
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioisostere-conjugated fluorescent probes for live-cell protein imaging without non-specific organelle accumulation†
Takuya
Kamikawa
 a,
Akari
Hashimoto
b,
Nozomi
Yamazaki
b,
Junya
Adachi
c,
Ayami
Matsushima
c,
Kazuya
Kikuchi
*bd and
Yuichiro
Hori
*c
a,
Akari
Hashimoto
b,
Nozomi
Yamazaki
b,
Junya
Adachi
c,
Ayami
Matsushima
c,
Kazuya
Kikuchi
*bd and
Yuichiro
Hori
*c
aGraduate School of Science, Kyushu University, 744 Motooka, Nishi, Fukuoka 819-0395, Japan
bGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
cFaculty of Science, Kyushu University, Fukuoka, Fukuoka 819-0395, Japan. E-mail: hori@chem.kyushu-univ.jp
dImmunology Frontier Research Center, Osaka University, Suita, Osaka 565-0871, Japan
First published on 1st May 2024
Abstract
Specific labeling of proteins using membrane-permeable fluorescent probes is a powerful technique for bioimaging. Cationic fluorescent dyes with high fluorescence quantum yield, photostability, and water solubility provide highly useful scaffolds for protein-labeling probes. However, cationic probes generally show undesired accumulation in organelles, which causes a false-positive signal in localization analysis. Herein, we report a design strategy for probes that suppress undesired organelle accumulation using a bioisostere for intracellular protein imaging in living cells. Our design allows the protein labeling probes to possess both membrane permeability and suppress non-specific accumulation and has been shown to use several protein labeling systems, such as PYP-tag and Halo tag systems. We further developed a fluorogenic PYP-tag labeling probe for intracellular proteins and used it to visualize multiple localizations of target proteins in the intracellular system. Our strategy offers a versatile design for undesired accumulation-suppressed probes with cationic dye scaffolds and provides a valuable tool for intracellular protein imaging.
Introduction
Selective protein labeling is an important technique for investigating the functions of proteins in a wide range of biological research areas.1–4 Fluorescent species are used for labeling to track the dynamics of proteins in live cells because fluorescent signals can be monitored in real-time through microscopic observation. Although fluorescent proteins can be fused with proteins of interest (POIs),5,6 a method has also been developed to fuse protein tags and use them in combination with synthetic fluorescent probes. This protein labeling technique has advantages in the availability of photostable fluorophores and the capability of conditional labeling at specific time points. In this approach, synthetic fluorescent probes containing various fluorescent dyes are designed to specifically bind to chemical tags, such as Halo tag,7,8 SNAP-tag,9 CLIP-tag,10 TMP-tag,11 FAST-tag,12 BL-tag13 and PYP-tag.14Cationic fluorescent dyes (e.g. rhodamine, cyanine, and pyridinium dye scaffolds) that show high fluorescence quantum yield, photobleaching resistance, and water solubility are highly useful components of fluorescent probes.15–18 In addition, some biocompatible cationic dyes are commercially available at a reasonable price.19 However, cationic dyes tend to be taken up by mitochondria due to the high negative potential of the inner membrane,20 generating non-specific fluorescent signals that cause serious artifacts for live cell imaging of non-mitochondrial proteins. This undesirable behavior limits the application of cationic fluorescent probes with protein tags.
The introduction of an anionic moiety to bring the net charge of the cationic fluorescent probe to zero is thought to suppress mitochondrial accumulation. In addition to suppressing the accumulation, in terms of the bioavailability, zwitterionic probes are also advantageous due to their good water solubility. Indeed, sulfonated zwitterionic probes based on cyanine or pyridinium dye scaffolds were known to work in cells, tissue samples, and in vivo system.21,22 However, the introduction of anionic groups, such as carboxylate, sulfonate, and phosphonate groups, into the probes mostly causes loss of membrane permeability of the probes.23–27 To keep permeability, there are reports of the development of membrane-permeable cationic probes with precursors of the anionic group, which are hydrolyzed by endogenous esterases in living cells.28–34 However, this approach has the problem of mitochondrial localization owing to the cationic and lipophilic nature of the fluorescent scaffold.28,33,34
A limited number of zwitterionic protein-labeling probes without modifying cationic fluorescent scaffolds, namely rhodamine dye scaffolds linked with a sulfonated alkyl chain, are available to visualize intracellular proteins.35 While these probes do not face the previously mentioned problems regarding the intracellular behavior of cationic probes, this method has several other limitations. First, a complicated washing step is required to visualize the localization of POIs, because fluorescent always-on probes remain as a background signal before washing. In this point, the development of fluorogenic probes is desired for tracking the movements of POIs in real-time since fluorogenic probes exhibit only low background signals from unbound states.36–38 Second, the molecular design of protein labeling probes utilizing sulfonate groups has been limited to the application of tetramethylrhodamine (TMR) and siliconrhodamine (SiR).33 Despite their favorable optical properties, the advancement of novel probes with these derivatives is hindered due to their extremely high cost from commercial sources (typically >$10,000 per g)20 and the requirement for more than seven steps from commercial sources in synthesis of SiR scaffolds.35,39 Thus, novel approaches on probe design using cationic dyes are required for intracellular protein imaging.
To address these limitations, we established a novel design for protein-labeled fluorescent probes based on cationic fluorescent scaffolds (Fig. 1). We considered that the loss of membrane permeability by introducing an anionic moiety was caused by the low hydrophobicity of the carboxylic or sulfonate groups and anticipated that a relatively hydrophobic anionic group would be preferable. In this study, we focused on bioisosteres of carboxylic acids. Bioisosteres are structural mimics with tunable physical and chemical properties of molecules or functional groups that exhibit similar or improved biological activity. In addition, bioisosteres have been broadly employed in medicinal chemistry as bioisosteric replacements for bioactive compounds. This approach has been used to extend the structure–activity relationship and enhance activity, selectivity, and biocompatibility.40,41 In particular, hydrophobic bioisosteres have been applied to improve membrane permeability in various bioactive compounds.42,43 However, a design strategy based on hydrophobic bioisosteres has not been reported for the improvement of membrane permeability in fluorescent probes. We believe that replacing anionic groups with hydrophobic bioisosteres is a promising strategy for developing intracellular protein-labeling probes.
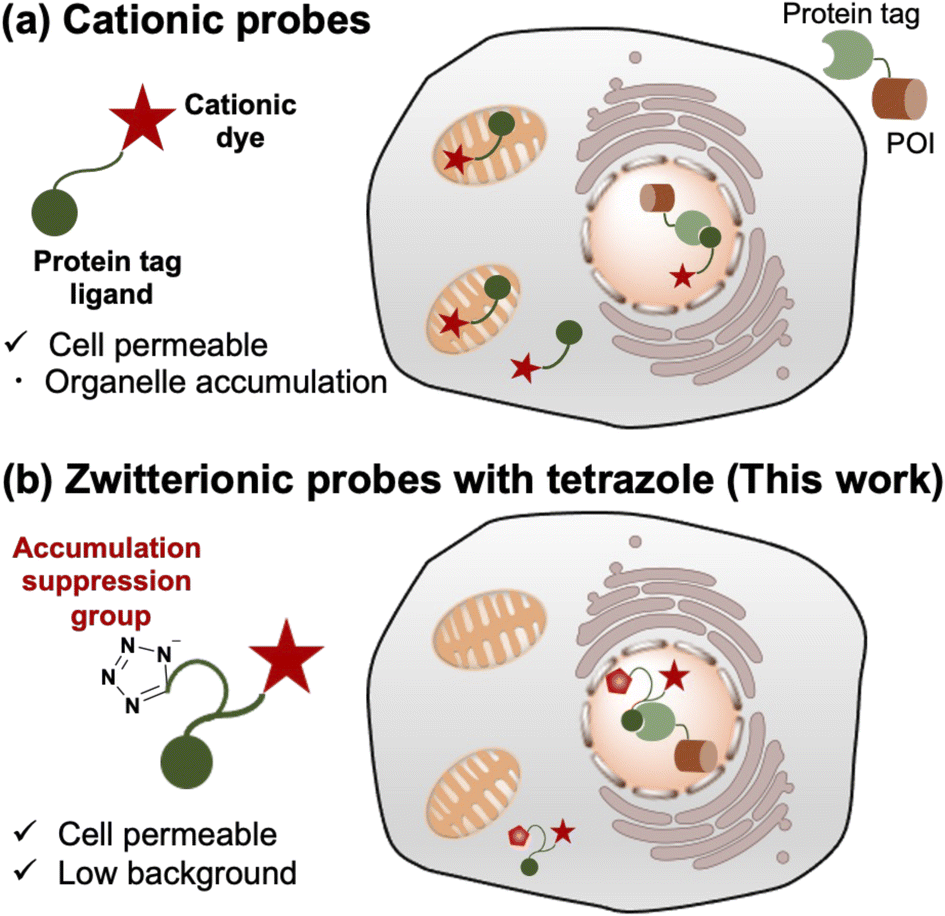 | ||
| Fig. 1 Schematic illustration of probe design strategy for intracellular protein imaging using various cationic scaffolds and protein tag labeling system. (a) Cationic probes for protein imaging are cell permeable but show organelle accumulation. (b) Zwitterionic probes with tetrazole as an organelle accumulation suppression group show cell permeability and low background. POI denotes protein of interest. | ||
Based on this discussion, we selected a tetrazole group, known as a bioisostere of carboxylic acid, as the anionic moiety to satisfy sufficient hydrophobicity.43 As a result, we demonstrated that fluorescent probes containing cationic dye scaffolds, rhodamine B tertiary amides (RB), and styrylpyridinium (SP) derivatives bearing tetrazole can function in living cells for the visualization of intracellular proteins without undesired mitochondrial accumulation. In particular, fluorogenic SP-based probes enable protein visualization without washing. All probes with tetrazole were easily prepared by concise conjugation using an amino acid derivative without modifying the cationic dye scaffolds. In addition, an amino acid derivative of tetrazole can be synthesized in one step using a commercial source. In the preparation of cationic dye scaffolds in this study, SP and RB dye scaffolds were synthesized in three steps from commercial sources (<$2 per g).20,44 We expect that our design will provide a versatile strategy for fluorescence imaging using protein-tag systems.
Results and discussion
Molecular design of protein labeling probes
To investigate the effect of anion introduction on the localization of free and bound probes, we focused on RB as a cationic dye scaffold because RB always shows fluorescence; RB is one of the commonly used dyes for cell imaging and exhibits strong fluorescence.20 We designed two RB-based probes consisting of PCAF as PYP-tag ligand:45PCAF-RB and PCAF-RBis (Fig. 2a). PCAF-RB was a cationic probe (+1) that contains RB linked to PCAF through the PEG5 linker. PCAF-RBis was a neutral probe (0) formed by the introduction of a tetrazole ring between the PEG linker and PCAF. The ligands form a covalent bond with Cys69 in the binding pocket of PYPWT, a small (125 amino acids) water-soluble bacterial protein.46,47 PCAF has previously been developed as a ligand that allows highly efficient and rapid labeling.45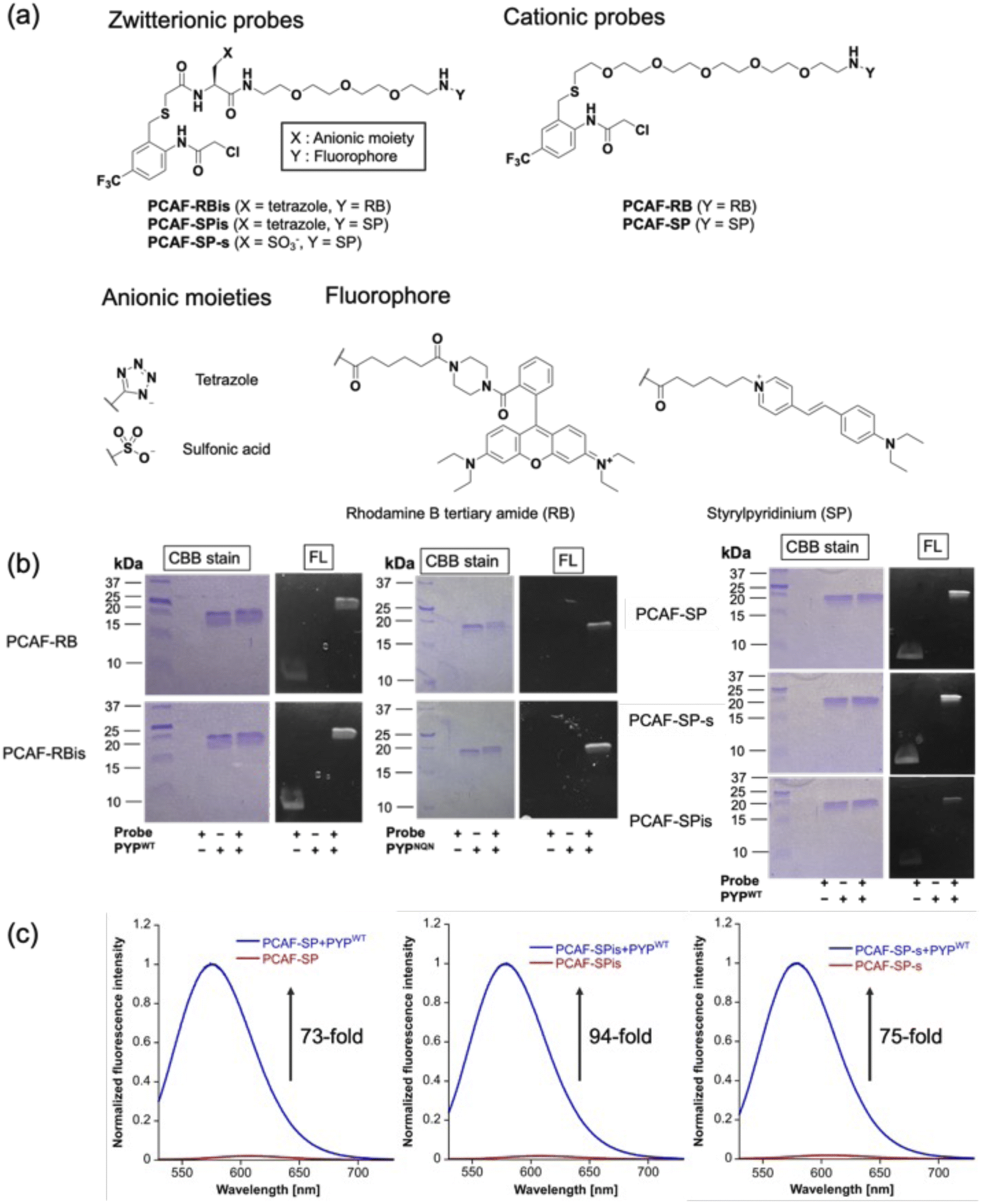 | ||
| Fig. 2 (a) Molecular structures of PYP labeling probes (PCAF-RB, PCAF-RBis, PCAF-SP, PCAF-SPis, and PCAF-SP-s). (b and c) The result of CBB staining and fluorescence images of the SDS-PAGE analysis. (b) PYPWT (2.0 μM) and PYPNQN (2.0 μM) incubated with PCAF-RB (1.0 μM) or PCAF-RBis (1.0 μM) for 30 min. PYPWT (2.0 μM) and PYPNQN (2.0 μM) incubated with PCAF-SP (1.0 μM), PCAF-SPis (1.0 μM), or PCAF-SP-s (1.0 μM) for 20 min. The SDS analysis images were obtained with the excitation at 470 nm. CBB and FL denote Coomassie brilliant blue staining and fluorescence images, respectively. Experiments were performed twice with similar results. (c) Normalized fluorescence spectra of PYP-tag labeling probes (PCAF-SP, PCAF-SPis, and PCAF-SP-s) reacted with/without PYPWT. PCAF-SP, PCAF-SPis, and PCAF-SP-s (5.0 μM) were incubated with/without PYPWT (6.0 μM) for 30 min in the solution of 20 mM HEPES, 150 mM NaCl, and 0.1% DMSO buffered to pH 7.4 at 37 °C. All spectra were obtained with the excitation at 484 nm. | ||
To develop fluorogenic probes, we focused on SP as a cationic dye scaffold.48,49 While SP shows almost no fluorescence under physiological conditions due to the TICT process,44 the fluorescence intensity is increased due to the ICT process, which is induced by a low polar environment or restriction of intramolecular rotation.50–52 Based on these properties, SP has been applied to develop fluorogenic protein tag labeling probes, which show an increase in fluorescence intensity upon binding to the protein tag. Indeed, Halo tag probe (F1) based on SP was reported to increase 27-fold upon binding to the in vitro Halo tag protein system.44,52 However, F1 has important limitations in live-cell imaging, namely undesired mitochondrial accumulation and the requirement for a washing step.52 Following the molecular designs of PCAF-RB and PCAF-RBis, two analogs, PCAF-SP and PCAF-SPis, were also prepared to study the effect of introducing anions (Fig. 2a). In addition, because the Gibbs group reported sulfonated zwitterionic probe designs for suppressing organelle accumulation of cationic dyes,30PCAF-SP-s were also prepared to evaluate the influence of the type of anionic moiety on membrane permeability (Fig. 2a). During the synthesis of the probes, the tetrazole moiety remained stable under the liquid-phase reaction conditions involving Boc or Fmoc deprotection, amino acid conjugation, and acylation reactions.
In vitro labeling reaction and photophysical properties
To examine whether the probes bind to the PYP-tag (PYPWT and PYPNQN), we conducted SDS-PAGE analysis followed by in-gel fluorescence detection and Coomassie Brilliant Blue (CBB) staining after the labeling reactions of the PYP-tag. In PYPNQN, the three key negative surface residues (Asp71, Glu74, and Asp97) are replaced with the neutral amino acids (Asn, Gln, and Asn (NQN mutations)).53 A fluorescent band was observed at a position corresponding to PYP-tag, indicating that covalent bonds were formed between the probe and PYP-tag (Fig. 2b).The photophysical properties of the probes were also evaluated (Table 1). The extinction coefficients (ε) of PCAF-RBis and PCAF-SPis were higher than that of PCAF-RB and PCAF-SP, respectively, in aqueous buffer at physiological pH. In addition, we performed fluorescence analysis to reveal the brightness of the probes. PCAF-RB and PCAF-RBis emitted fluorescence in the free state (Table S1†). Fluorescence analysis revealed an increase in the quantum yield of each probe by labeling to PYPWT. In contrast, PCAF-SP, PCAF-SPis, and PCAF-SP-s exhibited almost no fluorescence in their free states under physiological conditions. Upon the addition of PYPWT, the fluorescence intensity of all SP probes substantially increased. In particular, a 94-fold increase was observed for PCAF-SPis (Fig. 2c). The increase of their quantum yield (Φfl) after binding to PYPWT was similar (Table 1, PCAF-SP: 0.01 → 0.27, PCAF-SPis: 0.01 → 0.28, PCAF-SP-s: 0.01 → 0.29). Based on these results, the SP-based probes showed a fluorogenic response upon binding to the PYP-tag. To evaluate the effect of the NQN mutations on the photophysical properties of the probes, we examined the brightness of the SP-based probes bound to PYPNQN. As a result, the brightness of PCAF-SPis and PCAF-SP were slightly decreased by the NQN mutations (PCAF-SPis: 1.1 × 104 → 0.87 × 104, PCAF-SP: 0.84 × 104 → 0.75 × 104). On the other hand, PCAF-SP-s labeled PYPNQN showed similar brightness with PYPWT. These results suggest that the interaction pattern between SP and protein surface was affected by the surface amino acids of PYP-tag and the anionic groups.
| Probe | ε [104 M−1 s−1] | Φ fl | Brightness [103 M−1 s−1] | k 2 [103 M−1 s−1] |
|---|---|---|---|---|
| a All the experiments were conducted in the solution of 20 mM HEPES, 150 mM NaCl, and 0.1% containing DMSO buffered to pH 7.4 at 37 °C. b ε is extinction coefficient at λabs. c All data were obtained in triplicate experiments. d Data obtained after labeling reactions of PYP-tag proteins with probes were complete. | ||||
| PCAF-RBis | 9.2 | 0.29 | 2.7 | — |
| PCAF-RBis + PYPWT | 10d | 0.61d | 6.1 | 3.4 (±0.5) |
| PCAF-RBis + PYPNQN | 11d | 0.70d | 7.7 | 7.0 (±0.7) |
| PCAF-RB | 7 | 0.28 | 2 | — |
| PCAF-RB + PYPWT | 7.2d | 0.54d | 3.9 | 6.3 (±0.7) |
| PCAF-RB + PYPNQN | 7.6d | 0.69d | 5.2 | 5.7 (±0.9) |
| PCAF-SPis | 3.5 | 0.01 | 0.035 | — |
| PCAF-SPis + PYPWT | 3.8d | 0.28d | 1.1 | 9.1 (±0.9) |
| PCAF-SPis + PYPNQN | 3.0d | 0.29d | 0.87 | 4.1 (±0.4) |
| PCAF-SP-s | 3.1 | 0.01 | 0.31 | — |
| PCAF-SP-s + PYPWT | 3.4d | 0.29d | 0.99 | 4.4 (±0.2) |
| PCAF-SP-s + PYPNQN | 3.4d | 0.30d | 1 | 3.8 (±0.2) |
| PCAF-SP | 3.1 | 0.01 | 0.031 | — |
| PCAF-SP + PYPWT | 3.1d | 0.27d | 0.84 | 5.9 (±0.4) |
| PCAF-SP + PYPNQN | 3.0d | 0.25d | 0.75 | 21 (±1.0) |
The second-order rate constant (k2) was also determined using fluorescence analysis in triplicate. k2 values of all probes prepared in this study were in the order of 103 M−1 s−1, which is consistent with our previous study on PCAF ligands.47 When focusing on the effect of the net charge of the probes, RB-based probes decreased the reaction rate by introducing tetrazole (k2 = 6.3 × 103 to 3.4 × 103 M−1 s−1). Furthermore, to improve the reaction rate of zwitterionic probes, we employed PYPNQN. In a previous report, compared to PYPWT, PYPNQN increased the reaction rate between the PYP-tag and its anionic probe.53
The rate constant of PCAF-RBis with PYPNQN (k2 = 7.0 × 103 M−1 s−1) was higher than that of PCAF-RBis with PYPWT (k2 = 3.4 × 103 M−1 s−1). In contrast, PCAF-RB binds to PYPNQN (k2 = 5.7 × 103 M−1 s−1) as fast as PYPWT (k2 = 6.3 × 103 M−1 s−1). As shown in Table 1, while SP-based probes showed a moderate acceleration of the reaction rate as a result of the introduction of the tetrazole group (k2 = 5.9 × 103 to 9.1 × 103 M−1 s−1), PCAF-SP and its sulfonate-conjugate showed a similar reaction rate with PYPWT (k2 = 5.9 × 103 for PCAF-SP to 4.4 × 103 M−1 s−1 for PCAF-SP-s). In addition, we examined the labeling kinetics of the SP-based probes with PYPWT and PYPNQN. While PCAF-RBis binds to PYPNQN more rapidly than PYPWT, the rate constant of PCAF-SPis with PYPNQN was decreased as a result of the mutations (PCAF-SPis: k2 = 9.1 × 103 M−1 s−1 to 4.1 × 103 M−1 s−1). These results indicate that the cationic dye scaffolds and surface amino acids of PYP-tag affected the labeling constant. On the other hand, the rate constant of PCAF-SP-s, PCAF-SPis, or PCAF-SP with PYPNQN was similar, decreased, or increased, respectively, compared to those of PYPWT (PCAF-SP-s: k2 = 4.4 × 103 M−1 s−1 to 3.8 × 103 M−1 s−1, PCAF-SPis: k2 = 9.1 × 103 M−1 s−1 to 4.1 × 103 M−1 s−1, PCAF-SP: k2 = 5.9 × 103 M−1 s−1 to 21 × 103 M−1 s−1). These results indicate that the structure of an anionic group and surface amino acids of PYP-tag affected the labeling kinetics. Based on the above observations, the interaction mode of SP-containing probes with the protein seems to be different from that of RB-based probes. It is possible that the mutations might lead to alteration of not only electrostatic interaction but also the protein surface structure that affects the interaction with the anionic group or the fluorophore moieties of SP-containing probes, causing the change in the labeling rate.
Live cell imaging of intracellular proteins
We investigated the membrane permeability and intracellular distribution of the RB-based probes (PCAF-RB and PCAF-RBis) in HEK293T cells (purchased from RIKEN BRC) expressing the PYP-tag. Based on the photophysical properties of these RB-based probes in vitro, we selected PYPNQN for live-cell imaging experiments. HEK293T cells were transfected with a gene construct encoding nuclear localization signal (NLS)-fused PYPNQN (PYPNQN-NLS) for expressing the PYP-tag in the nucleus.29 The cells were first incubated with PCAF-RBis for 30 min and then washed to remove free probes. As shown in Fig. 3a, the fluorescence signals were observed only in the nuclei. Mock cells (transfected with an empty vector) in the presence of PCAF-RBis exhibited no fluorescence under the same conditions compared to the cells expressing PYPNQN-NLS. These results revealed that PCAF-RBis can successfully visualize intracellular proteins in living cells without nonspecific illumination. In contrast, incubation with PCAF-RB resulted in fluorescence signals that were not consistent with the distribution of PYP-tag (Fig. 3b). The staining pattern of PCAF-RB was consistent with that of MitoTracker Green FM, indicating that PCAF-RB accumulated in the mitochondria (Fig. S3†). These results demonstrated that the tetrazole group introduced into the probe suppressed undesired mitochondrial accumulation.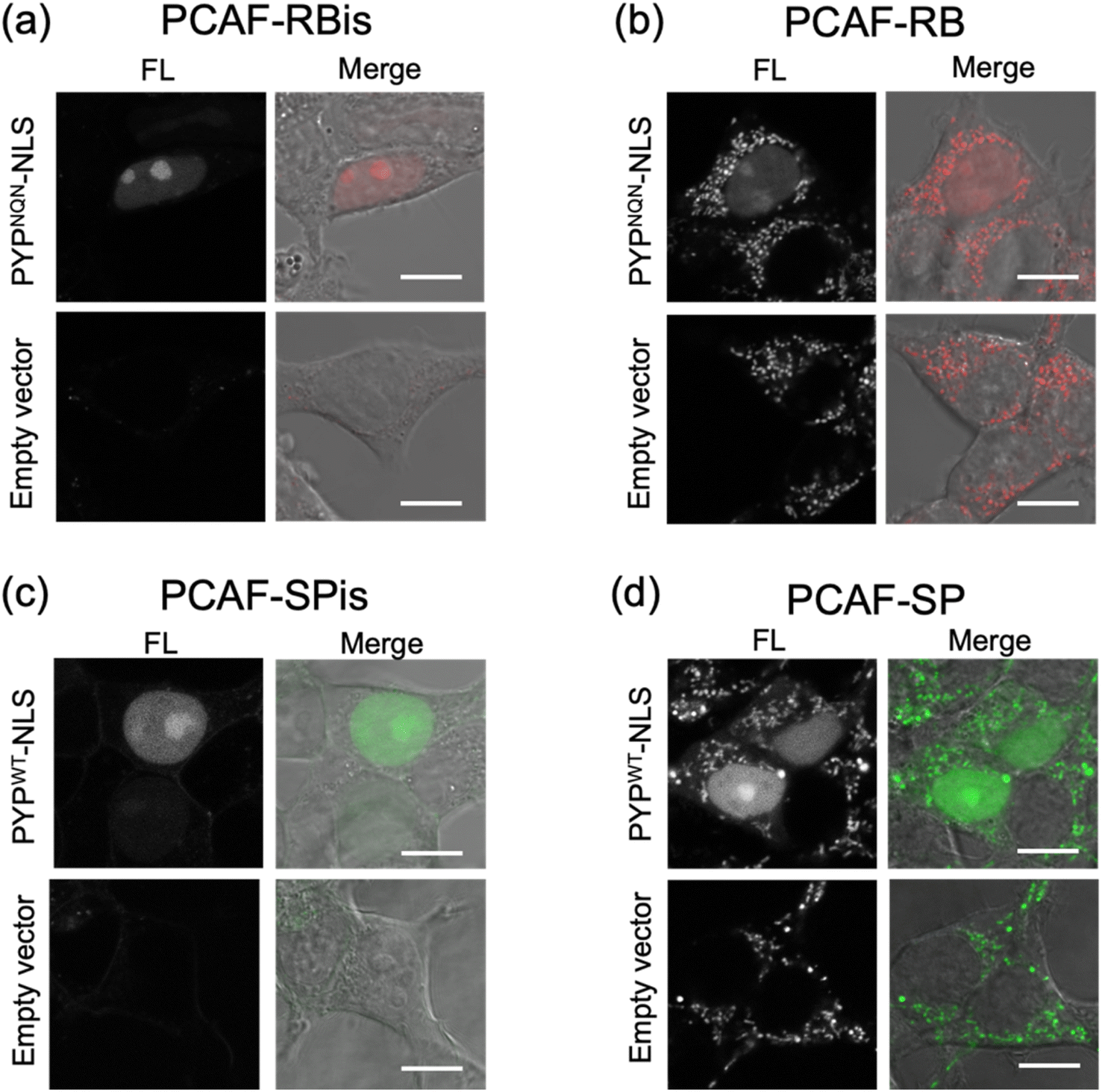 | ||
| Fig. 3 Fluorescence and phase contrast-merged images of HEK293T cells expressing HA-PYPNQN-NLS or mock cells (transfected with empty vector) stained with (a) PCAF-RBis (5.0 μM) and (b) PCAF-RB (5.0 μM). The images were measured at 566–685 nm emission with the excitation at 561 nm. (c) HA-PYPWT-NLS or mock cells stained with (c) PCAF-SPis (1.25 μM) or (d) PCAF-SP (1.25 μM). The images were measured at 550–650 nm emission with the excitation at 488 nm. Experiments were performed twice with similar results. Scale bars, 10 μm. FL denotes fluorescence image. | ||
In addition, to evaluate the increase in the fluorescence intensity of PCAF-SP and PCAF-SPis upon binding to the PYP-tag in the cellular system, we performed no-wash imaging experiments using these probes. HEK293T cells were transfected with a gene construct encoding NLS-fused PYPWT (PYPWT-NLS) to express a PYP-tag in the nucleus. As shown in Fig. 3c, the nuclei of PCAF-SPis-treated cells expressing PYPWT-NLS showed fluorescence signals similar to those of PCAF-RBis treated cells. In contrast, PCAF-SP showed non-specific illumination, which was not consistent with the distribution of PYP-tag (Fig. 3d). PCAF-RB showed higher brightness in the cytoplasmic signals than in the nuclear signals, whereas PCAF-SP showed similar brightness in the nucleus and cytoplasm. These results suggest that fluorescent probes with fluorogenicity can reduce non-specific illumination, which is caused by organelle accumulation, compared to always-on probes, although the fluorogenic property is not sufficient to completely suppress non-specific organelle illumination. In addition, we examined the labeling efficiencies of PCAF-SP and PCAF-SPis in live cells expressing HA-tag fused PYP-NLS. We modified a previously reported method,45 and used the method to estimate the labelling efficiencies. We performed pull-down and SDS-PAGE analyses, in which the labeling reaction induced band shift. Then, we determined the ratio of the band shift, giving labeling efficiencies of PCAF-SP and PCAF-SPis, which are approximately 10% and 20%, respectively (Fig. S4(b), ESI S1†).
We conducted dual-color imaging using PCAF-SP-s and PCAFred (ref. 45) in live cells co-expressing PYPWT-NLS and PYPWT-fused EGFR54 (expressing a PYP-tag on the cell surface) or mock cells (transfected with an empty vector) (Fig. S5†). In a previous study, PCAFred showed membrane permeability and functioned as an intracellular protein-labeling probe.45 In this study, the results showed that PCAF-SP-s selectively labeled PYP-tags on cell surfaces in live cells co-expressing PYPWT-NLS and PYPWT-fused EGFR (Fig. S5†). We then performed PYPWT-fused MBP55 (expressing PYP-tag in cytosol), PYPWT-NLS, and PYPWT-fused EGFR imaging experiments labeled with PCAF-SP-s under identical conditions (Fig. S6†). These results indicated that PCAF-SP-s is suitable for labeling membrane proteins (Fig. S5 and S6†).
Overall, PCAF-RBis and PCAF-SPis (tetrazole-conjugated zwitterionic probes) were used as membrane-permeable probes to visualize intracellular proteins without nonspecific illumination, whereas PCAF-RB and PCAF-SP (cationic probes) showed nonspecific illumination owing to organelle accumulation. Importantly, the SP-based probe images were obtained without a cell-washing step, indicating that PCAF-SPis works as a fluorogenic probe for the visualization of intracellular proteins.
Multicolor imaging for visualized multiple localizations of glucose transporter 4 (GLUT4) by co-staining with PYP-tag labeling probes
In addition to intracellular protein imaging, we used PCAF-RBis and PCAF-SP-s for the multicolor imaging of GLUT4. GLUT4 is known to be dynamically translocated to the cell membrane by insulin stimulation and then internalized via endocytosis.45,56,57 Our previous reports achieved staining with different colors depending on the location of GLUT4 in living cells.45,56 GLUT4 localized on the cell membrane and in the cytoplasmic region can be specifically stained using membrane-impermeable and permeable probes, respectively. Based on these GLUT4 observations, we anticipated that GLUT4-expressed cells would be the preferred system to demonstrate the versatility of the probes because the binding specificity of the probes used in this study can be evaluated by membrane-localized and intracellular-localized POIs. Since PCAF-SP-s can selectively label membrane proteins in live cells with co-expression of intracellular and membrane proteins (Fig. S5†), PCAF-SP-s was used to selectively stain membrane-localized GLUT4. However, since PCAF-RBis is membrane-permeable (Fig. 3a), PCAF-RBis was used to stain intracellular GLUT4.Based on our previous GLUT4 imaging studies,45,56 starved stable cell lines expressing PYPWT-GLUT4 were stimulated with insulin to translocate GLUT4 to the cell membrane. Upon the addition of PCAF-SP-s, fluorescence signals appeared on the cell membrane. These images indicated that PCAF-SP-s selectively visualized GLUT4 localized on the cell membrane (Fig. 4b). Moreover, signals of membrane-localized GLUT4 were observed using both PCAF-SP-s labeling and immunostaining (Fig. S7†). The fluorescence derived from GLUT4 labeled with PCAF-SP-s in the cell membrane overlaps with that detected using antibody. Fluorescence signals were not observed in HeLa cells (purchased from RIKEN BRC) that do not express PYP-tag. These results indicate that PCAF-SP-s can be applied to the imaging of membrane-localized GLUT4 (ESI S2†).
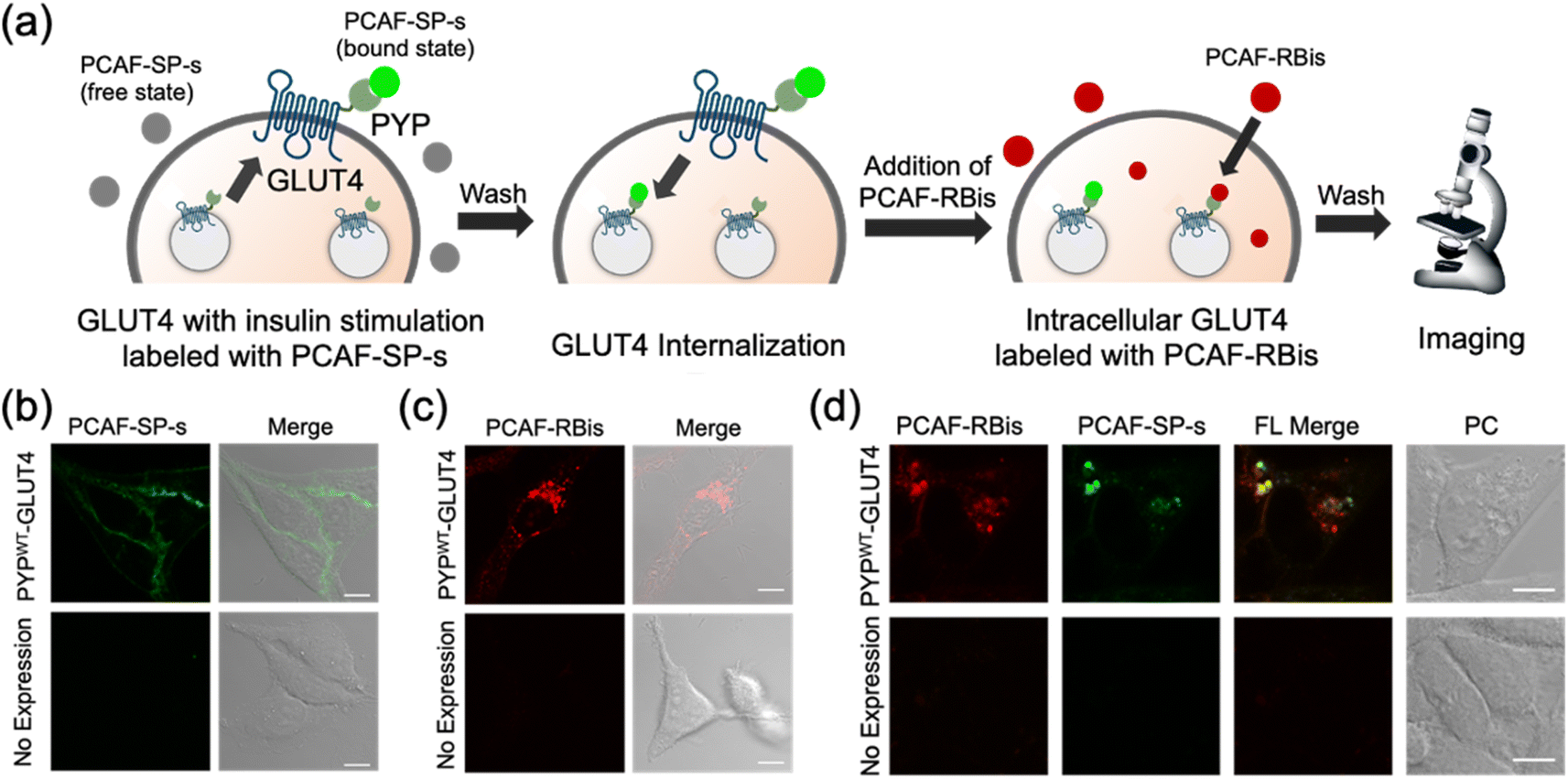 | ||
| Fig. 4 (a) Schematic view of GLUT4 multiple localization imaging with PCAF-SP-s and PCAF-RBis. (b and c) Fluorescence and phase contrast-merged images of HeLa cells stably expressing PYPWT-GLUT4 or HeLa cells (non-expressed PYP-tag) stained with (b) PCAF-SP-s (1.5 μM) in the presence of insulin or (c) PCAF-RB (1.25 μM) in the absence of insulin. (d) Multicolor imaging of HeLa cells stably expressing PYPWT-GLUT4 or HeLa cells (non-expressed PYP-tag) stained with PCAF-RBis (1.25 μM) and PCAF-SP-s (1.5 μM) for visualization of multiple localizations of GLUT4. All images were obtained with the excitation at 488 and 561 nm and detection at 500–630 (green) and 635–705 (red) nm emission, respectively. Scale bars, 10 μm. FL and PC denote fluorescence image and phase contrast image, respectively. FL merge denotes FL red image merged with FL green image. Experiments were performed twice with similar results. | ||
In addition, starved cells expressing PYPWT-GLUT4 were stained with PCAF-RBis without insulin stimulation, which resulted in fluorescent puncta in the cytoplasm. This result indicated that PCAF-RBis could stain intracellular GLUT4 (Fig. 4c). In addition, dual color imaging was performed in HeLa cells as negative control in same condition. As a result, fluorescence signals were not observed in each emission. These results indicated that PCAF-RBis could be applied with intracellular GLUT4 labeling without undesired signals by off-target labeling. Moreover, the fluorescence signals of intracellular GLUT4 were observed using both PCAF-RBis labeling and immunostaining (Fig. S8†). The localization of GLUT4 labeled with PCAF-RBis overlaps with that observed using antibody that stains GLUT4. In contrast, fluorescence signals were not observed in HeLa cells that do not express PYP-tag. These results indicate that PCAF-RBis can specifically image intracellular GLUT4 without undesired signals derived from off-target labeling.
Encouraged by the successful staining of GLUT4, depending on its location, we applied the probes to multicolor imaging of GLUT4 using PCAF-SP-s and PCAF-RBis. GLUT4, which is translocated to the membrane upon insulin stimulation, was first stained with PCAF-SP-s using the same protocol. After removing insulin and PCAF-SP-s from the culture medium for the internalization of GLUT4, intracellular GLUT4 was stained with PCAF-RBis. After washing the medium, the cells were observed under a microscope. The localization of GLUT4-labeling PCAF-SP-s did not partially overlap with the staining pattern of GLUT4-labeling PCAF-RBis (Fig. 4d).45 We also confirmed that bleed-through from one channel to the others is in the negligible level (Fig. S9†). Considering that there are two types of GLUT4, insulin-responsive and insulin-unresponsive, which show a difference in distribution according to a previous report,45 this fluorescence pattern that does not correspond to the distribution of PCAF-SP-s and PCAF-RBis indicates successful visualization of multiple localization of GLUT4. Overall, the versatility of our probe design was established by the fact that tetrazole-introduced membrane-permeable PCAF-RBis could be applied for multicolor imaging of targeted proteins with multiple localizations in living cells.
Extending application scopes to Halo tag system
Finally, our probe design was extended to Halo tag-labeling probes to further enhance its versatility. We designed RB-based probes connected to chloroalkanes as the Halo tag ligand (HTL), termed HTL-RBis (Fig. 5a). Because HTL-RBis had a tetrazole anionic moiety, its net charge was zero under physiological conditions. To examine the effect of the net molecular charge, we also synthesized HTL-RB, which does not have a tetrazole group, as a cationic probe (Fig. 5a). Fluorescence analysis was performed to determine the quantum yields of the probes. HTL-RB and HTL-RBis emitted fluorescence in the free state (Fig. S10 and Table S2;†HTL-RBis: Φfl = 0.24; HTL-RB:Φfl = 0.20). We then evaluated the specific labeling ability of the probes in live cells expressing the nuclear-localized Halo tag (Halo-NLS).32HTL-RBis successfully stained Halo-NLS in expressed cells and little fluorescence signals in mock cells (transfected with empty vector) (Fig. 5b). In contrast, HTL-RB staining revealed undesired cytoplasmic signals (Fig. 5c). These results indicate that this strategy, which introduces tetrazole, can also be applied to the Halo tag system, as well as the PYP-tag system.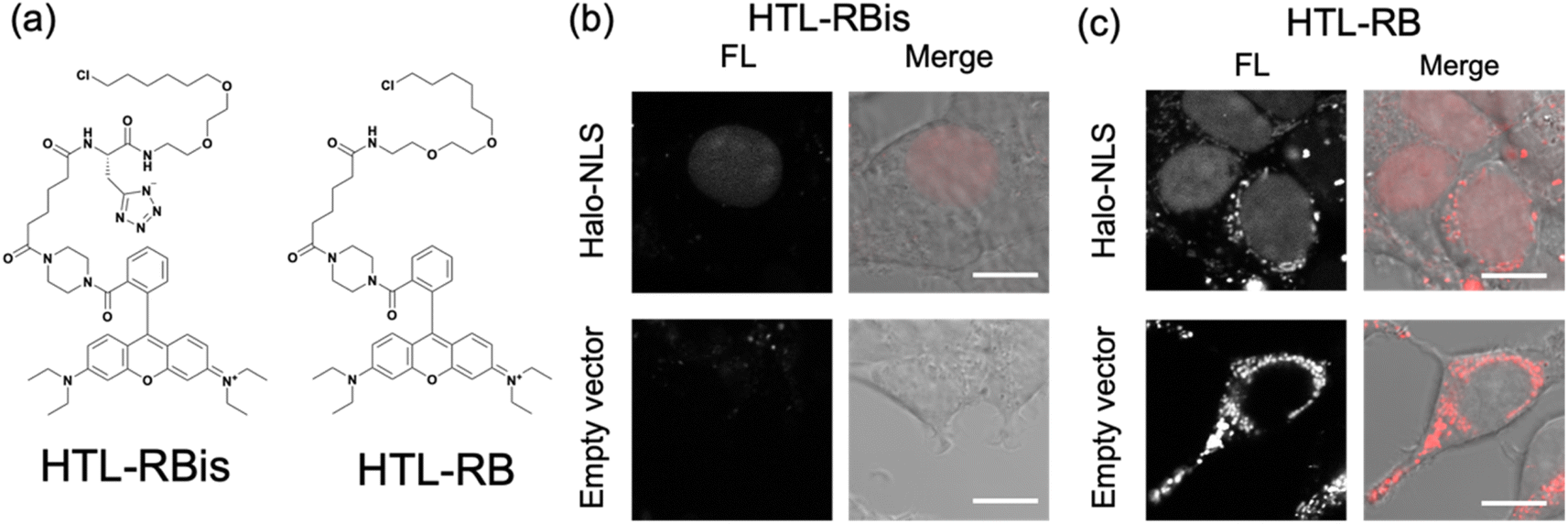 | ||
| Fig. 5 (a) Molecular structures of Halo tag labeling probe with or without tetrazole. (b and c) Fluorescence and phase contrast-merged images of HEK293T cells expressing HA-Halo-NLS or mock cells (transfected empty vector) stained with (b) HTL-RBis (2.5 μM) and (c) HTL-RB (2.5 μM). The images were measured with 566–685 nm emission and excitation at 561 nm. Scale bars, 10 μm. FL denotes Fluorescence image. Experiments were performed twice with similar results. | ||
Conclusions
We report a novel probe design strategy to suppress undesired organelle accumulation by using a tetrazole group for intracellular protein imaging in living cells. Comprehensive cell imaging experiments compared to control molecules demonstrated that tetrazole-introduced PCAF-RBis and PCAF-SPis are membrane-permeable and can successfully visualize intracellular proteins in living cells without undesired accumulation, which is a typical problem in the use of cationic dyes. Notably, PCAF-SPis can be applied to no-wash imaging of nuclear-localized proteins without false-positive signals caused by non-targeting accumulation or adhesion in the organelle. In addition, the combination of sulfonate-introduced PCAF-SP-s and tetrazole-introduced PCAF-RBis enabled the selective visualization of multiple GLUT4 localizations by taking advantage of their difference in membrane permeability. Finally, we demonstrated that this strategy could be expanded to live-cell protein imaging using the Halo tag system.We showed that our strategy could provide membrane-permeable probes through concise conjugation steps using amino acid derivatives, without modifying the cationic fluorescent scaffolds. Our strategy opens up new possibilities for the development of membrane-permeable probes, namely, novel probes with bioisosteres and cationic dye scaffolds, which could be provided by conjugation from arbitrary pairs. We believe that our strategy will find widespread application in the molecular design of bioimaging probes using cationic dye scaffolds.
Data availability
The data supporting the findings of this research can be found in both the main article and the ESI.†Author contributions
T. K., A. H., Y. Z. and A. M. synthesized and characterized the molecules. T. K. measured optical spectra. T. K. and A. H. performed cell imaging experiments. T. K. and J. A. co-wrote the initial draft. The project was directed by K.·K., Y.·H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI (Grant Numbers: JP20H02879, JP21K19048, and JP21H05075 to Y. H., JP17H06409 “Frontier Research on Chemical Communications”, JP18H03935, JP19K22255, and JP21H04706 to K. K., and JP23K19250 to J. A.), JSPS A3 Foresight Program, JSPS Asian CORE Program, “Asian Chemical Biology Initiative”, AMED-CREST, Grant-in-Aid for JSPS Fellows (JP22KJ1312 to J. A.), Grant-in-Aid for Transformative Research Area (A) “Latent Chemical Space” (23H04880 and 23H004881 to K. K.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and JST, the establishment of university fellowships towards the creation of science technology innovation (Grant Number: JPMJFS2132 to T. K.). Y. H. also acknowledges financial support to this work by Toray Science Foundation (19-6008), the Uehara Memorial Foundation, Yamada Science Foundation, and the research grant of Astellas Foundation for Research on Metabolic Disorders, TERUMO LIFE SCIENCE FOUNDATION and NAKATANI Foundation. J. A. also thanks R5 Young Researchers Support Project, Faculty of Science, KYUSHU UNIVERSITY. The authors appreciate Prof. Tohru Oishi and Dr Yoko Yasuno for ESI-MS measurement.Notes and references
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- X. Chena and Y. W. Wu, Org. Biomol. Chem., 2016, 14, 5417–5439 RSC.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. Mcdougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- J. Kompa, J. Bruins, M. Glogger, J. Wilhelm, M. S. Frei, M. Tarnawski, E. D'Este, M. Heilemann, J. Hiblot and K. Johnsson, J. Am. Chem. Soc., 2023, 145, 3075–3083 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, ACS Chem. Biol., 2013, 8, 1704–1712 CrossRef CAS PubMed.
- A. Gautier, Acc. Chem. Res., 2022, 55, 3125–3135 CrossRef CAS PubMed.
- S. Mizukami, S. Watanabe, Y. Hori and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 5016–5017 CrossRef CAS PubMed.
- Y. Hori and K. Kikuchi, Curr. Opin. Chem. Biol., 2013, 17, 644–650 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14026–14043 CrossRef CAS PubMed.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS PubMed.
- M. Heilemann, S. v. d. Linde, A. Mukherjee and M. Sauer, Angew. Chem., Int. Ed., 2009, 48, 6903–6908 CrossRef CAS PubMed.
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS PubMed.
- W. Xu, Z. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- (a) D. H. Li, R. S. Gamage, A. G. Oliver, N. L. Patel, S. Muhammad Usama, J. D. Kalen, M. J. Schnermann and B. D. Smith, Angew. Chem., Int. Ed., 2023, 62, e202305062 CrossRef CAS PubMed; (b) D. Su, C. L. Teoh, A. Samanta, N. Y. Kang, S.-J. Parka and Y. T. Chang, Chem. Commun.,, 2015, 51, 3989–3992 RSC; (c) D. Su, C. L. Teoh, S. J. Park, J. J. Kim, A. Samanta, R. Bi, U. S. Dinish, M. Olivo, M. Piantino, F. Louis, M. Matsusaki, S. S. Kim, M. A. Bae and Y. T. Chang, Chem, 2018, 4, 1128–1138 CrossRef CAS.
- M. Collot, E. Boutant, K. T. Fam, L. Danglot and A. S. Klymchenko, Bioconjugate Chem., 2020, 31, 875–883 CrossRef CAS PubMed.
- T. Komatsu, K. Johnsson, H. Okuno, H. Bito, T. Inoue, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2011, 133, 6745–6751 CrossRef CAS PubMed.
- K. Hanaoka, S. Iwaki, K. Yagi, T. Myochin, T. Ikeno, H. Ohno, E. Sasaki, T. Komatsu, T. Ueno, M. Uchigashima, T. Mikuni, K. Tainaka, S. Tahara, S. Takeuchi, T. Tahara, M. Uchiyama, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2022, 144, 19778–19790 CrossRef CAS PubMed.
- J. L. Turnbull, B. R. Benlian, R. P. Golden and E. W. Miller, J. Am. Chem. Soc., 2021, 143, 6194–6201 CrossRef CAS PubMed.
- R. Birke, J. Ast, D. A. Roosen, J. Lee, K. Robmann, C. Huhn, B. Mathes, M. Lisurek, D. Bushiri, H. Sun, B. Jones, M. Lehmann, J. Levitz, V. Haucke, D. J. Hodson and J. Broichhagen, Org. Biomol. Chem., 2022, 20, 5967–5980 RSC.
- J. Ast, D. Nasteska, N. H. F. Fine, D. J. Nieves, Z. Koszegi, Y. Lanoiselée, F. Cuozzo, K. Viloria, A. Bacon, N. T. Luu, P. N. Newsome, D. Calebiro, D. M. Owen, J. Broichhagen and D. J. Hodson, Nat. Commun., 2023, 14, 301 CrossRef CAS PubMed.
- A. Minta, J. P. Y. Kao and R. Y. Tsien, J. Biol. Chem., 1989, 264, 8171–8178 CrossRef CAS PubMed.
- S. M. Ward, T. Ördçg, S. D. Koh, S. A. Baker, J. Y. Jun, G. Amberg, K. Monaghan and K. M. Sanders, J. Physiol., 2000, 525, 355–361 CrossRef CAS PubMed.
- B. Rotman and B. W. Papermaster, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 134–141 CrossRef CAS PubMed.
- Y. Kamikawa, Y. Hori, K. Yamashita, L. Jin, S. Hirayama, D. M. Standley and K. Kikuchi, Chem. Sci., 2016, 7, 308–314 RSC.
- S. I. Reja, Y. Hori, T. Kamikawa, K. Yamasaki, M. Nishiura, S. D. Bull and K. Kikuchi, Chem. Sci., 2022, 13, 1419–1427 RSC.
- S. L. Sensi, D. Ton-That, J. H. Weiss, A. Rothe and K. R. Gee, Cell Calcium, 2003, 34, 281–284 CrossRef CAS PubMed.
- T. Egawa, K. Hirabayashi, Y. Koide, C. Kobayashi, N. Takahashi, T. Mineno, T. Terai, T. Ueno, T. Komatsu, Y. Ikegaya, N. Matsuki, T. Nagano and K. Hanaoka, Angew. Chem., Int. Ed., 2013, 52, 3874–3877 CrossRef CAS PubMed.
- L. G. Wang, A. R. Montaño, J. R. Combs, N. P. McMahon, A. Solanki, M. M. Gomes, K. Tao, W. H. Bisson, D. A. Szafran, K. S. Samkoe, K. M. Tichauer and S. L. Gibbs, Nat. Chem., 2023, 15, 729–739 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360–12365 CrossRef CAS PubMed.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165–172 CrossRef CAS PubMed.
- G. Lukinavicius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef CAS PubMed.
- C. W. Thornber, Chem. Soc. Rev., 1979, 8, 563–580 RSC.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- P. Lassalas, B. Gay, C. Lasfargeas, M. J. James, V. Tran, K. G. Vijayendran, K. R. Brunden, M. C. Kozlowski, C. J. Thomas, A. B. Smith, D. M. Huryn and C. Ballatore, J. Med. Chem., 2016, 59, 3183–3203 CrossRef CAS PubMed.
- S. A. Clark, V. Singh, D. V. Mendoza, W. Margolin and E. T. Kool, Bioconjugate Chem., 2016, 27, 2839–2843 CrossRef CAS PubMed.
- M. Nishiura, Y. Hori, M. Umeno and K. Kikuchi, Chem. Sci., 2023, 14, 5925–5935 RSC.
- A. Kroon, W. Hoff, H. Fennema, J. Gijzen, G.-J. Koomen, J. W. Verhoeven, W. Crielaard and K. J. Hellingwerf, J. Biol. Chem., 1996, 271, 31949–31956 CrossRef CAS PubMed.
- Y. Imamoto and M. Kataoka, Photochem. Photobiol., 2007, 83, 40–49 CrossRef CAS PubMed.
- W. J. Betz, F. Mao and C. B. Smith, Curr. Opin. Neurobiol., 1996, 6, 365–371 CrossRef CAS PubMed.
- W. J. Betz and G. S. Bewick, Science, 1992, 255, 200–203 CrossRef CAS PubMed.
- C. S. Abeywickrama, K. J. Wijesinghe, R. V. Stahelin and Y. Pang, Sens. Actuators, B Chem., 2019, 285, 76–83 CrossRef CAS PubMed.
- N. I. Wickramasinghe, B. Corbin, D. Y. Kanakarathna, Y. Pang, C. S. Abeywickrama and K. J. Wijesinghe, Biosensors, 2023, 13, 799 CrossRef CAS PubMed.
- C. Miró-vinyals, A. Stein, S. Fischer, T. R. Ward and A. Deliz Liang, ChemBioChem, 2021, 22, 3398–3401 CrossRef PubMed.
- J. Gao, Y. Hori, M. Nishiura, M. Bordy, J. Hasserodt and K. Kikuchi, Chem. Lett., 2020, 49, 232–235 CrossRef CAS.
- Y. Hori, K. Nakaki, M. Sato, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed., 2012, 51, 5611–5614 CrossRef CAS PubMed.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610–16611 CrossRef CAS PubMed.
- S. Hirayama, Y. Hori, Z. Benedek, T. Suzuki and K. Kikuchi, Nat. Chem. Biol., 2016, 12, 853–859 CrossRef CAS PubMed.
- Y. Haga, K. Ishii and T. Suzuki, J. Biol. Chem., 2011, 286, 31320–31327 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06957e |
| This journal is © The Royal Society of Chemistry 2024 |