
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic utility of functionalized alkylsilyl peroxides for Fe-catalyzed and visible-light-promoted radical transformation†
Jiahao
Liu
 ab,
Shiyong
Liu
ab,
Zhe
Wang
ab,
Terumasa
Kato
*abc,
Yan
Liu
*ab and
Keiji
Maruoka
*abc
ab,
Shiyong
Liu
ab,
Zhe
Wang
ab,
Terumasa
Kato
*abc,
Yan
Liu
*ab and
Keiji
Maruoka
*abc
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China. E-mail: terumasa.kato.j97@kyoto-u.jp; yanliu@gdut.edu.cn; maruoka.keiji.4w@kyoto-u.ac.jp
bGuangdong Provincial Key Laboratory of Plant Resources Biorefinery, Guangdong University of Technology, Guangzhou 510006, China
cGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo, Kyoto 606-8501, Japan
First published on 22nd February 2024
Abstract
α-Keto-, β-acetoxy- and β-amidoalkylsilyl peroxides are prepared from various precursors and utilized for Fe-catalyzed and visible-light-promoted radical functionalization with coupling partners under mild conditions with a broad substrate scope.
Introduction
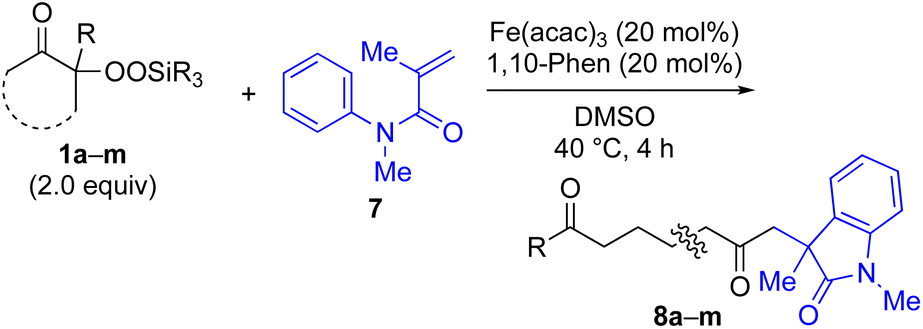
The generation of unstabilized, reactive alkyl radicals from appropriate organic precursors under mild conditions is very important and challenging in modern radical chemistry.1 One of the most reliable approaches to generate such alkyl radicals is the β-scission of alkoxy radicals, which are generally prepared from the corresponding alkanols under strongly oxidative conditions.2 However, due to the use of strong oxidants in this method, the choice of substrates is limited by the functional group tolerance and reaction conditions. Recently, the photocatalyzed generation of alkoxy radicals has also been reported with limited success.3 In this context, we have recently reported the successful generation of alkyl radicals from alkylsilyl peroxides4 under mildly reductive conditions (reductive β-scission strategy) using Cu, Fe or Ni catalysts; the in situ-generated alkyl radicals were then reacted with various coupling partners to furnish new C(sp3)–N,5 C(sp3)–C(sp),6 C(sp3)–B,7 C(sp3)–Si,7 C(sp3)–O,8 C(sp3)–C(sp2),9 and C(sp3)–C(sp3)10 bonds.11 Thus far, we have developed this radical chemistry using alkylsilyl peroxides without any functional groups. However, if various functional groups could be introduced into the carbon skeletons of the alkylsilyl peroxides, this radical chemistry would be further enhanced to a synthetically more useful level. In this work, α-keto-substituted alkylsilyl peroxides of type 1 are prepared, and transformed into the more-stable acyl radicals 2,12,13 rather than the alkyl radicals 2′ using the reductive β-scission strategy (Fig. 1a). In a similar manner, β-acetoxy- and β-amido-substituted alkylsilyl peroxides of types 3 and 5 are utilized for the generation of α-acetoxyalkyl and α-amidoalkyl radicals 4 and 6, respectively (Fig. 1b and c).14,15 These functionalized carbon radicals, thus generated, are then utilized for subsequent transformation with various types of coupling partners (F–H), thereby providing more synthetically valuable products. | ||
| Fig. 1 Transformation of α-ketoalkylsilyl peroxides 1 and β-acetoxyalkyl and β-amidoalkyl peroxides 3 and 5 for metal-catalyzed and visible-light-promoted functionalization. | ||
Results and discussion
α-Ketoalkylsilyl peroxide 1a was conveniently prepared from 2-methylcyclohexanone via an initial hydroperoxylation and subsequent trimethylsilylation using a literature procedure (see ESI†). First, based on our previously reported conjugate addition–cyclization sequence,16,17 reaction of α-ketoalkylsilyl peroxide 1a as an alkyl radical source was carried out with methacrylamide 7 as a coupling partner. Thus, the reaction of α-ketoalkylsilyl peroxide 1a (1.2 equiv.) with methacrylamide 7 in dioxane in the presence of 20 mol% each of CuI and 1,10-phenanthroline (1,10-phen) at 80 °C for 4 h gave rise to desired conjugate addition–cyclization product 8a exclusively in 11% yield (entry 1 in Table 1). The use of more-Lewis-acidic Cu(MeCN)4BF4 under similar conditions afforded 8a in slightly higher yield (entry 2). Replacing the Cu catalysts with Ni(OAc)2·4H2O catalyst exhibited a similar low reactivity (entry 3). Interestingly, the addition of Fe catalysts such as FeCl2 and Fe(acac)2 enhanced the yield of 8a to 42–53% yield (entries 4 and 5), although the use of FeCl3 gave less satisfactory results (entry 6). Finally, the use of Fe(acac)3 catalyst under similar conditions exhibited good yield (entry 7). Having identified Fe(acac)3 as a suitable catalyst, the solvent effect was then examined. The use of DMSO solvent at low temperature afforded product 8a in higher yield (69%) than MeCN, DCE, or benzene (entry 12 vs. 8–11). Furthermore, 95% of 8a was obtained by using excess 1a (2 equiv.) (entry 13). For more details of the reaction optimization, see Tables S1 and S2 in the ESI.†|
|
||||
|---|---|---|---|---|
| Entry | Metal catalyst | Solvent | Temp. (°C) | Yieldb (%) |
| a The reactions of 7 (0.2 mmol) and 1a (0.24 mmol) were carried out in the presence of metal catalyst (0.04 mmol) and 1,10-phen ligand (0.04 mmol) in solvent (1 mL) at the indicated temperature for 4 h. b The yield of 8a was determined by 1H NMR spectroscopy using nitromethane as an internal standard. c 1a (2.0 equiv.). d Isolated yield of 8a. | ||||
| 1 | CuI | Dioxane | 80 | 11 |
| 2 | Cu(MeCN)4BF4 | Dioxane | 80 | 19 |
| 3 | Ni(OAc)2·4H2O | Dioxane | 80 | 18 |
| 4 | FeCl2 | Dioxane | 80 | 42 |
| 5 | Fe(acac)2 | Dioxane | 80 | 53 |
| 6 | FeCl3 | Dioxane | 80 | 28 |
| 7 | Fe(acac)3 | Dioxane | 80 | 59 |
| 8 | Fe(acac)3 | MeCN | 80 | 50 |
| 9 | Fe(acac)3 | DCE | 80 | 42 |
| 10 | Fe(acac)3 | Benzene | 80 | 50 |
| 11 | Fe(acac)3 | DMSO | 80 | 65 |
| 12 | Fe(acac)3 | DMSO | 40 | 69 |
| 13c | Fe(acac)3 | DMSO | 40 | 95d |

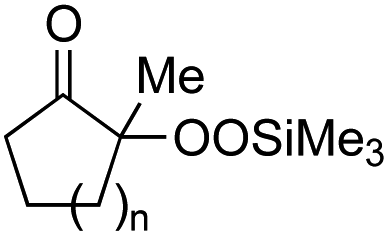
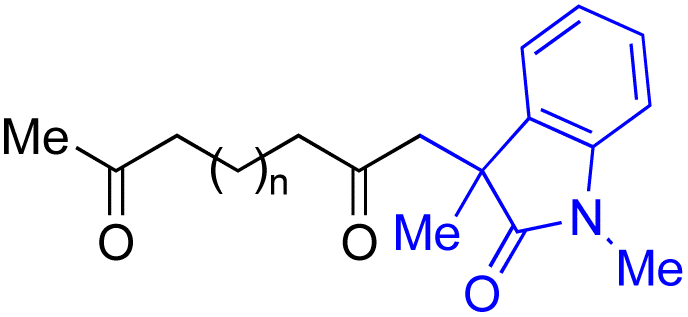
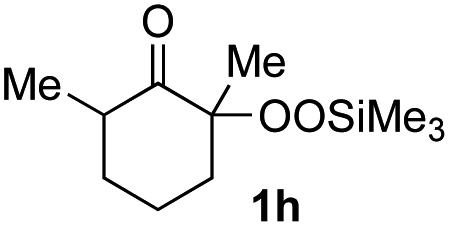
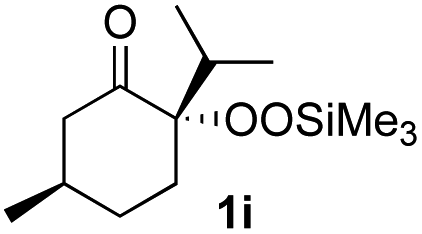
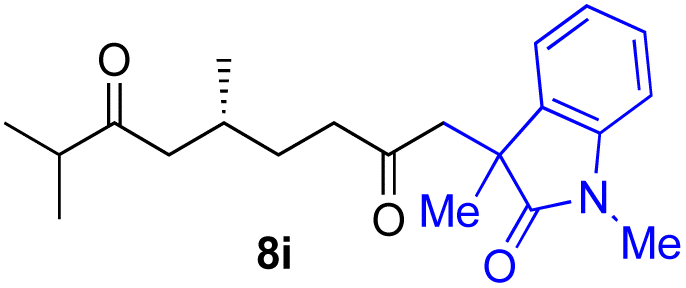
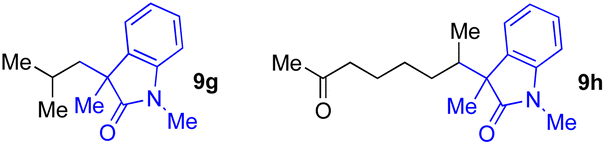
With the optimized conditions for the Fe(acac)3-catalyzed conjugate addition–cyclization sequence of 1a in hand, we subsequently examined the substrate scope of the Fe(acac)3-catalyzed radical functionalization of various α-ketoalkylsilyl peroxides 1a–m as shown in Table 2. Thus, the Fe(acac)3-catalyzed reaction of 5–8-membered α-ketoalkylsilyl peroxides 1a–d with methacrylamide 7 furnished conjugate addition–cyclization products 8a–d in high-to-excellent yield (entries 1–4). In a similar manner, aromatic-substituted α-ketoalkylsilyl peroxide 1e reacted with methacrylamide 7 to give product 8e in good yield (entry 5). Acyclic α-ketoalkylsilyl peroxide 1f also worked well (entry 6), but decarbonylation of the intermediary acyl radical took place in the case of the more-substituted substrate 1g to furnish a mixture of 8g and decarbonylated 9g in 20% and 15% yields, respectively (entry 7). Longer reaction time (24 h) enhanced the product yield of 8g (entry 8). Similarly, facile decarbonylation was observed with more-substituted substrate 1h (entries 9 and 10). Furthermore, the separate treatment of the diastereomers 1i and 1j of L-menthone-derived α-ketoalkylsilyl peroxides with methacrylamide 7 afforded the same coupling product 8i in high yield (entries 11 and 12). Ethyl-substituted 1k afforded the corresponding ethyl ketone 8k in good yield (entry 13), though phenyl-substituted 1m provided phenyl ketone 8m in low yield (entry 14).
|
|
|||
|---|---|---|---|
| Entry | α-Ketoalkylsilyl peroxide 1 | Product 8 | Yieldb (%) |
| a Unless otherwise specified, the reactions were carried out in the presence of 1 (0.4 mmol), 7 (0.2 mmol), Fe(acac)3 catalyst (0.04 mmol), 1,10-phen ligand (0.04 mmol) in DMSO (1 mL) at 40 °C for 4 h. b Isolated yield. c The yield of decarbonylation product 9g or 9h. d For 24 h. e For 12 h. f At 80 °C for 16 h. | |||
|
|
||
| 1 | 1b (n = 1) | 8b | 77 |
| 2 | 1a (n = 2) | 8a | 95 |
| 3 | 1c (n = 3) | 8c | 90 |
| 4 | 1d (n = 4) | 8d | 84 |
| 5 |
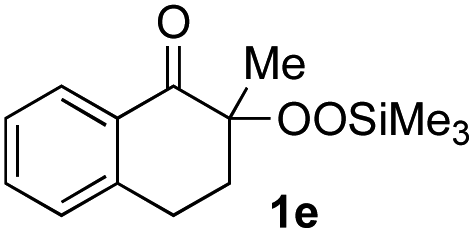
|
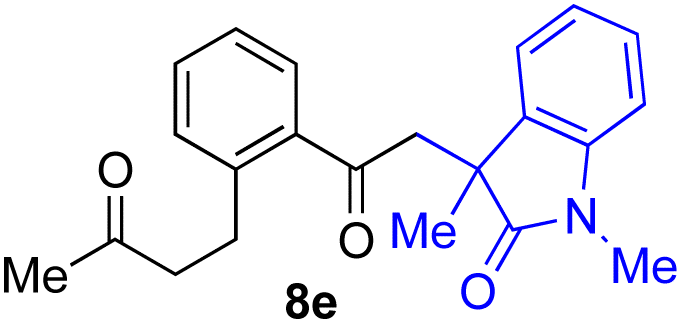
|
65 |

|

|
||
| 6 | 1f (R = H) | 8f | 89 |
| 7 | 1g (R = Me) | 8g | 20 (15)c |
| 8 | 1g (R = Me) | 8g | 57d (40)c,d |
| 9 |
|

|
18 (0)c |
| 10 | 37e (43)c,e | ||
| 11 |
|
|
46 (99)e |
| 12 |

|
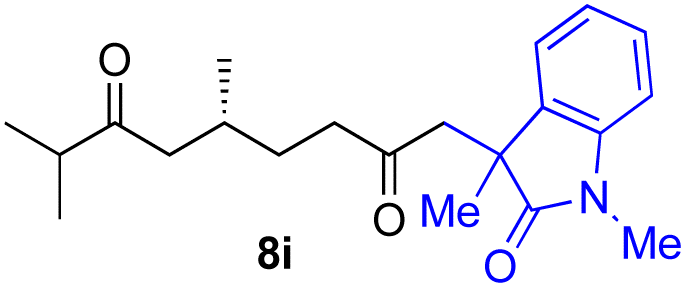
|
52 (86)e |
| 13 |
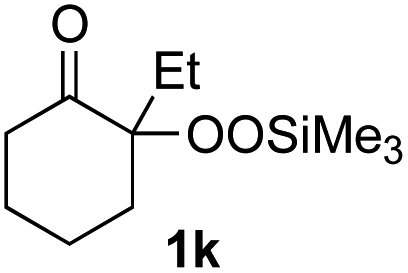
|
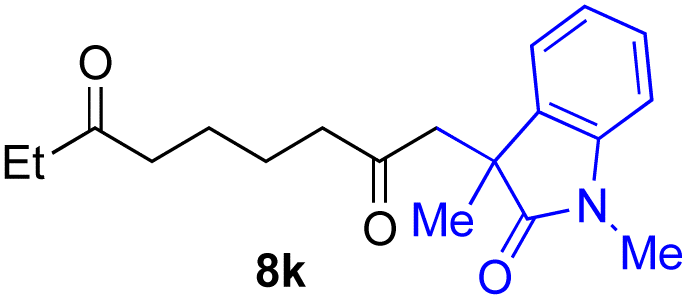
|
73 |
| 14 |
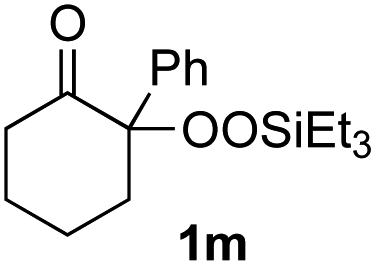
|
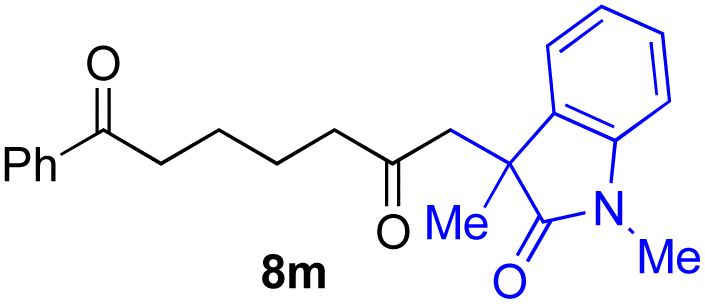
|
0 (27)f |
|
|||
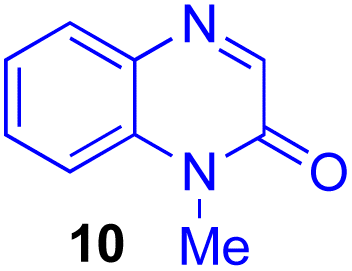
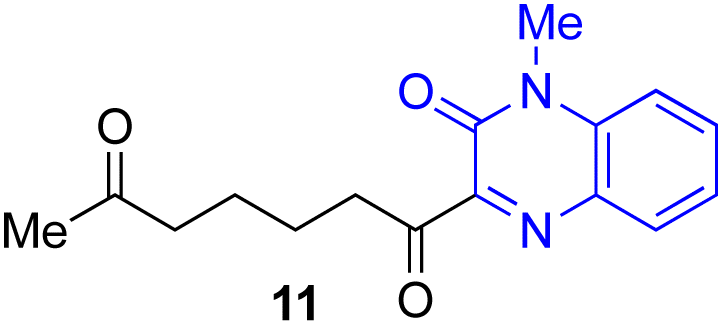
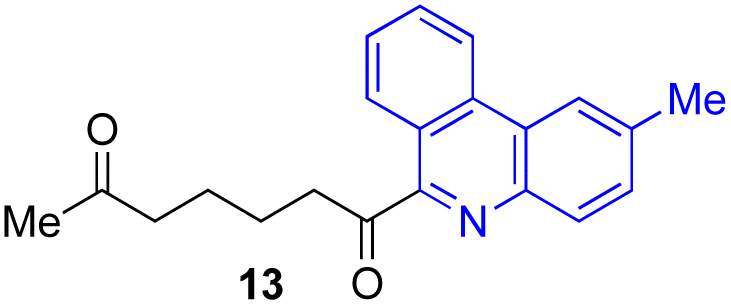
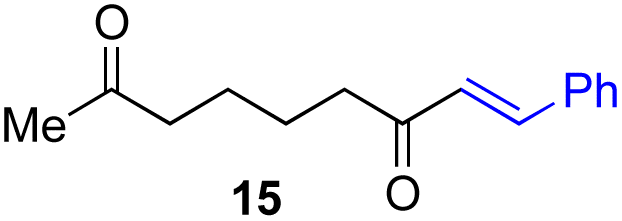
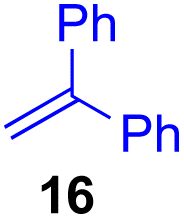
Next, we examined the substrate scope of various coupling partners (F–H) with α-ketoalkylsilyl peroxide 1a, as shown in Table 3. The Fe(acac)3-catalyzed reaction of α-ketoalkylsilyl peroxide 1a with 1-methylquinoxalin-2(1H)-one 10 furnished addition–rearomatization product 11 in high yield (entry 1). Although treatment of 1a with 2-isocyano-5-methyl-1,1′-biphenyl 12 gave addition–cyclization product 13 in very low yield under the standard conditions, the use of DMF in place of DMSO afforded 13 in high yield (entry 2). In a similar manner, while the initial reaction of 1a with cinnamic acid 14 provided the decarboxylated coupling product 15 in low yield, the use of excess 14 without the ligand 1,10-phen under otherwise similar conditions afforded 15 in good yield (entry 3). The reaction of 1a with 1,1-diphenylethylene 16 under the standard condition also gave poor results, but the FeSO4·7H2O-catalyzed reaction in DMF at 80 °C with excess 16 (3.0 equiv.) afforded 17 in good yield (entry 4). Furthermore, treatment of 1a with diethyl 2-benzylidenemalonate 18 produced the conjugate addition product 19 in moderate yield (entry 5).
|
|
|||
|---|---|---|---|
| Entry | Coupling partner (F–H) | Product | Yieldb (%) |
| a Unless otherwise specified, the reactions were carried out in the presence of 1a (0.4 mmol), coupling partner (0.2 mmol), Fe(acac)3 (0.04 mmol), 1,10-phen (0.04 mmol) in DMSO (1 mL) at 40 °C for 4 h under argon atmosphere. b Isolated yield. c In DMF. d 1,10-Phen ligand was not used. e Use of 14 (5 equiv.) at 80 °C for 24 h. f NMR yield using nitromethane as an internal standard. g FeSO4·7H2O was used instead of Fe(acac)3. h In DMF at 80 °C. i Use of 1a (0.2 mmol) and 16 (3.0 equiv.). j Use of 1a (0.2 mmol) and 18 (2.0 equiv.). | |||
| 1 |
|
|
91 |
| 2 |

|
|
9 (78)c |
| 3 |
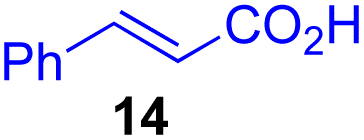
|
|
19 (56)c,d,e |
| 4 |
|

|
<5f (66)d,g,h,i |
| 5 |

|

|
0 (43)d,g,h,j |
| 6 |
|

|
<5h (34)d,g,h |
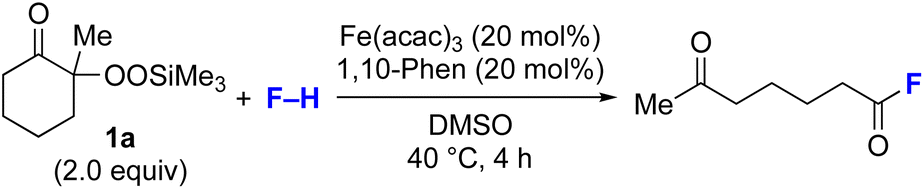
By taking advantage of the generation of reactive acyl chloride intermediate 22 in a practical manner, several synthetically useful transformations were accomplished in a highly efficient manner, as shown in Fig. 2. Thus, treatment of the intermediary 6-oxoheptanoyl chloride (22) with BnNH2/NEt3 or BnOH/DMAP afforded the corresponding amide 23 and ester 24, respectively, in excellent yields. Even a one-mmol-scale experiment using 1a afforded 23 in 84% yield. Friedel–Crafts acylation of 22 afforded the desired phenyl ketone 25 in moderate yield. These results demonstrate that our strategy is highly versatile due to the high synthetic utility of the acyl chloride intermediates 22. A control experiment for the conjugate addition–cyclization reaction with 7 was carried out to obtain insight into the reaction mechanism: the results supported the hypothesized generation of acyl radical intermediates. Specifically, when the reaction of α-ketoalkylsilyl peroxide 1a and methyacrylamide 7 with 20 mol% each of Fe(acac)3 and 1,10-phen in DMSO was conducted at 40 °C for 24 h in the presence of a radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxy, TEMPO, the conjugate addition–cyclization reaction was significantly inhibited, and the acyl radical/TEMPO adduct 26 was obtained in 40% NMR yield (Fig. 3). This observation provides evidence that the in situ-generated acyl radical is most likely involved in this sequential transformation.
 | ||
| Fig. 2 Synthetic transformations of acyl chloride 22 derived from 1a. aGeneration of 22 with Me3SiCl (3.0 equiv.), Fe(acac)3 (1 mol%), 1,10-phen (1 mol%) in CH2Cl2 at 40 °C for 1 h. | ||
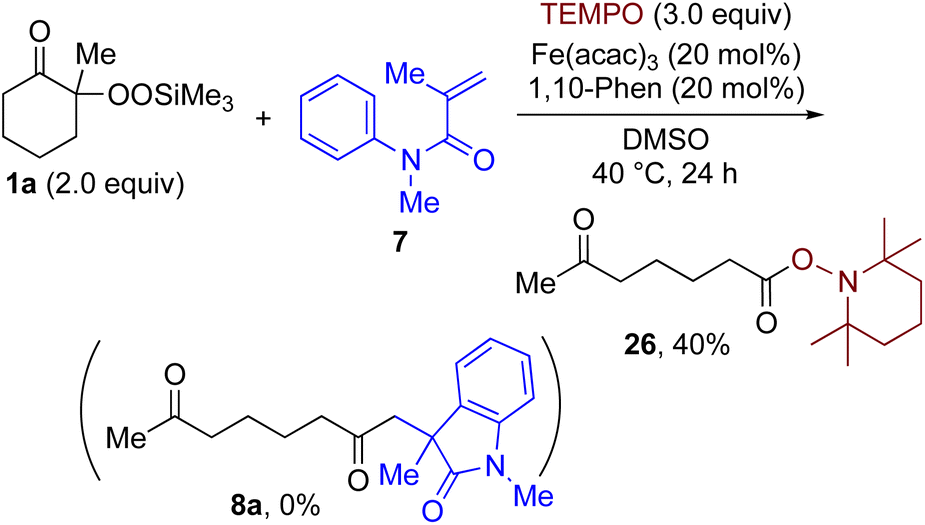 | ||
| Fig. 3 Radical trapping experiment of 1a with TEMPO. | ||
Based on our experimental results, a plausible reaction mechanism has been proposed for the Fe(acac)3-catalyzed conjugate addition–cyclization sequence of methacrylamide 9 with 1a (Fig. 4). The use of Fe(II) salts such as FeCl2, Fe(acac)2, and FeSO4·7H2O provided good to better results in the radical cleavage reaction of 1a (entries 4 and 5 in Table 1; entries 4–6 in Table 3).16 Thus, 1,10-phen-coordinated Fe(II) species would cleave the O–O bond of 1avia single-electron transfer (SET) process, leading to alkoxy radical 27 and trimethylsilanoxide. Oxy radical 27 then easily undergoes β-scission to generate the functionalized acyl radical 28. This acyl radical 28 subsequently reacts with 7 to afford the intermediary carbon radical 29, which further adds to the benzene ring to furnish the radical intermediate 30. This radical is then oxidized by the Fe(III) catalyst to give the corresponding carbocation species 31, which is deprotonated by trimethylsilanoxide to afford the final product 8a.
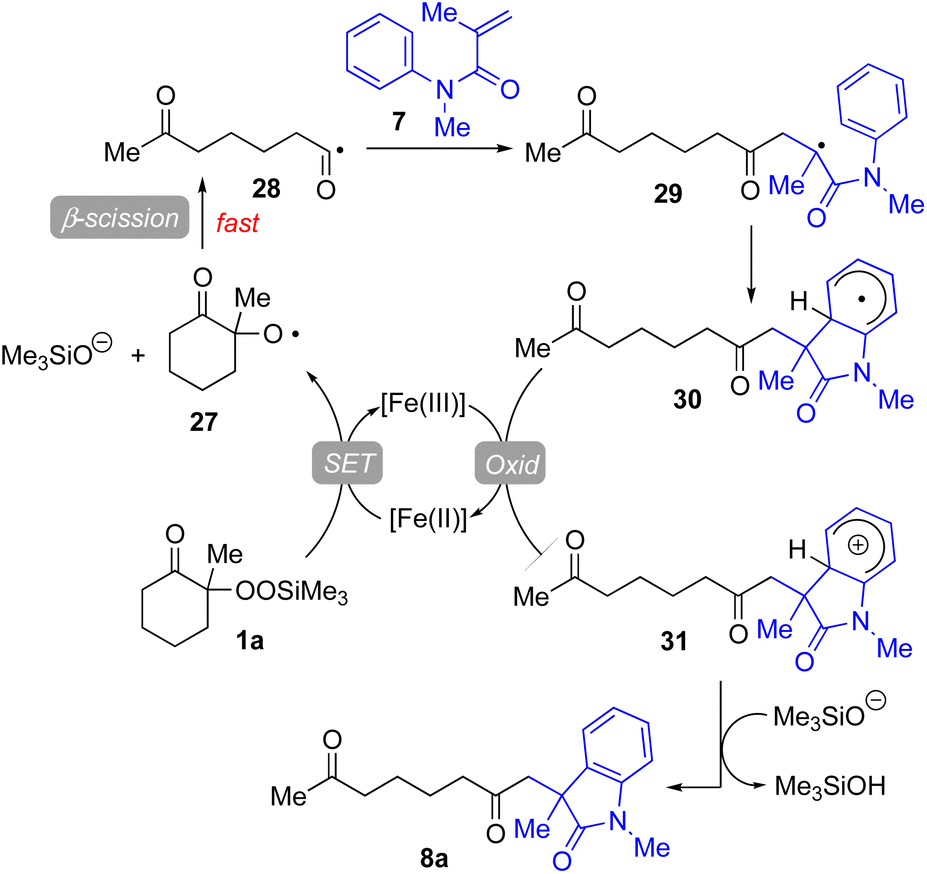 | ||
| Fig. 4 Proposed reaction mechanism for Fe-catalyzed reaction of 1a with 7. | ||
Attempted reactions of β-acetoxyalkylsilyl peroxides 32 with various coupling partners such as 7, 10, 12, 16 and 20 resulted in producing none or very low yields of desired coupling products. In contrast, the choice of Me3SiN3 as coupling partner gave the corresponding coupling product 33 in 85% yield (Fig. 5). In addition, Fe-catalyzed reactions of β-amidoalkylsilyl peroxides 34 with various coupling partners afforded none of desired coupling products. However, the Cu-catalyzed reaction of 34 with Me3SiCN as coupling partner gave the corresponding coupling product 35 in 88% yield.
 | ||
| Fig. 5 Metal-catalyzed reactions of β-acetoxy, and β-amidoalkylsilyl peroxides. | ||
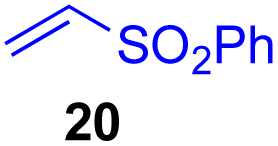
This approach is also applicable to our recently developed visible-light-promoted alkylation of electron-deficient alkenes with alkylsilyl peroxides.18 Treatment of α-ketoalkylsilyl peroxide 1a with phenyl vinyl sulfone (20) and an equimolar amount of Hantzsch ester in DMSO under blue light irradiation at room temperature for 3 h afforded the desired conjugate addition product 21 in 93% yield (Fig. 6). This approach can be further expanded to the visible-light-promoted alkylation of other functionalized alkylsilyl peroxides. For example, the reaction of β-acetoxy- and β-amidoalkylsilyl peroxides 32 and 34 (1.5 equiv.) with phenyl vinyl sulfone (20) and Hantzsch ester (1.5 equiv.) in DMSO under blue light irradiation at room temperature for 4–8 h gave rise to conjugate adducts 36 and 37, respectively, in 87% and 64% yields (Fig. 6).19
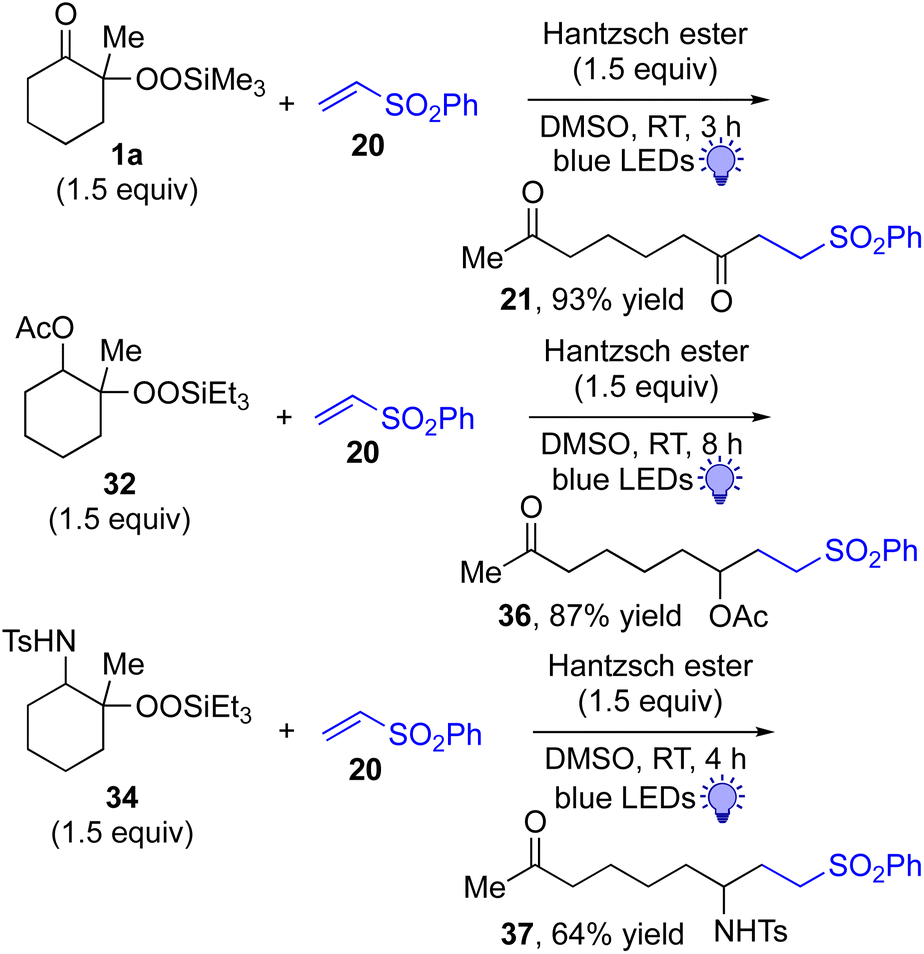 | ||
| Fig. 6 Visible light-promoted reaction of α-keto, β-acetoxy, and β-amidoalkylsilyl peroxides. | ||
Conclusions
In summary, we have developed an Fe-catalyzed and visible-light-promoted radical transformations for functionalized alkylsilyl peroxides, such as α-keto-, β-acetoxy-, and β-amidoalkylsilyl peroxides with several coupling partners under mild conditions and with a broad substrate scope. The synthetic utility of our approach is demonstrated by the facile generation of reactive acyl chloride intermediates, which can be easily transformed to the corresponding amides, esters, and phenyl ketones. A mechanistic study suggested the participation of intermediary acyl, α-acetoxyalkyl, and α-amidoalkyl radical species in the radical-promoted coupling reactions.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
K. M. conceptualized the research. J. L. and S. L. performed the experiments. T. K. and K. M. prepared the manuscript and the ESI.† Z. W. and Y. L. edited the ESI.† K. M. supervised the project and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21977019, 22101053, 22050410279, 22250710134) and JSPS KAKENHI Grant JP21H05026 and JP23H04910.Notes and references
- Selected reviews; (a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (b) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed; (c) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (d) Y. Sumida and H. Ohmiya, Chem. Soc. Rev., 2021, 50, 6320–6332 RSC; (e) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC.
- (a) M. Murakami and N. Ishida, Chem. Lett., 2017, 46, 1692–1700 CrossRef CAS; (b) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC; (c) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- Selected examples: (a) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef CAS PubMed; (b) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514–1517 CrossRef CAS PubMed; (c) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed; (d) A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed; (e) Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed; (f) Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS PubMed; (g) T. Xue, Z. Zhang and R. Zeng, Org. Lett., 2022, 24, 977–982 CrossRef CAS PubMed; (h) Q. An, Y.-Y. Xing, R. Pu, M. Jia, Y. Chen, A. Hu, S.-Q. Zhang, N. Yu, J. Du, Y. Zhang, J. Chen, W. Liu, X. Hong and Z. Zuo, J. Am. Chem. Soc., 2023, 145, 359–376 CrossRef CAS PubMed.
- A. Matsumoto and K. Maruoka, Bull. Chem. Soc. Jpn., 2021, 94, 513–524 CrossRef CAS.
- (a) S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Tetrahedron, 2019, 75, 172–179 CrossRef CAS; (b) W. Xu, Y. Liu, T. Kato and K. Maruoka, Org. Lett., 2021, 23, 1809–1813 CrossRef CAS PubMed.
- R. Sakamoto, T. Kato, S. Sakurai and K. Maruoka, Org. Lett., 2018, 20, 1400–1403 CrossRef CAS PubMed.
- T. Seihara, S. Sakurai, T. Kato, R. Sakamoto and K. Maruoka, Org. Lett., 2019, 21, 2477–2481 CrossRef CAS PubMed.
- S. Sakurai, T. Kano and K. Maruoka, Chem. Commun., 2021, 57, 81–84 RSC.
- S. Tsuzuki, S. Sakurai, A. Matsumoto, T. Kano and K. Maruoka, Chem. Commun., 2021, 57, 7942–7945 RSC.
- Recent reports: (a) W. Xu, T. Kato, Y. Liu, A. Matsumoto and K. Maruoka, Org. Lett., 2022, 24, 2641–2645 CrossRef CAS PubMed; (b) H. Lu, C. Zhou, Z. Wang, T. Kato, Y. Liu and K. Maruoka, J. Org. Chem., 2022, 87, 8824–8834 CrossRef CAS PubMed; (c) M. Zhou, H. Lu, Z. Wang, T. Kato, Y. Liu and K. Maruoka, Tetrahedron Lett., 2022, 110, 154176 CrossRef CAS.
- Recent reports by other groups: (a) J. Wei, Y. Tang, Q. Yang, H. Li, D. He and Y. Cai, Org. Lett., 2022, 24, 7928–7933 CrossRef CAS PubMed; (b) P.-Z. Wang, Y.-J. Liang, X. Wu, W. Guan, W.-J. Xiao and J.-R. Chen, ACS Catal., 2022, 12, 10925–10937 CrossRef CAS; (c) C. Liu, J. Wang, X. Liu, J. Feng and D. Du, Chem. Commun., 2023, 59, 13175–13178 RSC; (d) X. Tian, L. Chen, T. Zhu and J. Wu, Org. Chem. Front., 2023, 10, 4821–4826 RSC.
- Reviews on acyl radicals: (a) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed; (b) A. Banerjee, Z. Lei and M.-Y. Ngai, Synthesis, 2019, 51, 303–333 CrossRef CAS PubMed; (c) Y.-L. Liu, Y.-J. Ouyang, H. Zheng, H. Liu and W.-T. Wei, Chem. Commun., 2021, 57, 6111–6120 RSC; (d) H. Zhang, S. Liang, D. Wei, K. Xu and C. Zeng, Eur. J. Org Chem., 2022, 2022, e202200794 CrossRef CAS.
- Recent examples on the reactions using acyl radicals: (a) M. D. Vu, M. Das and X.-W. Liu, Chem.–Eur. J., 2017, 23, 15899–15902 CrossRef CAS PubMed; (b) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Chem.–Eur. J., 2018, 24, 1726–1731 CrossRef CAS PubMed; (c) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed; (d) S. Paul and J. Guin, Chem.–Eur. J., 2021, 27, 4412–4419 CrossRef CAS PubMed; (e) Y. Wang, X. Meng, C. Cai, L. Wang and H. Gong, J. Org. Chem., 2022, 87, 15042–15049 CrossRef CAS PubMed; (f) Z.-T. Luo, J.-H. Fan, B.-Q. Xiong, Y. Liu, K.-W. Tang and P.-F. Huang, Eur. J. Org Chem., 2022, 2022, e202200793 CrossRef CAS; (g) S. A. Paveliev, O. O. Segida, O. M. Mulina, I. B. Krylov and A. O. Terent'ev, Org. Lett., 2022, 24, 8942–8947 CrossRef CAS PubMed; (h) A. Chinchole, M. A. Henriquez, D. Cortes-Arriagada, A. R. Cabrera and O. Reiser, ACS Catal., 2022, 12, 13549–13554 CrossRef CAS; (i) Z.-L. Yu, Y.-F. Cheng, J.-R. Liu, W. Yang, D.-T. Xu, Y. Tian, J.-Q. Bian, Z.-L. Li, L.-W. Fan, C. Luan, A. Gao, Q.-S. Gu and X.-Y. Liu, J. Am. Chem. Soc., 2023, 145, 6535–6545 CrossRef CAS PubMed; (j) N. Guo, Y. Luo, L. Feng, Z. Liu, W. Cao and X. Feng, Asian J. Org. Chem., 2023, 12, e202300164 CrossRef CAS.
- Recent examples on the reactions with α-alkoxyalkyl radicals: (a) L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed; (b) X.- Zi Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef PubMed; (c) E. Voutyritsa, M. Garreau, M. G. Kokotou, I. Triandafillidi, J. Waser and C. G. Kokotos, Chem.–Eur. J., 2020, 26, 14453–14460 CrossRef CAS PubMed; (d) G. N. Papadopoulos, M. G. Kokotou, N. Spiliopoulou, N. F. Nikitas, E. Voutyritsa, D. I. Tzaras, N. Kaplaneris and C. G. Kokotos, ChemSusChem, 2020, 13, 5934–5944 CrossRef CAS PubMed; (e) X. Chen, X. Gong, Z. Li, G. Zhou, Z. Zhu, W. Zhang, S. Liu and X. Shen, Nat. Commun., 2020, 11, 2756 CrossRef CAS PubMed; (f) J.-T. Yu, Y. Li, R. Chen, Z. Yang and C. Pan, Org. Biomol. Chem., 2021, 19, 4520–4528 RSC; (g) H. Ji, D. Lin, L. Tai, X. Li, Y. Shi, Q. Han and L.-A. Chen, J. Am. Chem. Soc., 2022, 144, 23019–23029 CrossRef CAS PubMed; (h) N. Ahmed, R. J. Spears, T. D. Sheppard and V. Chudasama, Chem. Sci., 2022, 13, 8626–8633 RSC; (i) A. Wang, Y.-Y. Yin, Rukhsana, L.-Q. Wang, J.-H. Jin and Y.-M. Shen, J. Org. Chem., 2023, 88, 13871–13882 CrossRef CAS PubMed.
- Recent examples on the reactions using α-amidoalkyl radicals: (a) J. Y. Kim, Y. S. Lee, Y. Choi and D. H. Ryu, ACS Catal., 2020, 10, 10585–10591 CrossRef CAS; (b) M. Li, L. Zheng, L. Ma and Y. Chen, J. Org. Chem., 2021, 86, 3989–3998 CrossRef CAS PubMed; (c) R. S. J. Proctor, P. Chuentragool, A. C. Colgan and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 4928–4934 CrossRef CAS PubMed; (d) S. M. Cho, J. Y. Kim, S. Han and D. H. Ryu, J. Org. Chem., 2022, 87, 11196–11203 CrossRef CAS PubMed; (e) J. Y. Hwang, S. H. Lee, Y. Kim, M. Jin, K. Kang and E. J. Kang, Org. Lett., 2023, 25, 7359–7363 CrossRef CAS PubMed.
- Y. Shiozaki, S. Sakurai, R. Sakamoto, A. Matsumoto and K. Maruoka, Chem.–Asian J., 2020, 15, 573–576 CrossRef CAS PubMed.
- Recent examples on radical cascade conjugate addition-cyclization reactions; (a) N. Wang, Q.-S. Gu, Z.-L. Li, Z. Li, Y.-L. Guo, Z. Guo and X.-Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 14225–14229 CrossRef CAS PubMed; (b) Z. Luo and G. C. Tsui, Org. Chem. Front., 2022, 9, 4969–4974 RSC; (c) M. Zhu, Y. Tian, J. Sha and W. Fu, ChemistrySelect, 2022, 7, e202203986 CrossRef CAS; (d) Q. Liu, Q. Nia, Y. Zhou, L. Chen, S. Xiang, L. Zheng and Y. Liu, Org. Biomol. Chem., 2023, 21, 7960–7967 RSC.
- S. Nagano, N. Maeda, T. Kato, A. Matsumoto and K. Maruoka, Tetrahedron Lett., 2023, 122, 154486 CrossRef CAS.
- Attempted reaction of β-acetoxyalkylsilyl peroxides 32 and phenyl vinyl sulfone (20) with 20 mol% of FeSO4·7H2O in DMF at 40 °C for 4 h resulted in none of the desired conjugate adduct.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06857a |
| This journal is © The Royal Society of Chemistry 2024 |