
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide-free synthesis of metastable graphitic BC3†
Devin
McGlamery
a,
Charles
McDaniel
a,
Dylan M.
Ladd‡
 a,
Yang
Ha
b,
Martín A.
Mosquera
a,
Michael T.
Mock
a and
Nicholas P.
Stadie
*a
a,
Yang
Ha
b,
Martín A.
Mosquera
a,
Michael T.
Mock
a and
Nicholas P.
Stadie
*a
aDepartment of Chemistry & Biochemistry, Montana State University, Bozeman, Montana 59717, USA. E-mail: nstadie@montana.edu
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 15th February 2024
Abstract
Layered BC3, a metastable phase within the binary boron–carbon system that is composed of graphite-like sheets with hexagonally symmetric C6B6 units, has never been successfully crystallized. Instead, poorly-crystalline BC3-like materials with significant stacking disorder have been isolated, based on the co-pyrolysis of a boron trihalide precursor with benzene at around 800 °C. The halide leaving group (–X) is a significant driving force of these reactions, but the subsequent evolution of gaseous HX species at such high temperatures hampers their scaling up and also prohibits their further use in the presence of hard-casting templates such as ordered silicates. Herein, we report a novel halide-free synthesis route to turbostratic BC3 with long-range in-plane ordering, as evidenced by multi-wavelength Raman spectroscopy. Judicious pairing of the two molecular precursors is crucial to achieving B–C bond formation and preventing phase-segregation into the thermodynamically favored products. A simple computational method used herein to evaluate the compatibility of bottom-up molecular precursors can be generalized to guide the future synthesis of other metastable materials beyond the boron–carbon system.
Boron and carbon each exhibit uniquely complex elemental chemistry.1 In particular, boron has been crystallized in a plethora of allotropic forms owing to the rich chemistry that results from its lack of electrons compared to its number of ground state atomic orbitals.2 The vast number of carbon allotropes and their exceptional properties are well-known.3 When combined, boron and carbon give rise to a phase space that is both enormous and often difficult to accurately characterize, owing to the nearly identical size and hybridization states of the two elements. The single thermodynamically-stable binary phase at ambient conditions is boron carbide (nominally B4C), a structure comprised of the dominant B12 icosahedral unit prevalent in boron's elemental phases. However, as might be expected, this phase exhibits wide compositional variability and there remains disagreement as to its formal description as B12C3 or B13C2 in its most carbon-rich form.4,5
Numerous theoretical studies6–9 support growing experimental evidence10–12 for the existence of a high symmetry graphite-like phase of composition BC3 (Fig. 1). Indeed, the equilibrium solubility of boron in graphite is established to be relatively high: 2.35 at% at 2375 °C.13 The difference in formation energies between BC3 and equivalent quantities of graphite and boron carbide is not currently known, but is likely small compared to the thermal energies present at the temperatures characteristic of graphite and carbide synthesis. Hence, many researchers have reported strategies to isolating metastable BC3 at ambient conditions,12,14–20 an effort that has been intensified owing to the promising theoretical applications of BC3 for energy storage applications.21–26
 | ||
| Fig. 1 (a) Binary phase diagram of the boron–carbon system.27 The region of interest for the formation of metastable BC3 is highlighted. (b) The proposed in-plane structure and unit cell of BC3.7,10 (c) Thermochemical ranking of boron and carbon precursors based on their estimated temperature of decomposition (Td). | ||
Our previous work12 culminated in the synthesis of a bulk (free-standing) graphite-like material of composition BC3via the bottom-up reaction of two molecular precursors: boron tribromide (BBr3) and benzene (C6H6). At that time, previous efforts had only succeeded to produce either thin-films (led by Kouvetakis, Bartlett and coworkers)10,15 or bulk flakes via the laborious synthesis and subsequent pyrolysis of 1,3-bis-(dibromoboryl) benzene (led by King, Wright, and coworkers)11. While the earlier approach benefits from the direct pyrolysis of high-purity, commercially available precursors, only small quantities of BC3 could be produced. The latter approach produces bulk BC3-like material yet it suffers in terms of complexity and from the presence of residual contaminants due to the complex, multi-step organic synthesis of the boronated benzene; our approach combined these two strategies by leveraging the high-purity nature of combining only two commercially-available precursors (similar to the BCl3 and benzene chosen for the thin-film synthesis) and the liquid miscibility associated with a Br-terminated boron precursor (as inspired by the 1,3-bis-(dibromoboryl)benzene precursor chosen to produce the first free-standing BC3-like flakes). The crystalline quality of the resulting products is low, especially with respect to ordering in the stacking direction, but our direct synthesis approach has been shown to yield long-range ordered in-plane structures with a vibrational structure that is consistent with BC3.20
The investigation of directly-synthesized BC3-like materials for hydrogen storage and for electrochemical energy storage applications has remained severely limited by its inherent release of HBr at elevated temperatures, typically 800 °C, in closed-reactor conditions, preventing the use of stainless steel reaction vessels or their equivalent. Therefore, two important synthetic goals of research in this area became (1) to achieve higher crystallinity of bulk BC3 and (2) to devise halide-free routes to the synthesis of bulk BC3 in order to prevent the release of corrosive HX or X2 gases (X = halogen) at elevated temperatures. Our natural hypothesis was that diborane (B2H6), the smallest borane species and a reactive gas at ambient pressure,§ would be well-suited to reactions with benzene to produce bulk BC3 with only the release of H2 as a side-product.
The resulting studies of the reaction between diborane and benzene within a custom-designed apparatus comprising a closed, stainless steel reaction vessel at up to 800 °C (see ESI† for details) consistently revealed a heterogeneous mixture of products, a hodgepodge compounded by the presence of metal particle-catalyzed graphitic fibers which are known to be characteristic of carbon soots produced under similar conditions.28,29 Control reactions (e.g., of liquid mixtures of condensed diborane and benzene) in the absence of catalytic effects from the metal-walled reactor were not safe enough to conduct within the scope of this work. Meanwhile, benzene remained the optimal hydrocarbon precursor owing mainly to the desire to preserve its ring structure within the planar layers of BC3 (Fig. 1b), and hence we set out to identify a compatible, halide-free boron precursor to pair it with in our efforts to achieve the goals listed above.
Two additional halide-free molecular boron precursors were then explored as candidates toward elucidation of the synthesis of bulk BC3: decaborane (B10H14) and a bespoke polyaromatic carborane species (1,8-naphthalenediyl-bridged diborane, NDB).30 The former, a readily available commercial compound, is obtainable as a high-purity crystalline solid at ambient conditions and is miscible in benzene up to the desired B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio of 1:3. However, reactions between decaborane and benzene at up to 800 °C always generated carbon-rich flakes with a colorful (iridescent) thin surface layer of amorphous boron, and with no discernible content of BC3 (see ESI†). The latter precursor, NDB, itself requires a multi-step organic synthesis that, even under strict air-free handling and after judicious purification, inevitably leads to the introduction of impurities such as oxygen, chlorine, and aluminum in the eventual high-temperature pyrolysis product (see ESI†). Scanning electron microscopy (SEM) studies of this product revealed the existence of a rippled, carbide-like film on the surface of carbon-rich flakes. The overall maximum boron content of such products is in any case limited to ∼17 at% owing to its stoichiometry of B2C10H10 (B:C of 1:5), preventing the realization of BC3. No other planar carboranes exist with a higher content of boron, and hence a halide-free single precursor “tiling” route to bulk BC3 was also abandoned. The products of the attempted syntheses of bulk BC3 from diborane(II), decaborane(III), and NDB(IV) are compared to that from boron tribromide(I), the previously established route, in Fig. 2. In summary, a systematic approach to identifying a compatible halide-free boron precursor for reactions with benzene at near 800 °C was needed.
:C ratio of 1:3. However, reactions between decaborane and benzene at up to 800 °C always generated carbon-rich flakes with a colorful (iridescent) thin surface layer of amorphous boron, and with no discernible content of BC3 (see ESI†). The latter precursor, NDB, itself requires a multi-step organic synthesis that, even under strict air-free handling and after judicious purification, inevitably leads to the introduction of impurities such as oxygen, chlorine, and aluminum in the eventual high-temperature pyrolysis product (see ESI†). Scanning electron microscopy (SEM) studies of this product revealed the existence of a rippled, carbide-like film on the surface of carbon-rich flakes. The overall maximum boron content of such products is in any case limited to ∼17 at% owing to its stoichiometry of B2C10H10 (B:C of 1:5), preventing the realization of BC3. No other planar carboranes exist with a higher content of boron, and hence a halide-free single precursor “tiling” route to bulk BC3 was also abandoned. The products of the attempted syntheses of bulk BC3 from diborane(II), decaborane(III), and NDB(IV) are compared to that from boron tribromide(I), the previously established route, in Fig. 2. In summary, a systematic approach to identifying a compatible halide-free boron precursor for reactions with benzene at near 800 °C was needed.
 | ||
| Fig. 2 SEM images of the graphitic BCx pyrolysis products resulting from five different synthesis routes: (a) route I via boron tribromide, (b) route II via diborane, (c) route III via decaborane, (d) route IV via NDB, and (e) route V via sodium borohydride. | ||
The heterogeneity of each of the products of the aforementioned reactions can likely be explained by a mismatch in the decomposition or cracking temperatures of the boron and carbon species within each reaction. In order to realize a homogeneous metastable phase within a binary system, the thermodynamically favored phases (enriched in one or the other of the two elements, in this case graphite and boron carbide) must be prevented from forming, which requires the simultaneous decomposition of the corresponding precursors: i.e., co-pyrolysis. We undertook to estimate the relative decomposition temperatures (Td) of an expansive library of boron and carbon precursors using a simple density functional theory (DFT) method, described in detail in the ESI.† In short, the change in energy of simplified decomposition reactions were calculated at the MN15/def2-QZVPP31,32 level (recently benchmarked for studies of small molecular systems of composed of boron, carbon, and hydrogen).33,34 The normalized change in electronic energy, ΔESCF, where the Δ refers to decomposition into B or C and the relevant diatomic gases, was found to be highly correlated with the experimental decomposition temperature of each precursor (Fig. S24†). The results revealed that among all of the precursors investigated, borohydride (BH4−) exhibited the closest energetic match to benzene, without the presence of borohalide (B–X) bonds (Fig. 1c).
Borohydride salts are widely available in high purity, and their decomposition temperatures are known to be directly related to the electronegativity of the counter ion (M+).35 However, no previous studies of even common borohydrides (M = Li, Na, or Mg) with simple hydrocarbon precursors such as benzene have been reported. Many of the less-stable borohydrides are known to emit diborane at modest temperatures (e.g., as low as ∼61 °C for Al(BH4)3) during decomposition.35,36 On the other hand, the stable borohydrides such as Li(BH4) do not produce detectable diborane upon decomposition, leading to polymeric B–B bond formation and the eventual formation of higher boranes such as B12H122−.37 The similar decomposition temperatures of NaBH4 (∼600 °C)35 and benzene (∼750 °C)38 led us to predict that reactions between these two precursors could result in the formation of BC3.
A stoichiometric mixture of benzene and sodium borohydride (with a B:C ratio of 1:3) was charged into a quartz ampule under argon, solidified by submersion in liquid nitrogen, and then flame-sealed within the ampule under active vacuum (referred to herein as “synthesis route V,” see ESI†). Pyrolysis was carried out at 800 °C for 1 h, and the sample was then collected in air at room temperature. To prevent the formation of sodium metal and/or reactions of the sodium with the walls of the quartz reactor, a sodium-ion-trapping agent should also be added to the mixture (e.g., I2, Se, or Te) to form a water-soluble salt that can be easily separated during workup of the product. Dark lustrous flakes were collected, showing very little tendency to adhere to the inner walls of the ampule, a characteristic feature of bulk polycrystalline BC3 derived from BBr3 (referred to herein as “synthesis route I”).12 SEM imaging further supports that sodium borohydride-derived BC3 (route V) exhibits a homogeneous, flake-like morphology (∼2 μm in width) with the same “cratered” texture (Fig. 2e) as that derived from boron tribromide (Fig. 2a).
Further materials characterization (Fig. 3) reveals borohydride-derived bulk BC3 (route V) to be similar in structure and composition to that derived from boron tribromide (route I). The underlying structure is exclusively graphitic, as evidenced by powder X-ray diffraction (XRD), with significant misalignment (turbostratic disorder) along the stacking direction as shown by the broad reflection at 2θ = 25.4° corresponding to an interlayer spacing of 3.5 Å. This broadness is in agreement with theoretical predictions for crystalline BC3 which show that its various high-symmetry stacking sequences all lie within a narrow range of formation energies (±0.011 eV per atom),7 indicating the strong likelihood of stacking misalignment. The in-plane structure is revealed to be more ordered, with an average domain size of 42 Å according to a Scherrer analysis of the (10l) peak at 2θ = ∼42°. The elemental composition measured using energy dispersive X-ray spectroscopy (EDX) is estimated to be ∼BC5, identical to equivalent measurements of the products of synthesis route I. It must be noted that past work has shown EDX to underestimate the boron content compared to more sensitive methods such as elastic recoil detection analysis (ERDA).12
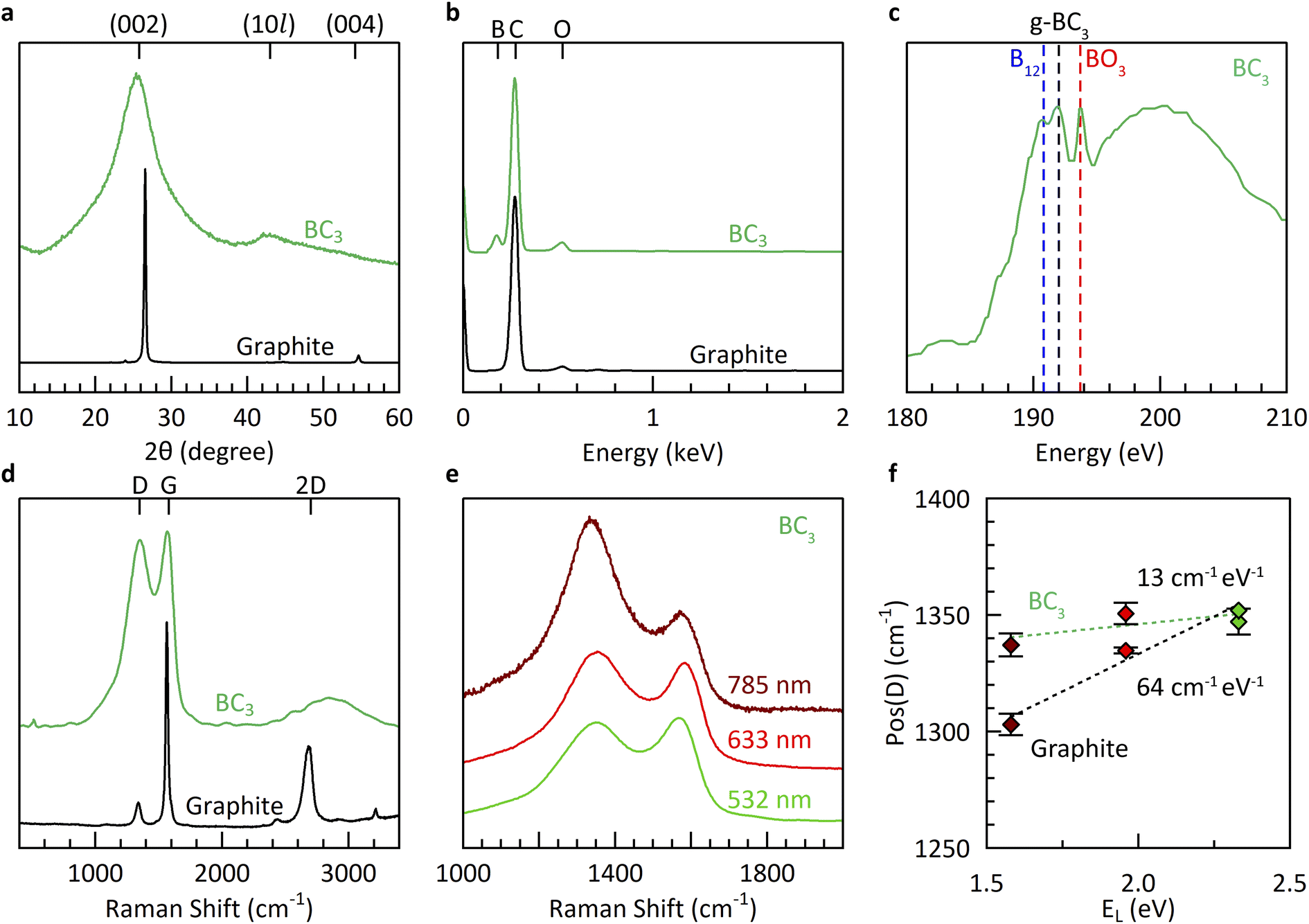 | ||
| Fig. 3 Materials characterization of borohydride-derived BC3 compared to graphite: (a) XRD patterns, (b) EDX spectra, (c) XAS spectra at the B K-edge, and (d) Raman spectra at 532 nm. (e and f) Dispersion relation of the D peak as revealed by multi-wavelength Raman spectroscopy. | ||
The boron chemical environments present in borohydride-derived BC3 were investigated by a combination of X-ray absorption spectroscopy (XAS) and multi-wavelength Raman spectroscopy (Fig. 3c–f). The presence of planar g-BC3 units in long-range ordered, hexagonal symmetry is evidenced by a dominant absorption feature in the B K-edge at 192.0 eV.39 Some icosahedral B12 environments are also detected at 190.8 eV, likely due to the small degree of mismatch between the decomposition temperatures of benzene and BH4− which favors the slightly earlier formation of boron rich phases prior to the desired reaction to form BC3. Most importantly, the Raman spectrum exhibits the characteristic features of bulk BC3: a broad and intense D peak at Pos(D) = ∼1350 cm−1, exhibiting almost no dispersion as a function of the energy of irradiation (EL), and a broad, intense, and modulated 2D region associated with two-phonon intervalley scattering.40 Previous work revealed that the dispersion relation of the D peak, in particular, is highly sensitive to the presence of hexagonally ordered flower-like B6C6 units, originating from the flattening of the highest optical phonon branches around the K point in BC3.20 The non-dispersive nature of the D peak of borohydride-derived BC3 coupled with the presence of an intense feature in the XAS spectrum at 192 eV is the ultimate evidence for the structure of BC3 (in the absence of long-range crystalline order).20
In conclusion, we report a novel borohalide-free route to turbostratic BC3 with exceptional in-plane ordering via the reaction of benzene with sodium borohydride at 800 °C. The computational method developed herein can be extended to identify candidate precursors for other bottom-up synthesis efforts toward binary metastable phases. The limited set of experimental conditions explored herein compels future work to tune the decomposition profile of the borohydride precursor by varying the counter ion and/or matching a given salt with a hydrocarbon of similar stability, opening an abundance of possible chemical routes to more crystalline forms of graphite-like BC3.
Data availability
The associated experimental and computational data are available in the ESI† and further details are available by request from the corresponding author.Author contributions
The manuscript was written through contributions of all authors. D. M. developed the hypothesis, performed the primary experiments, analyzed all of the data, and wrote the first draft of the manuscript. C. M. and M. A. M. performed computational experiments and analysis. C. M. and D. L. assisted with pyrolysis experiments. D. M. and Y. H. performed XAS measurements. N. P. S. and M. T. M. supervised the experimental work and M. A. M. supervised the computational work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the donors of the American Chemical Society Petroleum Research Fund (PRF) as well as support from the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Hydrogen and Fuel Cell Technologies and Vehicle Technologies Offices (award DE-EE0008815). The XAS experiments were performed at beam line BL8.0.1.4 at the Advanced Light Source (ALS), which is a DOE Office of Science User Facility (contract DE-AC02-05CH11231). The NMR experiments were performed using equipment supported by the National Science Foundation (awards MRI:DBI-1532078 and MRI:CHE-2018388) and the Murdock Charitable Trust Foundation (2015066:MNL). We thank Tom Autrey for many exciting and helpful conversations. We also thank Steven Christensen for assistance with developing the safety protocols related to handling diborane.References
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, 2nd edn, 1997, DOI:10.1016/B978-0-7506-3365-9.50012-2.
- B. Albert and H. Hillebrecht, Boron: Elementary Challenge for Experimenters and Theoreticians, Angew. Chem., Int. Ed., 2009, 48(46), 8640–8668, DOI:10.1002/anie.200903246.
- A. Hirsch, The Era of Carbon Allotropes, Nat. Mater., 2010, 9(11), 868–871, DOI:10.1038/nmat2885.
- G. Will and K. H. Kossobutzki, An X-Ray Diffraction Analysis of Boron Carbide, B13C2, J. Less-Common Met., 1976, 47, 43–48, DOI:10.1016/0022-5088(76)90072-2.
- F. Thévenot, Boron Carbide—A Comprehensive Review, J. Eur. Ceram. Soc., 1990, 6(4), 205–225, DOI:10.1016/0955-2219(90)90048-K.
- Q. Wang, L.-Q. Chen and J. F. Annett, Ab Initio Calculation of Structural Properties of C3B and C5B Compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55(1), 8–10, DOI:10.1103/PhysRevB.55.8.
- H. Sun, F. J. Ribeiro, J.-L. Li, D. Roundy, M. L. Cohen and S. G. Louie, Ab Initio Pseudopotential Studies of Equilibrium Lattice Structures and Phonon Modes of Bulk BC3, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(2), 024110, DOI:10.1103/PhysRevB.69.024110.
- X. Luo, J. Yang, H. Liu, X. Wu, Y. Wang, Y. Ma, S.-H. Wei, X. Gong and H. Xiang, Predicting Two-Dimensional Boron–Carbon Compounds by the Global Optimization Method, J. Am. Chem. Soc., 2011, 133(40), 16285–16290, DOI:10.1021/ja2072753.
- T. M. Dieb, Z. Hou and K. Tsuda, Structure Prediction of Boron-Doped Graphene by Machine Learning, J. Chem. Phys., 2018, 148(24), 241716, DOI:10.1063/1.5018065.
- J. Kouvetakis, R. B. Kaner, M. L. Sattler and N. Bartlett, A Novel Graphite-like Material of Composition BC3, and Nitrogen–Carbon Graphites, J. Chem. Soc., Chem. Commun., 1986, 24, 1758–1759, 10.1039/C39860001758.
- T. C. King, P. D. Matthews, H. Glass, J. A. Cormack, J. P. Holgado, M. Leskes, J. M. Griffin, O. A. Scherman, P. D. Barker, C. P. Grey, S. E. Dutton, R. M. Lambert, G. Tustin, A. Alavi and D. S. Wright, Theory and Practice: Bulk Synthesis of C3B and Its H2- and Li-Storage Capacity, Angew. Chem., Int. Ed., 2015, 54(20), 5919–5923, DOI:10.1002/anie.201412200.
- N. P. Stadie, E. Billeter, L. Piveteau, K. V. Kravchyk, M. Döbeli and M. V. Kovalenko, Direct Synthesis of Bulk Boron-Doped Graphitic Carbon, Chem. Mater., 2017, 29(7), 3211–3218, DOI:10.1021/acs.chemmater.7b00376.
- C. E. Lowell, Solid Solution of Boron in Graphite, J. Am. Ceram. Soc., 1967, 50(3), 142–144, DOI:10.1111/j.1151-2916.1967.tb15064.x.
- R. B. Kaner, J. Kouvetakis, C. E. Warble, M. L. Sattler and N. Bartlett, Boron-Carbon-Nitrogen Materials of Graphite-like Structure, Mater. Res. Bull., 1987, 22(3), 399–404, DOI:10.1016/0025-5408(87)90058-4.
- J. Kouvetakis, T. Sasaki, C. Shen, R. Hagiwara, M. Lerner, K. M. Krishnan and N. Bartlett, Novel Aspects of Graphite Intercalation by Fluorine and Fluorides and New B/C, C/N and B/C/N Materials Based on the Graphite Network, Synth. Met., 1989, 34(1), 1–7, DOI:10.1016/0379-6779(89)90355-X.
- B. M. Way, J. R. Dahn, T. Tiedje, K. Myrtle and M. Kasrai, Preparation and Characterization of BxC1-x Thin Films with the Graphite Structure, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46(3), 1697–1702, DOI:10.1103/PhysRevB.46.1697.
- W. Cermignani, T. E. Paulson, C. Onneby and C. G. Pantano, Synthesis and Characterization of Boron-Doped Carbons, Carbon, 1995, 33(4), 367–374, DOI:10.1016/0008-6223(94)00160-2.
- J. G. Naeini, B. M. Way, J. R. Dahn and J. C. Irwin, Raman Scattering from Boron-Substituted Carbon Films, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(1), 144–151, DOI:10.1103/PhysRevB.54.144.
- T. Shirasaki, A. Derré, M. Ménétrier, A. Tressaud and S. Flandrois, Synthesis and Characterization of Boron-Substituted Carbons, Carbon, 2000, 38(10), 1461–1467, DOI:10.1016/S0008-6223(99)00279-1.
- D. McGlamery, A. A. Baker, Y.-S. Liu, M. A. Mosquera and N. P. Stadie, Phonon Dispersion Relation of Bulk Boron-Doped Graphitic Carbon, J. Phys. Chem. C, 2020, 124(42), 23027–23037, DOI:10.1021/acs.jpcc.0c06918.
- C. Zhang and A. Alavi, Hydrogen Absorption in Bulk BC3: A First-Principles Study, J. Chem. Phys., 2007, 127(21), 214704, DOI:10.1063/1.2802338.
- X. Sha, A. C. Cooper, W. H. I. Bailey and H. Cheng, Revisiting Hydrogen Storage in Bulk BC3, J. Phys. Chem. C, 2010, 114(7), 3260–3264, DOI:10.1021/jp910356v.
- R. P. Joshi, B. Ozdemir, V. Barone and J. E. Peralta, Hexagonal BC3: A Robust Electrode Material for Li, Na, and K Ion Batteries, J. Phys. Chem. Lett., 2015, 6(14), 2728–2732, DOI:10.1021/acs.jpclett.5b01110.
- A. A. Kuzubov, A. S. Fedorov, N. S. Eliseeva, F. N. Tomilin, P. V. Avramov and D. G. Fedorov, High-Capacity Electrode Material BC3 for Lithium Batteries Proposed by Ab Initio Simulations, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(19), 195415, DOI:10.1103/PhysRevB.85.195415.
- K. Belasfar, M. Houmad, M. Boujnah, A. Benyoussef and A. E. Kenz, First-Principles Study of BC3 Monolayer as Anodes for Lithium-Ion and Sodium-Ion Batteries Applications, J. Phys. Chem. Solids, 2020, 139, 109319, DOI:10.1016/j.jpcs.2019.109319.
- L. Zhao, Y. Li, G. Zhou, S. Lei, J. Tan, L. Lin and J. Wang, First-Principles Calculations of Stability of Graphene-like BC3 Monolayer and Its High-Performance Potassium Storage, Chin. Chem. Lett., 2021, 32(2), 900–905, DOI:10.1016/j.cclet.2020.07.016.
- H. B.-C. Okamoto, Boron-Carbon, J. Phase Equilib., 1992, 13(4), 436, DOI:10.1007/BF02674994.
- M. Toebes, Impact of the Structure and Reactivity of Nickel Particles on the Catalytic Growth of Carbon Nanofibers, Catal. Today, 2002, 76(1), 33–42, DOI:10.1016/S0920-5861(02)00209-2.
- K. P. De Jong and J. W. Geus, Carbon Nanofibers: Catalytic Synthesis and Applications, Catal. Rev., 2000, 42(4), 481–510, DOI:10.1081/CR-100101954.
- A. S. Scholz, J. G. Massoth, M. Bursch, J.-M. Mewes, T. Hetzke, B. Wolf, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, BNB-Doped Phenalenyls: Modular Synthesis, Optoelectronic Properties, and One-Electron Reduction, J. Am. Chem. Soc., 2020, 142(25), 11072–11083, DOI:10.1021/jacs.0c03118.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn–Sham Global-Hybrid Exchange–Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions, Chem. Sci., 2016, 7(8), 5032–5051, 10.1039/C6SC00705H.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305, 10.1039/B508541A.
- R. Rowsey, E. E. Taylor, S. Irle, N. P. Stadie and R. K. Szilagyi, Methane Adsorption on Heteroatom-Modified Maquettes of Porous Carbon Surfaces, J. Phys. Chem. A, 2021, 125(28), 6042–6058, DOI:10.1021/acs.jpca.0c11284.
- R. Rowsey, E. E. Taylor, R. W. Hinson, D. Compton, N. P. Stadie and R. K. Szilagyi, Does Boron or Nitrogen Substitution Affect Hydrogen Physisorption on Open Carbon Surfaces?, Phys. Chem. Chem. Phys., 2022, 24(46), 28121–28126, 10.1039/D2CP03975C.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. Li, N. Ohba, S. Towata, A. Züttel and S. Orimo, Correlation between Thermodynamical Stabilities of Metal Borohydrides and Cation Electronegativites: First-Principles Calculations and Experiments, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(4), 045126, DOI:10.1103/PhysRevB.74.045126.
- E. Callini, A. Borgschulte, A. J. Ramirez-Cuesta and A. Züttel, Diborane Release and Structure Distortion in Borohydrides, Dalton Trans., 2012, 42(3), 719–725, 10.1039/C2DT31608K.
- M. P. Pitt, M. Paskevicius, D. H. Brown, D. A. Sheppard and C. E. Buckley, Thermal Stability of Li2B12H12 and Its Role in the Decomposition of LiBH4, J. Am. Chem. Soc., 2013, 135(18), 6930–6941, DOI:10.1021/ja400131b.
- J. E. Zanetti and G. Egloff, The Thermal Decomposition of Benzene, J. Ind. Eng. Chem., 1917, 9(4), 350–356, DOI:10.1021/ie50088a009.
- C. Pallier, J.-M. Leyssale, L. A. Truflandier, A. T. Bui, P. Weisbecker, C. Gervais, H. E. Fischer, F. Sirotti, F. Teyssandier and G. Chollon, Structure of an Amorphous Boron Carbide Film: An Experimental and Computational Approach, Chem. Mater., 2013, 25(13), 2618–2629, DOI:10.1021/cm400847t.
- A. C. Ferrari and D. M. Basko, Raman Spectroscopy as a Versatile Tool for Studying the Properties of Graphene, Nat. Nanotechnol., 2013, 8(4), 235–246, DOI:10.1038/nnano.2013.46.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis details, experimental methods, materials characterization of all reaction products from routes I–V, computational methods, and thermochemical correlations between computation and experiments. See DOI: https://doi.org/10.1039/d3sc06837d |
| ‡ Present Addresses: Department of Materials Science & Engineering, University of Colorado, Boulder, Colorado, 80309, United States. |
| § Important safety precautions associated with the handling of gaseous diborane at high pressures are detailed in the ESI.† |
| This journal is © The Royal Society of Chemistry 2024 |