential fit that the data are not accurately represented with only two components (Fig. S5b†).
Fig. 3c shows the derived decay-associated spectra (DAS, σi, i = 1, 2, 3) and their associated time constants (τi). The 14 ps time constant of σ1 is assigned to the decay of the S1 state as there are clear SE and ESA features at 475 and 535 nm respectively. Now, when a second species is generated by a decay, its spectrum will be imprinted on the decay-associated spectrum with an inverted sign. This leads to spectral anti-correlation when this species decays, there is such an anti-correlation between σ1 and σ2 (see inset to Fig. 3c), which indicates that the species that decays with time constant τ2 is generated by the decay with time-constant τ1. However, there is still clearly S1 character in σ2, evidenced by the ESA and SE peaks, indicating that the singlet does not entirely decay by τ1. This is strongly suggestive of a quasi-equilibrium between S1 and a state that is not available to the monomer in solution. The leading candidate for the equilibrium partner of S1 is the triplet pair state 1(TT), which may not be distinguishable from the weakly coupled pair state, l(T⋯T) (where l = 1, 3, 5). σ3 will only reflect the longest-lived species, which we show to be free triplets.
We, therefore, proceed in the analysis by proposing that a quasi-equilibrium is rapidly established between A and B (S1 and 1(TT)), and that this decays to a third species, C (free triplets).
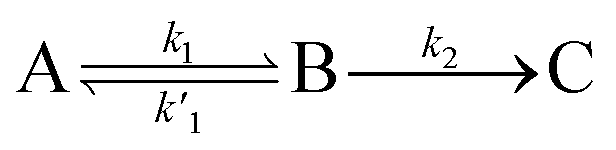 | (1) |
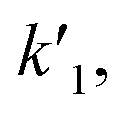 k2 are the rate constants (see ESI S4,† and Fig. 3d). The quasi-equilibrium is described by the equilibrium constant:
k2 are the rate constants (see ESI S4,† and Fig. 3d). The quasi-equilibrium is described by the equilibrium constant: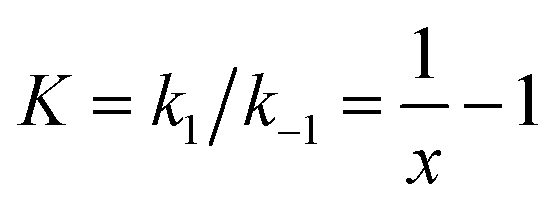 | (2) |
log(5.25), about −0.04 eV. This number reflects both the energetic and entropic contributions to singlet fission.36σ3 is identified with the long-lived free triplets, σC.
To further confirm the presence of triplets in thin films of TIPS-Ac, as opposed to the formation of other long-lived species, long-time TA and sensitization experiments were performed. Sensitization of TIPS-Ac was undertaken by preparing a 1:10 molar ratio of Pd(II) octaethyl porphyrin (PdOEP) to TIPS-Ac in toluene. The sensitizer was selectively excited at 532 nm. The formation of a spectrum that matches species C is observed (Fig. 4), confirming that species C belongs to the T1 state of TIPS-Ac. A slight hypsochromic shift in the bleach is due to the measurement being performed in solution. Further confirmation is provided by the time-slice taken at 1 μs of TIPS-Ac, which overlays well with species C (Fig. 4). The broad feature in species C that starts at 475 nm is assigned to the excimer state. This feature is not observed in the sensitized spectrum or the 1 μs timeslice due to its 6.2 ns lifetime (Fig. 6). The longtime transient absorption kinetics tracked at 441 nm shows two decay constants τ1 = 100 ± 20 ns and τ2 > 4 μs. These results reflect the polycrystalline nature of the TIPS-Ac film and likely arise due to distinct triplet populations (Fig. S6†). The 100 ns lifetime likely arises due to geminate annhilation in regions where SF generated pairs cannot escape one another.37 Similar time constants are observed for thin films of BPEA.18 With the formation of the T1 state confirmed, the triplet yield is estimated from the TA spectrum (Fig. 3a).
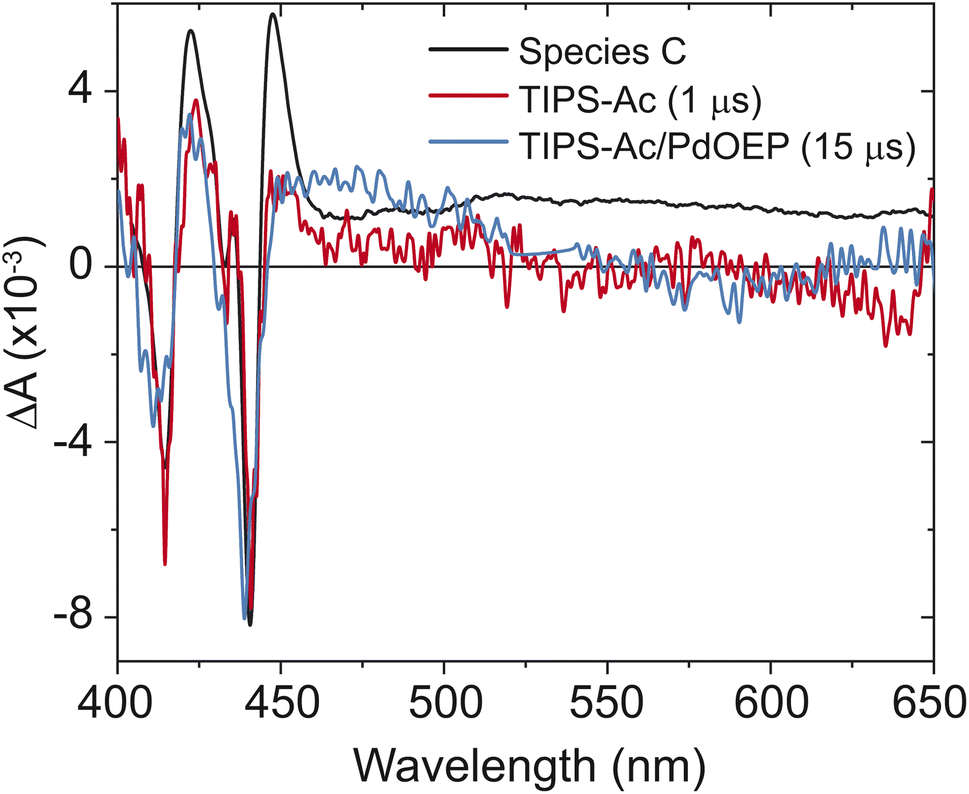 | ||
| Fig. 4 Overlay of species C (black) and time slices of the long-time spectrum of TIPS-Ac thin films at 1 μs (red) and the sensitized spectrum in solution at 15 μs (blue). The triplet state was sensitized with PdOEP. The time slices are scaled to the GSB at 442 nm. | ||
The triplet yield may be estimated by the GSB contribution as a function of time delay. SF results in a prolonged GSB lifetime with potential enhancement due to exciton multiplication. Therefore, an estimation of the triplet yield is undertaken by dividing late time by early time GSB contributions (Fig. S7a–f†). The contribution of the ground state absorption spectrum to the species-associated spectra, σα (α = A, B, C), is a measure of the relative number of excited ground state chromophores in the spectrum. The singlet spectrum, σA, is well fit up to 442 nm by the negative-going ground state spectrum (−σ0), plus a broad Gaussian peaking in the near ultraviolet (Fig S7a†). The fit is truncated at 442 nm as the ground state absorption spectrum cannot account for the SE. However, the fit shows the SE contribution to have mirror symmetry to the GSB (Fig. S7a†).
The fit to the triplet-pair spectrum, σB, shows that its GSB is of a similar magnitude to σA (Fig. S7a and b†). In the absence of significant excitonic coupling in the singlet manifold, a triplet pair is expected to exhibit twice the GSB contribution of the S1 state.38 As such, there must be a mechanism responsible for the loss of excited chromophores during the initial decay. Further, only 30% of σB forms σC indicating additional loss mechanisms in the triplet manifold (Fig. S7b and c†).
The GSB contribution as a function of time delay is seen to decay to 39% at 100 ps, and 19% at 1000 ps compared to the GSB at 0.5 ps (Fig. S7d–f†). As such, the singlet fission yield is clearly very low. This low yield agrees with the previously estimated 40%.22 Indeed, it implies that SF is subject to loss mechanisms. To investigate further, magnetic field effect and transient photoluminescent measurements were undertaken.
3.2 Magnetic field effect
The effect of a magnetic field on the photoluminescence (MPL) of an organic semiconductor is a well-documented phenomenon for singlet fission and triplet–triplet annihilation.39–42 By applying a magnetic field to parallel chromophores, the number of triplet pair states with singlet character is reduced from three to two, resulting in a decrease in entropy of the weakly coupled triplet pair l(T⋯T). This decrease in entropy increases the free energy of the l(T⋯T) state, creating an effective energy barrier between the singlet and triplet manifolds. Therefore, for singlet fission, an increase in the magnetic field will hinder the formation of free triplets and retain the emissive excited state population in the singlet manifold. The overall result is an increase in the PL. Conversely, for TTA the excited state population will be detained in the triplet manifold, resulting in a decrease in PL.Fig. 5 depicts the magnetic field effect (MPL) on thin films of TIPS-Ac and TIPS-tetracene (TIPS-Tc), a well-known SF material with a reported yield of 120% in concentrated solutions.9 At magnetic field strengths less than 50 mT the ΔPL/PL drops below zero before slightly increasing at greater magnetic fields. Such behavior is characteristic of singlet fission, as the number of triplet pair states with singlet character increases to four at low field strengths.
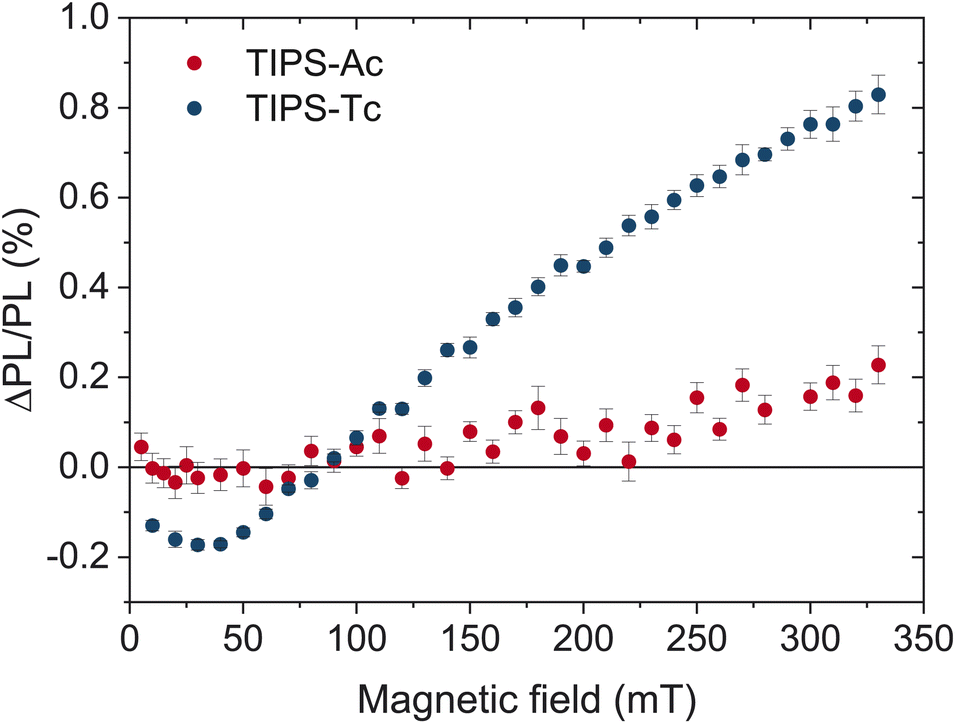 | ||
| Fig. 5 Magnetic field effect on the PL of TIPS-Ac films. The MPL of TIPS-Ac (red) is compared to TIPS-Tc (blue) which is efficient at singlet fission. Error bars represent the standard error of the mean. | ||
To confirm that the MPL is not swamped by a morphology that favors emission, time-correlated single photon counting (TCSPC) was performed by exciting TIPS-Ac at 405 nm and collecting all the emission longwards of 450 nm (Fig. 6). The exponential fit produced three-time constants which are reported with their integrated fluorescence contribution in parentheses: 115 ± 3 ps (0.5), 1.14 ± 0.05 ns (0.25), and 6.23 ± 1.21 ns (0.25) which we assign respectively to SF, singlet emission and excimer emission. From the TCSPC it is evident that 95% of the initial excitation decays rapidly, therefore it is determined that the SF regions comprise >95% of the film (Fig. 6). This rapid decay accounts for half of the integrated fluorescence with the other half collectively belonging to singlet and excimer emission. As such, the highly emissive regions dampen the MPL by a factor of two, and cannot completely account for the low MPL or SF yield.
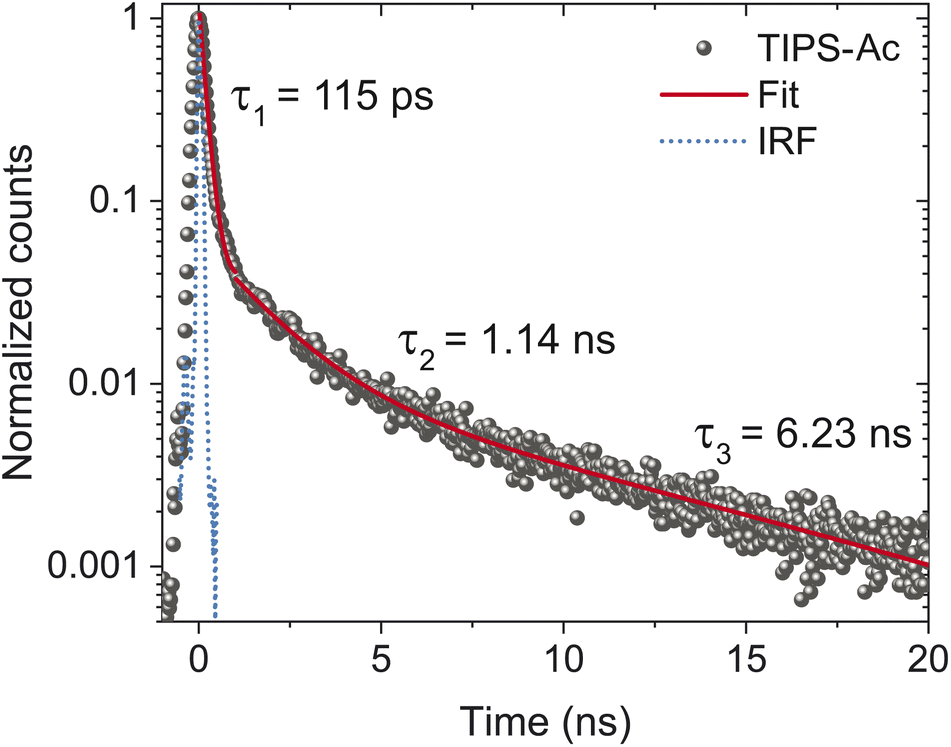 | ||
| Fig. 6 Time-correlated single photon counting of TIPS-Ac thin films. Films were excited at 405 nm (1 μJ cm−2). All emission was collected after the 450 nm short-pass filter. The red line represents the fit of the data with τ values displayed in the fit. The values with their corresponding errors and integration in parenthesis are τ1 = 115 ± 3 ps (0.14), τ2 = 1.14 ± 0.05 ns (0.07), τ3 = 6.23 ± 1.21 ns (0.07). | ||
For further insight, a previously reported model derived by Clark and co-workers was modified to investigate the effects of various parameters on the MPL of TIPS-Ac (see ESI†).25 The model integrates the PL to 1 ns and thus captures the whole singlet fission process which occurs within 100 ps (Fig. 3c). Fig. S8† shows a full representation of the model, which is constrained by observed kinetics, triplet yield, and MPL. The singlet character of weakly-coupled triplet-pairs was calculated from the molecular geometries of the polymorph which favors SF.32 The zero-field splitting parameters were estimated as D = 2063 MHz and E = −234 MHz.43
The model provides two competing routes to triplet formation, namely SF and high-level ISC from S1 to T2 which then decays to T1. SF results in the formation of two free triplets whereas in high-level ISC, via the T2 state, only one triplet is formed. The TA experiments are used to inform the model. From the DAS it is known that the quasi-equilibrium is formed after 15 ps. Therefore the total rate for kIC, kSF, kR and kISC may not exceed 6.67 × 1010 s−1. To match experimental observations regarding kinetics and triplet yield, kSF was adjusted to 1.7 × 1010 s−1, and the relatively small radiative rate is set to kR = 1 × 108 s−1. Initially, kISC was set to zero, with internal conversion acting as the major decay pathway. However, this results in a triplet yield about twice what is observed. To match the experimentally observed triplet yield (∼20%), a non-radiative decay from the 1(TT) was introduced: kNR = 9.0 × 109 s−1. The observation emission from 1(TT) states shows that they exhibit significant S1 character and thus should also be plagued by non-radiative decay.44
To tune the model to the observed MPL, the effect of the triplet dissociation rate (kD) was explored. Fig. 7b depicts the effect an increase in kD has on the MPL and yield of free triplets. At 7 × 109 s−1 the MPL is high and the triplet yield is just 3%. This is because at low kD the triplets struggle to escape the triplet manifold to form free triplets, so with increasing magnetic field the triplet manifold population is converted back to the emissive S1 state, increasing PL. Now if the rate is increased to 1 × 1011 s−1 the majority of the triplet population is converted to form free triplets, which drives down the MPL but increases the triplet yield. At a dissociation rate of 3 × 1010 s−1 an MPL response and triplet yield that matches the experimental observation is obtained (Fig. 7b). The population dynamics (Fig. 7c) at this rate match the expectation from the DAS (Fig. 3c). The 1(TT) population peaks at 10 ps and forms a quasi-equilibrium with S1. The free triplet population also appears as the major species at 100 ps which agrees with the 110 ps lifetime of the 1(TT) state.
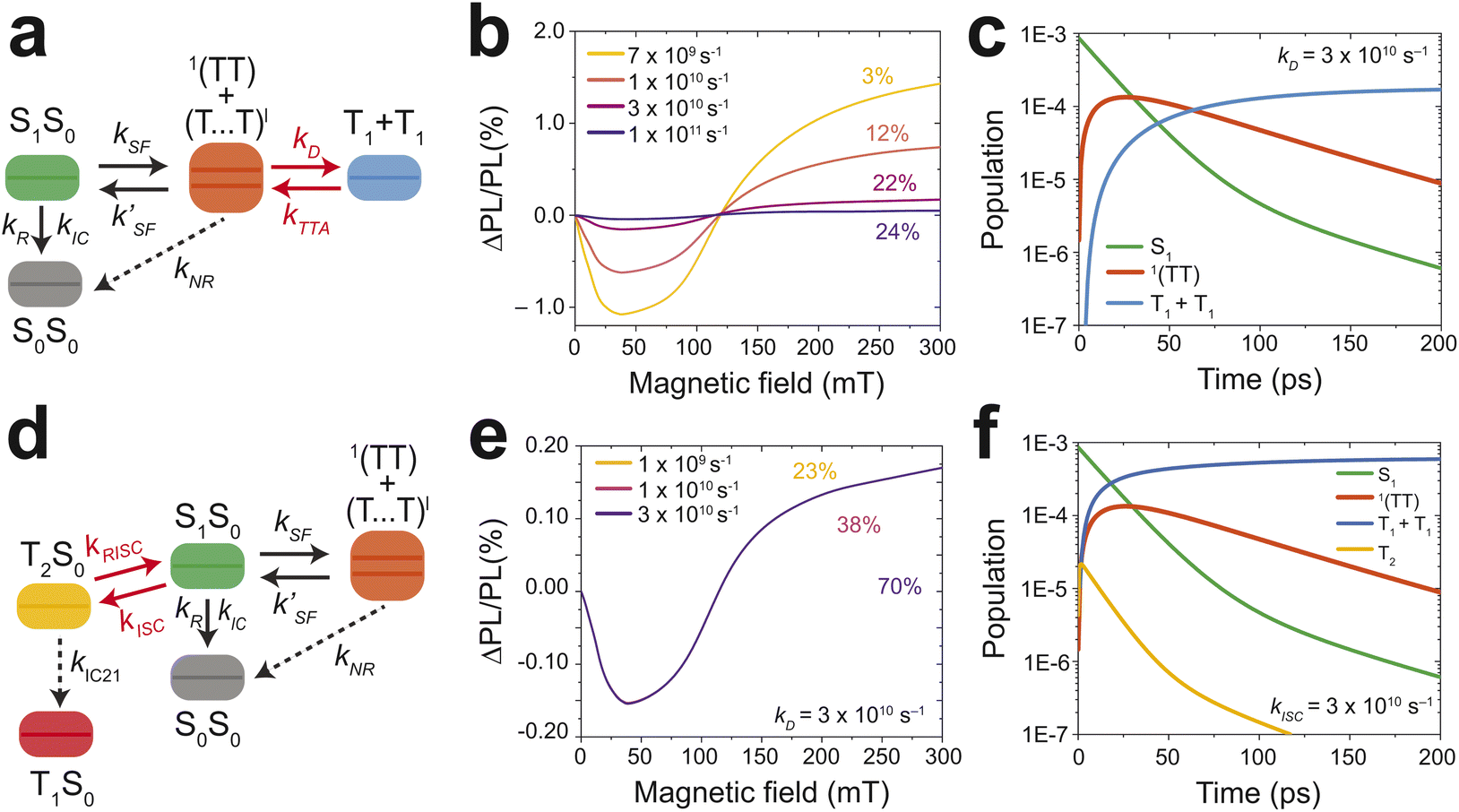 | ||
| Fig. 7 Kinetic modeling of the magnetic field effect on photoluminescence. (a) Simplified schematic of the model used to investigate dissociation's (kD) effect on the MPL. (b) The effect of dissociation on the MPL, the numbers above the lines indicate the free triplet yields. (c) Population dynamics of the involved species with kD set at 3 × 1010 s−1. (d) Simplified schematic of the model used to investigate intersystem crossings (kISC) effect on the MPL. The model is the same as (a), but with the addition of the T2 state. (e) The effect of ISC on the MPL with kD set at 3 × 1010 s−1. MPL curves are overlayed as changing the rate of ISC has no effect. (f) Population dynamics of the involved species with kISC set at 3 × 1010 s−1. More information on the kinetic model may be found in the ESI with Fig. S8† showing the complete model. The rates applied to the model are summarised in Table S1.† | ||
What is apparent, is that the observed kinetics, triplet yield, and MPL can be accounted for without introducing intersystem crossing to the T2 state. High-level intersystem crossing from S1 to T2 is a possible loss mechanism that has previously been reported for anthracene and some of its derivatives.45–47 Sfeir and coworkers have reported ISC via T2 taking place in anthracenothiophene dimers.48 These results were confirmed by the polarization patterns from electron-paramagnetic resonance spectroscopy and not resolvable in their TA measurements due to the rapid internal conversion of T2 → T1. ISC and its effects on the MPL are further investigated here with the model (Fig. 7d). An increase in the rate from 1 × 109 s−1 to 3 × 1010 s−1 has a minimal effect on the MPL but increases the yield of the free triplets from 23% to 70%. The population dynamics are also affected by increasing rates of kISC resulting in the formation of free triplets at around 25 ps (Fig. 7f). These results contradict σ3 which forms at 110 ps (Fig. 3c). Therefore if ISC is taking place it is expected to occur at a rate of ≤1 × 109 s−1 and is therefore not the major loss mechanism for SF in TIPS-Ac.
To summarise, the model can rule out high-level ISC to the T2 state as the major loss pathway for SF. Therefore the low SF-yield in TIPS-Ac is attributed to a loss of the singlet population due to internal conversion back to the ground state within both the S1 state and the 1(TT) state. The latter has been observed in covalent acene dimers.49 ISC does not occur in the isolated monomer and thus we propose that it is enabled in the solid state by virtual a charge-transfer intermediate state. The loss from S1 occurs mostly within the initial 15 ps. An additional loss of the triplet population is incurred within 1 ns from the triplet-pair manifold. Finally, it was observed that the rate of triplet dissociation has a profound effect on the MPL, with larger rates resulting in a dampening of the effect. It is noted that other combinations of rate constants which satisfy the kinetics can satisfy the triplet yield or the observed MPL, but not both. As such, we find that our conclusions are robust in the face of the available data.
4 Conclusions
TIPS-Ac thin films were confirmed to undergo singlet fission with transient absorption spectroscopy and magnetic photoluminescent measurements. The resulting yield of free triplets was 19%. The reasons behind the low yield were further investigated by kinetically modeling the magnetic photoluminescence. From this kinetic modeling, it was determined that singlet fission in TIPS-Ac thin films is plagued by non-radiative internal conversion of the S1 state and that the low magnetic photoluminescence response is likely due to fast dissociation of the weakly coupled triplet pair into free triplets. Furthermore, intersystem crossing to a T2 state was investigated and determined to not be a major competing loss mechanism for singlet fission in TIPS-Ac as it could not account for the low triplet yield.Data availability
Data is available upon request from the corresponding author.Author contributions
D. M. d. C. prepared the films performed steady state absorption and PL, time resolved measurements, magnetic field measurements, wrote the first draft and prepared the figures. M. I. C. calculated the singlet character of the weakly coupled triplet pair. N. P. S. performed X-ray diffraction measurements. J. F. assisted D. M. d. C. with the MPL measurements. D. R. M. supervised the X-ray and singlet character calculations. M. J. Y. T. and M. P. N. supervised the TA experiments and provided feedback on the draft manuscript. T. W. S. conceived the project and assisted D. M. d. C. with the MPL model.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. W. S., M. P. N., J. F., D. M. d. C. acknowledge the Australian Research Council (ARC) Centre of Excellence in Exciton Science (project No. CE17100026). D. M. d. C. acknowledges the Australian Government Research Training Program (RTP) for PhD funding. M. P. N. acknowledges the support of the UNSW Scientia Program. All authors thank John E. Anthony for providing the TIPS-tetracene.References
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- R. W. MacQueen, M. Liebhaber, J. Niederhausen, M. Mews, C. Gersmann, S. Jäckle, K. Jäger, M. J. Y. Tayebjee, T. W. Schmidt, B. Rech and K. Lips, Mater. Horiz., 2018, 5, 1065–1075 RSC.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS.
- A. J. Baldacchino, M. I. Collins, M. P. Nielsen, T. W. Schmidt, D. R. McCamey and M. J. Y. Tayebjee, Chem. Phys. Rev., 2022, 3, 021304 CrossRef CAS.
- M. J. Y. Tayebjee, D. R. McCamey and T. W. Schmidt, J. Phys. Chem. Lett., 2015, 6, 2367–2378 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, A. A. Gray-Weale and T. W. Schmidt, J. Phys. Chem. Lett., 2012, 3, 2749–2754 CrossRef CAS.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. E. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7656–7661 CrossRef CAS.
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305–310 CrossRef CAS.
- K. Broch, J. Dieterle, F. Branchi, N. J. Hestand, Y. Olivier, H. Tamura, C. Cruz, V. M. Nichols, A. Hinderhofer, D. Beljonne, F. C. Spano, G. Cerullo, C. J. Bardeen and F. Schreiber, Nat. Commun., 2018, 9, 954 CrossRef CAS PubMed.
- C. Grieco, G. S. Doucette, K. T. Munson, J. R. Swartzfager, J. M. Munro, J. E. Anthony, I. Dabo and J. B. Asbury, J. Chem. Phys., 2019, 151, 154701 CrossRef PubMed.
- N. J. L. K. Davis, J. R. Allardice, J. Xiao, A. J. I. Petty, N. C. Greenham, J. E. Anthony and A. Rao, J. Phys. Chem. Lett., 2018, 9, 1454–1460 CrossRef CAS PubMed.
- S. N. Sanders, A. B. Pun, K. R. Parenti, E. Kumarasamy, L. M. Yablon, M. Y. Sfeir and L. M. Campos, Chem, 2019, 5, 1988–2005 CAS.
- B. D. Folie, J. B. Haber, S. Refaely-Abramson, J. B. Neaton and N. S. Ginsberg, J. Am. Chem. Soc., 2018, 140, 2326–2335 CrossRef CAS.
- M. Dvořák, S. K. K. Prasad, C. B. Dover, C. R. Forest, A. Kaleem, R. W. MacQueen, A. J. I. Petty, R. Forecast, J. E. Beves, J. E. Anthony, M. J. Y. Tayebjee, A. Widmer-Cooper, P. Thordarson and T. W. Schmidt, J. Am. Chem. Soc., 2021, 143, 13749–13758 CrossRef PubMed.
- Y. Matsui, S. Kawaoka, H. Nagashima, T. Nakagawa, N. Okamura, T. Ogaki, E. Ohta, S. Akimoto, A. Sato-Tomita, S. Yagi, Y. Kobori and H. Ikeda, J. Phys. Chem. C, 2019, 123, 18813–18823 CrossRef CAS.
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- J. K. H. Pun, J. K. Gallaher, L. Frazer, S. K. K. Prasad, C. B. Dover, R. W. MacQueen and T. W. Schmidt, J. Photonics Energy, 2018, 8, 1 Search PubMed.
- N. Nishimura, V. Gray, J. R. Allardice, Z. Zhang, A. Pershin, D. Beljonne and A. Rao, ACS Mater. Lett., 2019, 1, 660–664 CrossRef CAS.
- J. Wong, S. Wei, R. Meir, N. Sadaba, N. A. Ballinger, E. K. Harmon, X. Gao, G. Altin-Yavuzarslan, L. D. Pozzo, L. M. Campos and A. Nelson, Adv. Mater., 2023, 35, year Search PubMed.
- A. Nandi, B. Manna and R. Ghosh, J. Phys. Chem. C, 2023, 127, 12621–12630 CrossRef CAS.
- K. Bhattacharyya and A. Datta, J. Phys. Chem. C, 2017, 121, 1412–1420 CrossRef CAS.
- J. Feng, J. Alves, D. M. de Clercq and T. W. Schmidt, Annu. Rev. Phys. Chem., 2023, 74, 145–168 CrossRef CAS PubMed.
- D. G. Bossanyi, Y. Sasaki, S. Wang, D. Chekulaev, N. Kimizuka, N. Yanai and J. Clark, JACS Au, 2021, 1, 2188–2201 CrossRef CAS.
- Y. Luo, K. Zhang, Z. Ding, P. Chen, X. Peng, Y. Zhao, K. Chen, C. Li, X. Zheng, Y. Huang, X. Pu, Y. Liu, S.-J. Su, X. Hou and Z. Lu, Nat. Commun., 2022, 13, 6892 CrossRef CAS PubMed.
- X. Tang, R. Pan, X. Zhao, H. Zhu and Z. Xiong, J. Phys. Chem. Lett., 2020, 11, 2804–2811 CrossRef CAS PubMed.
- T. Miyashita, P. Jaimes, A. Mardini, M. Fumanal and M. L. Tang, J. Phys. Chem. Lett., 2023, 14, 6119–6126 CrossRef CAS.
- J. Chen, H. Liu, J. Guo, J. Wang, N. Qiu, S. Xiao, J. Chi, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116810 CrossRef CAS PubMed.
- S. A. Odom, S. R. Parkin and J. E. Anthony, Org. Lett., 2003, 5, 4245–4248 CrossRef CAS PubMed.
- A. S. Davydov, J. Exp. Theor. Phys., 1948, 18, 210–218 CAS.
- A. Moliterni, D. Altamura, R. Lassandro, V. Olieric, G. Ferri, F. Cardarelli, A. Camposeo, D. Pisignano, J. E. Anthony and C. Giannini, Acta Crystallogr., Sect. B: Struct. Sci., 2020, 76, 427–435 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, R. G. C. R. Clady and T. W. Schmidt, Phys. Chem. Chem. Phys., 2013, 15, 14797–14805 RSC.
- B. S. Basel, C. Hetzer, J. Zirzlmeier, D. Thiel, R. Guldi, F. Hampel, A. Kahnt, T. Clark, D. M. Guldi and R. R. Tykwinski, Chem. Sci., 2019, 10, 3854–3863 RSC.
- C. Zeiser, L. Moretti, D. Lepple, G. Cerullo, M. Maiuri and K. Broch, Angew. Chem., Int. Ed., 2020, 59, 19966–19973 CrossRef CAS PubMed.
- H. Nagashima, S. Kawaoka, S. Akimoto, T. Tachikawa, Y. Matsui, H. Ikeda and Y. Kobori, J. Phys. Chem. Lett., 2018, 9, 5855–5861 CrossRef CAS PubMed.
- S. L. Bayliss, A. D. Chepelianskii, A. Sepe, B. J. Walker, B. Ehrler, M. J. Bruzek, J. E. Anthony and N. C. Greenham, Phys. Rev. Lett., 2014, 112, 238701 CrossRef PubMed.
- C. Wang and M. J. Tauber, J. Am. Chem. Soc., 2010, 132, 13988–13991 CrossRef CAS.
- R. C. Johnson, R. E. Merrifield, P. Avakian and R. B. Flippen, Phys. Rev. Lett., 1967, 19, 285–287 CrossRef CAS.
- R. E. Merrifield, J. Chem. Phys., 1968, 48, 4318–4319 CrossRef CAS.
- G. B. Piland, J. J. Burdett, R. J. Dillon and C. J. Bardeen, J. Phys. Chem. Lett., 2014, 5, 2312–2319 CrossRef CAS PubMed.
- R. Forecast, E. M. Gholizadeh, S. K. K. Prasad, S. Blacket, P. C. Tapping, D. R. Mccamey, J. Y. Tayebjee, D. M. Huang, J. H. Cole and T. W. Schmidt, J. Phys. Chem. Lett., 2023, 14, 24 CrossRef PubMed.
- D. Haarer, D. Schmid and H. C. Wolf, Phys. Status Solidi B, 1967, 23, 633–638 CrossRef CAS.
- D. G. Bossanyi, M. Matthiesen, S. Wang, J. A. Smith, R. C. Kilbride, J. D. Shipp, D. Chekulaev, E. Holland, J. E. Anthony and J. Zaumseil, et al. , Nat. Chem., 2021, 13, 163–171 CrossRef CAS PubMed.
- R. E. Kellogg, J. Chem. Phys., 1966, 44, 411–412 CrossRef CAS.
- N. I. Nijegorodov and W. S. Downey, Spectrochim. Acta, Part A, 1995, 51, 2335–2346 CrossRef.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- G. He, K. R. Parenti, P. J. Budden, J. Niklas, T. Macdonald, E. Kumarasamy, X. Chen, X. Yin, D. R. McCamey, O. G. Poluektov, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2023, 145, 22058–22068 CrossRef CAS PubMed.
- G. He, K. R. Parenti, L. M. Campos and M. Y. Sfeir, Adv. Mater., 2022, 34, 2203974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06774b |
| This journal is © The Royal Society of Chemistry 2024 |