DOI:
10.1039/D4SC00201F
(Edge Article)
Chem. Sci., 2024,
15, 5376-5384
Sequential radical and cationic reactivity at separated sites within one molecule in solution†
Received
10th January 2024
, Accepted 29th February 2024
First published on 1st March 2024
Abstract
Distonic radical cations (DRCs) with spatially separated charge and radical sites are expected to show both radical and cationic reactivity at different sites within one molecule. However, such “dual” reactivity has rarely been observed in the condensed phase. Herein we report the isolation of crystalline 1λ2,3λ2-1-phosphonia-3-phosphinyl-cyclohex-4-enes 2a,b˙+, which can be considered delocalized DRCs and were completely characterized by crystallographic, spectroscopic, and computational methods. These DRCs contain a radical and cationic site with seven and six valence electrons, respectively, which are both stabilized via conjugation, yet remain spatially separated. They exhibit reactivity that differs from that of conventional radical cations (CRCs); specifically they show sequential radical and cationic reactivity at separated sites within one molecule in solution.
Introduction
Stable organic radicals, such as the triphenylmethyl radical, already discovered in 1900,1 or Thiele's hydrocarbon – the first well-characterized hydrocarbon diradicaloid discovered shortly thereafter,2 have found numerous applications ranging from synthetic chemistry to materials sciences.3 In classical radical cations, the charge and spin density are localized on one atom or delocalized across a group of atoms. These radicals are classified as conventional radical cations (CRCs). The octet rule predicts that a neutral radical R3E˙, or cationic radical R3E˙+ with seven valence electrons will react with a second radical R1˙, under the formation of a shared two electron bond to give neutral R3E–R1 or cationic [R3E–R1]+.4 The same holds true when a six-valence electron cation R3E+ reacts with a nucleophile Nu− as a two electron donor. In contrast, a reaction between a CRC and Nu− will result in a high-energy radical with nine valence electrons, which is difficult to achieve when E is an element from the second and/or third period (Fig. 1a).
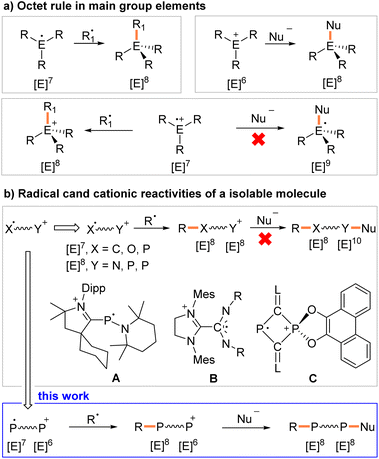 |
| | Fig. 1 Octet rule in controlling the reactivity of (a) neutral radicals, cations, and conventional radical cations, and (b) distonic radical cations. Dipp, 2,6-diisopropylphenyl; Mes, 2,4,6-trimethylphenyl; L, 1,3-bis(2,6-diisopropylphenyl)imidazoline-2-ylidene. | |
Distonic radical cations (DRCs) are a special class of radical cations with spatially separated charge and radical sites.5 A large number of DRCs have been studied in the gas phase,6 and so called “dual reactivity” has been observed with distonic acylium radicals. Specifically, radical coupling reactivity at the radical site and cycloaddition reactivity at the acylium cation site were demonstrated.7 But most DRCs are fleeting reactive intermediates in solution,8 or in crystalline or rigid matrices,9 and only a very limited number of crystallographically characterized DRCs are known.10 The group of Bertrand (A, Fig. 1b), Wang, and Schulz independently disclosed DRCs with a cationic carbene moiety and a radical center localized mainly on a phosphorus atom.10a,b,e Johnson reported a DRC with again a cationic imidazolium unit and a radical site delocalized over a carbodiimide moiety (B, Fig. 1b).10c Recently, we described the heterocyclic DRC C (Fig. 1b) with a two-coordinated radical λ2,σ2-P-center and four-coordinated cationic λ4,σ5-P-phosphonium center within a central C2P2 ring.10f But these DRCs show only radical reactivity.10a,e,f
To the best of our knowledge, dual reactivity within one molecule, including CRCs, DRCs, and charged nitroxide radicals,11 has not been reported in the condensed phase.12 We report here the synthesis and characterization of crystalline, room-temperature (RT) stable 1λ2,3λ2-1-phosphonia-3-phosphinyl-cyclohex-4-enes 2a,b˙+(BArF) (BArF = [B(3,5-(CF3)2C6H3)4]−). They contain formally a neutral two-coordinated phosphanyl P(II) radical site with seven and a two-coordinated phosphenium P(III) cation center with six valence electrons (Scheme 1). Hence both sites are electronically and coordinatively unsaturated. These features allow the study of the unique sequential radical and cationic reactivity at separated sites within one molecule (Fig. 1b).
 |
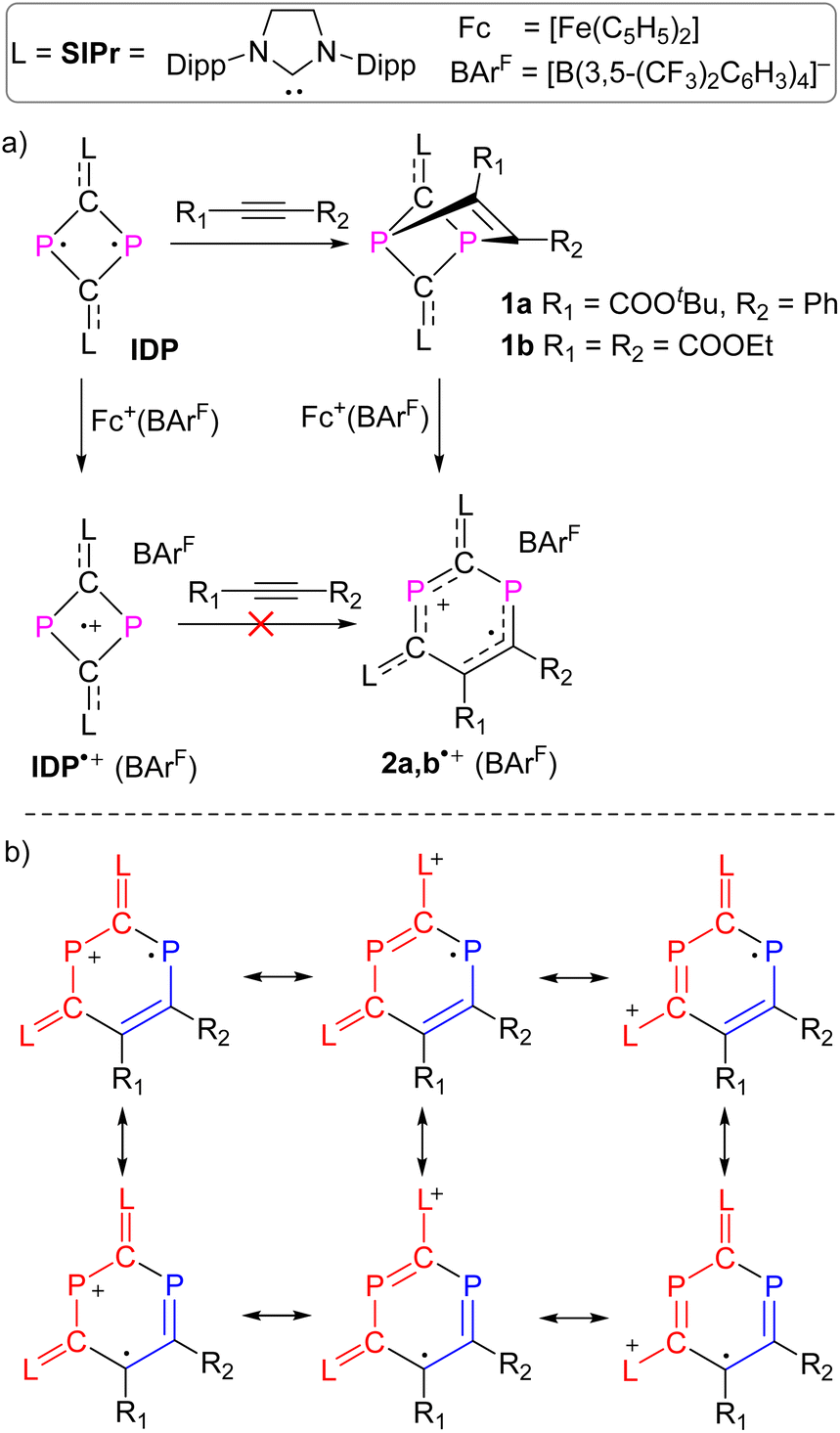
| | Scheme 1 (a) Synthesis of compounds 1 and 2˙+(BArF); (b) selected resonance structures of DRC 2˙+ in order to indicate delocalization of the positive charge in the red part of the molecule and delocalization of the spin in the blue area. | |
Results and discussion
Synthesis of DRCs 2a,b˙+
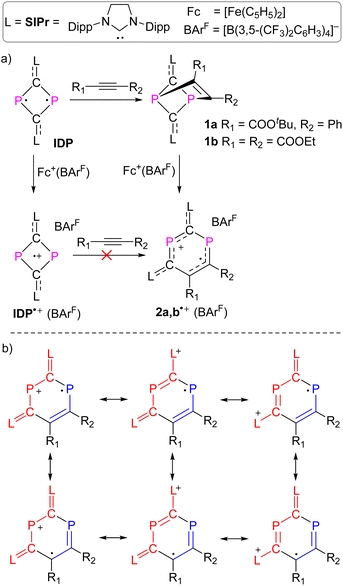
We recently reported the reaction of imidazolium-stabilized diphosphete-diide IDP (ref. 13) with one equivalent of a phosphaalkyne yielding tricyclic neutral L2(Ph3C)C3P3 derivatives (L = IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolium-2-ylidene), which upon oxidation rearranged cleanly into a radical cation [L2(Ph3C)C3P3]˙+ with an almost planar C3P3 cycle over which the spin density is more or less evenly delocalized.14 We now reacted IDP with alkynes, such as tert-butyl phenylpropiolate (PTP) or diethyl acetylenedicarboxylate,15 which led to yellow powders 1a,b in 90–95% yield, respectively (Scheme 1). Multinuclear NMR spectroscopy and single-crystal X-ray diffraction analyses unambiguously identify these compounds as [2 + 2] cycloaddition products [(L)2C2P2C(R1)C(R2)] (1a,b, L = SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazoline-2-ylidene). Monitoring the reaction process by 31P NMR spectroscopy indicated the formation of 1a as a sole new product with the appearance of two doublets at 158.8 and 153.8 ppm (2JPP = 6.5 Hz). The solid-state structure of 1a, obtained by X-ray diffraction (XRD) methods using single crystals, confirms the [2.1.1] bicyclic skeleton (Fig. 1a). The synthesis and characterization of 1b were reported by us.15 Similar reactivity between (RN)2P2 biradicaloids and alkynes has been observed previously by Schulz et al.16
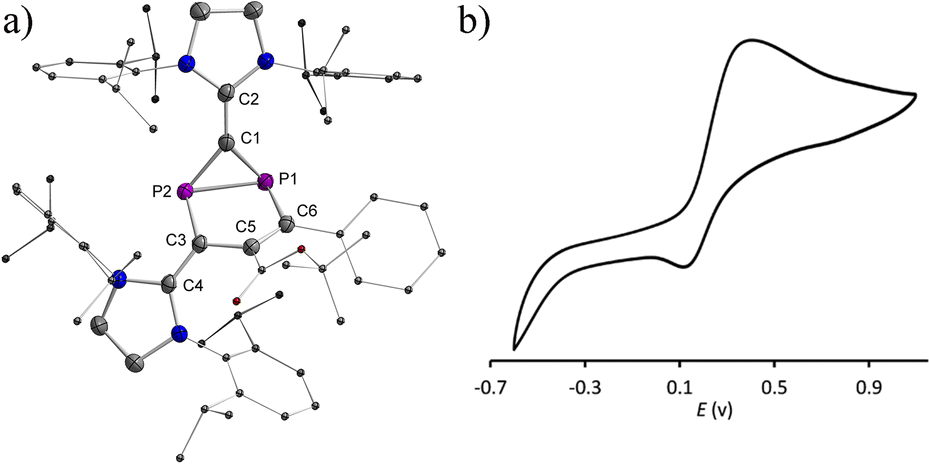
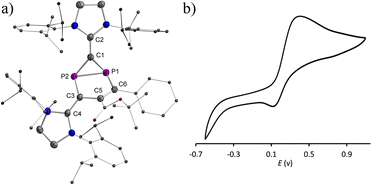
No decomposition or rearrangement was observed after heating toluene solutions of 1a,b at 100 °C for more than 4 hours. The cyclic voltammogram of 1a, selected as an example, shows an irreversible one electron oxidation at EP = +0.07 V vs. Fc+/Fc (Fig. 2b). Adding 0.8 equivalent of the ferrocenium salt Fc+(BArF), dissolved in diethyl ether to a diethyl ether solution of 1a,b at −30 °C, led to highly air sensitive, paramagnetic, green compounds 2a,b˙+(BArF) in high yield (Scheme 1). Excess Fc+(BArF) was found to react further with 2a,b˙+(BArF). Of note, the reactions between IDP and diphenylacetylene or methyl(phenyl)acetylene also gave the expected cycloaddition products but they show no clean redox chemistry. XRD methods and EPR spectroscopy confirm the structure of these compounds as 1λ2,3λ2-1-phosphonia-3-phosphinyl-cyclohex-4-ene radical cation salts 2a,b˙+(BArF) (Scheme 1). Note that no apparent decomposition was observed after heating the tetrahydrofuran solution of 2a,b˙+(BArF) at 60 °C for more than 48 hours, and the reaction of the stable radical cation IDP˙+(BArF)13 with alkynes does not allow the synthesis of 2a,b˙+(BArF).
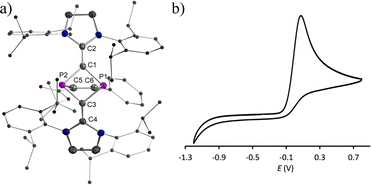 |
| | Fig. 2 (a) Plots of the molecular structure of 1a (ellipsoids are set to 50% probability; H atoms and solvents are omitted for clarity). Selected distances (Å): P1–C1 1.838(2), P1–C3 1.838(2), C3–P2 1.850(2), P2–C1 1.843(2), C1–C2 1.352(3), C3–C4 1.349(3), P1–C6 1.917(2), P2–C5 1.900(2), and C5–C6 1.353(3); (b) cyclic voltammogram for 10−3 M THF solution of 1a containing 0.1 M n-Bu4N(BArF) as electrolyte (potential versus Fc+/Fc) at scan rate 100 mV s−1 with platinum as the working and counter electrode and Ag/Ag+ as the reference electrode. | |
Characterization of DRCs 2a,b˙+
The EPR spectra at RT show a doublet of doublets due to a large and a very small isotropic 31P hyperfine coupling constant (HFC) (av. aiso(P1) = 108.0 MHz and aiso(P2) = 11.5 MHz) for 2a˙+(BArF) (Fig. 3a). For 2b˙+(BArF), only a doublet is observed due to a large isotropic 31P hyperfine interaction [av. aiso(P1) = 85.7 MHz and giso = 2.008] (Fig. S24†). The HFC with the P1 nucleus is smaller than the ones observed for comparable phosphorus centered radicals where the spin is predominantly localized on the P center (spin density > 0.5e, aiso > 150 MHz).10a,b,13,17 This indicates delocalization of the spin density but very little is localized on P2. In frozen solution at 100 K, a strongly anisotropic EPR spectrum is recorded (Fig. 3b). The simulation of the spectrum yielded the principal g values and 31P HFCs for 2a˙+(BArF) (g = [2.0042, 2.0091, 2.0130], A(31P1) = [−23.1, 5.05, 342.1] MHz, A(31P2) = [−25.3, 10.6, 49.2] MHz). The large anisotropic 31P HFC tensor confirms that the majority of spin density is indeed localized in a p-type orbital at P1 (Fig. 3c). The EPR spectrum of the frozen solution of 2b˙+(BArF) is too broad to be simulated (Fig. S25†).
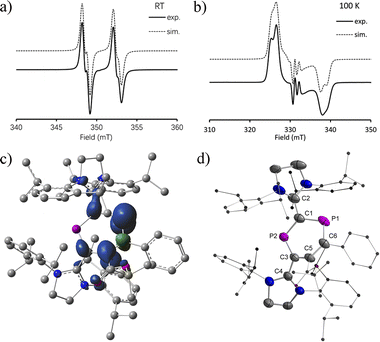 |
| | Fig. 3 (a) Continuous wave EPR spectra at RT and (b) at 100 K; (c) calculated spin density (density = 0.004); and (d) plots of the molecular structure of 2a˙+(BArF) (ellipsoids are set to 50% probability; H atoms and anions are omitted for clarity). Selected distances (Å): 2a˙+: P1–C1 1.785(5), P1–C6 1.725(5), C6–C5 1.412(6), C5–C3 1.455(6), C3–P2 1.719(4), P2–C1 1.735(4), C1–C2 1.444(6), and C3–C4 1.470(5). | |
Slow evaporation of a saturated diethyl ether/hexane solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at RT yielded green single crystals of 2a,b˙+(BArF) suitable for X-ray diffraction studies. Both compounds have very similar structures with a central six-membered C4P2 heterocycle with two λ2-phosphorus atoms in the 1,3-position as a result of a skeletal rearrangement (Fig. 3d and S24†). The exocyclic C–L bonds in 2a˙+ (av. 1.457 Å) and in 2b˙+ (av. 1.452 Å) are longer than those of 1a (av. 1.351 Å) and 1b (av. 1.356 Å),15 but rather similar to the ones observed in IDP˙+ (av. 1.41 Å), indicating a strong electron-donation from the SIPr to the C4P2 heterocycle but little back-donation.3a,c,18 On one hand, the lengths of the P1–C6 (av. 1.736 Å) and C6–C5 (av. 1.393 Å) bonds in 2a,b˙+ indicate a conjugated P1–C6–C5 unit over which the unpaired electrons are mainly delocalized. On the other hand, the P2–C1 (av. 1.751 Å) and P2–C3 (av. 1.722 Å) bonds are within the range observed in cyclic phosphenium cations (1.716–1.763 Å) and indicate delocalization of the positive charge in these units.17b,19 Both the P1–C6–C5 and C1–P2–C3 parts are connected via P1–C1 (av. 1.789 Å) and C3–C5 (av. 1.457), which are significantly longer. These structure parameters, in conjunction with the EPR analyses and theoretical calculations (vide infra), are in accord with the resonance structures shown in Scheme 1b, which indicate that the spin is mainly delocalized in the blue part and the positive charge is mainly in the red part of the molecule. Both halves are connected by elongated P1–C1 and C3–C5 bonds, respectively.
:1) at RT yielded green single crystals of 2a,b˙+(BArF) suitable for X-ray diffraction studies. Both compounds have very similar structures with a central six-membered C4P2 heterocycle with two λ2-phosphorus atoms in the 1,3-position as a result of a skeletal rearrangement (Fig. 3d and S24†). The exocyclic C–L bonds in 2a˙+ (av. 1.457 Å) and in 2b˙+ (av. 1.452 Å) are longer than those of 1a (av. 1.351 Å) and 1b (av. 1.356 Å),15 but rather similar to the ones observed in IDP˙+ (av. 1.41 Å), indicating a strong electron-donation from the SIPr to the C4P2 heterocycle but little back-donation.3a,c,18 On one hand, the lengths of the P1–C6 (av. 1.736 Å) and C6–C5 (av. 1.393 Å) bonds in 2a,b˙+ indicate a conjugated P1–C6–C5 unit over which the unpaired electrons are mainly delocalized. On the other hand, the P2–C1 (av. 1.751 Å) and P2–C3 (av. 1.722 Å) bonds are within the range observed in cyclic phosphenium cations (1.716–1.763 Å) and indicate delocalization of the positive charge in these units.17b,19 Both the P1–C6–C5 and C1–P2–C3 parts are connected via P1–C1 (av. 1.789 Å) and C3–C5 (av. 1.457), which are significantly longer. These structure parameters, in conjunction with the EPR analyses and theoretical calculations (vide infra), are in accord with the resonance structures shown in Scheme 1b, which indicate that the spin is mainly delocalized in the blue part and the positive charge is mainly in the red part of the molecule. Both halves are connected by elongated P1–C1 and C3–C5 bonds, respectively.
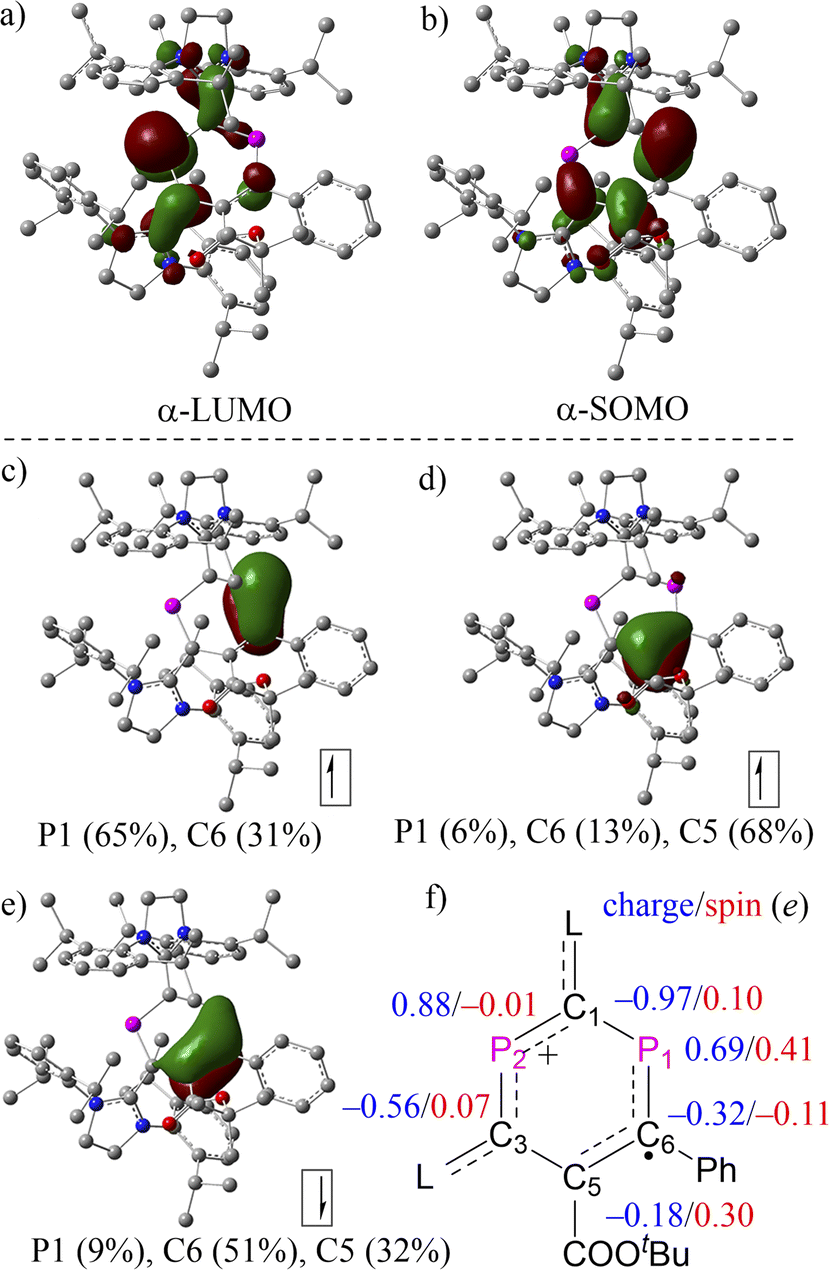
The molecular structures of 2a,b˙+ were further examined using density functional theory (DFT) calculations optimized at the level of (U)M06-2X/def2-SVP20 and natural bond orbital (NBO) analysis at the level of (U)M06-2X/def2-TZVP.21 The composition of molecular orbitals (MOs) and natural localized MOs (NLMOs) evolving from an NBO analysis supports the description of 2a,b˙+ as DRCs (Fig. 4 and S26†). In the case of 2a˙+, MO analysis revealed no apparent difference between the α-LUMO and β-LUMO+1 and between α-SOMO and β-LUMO, where the former two empty MOs comprise mainly a π-type lone pair at P2 with contributions from adjacent π-type bonding orbitals of the CL fragments (CL = C-SIPr, Fig. 4a). The latter involves mainly singly occupied and empty allyl-type MOs with a small contribution from adjacent π-type bonding orbitals of the CL fragments (Fig. 4b). These results imply the partial separation of charge and spin density in 2a˙+. The NLMOs disclose an α-electron at the π-type bonding orbital on P1(65%)–C6(31%), an α-electron delocalized over P1(6%)–C6(13%)–C5(68%), and an β-electron at the π-type bonding orbital P1(9%)–C6(51%)–C5(32%). These results indicate the presence of a delocalized allyl-type endocyclic P1–C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 radical fragment where two α-electrons and one β-electron are delocalized over the π-type bonding orbital. Natural population analysis (NPA) reveals that the spin density in 2a+˙ is localized mainly at P1 (0.41e), C6 (−0.11e), and C5 (0.30e), with very small contributions from C1 (0.10e) and C3 (0.07e), and virtually no spin density on P2 (−0.01e), which agrees with the analysis of EPR spectra. Additional spin density is delocalized over CL fragments (Fig. 3c). In addition, the P2 center within the phosphenium unit carries the highest positive charge (2a˙+: +0.69e for P1 and +0.88e for P2). An inspection of the Wiberg bond indices (WBIs) shows that the P1–C1 (1.00) and C5–C3 (1.13) connecting the two subunits P1–C6–C5 and C1–P2–C3 are nearly single bonds, and their bond orders are substantially smaller than the rest of the endocyclic bonds, such as P1–C6 (1.26), C6–C5 (1.40), C3–P2 (1.35), and P2–C1 (1.17). The WBIs of bonds linking the P2C4 cycle to the L substituents, C1–C2 (1.22) and C3–C4 (1.11), further support the strong electron-donation from SIPr. The presence of the two carboxylic ester units COOEt as stronger electron-withdrawing substituents renders the two phosphorus centers (+0.75e for P1 and +0.90e for P2) in 2b˙+ slightly more positively charged than those in 2a˙+, but otherwise the electronic structures of 2a˙+ and 2b˙+ are very similar (Fig. S26†).
C5 radical fragment where two α-electrons and one β-electron are delocalized over the π-type bonding orbital. Natural population analysis (NPA) reveals that the spin density in 2a+˙ is localized mainly at P1 (0.41e), C6 (−0.11e), and C5 (0.30e), with very small contributions from C1 (0.10e) and C3 (0.07e), and virtually no spin density on P2 (−0.01e), which agrees with the analysis of EPR spectra. Additional spin density is delocalized over CL fragments (Fig. 3c). In addition, the P2 center within the phosphenium unit carries the highest positive charge (2a˙+: +0.69e for P1 and +0.88e for P2). An inspection of the Wiberg bond indices (WBIs) shows that the P1–C1 (1.00) and C5–C3 (1.13) connecting the two subunits P1–C6–C5 and C1–P2–C3 are nearly single bonds, and their bond orders are substantially smaller than the rest of the endocyclic bonds, such as P1–C6 (1.26), C6–C5 (1.40), C3–P2 (1.35), and P2–C1 (1.17). The WBIs of bonds linking the P2C4 cycle to the L substituents, C1–C2 (1.22) and C3–C4 (1.11), further support the strong electron-donation from SIPr. The presence of the two carboxylic ester units COOEt as stronger electron-withdrawing substituents renders the two phosphorus centers (+0.75e for P1 and +0.90e for P2) in 2b˙+ slightly more positively charged than those in 2a˙+, but otherwise the electronic structures of 2a˙+ and 2b˙+ are very similar (Fig. S26†).
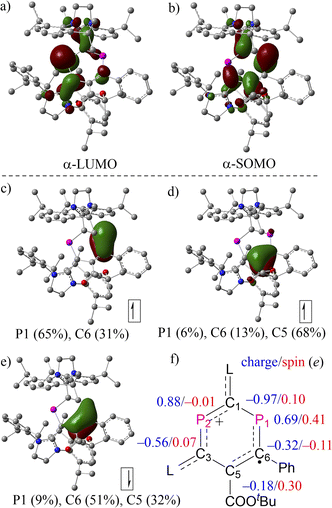 |
| | Fig. 4 Selected (a) α-LUMO; (b) β-SOMO, and NLMOs resulting from an NBO analysis of 2a˙+: (c) a π-bond α-electron; (d) a π-bond α-electron; (e) a π-bond β-electron; (f) selected NPA charges and spin populations. | |
Possible mechanism for the formation of DRCs 2˙+
The mechanism leading to 2a˙+ from the putative intermediate radical cation 1a˙+ – as a product of the one-electron oxidation of 1a – was investigated using DFT calculations ((U)SMD-M06-2X/def2-TZVP//(U)M06-2X/def2-SVP) for 2a˙+ as a selected example. A possible minimum energy reaction pathway (MERP) is shown in Fig. 5.20 The intramolecular rearrangement starts from the [2.1.1] bicyclic radical cation 1a˙+, which is at ΔG = 0.0 kcal mol−1 on the relative energy scale. The 1,2-shift of the CR1 group from a P center to the adjacent carbon center of the CL unit only requires 9.8 kcal mol−1 to reach the activated complex at TS1. Intermediate IN1 is formed in an exergonic reaction (ΔG = −12.5 kcal mol−1). This further rearranges again via an activated complex at an energetically low-lying transition state TS2 (10.4 kcal mol−1) to give the six-membered DRC 2a˙+ in an exergonic reaction (−12.8 kcal mol−1). We also investigated the alkyne dissociation from 2a˙+, which is the microscopically reverse reaction of the addition of an alkyne to the radical cation IDP˙+. As the left branch of the MERP shows, this dissociation is indeed a thermodynamically preferred process (ΔG = −23.1 kcal mol−1) but requires the formation of activated complexes at energetically higher lying transition states TS3 (19.1 kcal mol−1) and TS4 (19.2 kcal mol−1), respectively, which disfavours this process kinetically. Thus, the calculations are in good agreement with the experimental results, which show that (i) one-electron oxidation of 1a exclusively leads to 2a˙+ and (ii) that the reaction between IDP˙+ and PTP will not give 2a˙+.
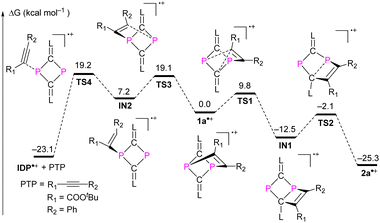 |
| | Fig. 5 Free energy profile for the formation of 2a˙+ or IDP˙+ and PTP from the transient radical cation intermediate 2a˙+. | |
Treatment of 2a˙+(BArF) with an excess of Mg powder as a reducing agent at RT under vigorous stirring overnight did not regenerate the neutral tricyclic species 1a, but yielded a red powder 3 with excellent yield 91% (Scheme 2). Attempts to reduce 2b˙+(BArF) under the same reaction conditions led to a mixture of unidentified products. The 31P spectrum of 3 shows two doublets at −154.1 ppm and −197.1 ppm (1JPP = 64.8 Hz), which is in the range of diphosphiranes (−117 – −176 ppm).22 Indeed, XRD analysis of a single crystal of 3 unambiguously confirms the formation of a [3.1.0] bicyclic skeleton and a CP2 phosphirane ring annealed to an unsaturated five-membered C3P2 ring (Fig. 6a). The P1–P2 bond [2.2685(6) Å] is slightly longer than standard P–P single bonds (2.214 Å vs. 3.124 Å in 2a˙+)23 and comparable with the P–P bonds (2.167–2.223 Å) in similar housane-type NP2 fragment reported by the group of Schulz.16a,b,24 The P–C bonds in the phosphirane moiety [P1–C1 1.8075(19); P2–C1 1.7911(19) Å] are notably shorter than the ones in the five-membered C3P2 ring [P1–C6 1.8408(17); P2–C1 1.7911(19) Å]. The C5C6 bond [1.376(2) Å] corresponds to a double bond, while the adjacent C5–C3 bond [1.468(2) Å] is significantly longer. The fold angle between the three- and five-membered rings amounts to 100°. The cyclic voltammogram of 3 shows a quasi-reversible redox wave at EP = +0.42 V vs. Fc+/Fc (Fig. 6b). Indeed, one-electron oxidation of 3 with Fc+(BArF) led cleanly to 2a˙+(BArF) suggesting that the conversion 2a˙+ + e ⇆ 3 is fully reversible.
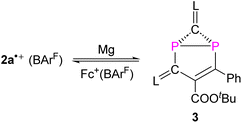 |
| | Scheme 2 Synthesis of compound 3. | |
 |
| | Fig. 6 (a) Plots of the molecular structure of 3 (ellipsoids are set to 50% probability; H atoms and solvents are omitted for clarity). Selected distances (Å): P1–P2 2.2685(6), P1–C1 1.8075(19), P1–C6 1.8408(17), C6–C5 1.376(2), C5–C3 1.468(2), C3–P2 1.8387(17), P2–C1 1.7911(19), C1–C2 1.365(3), and C3–C4 1.383(2); (b) cyclic voltammogram for 10−3 M THF solution of 3 containing 0.1 M n-Bu4N(BArF) as electrolyte (potential versus Fc+/Fc) at scan rate 100 mVs−1 with platinum as the working and counter electrode and Ag/Ag+ as the reference electrode. | |
Note that the reaction of IDP with trityl phosphaalkyne resulted in a bicyclic product similar to that of 3 (Scheme 2), and the subsequent one electron oxidation afforded a delocalized cyclic C3P3 radical cation instead of a DRC.14 Wang and co-workers recently demonstrated that electron-withdrawing groups in indeno[2,1-b]fluorenes favor a triplet ground state over a singlet ground state.25 This observation suggests that electron-withdrawing groups can impede electron delocalization to some extent and we therefore assume that the DRC character of 2a,b˙+ is caused by the carboxylate residues as electron withdrawing groups.
Reactivity of DRCs 2a,b˙+
To probe chemically the sequential radical and cationic reactivity of 2a,b˙+ as delocalized DRCs, 2a˙+(BArF) was selected to react with half an equivalent of diphenyl disulfide. The reaction between phosphorus radicals and disulfides to give compounds with a P–S bond is well documented,10f,15,26 and we expected that the radical center P1 would add to PhS˙ to give product 4+(BArF) as an EPR silent but an NMR active product. This is indeed the case and 4+(BArF) is obtained as red powder in 90% yield (Scheme 3). The 31P NMR spectrum of 4+(BArF) displays resonance signals at 309.2 ppm for the two-coordinated phosphenium center P2 and at 6.3 ppm for the three-coordinated λ3, σ3-P1 nucleus. The solid-state structure of 4+(BArF) is shown in Fig. 7a. The formation of a new P1–S1 bond [2.2123(16) Å; ∑rcov(P–S) = 2.14 Å]23 interrupts the electron delocalization mainly within the endocyclic P1–C6–C5 skeleton where the spin was delocalized in 2a˙+. For instance, a significantly shortened C5C6 bond [1.363(4) Å, vs. 1.412(6) Å in 2a˙+] and elongated P1–C6 bond [1.818(3) Å, vs. 1.725(5) Å in 2a˙+] are observed. Other structural changes are noticeable but smaller. On the other side, the C4–C3–P2–C1–C2 unit in 4+ has similar structural parameters to the one observed in the 2a˙+ underlining that this part is still best described as a delocalized phosphenium cation (Table S2†).
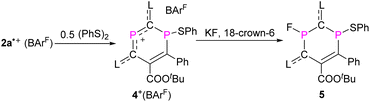 |
| | Scheme 3 Synthesis of compounds 4+(BArF) and 5. | |
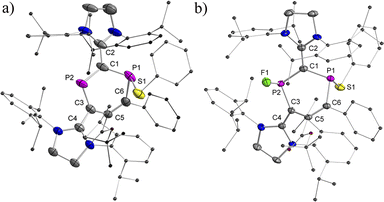 |
| | Fig. 7 Plots of the molecular structures of 4+(BArF) and 5 (ellipsoids are set to 50% probability; H atoms, anions, and solvents are omitted for clarity). Selected distances (Å): (a) 4+: P1–S1 2.2123(16), P1–C1 1.808(4), P1–C6 1.818(3), C6–C5 1.363(4), C5–C3 1.487(4), C3–P2 1.723(3), P2–C1 1.752(3), C1–C2 1.432(5), and C3–C4 1.473(4); (b) 5 (only the trans-isomer of 5 is shown): P1–S1 2.1241(10), P1–C1 1.798(3), P1–C6 1.825(3), C6–C5 1.361(4), C5–C3 1.489(3), C3–P2 1.798(3), P2–C1 1.801(3), P2–F1 1.622(2), C1–C2 1.393(4), and C3–C4 1.395(3). | |
The cation 4+ reacted cleanly with fluoride, but did not react with other halogen anions, azide, or cyanate. The treatment of 4+(BArF) with three equivalents of potassium fluoride in the presence of equimolar 18-crown-6 gave a yellow powder 5 in 73% yield (Scheme 3). In the 31P NMR spectrum, 5 shows only one doublet at 146.7 ppm (1JPF = 918.1 Hz) and one singlet at 13.5 ppm, while in the 19F NMR spectrum, one major isomer at −124.2 ppm (1JPF = 919.3 Hz) and a second minor isomer at −127.6 ppm (1JPF = 926.4 Hz) were observed. This agrees well with the result from an XRD study (Fig. 7b), which shows two isomers, which co-crystallized in one single crystal with a major trans-isomer [P2–F1 1.622(2) Å, 78%] and a cis-isomer (P2–F1A 1.574(6) Å, 22%) (trans- and cis-indicate the mutual position of the P–F and P–S bonds with respect to the ring plane). There is rather little structural change between 4+ and 5 and only the P2–C1 [1.801(3) Å] and P2–C3 [1.798(3) Å] bonds in 5 are evidently elongated.
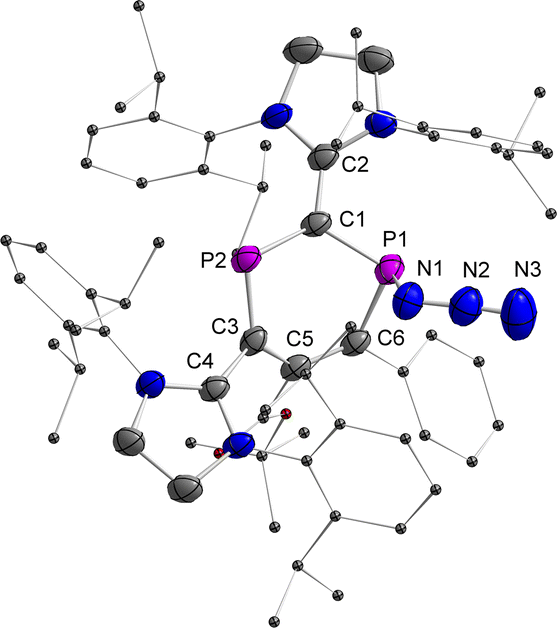
Finally, the reactivity of 2a,b˙+ toward various nucleophiles was probed. The reactions of 2a,b˙+(BArF) with KF, CsF, Et3NHF, potassium triethylborohydride, phenylmagnesium chloride, 1,3,4,5-tetramethylimidazol-2-ylidene or 4-dimethylaminopyridine afforded mixtures of compounds, which are NMR active but remained unidentified. Treatment of 2a˙+(BArF) with excess sodium azide (NaN3) afforded mainly compound 6+(BArF) along with minor amounts of 3 and other unidentified products (Scheme 4 and Fig. S21†). Recrystallization of the crude product led to 15% yield of crystalline reddish materials of 6+(BArF), which are still contaminated with minor impurities. We assume that the formation of 6+(BArF) is the result of the reaction of 2a˙+(BArF) with a transient azidyl radical (N3˙), which is generated via the oxidation of N3− by 2a˙+(BArF). Single electron reduction of 2a˙+(BArF) would lead to 3 in accordance with its observation. Note, in this context, that the generation of N3˙ by electrochemical or chemical oxidation of azide with 2,2,6,6-tetramethyl-1-oxopiperidinium (TEMPO+) has been studied extensively in synthetic chemistry.27 In the 31P NMR spectra, singlets at 305.7 and 42.7 ppm for 6+(BArF) were observed. And XRD studies confirmed the molecular structure of 6+(BArF) (Fig. 8). The azidyl group is bound to the radical site P1 leaving the cationic P2 site unreacted. The newly formed P1–N1 bond [1.775(3) Å] corresponds to a single bond. The other structural parameters of 6+(BArF) are comparable to the ones observed in 4+(BArF).
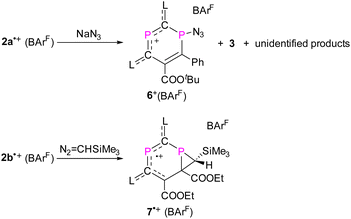 |
| | Scheme 4 Synthesis of compounds 6+(BArF) and 7˙+(BArF). | |
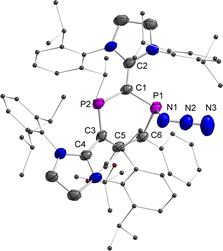 |
| | Fig. 8 Plots of the molecular structure of 6+(BArF) (ellipsoids are set to 50% probability; H atoms, anions, and solvents are omitted for clarity). Selected distances (Å): 6+: P1–N1 1.775(3), N1–N2 1.215(5), N2–N3 1.142(6), P1–C1 1.794(3), P1–C6 1.814(4), C6–C5 1.359(5), C5–C3 1.483(4), C3–P2 1.723(4), P2–C1 1.746(4), C1–C2 1.415(5), and C3–C4 1.470(5). | |
Treatment of 2b˙+(BArF) with one equivalent of (trimethylsilyl)diazomethane at RT led to a spontaneous colour change from green to blue. After stirring the solution for 1 hour, we observed the formation of an EPR active paramagnetic species 7˙+(BArF) resulting from a [2 + 1] cycloaddition reaction under the elimination of dinitrogen (Scheme 4). The EPR spectra of diethyl ether solutions of 7˙+(BArF) at RT and 100 K show HFC tensors to which clearly only one 31P nucleus contributes with a relatively large 31P hyperfine interaction g = [2.0041, 2.0165, 2.0081], giso = 2.010, A(31P1) = [−1.9, −2.8, 317.7] Mz, and aiso(P1) = 104.4 MHz (Fig. 9a and b), which is comparable with that of 2a,b˙+(BArF). The solid-state structure of 7˙+(BArF) is shown in Fig. 9d. The fold angle between the three-membered C2P and six-membered C4P2 ring within the [4.1.0] bicyclic skeleton is 104°. The P1–C6 (1.867(2) Å), P1–C7 (1.848(2) Å), and C6–C7 (1.532(3) Å) bonds in the phosphirane ring are in the range of classical single bonds (∑rcov(P–C) = 1.86 Å, ∑rcov(PC) = 1.69 Å, ∑rcov(C–C) = 1.50 Å, and ∑rcov(CC) = 1.34 Å).23 A new double bond is established between C3C5 (1.381(3) Å) while the adjacent C5–C6 bond is elongated to 1.487(3) Å [vs. C5–C6 1.373(3) Å in 2b˙+, Fig. S27†]. The structural metrics within the C2–C1–P2–C3–C4 unit (C2C1 1.414(3) Å, P2–C1 1.743(2) Å, P2–C3 1.789(2) Å, and C3–C4 1.498(3) Å) indicate delocalization of both charge and spin density, which however only involves one phosphorus center. An NPA analysis shows that the majority of the spin density is delocalized over an allyl-type endocyclic P2–C3–C5 unit [spin density: P2 (0.39e), C3 (−0.07e), and C5 (0.31e)] with a very small contribution from C1 (0.08e) and virtually no spin density on P1 and C6 (Fig. 9c). The formation of 7˙+(BArF) further supports the presence of an allyl-type delocalized radical fragment in 2a,b˙+(BArF).
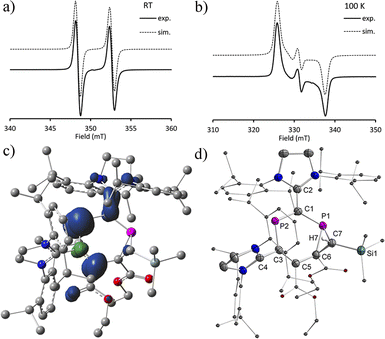 |
| | Fig. 9 a) Continuous wave EPR spectra at RT and (b) at 100 K; (c) spin density (density = 0.004) of 7˙+; (d) plots of the molecular structure of 7˙+(BArF) (ellipsoids are set to 50% probability; H atoms except H7 and anions are omitted for clarity). Selected distances (Å): 7˙+: P1–C1 1.743(2), P1–C6 1.867(2), P1–C7 1.848(2), C7–Si1 1.890(2), C7–C6 1.532(3), C6–C5 1.487(3), C5–C3 1.381(3), C3–P2 1.789(2), P2–C1 1.743(2), C1–C2 1.414(3), and C3–C4 1.498(3). | |
Conclusions
The octet rule predicts that CRCs only react with radical sites forming cations with eight valence electrons. In contrast, DRCs with spatially separated charge and radical sites are promising candidates to show both radical and cationic reactivity at different sites within one molecule. However, such dual reactivity has presently only been reported in the gas phase. In the condensed phase, a molecule with such dual reactivity has not been reported yet. In this work, delocalized DRCs 2a,b˙+(BArF) with an unsaturated radical and cationic site have been isolated and analysed by EPR spectroscopic analysis, XRD determination and DFT calculations. Indeed, 2a˙+(BArF), as an example, manifests sequential radical and cationic reactivity in the condensed phase. Attempts to demonstrate sequential cationic and radical reactivity of 2a,b˙+(BArF) failed, which may be attributed to the high instability of the resulting neutral radicals after reaction with a nucleophile. Furthermore, the reaction between 2a˙+(BArF) and azide anions indicates that the former may behave as a rather strong oxidant eventually generating transient azidyl radicals, which further complicates the chemistry of the DRCs reported here but also offers further applications in synthetic chemistry.
Data availability
All data associated with this article are available from ESI.†
Author contributions
S. Liu carried out most of the experimental work. J. Lin assisted with the HRMS measurements. Y. Li, Z. Ke, and Z. Li carried out the computational studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271315, 22171291, 21821003 and 21890380), Tip-top Scientific and Technical Innovative Youth Talents of Guangdong Special Support Program (2019TQ05C926), Guangdong Basic and Applied Basic Research Foundation (2021A1515012028 and 2023A1515010092), Science and Technology Planning Project of Guangzhou (202102080189) and National Key Research and Development Program of China (2021YFA1500401).
Notes and references
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
-
(a) C. D. Martin, S. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC;
(b) Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS;
(c) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed;
(d) L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC;
(e) Z. X. Chen, Y. Li and F. Huang, Chem, 2020, 7, 1–45 CrossRef;
(f) D. Wang, C. Zhai, Y. Chen, Y. He, X.-d. Chen, S. Wang, L. Zhao, G. Frenking, X. Wang and G. Tan, Nat. Chem., 2023, 15, 200–205 CrossRef CAS PubMed;
(g) Z. Feng, S. Tang, Y. Su and X. Wang, Chem. Soc. Rev., 2022, 51, 5930–5973 RSC;
(h) K. Hatakeyama-Sato and K. Oyaizu, Chem. Rev., 2023, 123, 11336–11391 CrossRef CAS PubMed;
(i) M. Ju, Z. Lu, L. F. T. Novaes, J. I. Martinez Alvarado and S. Lin, J. Am. Chem. Soc., 2023, 145, 19478–19489 CrossRef CAS PubMed;
(j) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC;
(k) Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CrossRef CAS;
(l) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed;
(m) Y. K. Loh, P. Vasko, C. McManus, A. Heilmann, W. K. Myers and S. Aldridge, Nat. Commun., 2021, 12, 7052 CrossRef CAS;
(n) T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 CrossRef CAS.
-
(a) T. Li, H. Wei, Y. Fang, L. Wang, S. Chen, Z. Zhang, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 632–636 CrossRef CAS;
(b) G. Tan and X. Wang, Chin. J. Chem., 2018, 36, 573–586 CrossRef CAS;
(c) Z. Feng, Y. Fang, H. Ruan, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 6769–6774 CrossRef CAS PubMed;
(d) B. Wang, Y. Li, R. Ganguly, R. D. Webster and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 7826–7829 CrossRef CAS PubMed.
-
(a) B. F. Yates, W. J. Bouma and L. Radom, J. Am. Chem. Soc., 1984, 106, 5805–5808 CrossRef CAS;
(b) B. F. Yates, W. J. Bouma and L. Radom, Tetrahedron, 1986, 42, 6225–6234 CrossRef CAS.
-
(a) S. Hammerum, Mass Spectrom. Rev., 1988, 7, 123–202 CrossRef CAS;
(b) K. M. Stirk, L. K. M. Kiminkinen and H. I. Kenttamaa, Chem. Rev., 1992, 92, 1649–1665 CrossRef CAS;
(c) H. I. Kenttämaa, Org. Mass Spectrom., 1994, 29, 1–10 CrossRef;
(d) D. M. Tomazela, A. A. Sabino, R. Sparrapan, F. C. Gozzo and M. N. Eberlin, J. Am. Soc. Mass Spectrom., 2006, 17, 1014–1022 CrossRef CAS PubMed;
(e) P. E. Williams, B. J. Jankiewicz, L. Yang and H. I. Kenttämaa, Chem. Rev., 2013, 113, 6949–6985 CrossRef CAS PubMed.
-
(a) L. A. B. Moraes and M. N. Eberlin, J. Am. Soc. Mass Spectrom., 2000, 11, 697–704 CrossRef CAS PubMed;
(b) L. A. B. Moraes and M. N. Eberlin, J. Am. Chem. Soc., 1998, 120, 11136–11143 CrossRef CAS.
-
(a) S. Tojo, S. Yasui, M. Fujitsuka and T. Majima, J. Org. Chem., 2006, 71, 8227–8232 CrossRef CAS PubMed;
(b) C. A. Marquez, H. Wang, F. Fabbretti and J. O. Metzger, J. Am. Chem. Soc., 2008, 130, 17208–17209 CrossRef CAS PubMed;
(c) L. A. Rios, W. R. Dolbier, R. Paredes, J. Lusztyk, K. U. Ingold and M. Jonsson, J. Am. Chem. Soc., 1996, 118, 11313–11314 CrossRef CAS;
(d) F. Widjaja, Z. Jin, J. J. Nash and H. I. Kenttämaa, J. Am. Chem. Soc., 2012, 134, 2085–2093 CrossRef CAS PubMed;
(e) B. B. Noble and M. L. Coote, J. Polym. Sci., 2020, 58, 52–61 CrossRef CAS;
(f) M. L. Grimm, N. K. Suleman, A. N. Hancock, J. N. Spencer, T. Dudding, R. Rowshanpour, N. Castagnoli and J. M. Tanko, J. Am. Chem. Soc., 2020, 142, 2640–2652 CrossRef CAS PubMed;
(g) J. H. Horner and M. Newcomb, J. Org. Chem., 2007, 72, 1609–1616 CrossRef CAS PubMed.
-
(a) R. P. Kohin and P. G. Nadeau, J. Chem. Phys., 1966, 44, 691–694 CrossRef CAS;
(b) E. A. C. Lucken and C. Mazeline, J. Chem. Soc. A, 1966, 1074–1077 RSC;
(c) J. Sinclair, J. Chem. Phys., 1971, 55, 245–251 CrossRef CAS;
(d) L. Bonazzola, C. Iacona, J. P. Michaut and J. Roncin, J. Chem. Phys., 1980, 73, 4175–4177 CrossRef CAS;
(e) I. Janovský, W. Knolle, S. Naumov and F. Williams, Chem.–Eur. J., 2004, 10, 5524–5534 CrossRef PubMed.
-
(a) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed;
(b) X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC;
(c) N. M. Gallagher, H. Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS;
(d) F. Biaso, T. Cantat, N. Mézailles, L. Ricard, P. Le Floch and M. Geoffroy, Angew. Chem., Int. Ed., 2006, 45, 7036–7039 CrossRef CAS;
(e) M. K. Sharma, S. Chabbra, C. Wolper, H. M. Weinert, E. J. Reijerse, A. Schnegg and S. Schulz, Chem. Sci., 2022, 13, 12643–12650 RSC;
(f) X. Chen, L. L. Liu, S. Liu, H. Grützmacher and Z. Li, Angew. Chem., Int. Ed., 2020, 59, 23830–23835 CrossRef CAS PubMed;
(g) R. J. Andrews and D. W. Stephan, Chem.–Eur. J., 2020, 26, 7194–7198 CrossRef CAS PubMed.
-
(a) C.-X. Miao, J.-Q. Wang, B. Yu, W.-G. Cheng, J. Sun, S. Chanfreau, L.-N. He and S.-J. Zhang, Chem. Commun., 2011, 47, 2697 RSC;
(b) Y. Liu, M.-A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chem, 2019, 5, 1861–1870 CrossRef CAS;
(c) B. Rahman, K.-i. Kanbara, H. Akutsu, J.-i. Yamada and S. i. Nakatsuji, Polyhedron, 2007, 26, 2287–2290 CrossRef CAS;
(d) S. Aonuma, H. Casellas, C. Faulmann, B. Garreau de Bonneval, I. Malfant, P. Cassoux, P. G. Lacroix, Y. Hosokoshi and K. Inoue, J. Mater. Chem., 2001, 11, 337–345 RSC;
(e) T. Hirashita, M. Nakanishi, T. Uchida, M. Yamamoto, S. Araki, I. W. C. E. Arends and R. A. Sheldon, ChemCatChem, 2016, 8, 2704–2709 CrossRef CAS;
(f) N. Jayaraj, M. Porel, M. F. Ottaviani, M. V. S. N. Maddipatla, A. Modelli, J. P. Da Silva, B. R. Bhogala, B. Captain, S. Jockusch, N. J. Turro and V. Ramamurthy, Langmuir, 2009, 25, 13820–13832 CrossRef CAS.
-
(a) T. Weiske, H. V. D. Wel, N. M. M. Nibbering and H. Schwarz, Angew. Chem., 1984, 96, 694–695 CrossRef CAS;
(b) P. Mourgues, H. E. Audier, D. Leblanc and S. Hammerum, Org. Mass Spectrom., 1993, 28, 1098–1100 CrossRef CAS;
(c) A. E. P. M. Sorrilha, F. C. Gozzo, R. S. Pimpim and M. N. Eberlincor, J. Am. Soc. Mass Spectrom., 1996, 7, 1126–1137 CrossRef CAS PubMed.
- Z. Li, X. Chen, D. M. Andrada, G. Frenking, Z. Benkö, Y. Li, J. R. Harmer, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 5744–5749 CrossRef CAS PubMed.
- P. Coburger, C. Schweinzer, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2023, 62, e202214548 CrossRef CAS PubMed.
- Z. Li, Y. Hou, Y. Li, A. Hinz and X. Chen, Chem.–Eur. J., 2018, 24, 4849–4855 CrossRef CAS PubMed.
-
(a) A. Hinz, A. Schulz, W. W. Seidel and A. Villinger, Inorg. Chem., 2014, 53, 11682–11690 CrossRef CAS PubMed;
(b) L. Chojetzki, A. Schulz, A. Villinger and R. Wustrack, Z. Anorg. Allg. Chem., 2020, 646, 614–624 CrossRef CAS;
(c) T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed.
-
(a) Z. Li, Y. Hou, Y. Li, A. Hinz, J. R. Harmer, C.-Y. Su, G. Bertrand and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 198–202 CrossRef CAS PubMed;
(b) X. Chen, A. Hinz, J. R. Harmer and Z. Li, Dalton Trans., 2019, 48, 2549–2553 RSC;
(c) L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259–267 CrossRef CAS;
(d) A. Brückner, A. Hinz, J. B. Priebe, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 7426–7430 CrossRef PubMed;
(e) Y. Su, X. Zheng, X. Wang, X. Zhang, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 6251–6254 CrossRef CAS PubMed;
(f) X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561–5564 CrossRef CAS PubMed;
(g) X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed;
(h) G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed;
(i) S. Ishida, F. Hirakawa and T. Iwamoto, J. Am. Chem. Soc., 2011, 133, 12968–12971 CrossRef CAS PubMed;
(j) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed;
(k) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
-
(a) X. Zhang, X. Chen, H. Zhai, S. Liu, C. Hu, L. L. Liu, S. Wang and Z. Li, Dalton Trans., 2020, 49, 6384–6390 RSC;
(b) X. Chen, C. Hu, X. Zhang, S. Liu, Y. Mei, G. Hu, L. L. Liu, Z. Li and C.-Y. Su, Inorg. Chem., 2021, 60, 5771–5778 CrossRef CAS PubMed;
(c) C. C. Chong, B. Rao, R. Ganguly, Y. Li and R. Kinjo, Inorg. Chem., 2017, 56, 8608–8614 CrossRef CAS PubMed.
-
M. J. Frisch, et al., Gaussian 16, Revision C.01, 2016 Search PubMed.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
-
(a) M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7578–7582 CrossRef CAS PubMed;
(b) C. Jones and M. Waugh, Dalton Trans., 2004, 1971–1979 RSC;
(c) L. E. Longobardi, C. A. Russell, M. Green, N. S. Townsend, K. Wang, A. J. Holmes, S. B. Duckett, J. E. McGrady and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 13453–13457 CrossRef CAS PubMed;
(d) S. Wang, K. Samedov, S. C. Serin and D. P. Gates, Eur. J. Inorg. Chem., 2016, 2016, 4144–4151 CrossRef CAS.
-
(a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed;
(b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 2, S1–S19 RSC.
-
(a) T. Völzer, H. Beer, A. Schulz, S. Lochbrunner and J. Bresien, Phys. Chem. Chem. Phys., 2021, 23, 7434–7441 RSC;
(b) H. Beer, J. Bresien, D. Michalik, A. Schulz and A. Villinger, Dalton Trans., 2020, 49, 13986–13992 RSC;
(c) J. Bresien, T. Kröger-Badge, S. Lochbrunner, D. Michalik, H. Müller, A. Schulz and E. Zander, Chem. Sci., 2019, 10, 3486–3493 RSC;
(d) A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 2776–2779 CrossRef CAS PubMed;
(e) A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed.
- Z.-Y. Wang, Y.-Z. Dai, L. Ding, B.-W. Dong, S.-D. Jiang, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2021, 60, 4594–4598 CrossRef CAS PubMed.
- P. Agarwal, N. A. Piro, K. Meyer, P. Müller and C. C. Cummins, Angew. Chem., Int. Ed., 2007, 46, 3111–3114 CrossRef CAS PubMed.
-
(a) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed;
(b) H. M. Nelson, J. C. Siu, A. Saha, D. Cascio, S. N. MacMillan, S.-B. Wu, C. Lu, J. A. Rodríguez, K. N. Houk and S. Lin, Org. Lett., 2021, 23, 454–458 CrossRef CAS PubMed;
(c) J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140, 12511–12520 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, and crystallographic and computational details. CCDC 2215136–2215138, 2215140–2215143 and 2314776. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00201f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Hansjörg
Grützmacher
b,
Hansjörg
Grützmacher