
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-temperature synthesis of cation-ordered bulk Zn3WN4 semiconductor via heterovalent solid-state metathesis†
Christopher L.
Rom
 *a,
Shaun
O'Donnell
ab,
Kayla
Huang
ac,
Ryan A.
Klein
ad,
Morgan J.
Kramer
de,
Rebecca W.
Smaha
a and
Andriy
Zakutayev
*a
*a,
Shaun
O'Donnell
ab,
Kayla
Huang
ac,
Ryan A.
Klein
ad,
Morgan J.
Kramer
de,
Rebecca W.
Smaha
a and
Andriy
Zakutayev
*a
aMaterials, Chemical, and Computational Science, National Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: christopher.rom@nrel.gov; andriy.zakutayev@nrel.gov
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA
cUniversity of Illinois Urbana-Champaign, Champaign, IL 61801, USA
dCenter for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA
eDepartment of Chemistry, Southern Methodist University, Dallas, TX 75275, USA
First published on 15th May 2024
Abstract
Metathesis reactions are widely used in synthetic chemistry. While state-of-the-art organic metathesis involves highly controlled processes where specific bonds are broken and formed, inorganic metathesis reactions are often extremely exothermic and, consequently, poorly controlled. Ternary nitrides offer a technologically relevant platform for expanding synthetic control of inorganic metathesis reactions. Here, we show that energy-controlled metathesis reactions involving a heterovalent exchange are possible in inorganic nitrides. We synthesized Zn3WN4 by swapping Zn2+ and Li+ between Li6WN4 and ZnX2 (X = Br, Cl, F) precursors. The in situ synchrotron powder X-ray diffraction and differential scanning calorimetry show that the reaction onset is correlated with the ZnX2 melting point and that product purity is inversely correlated with the reaction's exothermicity. Therefore, careful choice of the halide counterion (i.e., ZnBr2) allows the synthesis to proceed in a swift but controlled manner at a surprisingly low temperature for an inorganic nitride (300 °C). High resolution synchrotron powder X-ray diffraction and diffuse reflectance spectroscopy confirm the synthesis of a cation-ordered Zn3WN4 semiconducting material. We hypothesize that this synthesis strategy is generalizable because many Li–M–N phases are known (where M is a metal) and could therefore serve as precursors for metathesis reactions targeting new ternary nitrides. This work expands the synthetic control of inorganic metathesis reactions in a way that will accelerate the discovery of novel functional ternary nitrides and other currently inaccessible materials.
Introduction
Metathesis is a powerful synthetic tool used across a wide range of chemistries. In metathesis reactions, a desired product BQ is synthesized following the general form, AQ + BX → BQ + AX, where the units A and B trade partners and AX is an (ideally separable) byproduct. Initial reports of metathesis reactions date back to 1925, where binary sulfides were synthesized via inorganic solid-state reactions: e.g., ZnS + CdO → CdS + ZnO.1 Subsequently, catalyzed olefin metathesis was discovered in the 1950's, and decades of work led to deep mechanistic understanding, expansive synthetic control, and ultimately, to the 2005 Nobel Prize in Chemistry.2–6 Presently, the technique enables synthetic precision for organic and organometallic chemistry,7–11 metal–organic-frameworks,12–14 polymers,15–18 and beyond. In contrast, research on inorganic solid-state metathesis mostly focused on rapid, highly exothermic syntheses for binary compounds in the 1990's and early 2000's.19 Despite the historic head-start for inorganic solid-state metathesis, synthetic control is nascent.20,21Ternary nitrides provide a prime set of materials for expanding the synthetic control of metathesis reactions. Ternary nitrides are a promising class of semiconductors,22 yet relatively few are known. This dearth of nitrides is primarily due to the synthetic challenges of realizing these materials from elemental metal (or binary) precursors and dinitrogen gas.22–25 Molecular (di)nitrogen, N2, is highly stable, and high temperatures are needed to break the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond (945 kJ mol−1).26 High temperatures (>800 °C) are also needed to drive diffusion, as nitrides tend to have high cohesive energies (i.e., strong M–N bonds) and slow diffusion.27–29 Moreover, entropic penalties disfavor nitride incorporation in solids at high temperatures (i.e., gaseous N2 is favored). Finding a synthesis temperature that is hot enough for reactivity but cool enough to avoid decomposition is therefore challenging. Adding to the difficulty, O2 is more reactive towards most metals than N2, so syntheses must be conducted in rigorously air-free conditions to avoid the formation of oxide impurities. Consequently, the number of known ternary nitrides lags behind the ternary oxides by an order of magnitude.22–25 Developing new synthesis methods (e.g., controlled metathesis reactions) will help narrow this disparity, and in doing so, discover new materials upon which improved technologies can be built.
N triple bond (945 kJ mol−1).26 High temperatures (>800 °C) are also needed to drive diffusion, as nitrides tend to have high cohesive energies (i.e., strong M–N bonds) and slow diffusion.27–29 Moreover, entropic penalties disfavor nitride incorporation in solids at high temperatures (i.e., gaseous N2 is favored). Finding a synthesis temperature that is hot enough for reactivity but cool enough to avoid decomposition is therefore challenging. Adding to the difficulty, O2 is more reactive towards most metals than N2, so syntheses must be conducted in rigorously air-free conditions to avoid the formation of oxide impurities. Consequently, the number of known ternary nitrides lags behind the ternary oxides by an order of magnitude.22–25 Developing new synthesis methods (e.g., controlled metathesis reactions) will help narrow this disparity, and in doing so, discover new materials upon which improved technologies can be built.
Zn-containing ternary nitrides epitomize the promising applications and synthetic challenges of this class of materials. Fully nitridized compounds like ZnSnN2 and Zn3WN4 (with metals in the highest oxidation state) are of interest as semiconductors for their high earth abundance and tunable bandgaps (spanning ca. 1 eV for ZnSnN2 to 4 eV for Zn3WN4).30,31 However, the bulk synthesis techniques that have been reported for Zn–M–N phases are limited to traditional ceramic methods (i.e., metals + N2 or NH3 at high temperatures) or high-pressure solid state metathesis reactions (e.g., 2 Li3N + ZnF2 + SnF4 → ZnSnN2 + 6 LiF).32,33 These bulk methods have only produced fully nitridized phases when M is a main group element (i.e., LiZnN, Ca2ZnN2, Sr2ZnN2, Ba2ZnN2, ZnSiN2, ZnGeN2, ZnSnN2).28,30–40 When transition metals are used in bulk syntheses, they tend to form sub-nitrides: e.g., Ti3ZnN0.5, V3Zn2N, Ti2ZnN, Mn3ZnN, and Fe3ZnN.41–46 The nitrogen-poor nature of these materials stems from the challenges described above (i.e., N2 stability, slow diffusion). Synthesizing fully-nitridized Zn–M–N (where M is a transition metal) in bulk would advance technologies in which thin film nitrides have already shown promise, like photoelectrochemical energy conversion (ZnTiN2),47 transparent conducting oxides (ZnZrN2),48 and non-linear optics (Zn3WN4).49
Synthesizing Zn–M–N ternary nitrides via traditional methods is difficult. Many transition metals are highly refractory, meaning high temperatures would likely be needed for interdiffusion of reactants. However, Zn has a low melting point (419 °C) and a relatively low boiling point (907 °C),50 meaning that high temperatures would volatilize Zn away from the other metal unless special measures were taken (e.g., high pressure, closed vessels). Forming binary nitrides to use as precursors instead of metals is also challenging: Zn (like other late-transition metals) does not react with N2 at elevated temperatures, so Zn3N2 must be synthesized under ammonia.51 And as noted in thin film work, fully nitridized transition metal Zn–M–N phases have low decomposition temperatures on the order of 600–700 °C.24,47,48,52 These challenges mean that bulk synthesis of Zn–M–N from the elements or binaries would likely proceed only at low temperatures and extremely slowly, unless special high-pressure methods were employed (e.g., ammonothermal synthesis,33 diamond anvil cell synthesis,53etc.).
Metathesis reactions are one promising way to circumvent the challenge of diffusion in the solid state.54 To synthesize nitrides, this strategy starts with one nitrogen-containing precursor and one halide precursor, rather than elements or binary nitrides. The balanced reaction targets the desired phase along with a byproduct (often a halide salt). The formation of this byproduct provides a large thermodynamic driving force for the reaction and (ideally) can be washed away post-reaction. For example, Kaner et al. showed that mixing Li3N with metal chlorides would produce LiCl in highly exothermic metathesis reactions that yielded a range of binary nitrides55–63 and some ternary nitrides.64,65 Alternatively, less exothermic reactions can be conducted with greater synthetic control,66–70 including low-temperature topotactic reactions (Trxnca. 200–400 °C).71–73 As for Zn–M–N compounds, ZnSnN2 and ZnSiN2 have been made using high pressure metathesis reactions, where the pressure is necessary to the loss of avoid gaseous N2.32,40 Metathesis is well known for “turning down the heat” in solid-state synthesis74 but is underutilized for synthesizing nitrides.
Here, we synthesize Zn3WN4via a near-topotactic metathesis reaction between Li6WN4 and ZnX2 (X = Br, Cl, F) at 300 °C and low pressure. In situ synchrotron powder X-ray diffraction (SPXRD) paired with differential scanning calorimetry (DSC) measurements reveal the reaction pathways and show that using a ZnBr2 precursor is preferable over the fluoride or chloride analogs. High resolution SPXRD measurements indicate that the Zn3WN4 product is a mostly cation-ordered structure in space group Pmn21. We report some preliminary property characterizations for Zn3WN4, revealing optical absorption onsets near 2.5 eV and 4.0 eV, as well as paramagnetism consistent with some degree of disorder and off-stoichiometry. The reaction is near-topotactic, in that the structures of the Li6WN4 precursor and the Zn3WN4 product are related by a shift in anion layers but the [WN4] tetrahedral unit is preserved. Using this synthesis approach, we also synthesized Zn3MoN4, albeit with lower levels of purity in our un-optimized reactions. This work demonstrates the viability of Li–M–N phases as metathesis precursors to synthesize other ternary nitride compounds, expanding the toolkit for materials discovery.
Results and discussion
In situ SPXRD measurements
Zn3WN4 was successfully synthesized via metathesis reactions. The net reaction is:| Li6WN4 + 3 ZnX2 → Zn3WN4 + 6 LiX (X = Br, Cl, F) |
In situ SPXRD measurements reveal that Li6WN4 (synthesized by a ceramic method, Fig. S1†) directly converts to Zn3WN4via metathesis without intermediate nitride phases or solid solution behavior as a function of temperature (Fig. 1, S2 and S3†). However, the halide precursor exerts an influence on the reaction kinetics and thermodynamics, which ultimately impact the reaction pathway and final product purity.
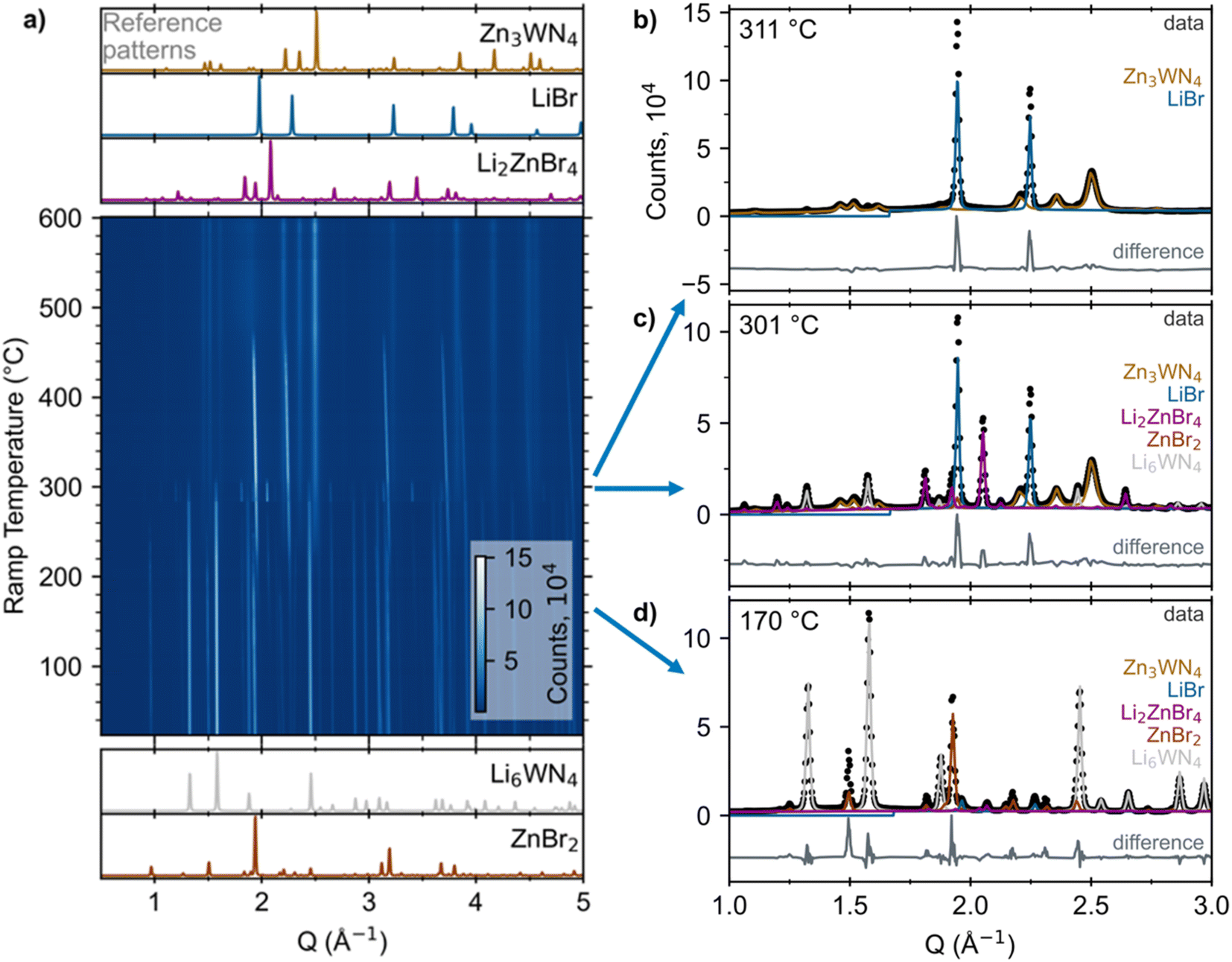 | ||
| Fig. 1 (a) Heatmap of in situ SPXRD data upon heating a sealed capillary of 3 ZnBr2 + Li6WN4 at +10 °C min−1. Reference patterns for the reactants and products/intermediates are simulated at the bottom and top, respectively (ICSD Col. Codes 30803 for ZnBr2, 66096 for Li6WN4, 73223 for Li2ZnBr4, and 53819 for LiBr).75–77 Analogous plots of the ZnCl2 and ZnF2 reactions are in Fig. S2 and S3.† Select patterns and fits are shown for ramp temperatures of (b) 311 °C, (c) 301 °C, and (d) 170 °C. Contributions from each phase (determined via Rietveld fitting) are displayed as colored lines. Difference traces are offset for clarity. | ||
In situ SPXRD measurements reveal that the reaction pathway proceeds without intermediate nitrides between Li6WN4 and Zn3WN4. Fig. 1a shows a heatmap for X = Br as an example; subsequent examination revealed that this anion leads to the most phase pure product. The reaction of Li6WN4 + 3 ZnBr2 → Zn3WN4 + 6 LiBr initiates near 170 °C and proceeds to completion within the 14 minutes of ramp time up to 310 °C (Fig. 1b). Near 170 °C, the Bragg peaks arising from crystalline Li6WN4 and ZnBr2 begin to gradually decline in intensity. Shortly thereafter, new sets of Bragg peaks that can be indexed to LiBr, Li2ZnBr4, and Zn3WN4 begin growing in intensity (Fig. 1d). The Bragg peaks corresponding to Li2ZnBr4 gradually decrease in intensity between 210 °C and 270 °C, increase dramatically in intensity at 275 °C, and then disappear entirely at 305 °C (Fig. 1c). Such fluctuations may stem from crystal nucleation and growth within the capillary, especially given the small spot size of the synchrotron X-ray beam, possibly combined with crystallite motion in a liquid-like medium. Diffraction images show spotty diffraction patterns, consistent with crystallite growth. These data indicate that the synthesis proceeds directly via Li6WN4 + 3 ZnBr2 → Zn3WN4 + 6 LiBr. While this process occurs, the metal halides also react with one another: 2 LiBr + ZnBr2 → Li2ZnBr4. We do not observe signs of a crystalline theoretically-predicted LiZn4W2N7 structure,78 although this does not rule out the presence or synthesizability of such a phase. Similar trends are noted with the ZnCl2 and ZnF2 reactions (Fig. S2 and S3†), as shown by sequential Rietveld analysis (Fig. 2).
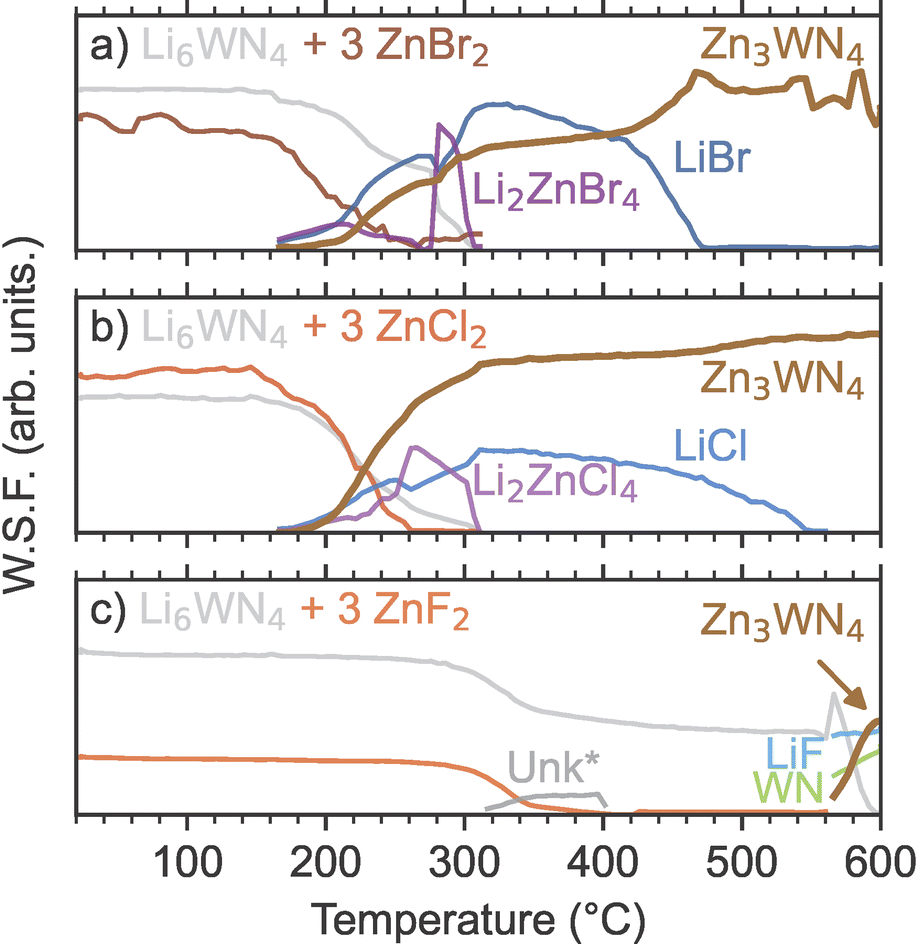 | ||
| Fig. 2 Sequential Rietveld analysis of in situ SPXRD patterns yields the weighted scale factors (W.S.F.), which are plotted as a function of temperature. The W.S.F. indicates the relative phase concentrations for the crystalline phases during reactions between Li6WN4 and (a) ZnBr2, (b) ZnCl2, and (c) ZnF2. One unknown phase (Unk*, likely a Li–Zn–F phase) is modeled using a peak fit to the most intense reflection, rather than full-pattern Rietveld analysis. | ||
Sequential Rietveld analysis of in situ variable temperature SPXRD measurements of the Li6WN4 + 3 ZnX2 reactions shows that the ZnBr2 and ZnCl2 reactions initiate at much lower temperatures than the ZnF2 reaction (Fig. 2). For both the ZnBr2 and ZnCl2 reactions (Fig. 2a and b), the concentrations of the precursor phases start decreasing near 170 °C, followed shortly thereafter by Zn3WN4 and LiX formation and growth. At the same time, ternary halides Li2ZnBr4 and Li2ZnCl4 form, and are then consumed or melt near 300 °C. In contrast, the precursors of the ZnF2-based reaction do not begin declining until over 300 °C (Fig. 2c). The concomitant decrease in Li6WN4 and ZnF2 suggests reactivity, but neither Zn3WN4 nor LiF are detected in our data at this temperature. Instead, very weak reflections for an unknown phase appear in the data (labeled as Unk*). This phase may be a Li–Zn–F ternary, but it does not index to any known ternary fluoride unit cells, including the reported Li2ZnF4 phase.79 An amorphous phase is likely present in the 400 to 570 °C region, given the decrease in precursor peaks and lack of new intermediate peaks. Zn3WN4 and LiF crystallize above 570 °C, along with a rocksalt phase (fit with WN, but the material may be a (Zn,W)Nx phase as observed with the Mo-based system, Fig. S4†). We did not study the ZnF2 reactions further, given that phase-pure Zn3WN4 did not crystallize and given the challenge associated with washing away LiF from the product. Instead, we focus on the ZnBr2 and ZnCl2 reactions.
DSC measurements
DSC was employed to study the ZnBr2- and ZnCl2-based reactions, with reaction mixtures sealed in aluminum pans under argon for the measurements. The lower exothermicity of the ZnBr2-based reaction leads to a more controlled release of heat and greater product purity, compared to the ZnCl2-based reaction. DSC measurements show that the ZnBr2 reaction has three small exotherms (Fig. 3a). A gradual exotherm starts near 190 °C (a-i), followed by two exotherms near 305 °C (a-ii) and 334 °C (a-iii). Peak a-i is likely a slow, solid–solid reaction of Li6WN4 + 3 ZnBr2 → Zn3WN4 + 6 LiBr, consistent with the in situ SPXRD measurement. Peak a-ii may be the reaction 2 LiBr + ZrBr2 → Li2ZnBr4. Li2ZnBr4 melts at 326 °C,77 so peak a-iii is likely a rapid completion of the Zn3WN4 formation facilitated by the presence of a liquid phase. This kind of rapid exothermic event is common in metathesis reactions; once a liquid phase forms, reaction kinetics accelerate and the heat release self-propagates.57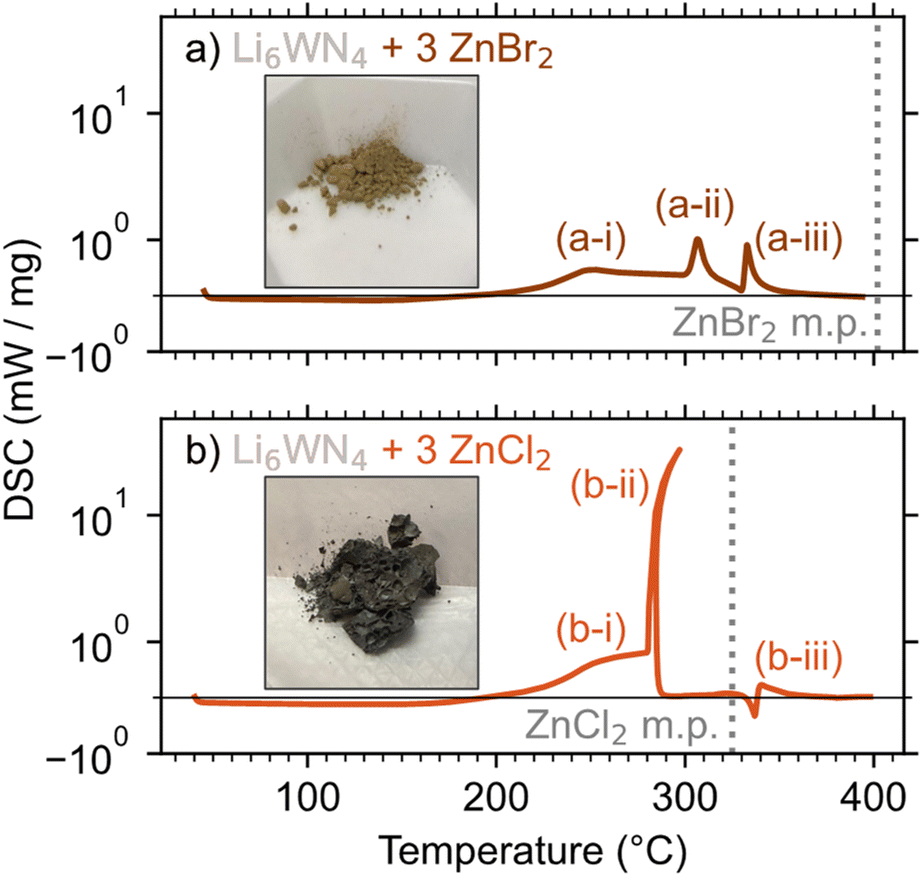 | ||
| Fig. 3 DSC measurements of Li6WN4 reacting with (a) 3 ZnBr2 and (b) 3 ZnCl2. Inset photos show the products from reactions heated at 300 °C for 1 h. | ||
The ZnCl2 reaction (Fig. 3b) starts similarly, with a gradual exotherm between 190 °C and 280 °C (b-i), a solid–solid reaction yielding Zn3WN4 and LiCl. Then at ∼280 °C, a massive exotherm (b-ii) initiates just below the melting point of ZnCl2 (325 °C). This event likely corresponds to the formation of a liquid phase, such as a LiCl–ZnCl2 eutectic (275 °C at 78% ZnCl2 and 287 °C at 91% ZnCl2).80 Peak b-ii has curvature because this event releases heat so quickly that the DSC stage increases in temperature by approximately 15 °C, after which the DSC pan cools slightly (Fig. S5†). Lastly, a small endotherm is observed at 336 °C (b-iii), consistent with the melting of Li2ZnCl4. These results are broadly consistent with the in situ SPXRD results, albeit shifted slightly in temperature owing to differences in experimental configuration.
These DSC results show why the ZnBr2 reaction yields the purest product while the ZnCl2 reaction exhibits a small Zn impurity. The rapid release of heat in the ZnCl2 reaction causes small portions of the material to decompose: Zn3WN4 → W + 3 Zn + 2 N2 or Zn3WN4 → WN + 3 Zn + 3/2 N2 (Fig. S6†). In contrast, the washed product of the ZnBr2 synthesis yielded a PXRD pattern with all Bragg peaks indexed to Pmn21 Zn3WN4. These differences can easily be seen in the color of the material (see insets, Fig. 3), where Zn impurities in the ZnCl2 reaction led to a grey color. The Zn3WN4 sample produced by the ZnBr2 reaction is light brown and was phase pure (as discussed subsequently). These differences stem from both thermodynamic and kinetic factors.
Thermodynamic and kinetic factors
Zn3WN4 is calculated to be thermodynamically stable at moderate temperatures, so differences in product purity reflect differences in kinetic control for the ZnX2 reactions. For each halide, the reactions are calculated to be enthalpically driven (i.e., negative ΔHrxn, Fig. 4). Furthermore, chemical potential diagrams show that Li6WN4 and Zn3WN4 share a border in chemical potential space (Fig. S7†), indicating that diffusion can occur between these phases without nucleating an intermediate phase at the interface,81 as previously shown for metathesis reactions towards ternary oxides and nitrides.70,82 However, Zn3WN4 is only stable down to a nitrogen chemical potential of μN = −0.26 eV when at 300 °C (Fig. S7a and b†), whereas these reactions were conducted in evacuated ampules (p < 0.03 torr, μN < −0.5 eV). This means thermodynamics favor some N2 (g) evolution from the reaction (with Zn and W as decomposition products). Therefore, the fact that the ZnBr2 reaction proceeds without detectable decomposition means the reaction is fully under kinetic control, while the ZnCl2 and ZnF2 reactions are not.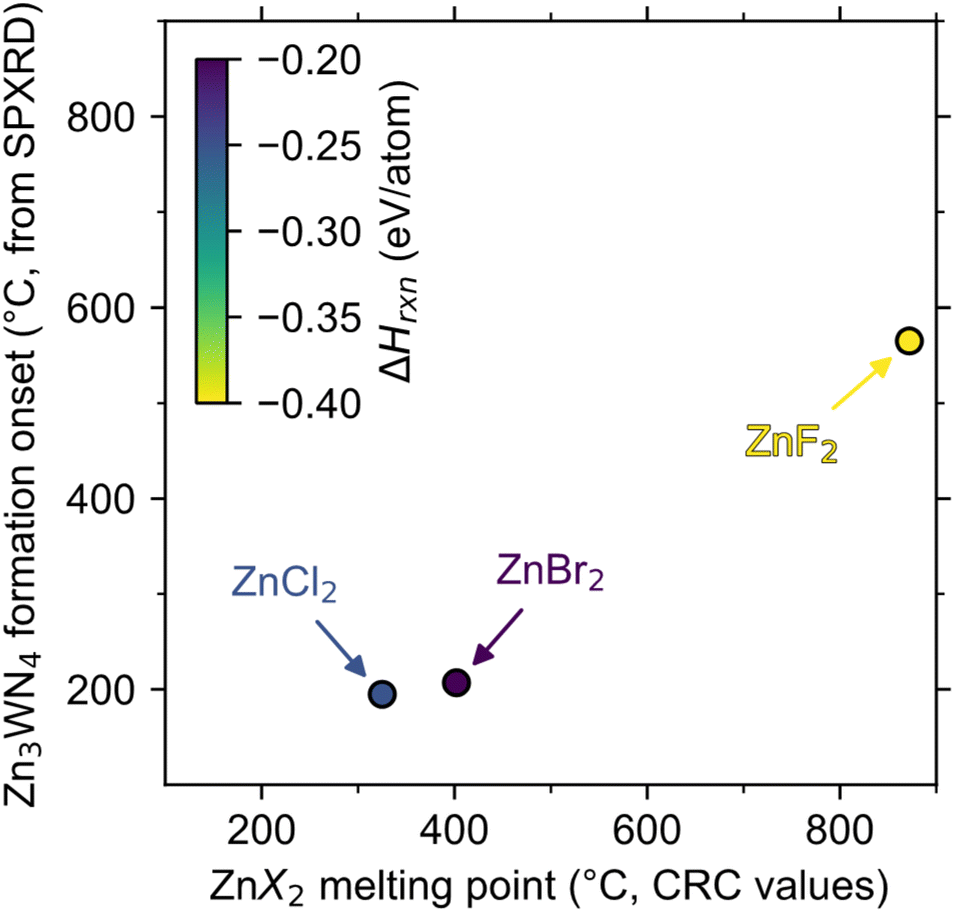 | ||
| Fig. 4 The initial formation of Zn3WN4 (as detected by in situ SPXRD) is correlated with the melting point of the halide salt (from CRC values)50 but is not correlated with calculated reaction enthalpy (ΔHrxn, color, from the materials project).84 The dashed line shows where melting point would equal onset temperature. | ||
The magnitude of the exothermicity combined with the melting points of the halide salts affect the degree of kinetic control. The lowest melting point we can identify in the LiBr–ZnBr2 system is 326 °C for Li2ZnBr4,77 meaning the reaction may be controlled by solid-state diffusion below this point. Additionally, the ΔHrxn of the ZnBr2 reaction (−0.20 eV per atom) is less exothermic than the ZnCl2 reaction (−0.25 eV per atom), which may limit local self-heating and avoid decomposition (Fig. 3a). In contrast, the liquidus line in the LiCl–ZnCl2 system extends as low as 275 °C,80 at which point diffusion accelerates rapidly (Fig. 3b, peak b-ii). Even if the temperature is kept lower (e.g., 250 °C), partial Zn3WN4 decomposition is observed in the ZnCl2 system (Fig. S6†). This decomposition indicates a loss of kinetic control. The ZnF2 reaction is worse. The high melting point of ZnF2 (872 °C)50 means the kinetics are sluggish until the reaction is perilously close to the decomposition temperature (estimated near 700 °C, Fig. S7c and d†), at which point the self-heating from the high exothermicity (ΔHrxn = −0.40 eV per atom)83 likely drives substantial decomposition (Fig. 2c). In contrast, the ZnBr2-based reaction retains kinetic control as the moderate melting point of Li–Zn–Br phases enable diffusion near 300 °C while the low ΔHrxn prevents excessive self-heating, thereby avoiding decomposition.
This type of reaction control has been explored in oxides but is less well studied for ternary nitrides. “Spectator ions” that are not incorporated into the final product still have substantial influence over reaction pathways and polymorph formation, as demonstrated for syntheses of Y–Mn–O phases.21,82,85–88 In particular, work on “co-metathesis” identified that when eutectic halide mixtures form in situ, these liquids decrease reaction onset temperatures relative to systems without eutectics.85,86 Similar eutectics are likely forming between ZnX2 and LiX in our syntheses of Zn3WN4. This thermodynamic analysis, along with in situ SPXRD and DSC measurements, guided our optimization of the synthesis for Zn3WN4.
Structural and composition analysis of Zn3WN4
The best conditions we found for the synthesis of Zn3WN4 were heating ZnBr2 with Li6WN4 at a ramp of +5 °C min−1 to 300 °C for a 1 h dwell, followed by natural cooling in the furnace. This reaction was scaled up to ca. 1 g of reactant mix for ex situ analysis. Washing with anhydrous methanol successfully removed byproduct LiBr and excess ZnBr2 while preserving the targeted phase. We subsequently learned that Zn3WN4 does not appear to be moisture or air sensitive, so washing with water may be viable in future work. We used a slight excess of ZnBr2 (3.1 ZnBr2 + Li6WN4) to ensure complete conversion and minimize reaction temperature by acting as a heat sink. XRF measurements show a Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W ratio of 3.165(3):1, slightly higher than the expected 3:1 ratio of Zn3WN4 (representative XRF spectrum shown in Fig. S8†), which may be a result of the excess ZnBr2. PXRD techniques confirmed that this synthesis of Zn3WN4 proceeded without the formation of decomposition products (i.e., Zn, (Zn,W)Nx phases).
:W ratio of 3.165(3):1, slightly higher than the expected 3:1 ratio of Zn3WN4 (representative XRF spectrum shown in Fig. S8†), which may be a result of the excess ZnBr2. PXRD techniques confirmed that this synthesis of Zn3WN4 proceeded without the formation of decomposition products (i.e., Zn, (Zn,W)Nx phases).
High resolution SPXRD measurements confirm the successful synthesis of Zn3WN4 (Fig. 5). Rietveld analysis of the SPXRD data (Fig. 5a) shows that Zn3WN4 crystallizes in space group Pmn21 with lattice parameters a = 6.5602(8) Å, b = 5.6813(7) Å, and c = 5.3235(2) Å. The presence of intensity at the (010), (110), (101), and (011) Bragg positions indicates a substantial degree of cation ordering (Fig. 5c and f). The peaks for the (210), (002), and (211) reflections are characteristic of wurtzite-derived structures; these correspond to the (100), (002), and (101) reflections in the prototypical wurtzite structure (P63mc), respectively (Fig. 5b and e). Rietveld-refined occupancies suggest a Zn:W ratio of 3.8:1, a higher ratio than that measured by XRF (3.2:1), with partial occupancy of Zn on the W site (Table S1†), indicating a composition of Zn3.17W0.83N4 (Fig. 5d). The occupancies of the N atoms refined to 1 within error and so were fixed at unity. Alternative structural models were also considered, as discussed further in the ESI (Table S2 and Fig. S9–S11).† We selected the single-phase model shown in Fig. 5 as it is the simplest model that effectively describes both the diffraction data (presented here) and the optical data (discussed subsequently).
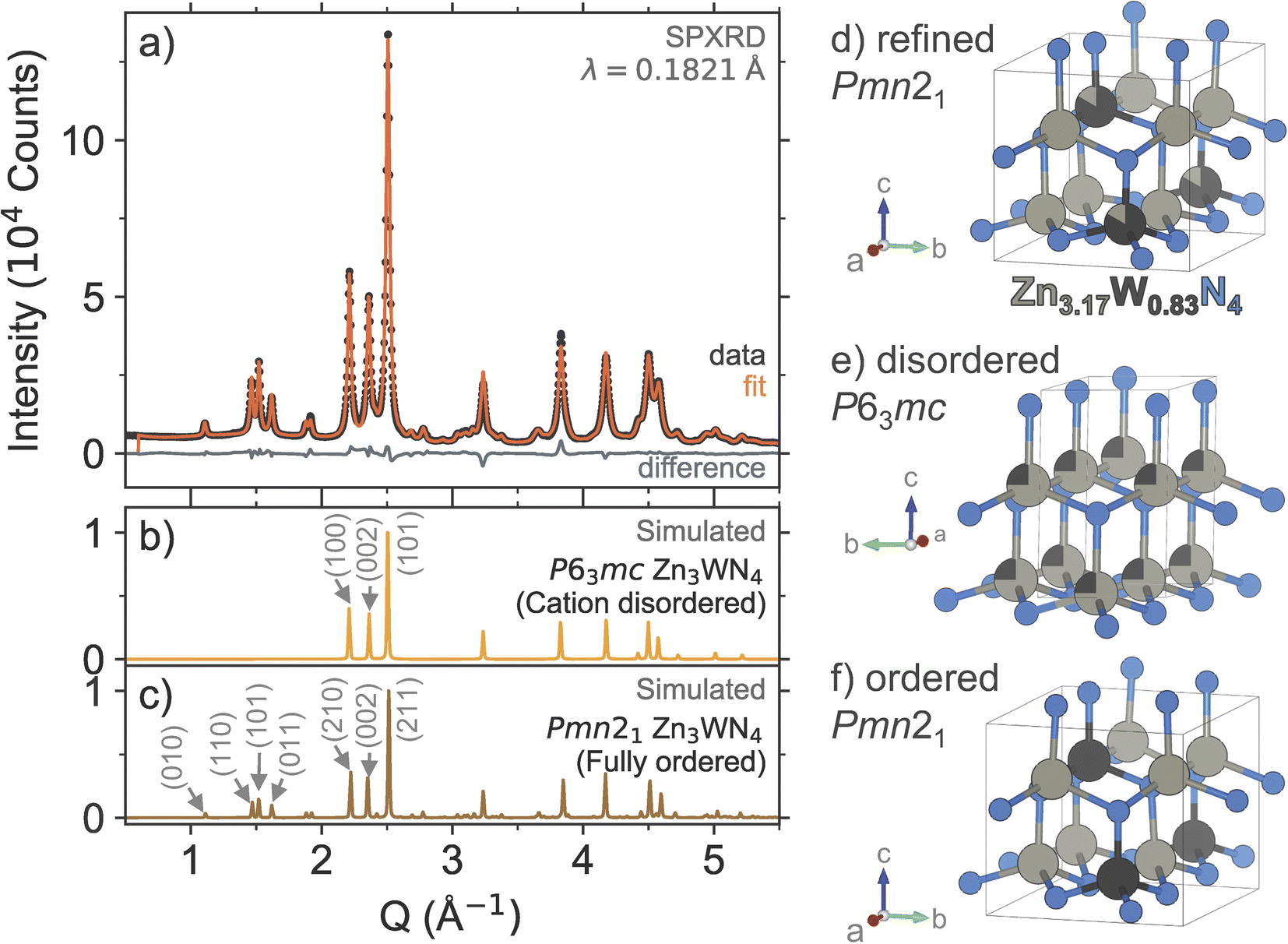 | ||
| Fig. 5 (a) High resolution SPXRD data (black dots) and Rietveld refinement (orange trace) of Zn3WN4 powder. Simulated patterns are shown for reference: (b) the cation-disordered P63mc model and (c) the fully ordered Pmn21 model. Visualizations of (d) the Pmn21 structure of Zn3WN4 refined from the SPXRD data, (e) the cation-disordered P63mc model, and (f) the fully ordered Pmn21 model. | ||
Our metathesis approach yielded a different polytype for Zn3WN4 compared to prior thin film syntheses. Metathesis between Li6WN4 and ZnBr2 successfully synthesized Zn3WN4 in space group Pmn21 (Fig. 5). In contrast, prior thin film sputtering work produced cation-disordered P63mc structures.23,52,89 While both the Pmn21 and P63mc structures are wurtzite-derived, the cation-ordered structure is expected to be the thermodynamic ground state.23,48 In thin film sputtering, high-energy plasma precursors deposit onto a substrate and quench rapidly in a local energy minimum, thus locking in the disordered cation arrangement.48 While bulk syntheses can sometimes lead to cation-disordered structures,28,32,66,67,70 the high charge on W(6+) likely encourages ordering to maximize the spacing between the hexavalent cations. The reaction pathway here proceeds in a way that avoids the local energy minimum of the disordered structure, instead forming a (mostly) ordered structure. This aspect of the bulk synthesis may be due to the ordered nature of W6+ in the Li6WN4 precursor. Further work is needed to assess the influence of reaction conditions (e.g., precursor ratios, halide choice, heating profiles, etc.) on cation ordering, as the degree of cation ordering affects the optical properties of the material.
Property measurements
Diffuse reflectance spectroscopy measurements reveal two absorption onsets for Zn3WN4: one near 2.5 eV and another near 4.0 eV (Fig. 6). The absorption feature near 4.0 eV is consistent with the expected bandgap for long-range cation-ordered Zn3WN4 (i.e., the Pmn21 space group). The GW-calculated indirect band gap for this phase is 3.96 eV, with a direct bandgap of 4.20 eV (NREL MatDB ID 287103; blue trace in Fig. 6).90,91 Other researchers using a hybrid functional, HSE06, calculated the bandgap to be 3.60 eV.49 The absorption feature near 2.5 eV is similar to the 2.0 eV to 2.4 eV absorption onset reported for cation disordered Zn3MoN4 and Zn3WN4 synthesized as thin films.52,89 Computational work on similar materials has shown that localized, mid-gap electronic states arise when N atoms have Zn-rich coordination.92 These two optical features are consistent with the Rietveld analysis of our Zn3WN4 powder sample (Fig. 5): long-range cation order is present (leading to the 4.0 eV absorption) along with some Zn anti-site defects on the W position (which lead to Zn-rich coordination for N and the 2.5 eV absorption). Magnetic susceptibility measurements are consistent with the presence of a paramagnetic impurity, as the material does not exhibit purely diamagnetic behavior (Fig. S12†). We note that the same batch of Zn3WN4 was used for the diffuse reflectance optical spectroscopy, magnetometry, XRF, and the high resolution SPXRD measurements.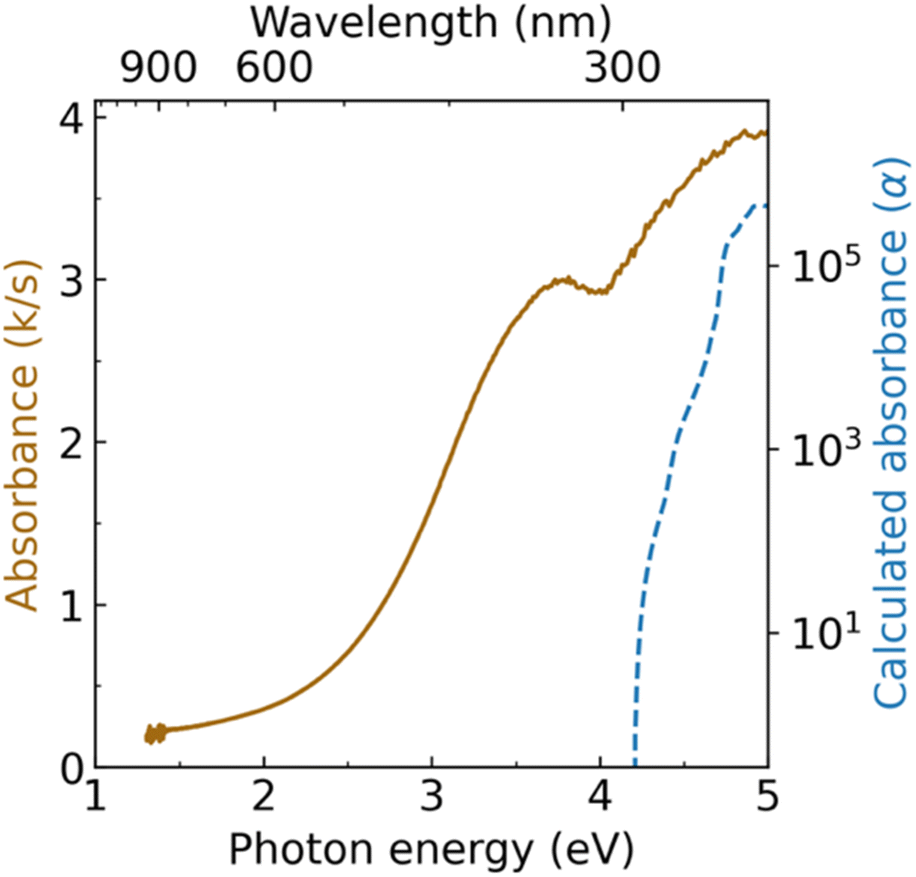 | ||
| Fig. 6 Diffuse reflectance spectrum of Zn3WN4 (solid brown trace) compared with GW-calculated absorbance (dashed blue trace). | ||
Structural relation of precursor and product
The transformation from Li6WN4 to Zn3WN4 involves slight structural rearrangements (Fig. 7). The W6+ retains its tetrahedral coordination and (for the most part) its oxidation state through the process, but the orientations of the polyhedra change. The fcc anion lattice of Li6WN4 converts to the hcp anion lattice of Zn3WN4. Visual inspection of the structures using VESTA software93 shows that one likely path for this transformation is for half of the W6+ ions in Li6WN4 to migrate through an octahedral site to a new tetrahedral site in Zn3WN4 (red annotations). However, various migration pathways may be occurring during the synthesis (e.g., W-migration between anion layers). Zn3WN4 has broader peaks in SPXRD data than Li6WN4 (Fig. 1 and 5), indicating shorter crystalline domain lengths. That difference may arise from the W in different sections of the Li6WN4 crystallites migrating in alternative directions (Fig. 7a, dashed orange arrow). The anion packing layers also decrease in spacing from 2.767(1) Å in Li6WN4 to 2.662(1) Å in Zn3WN4. The shortest W–W distance decreases from 4.927(1) Å to 4.646(1) Å. Lastly, the centrosymmetric structure of Li6WN4 (P42/nmc) converts to a polar structure of Zn3WN4 (Pmn21). We did not observe any signs of solid solution behavior (i.e., Li6−xZnx/2WN4) in the in situ SPXRD studies. However, solid solution behavior may be present but undetected by the in situ SPXRD data if it occurs on short timescales (<30 s) or small length scales (ca. 10 nm).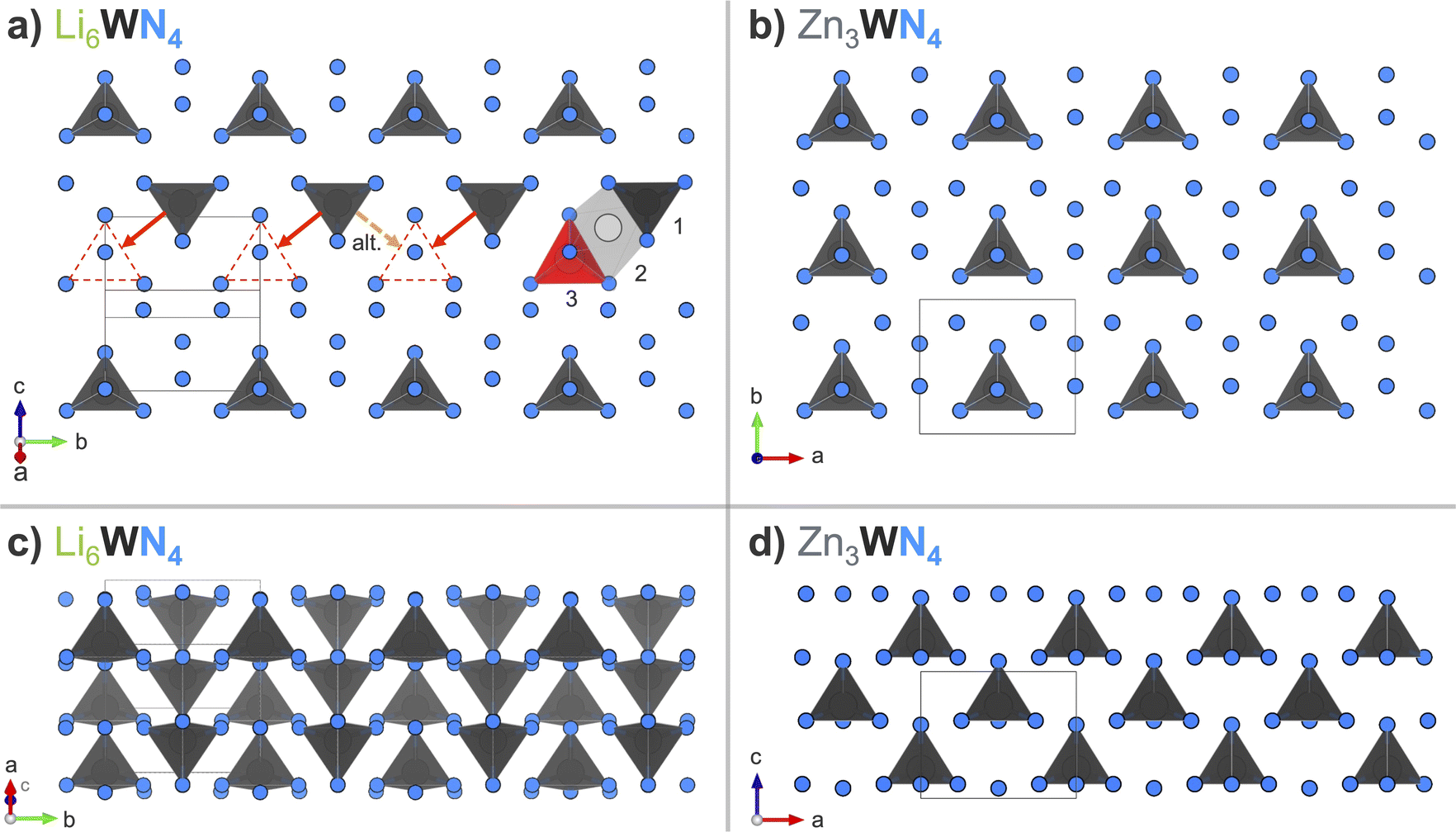 | ||
| Fig. 7 Arrangement of [WN4] tetrahedral units when looking down (a) on one layer of Li6WN4 (P42/nmc) in the (201) plane, and (b) on a layer of Zn3WN4 (Pmn21) in the (001) plane. Red annotations in (a) show the likely displacement undergone by some W6+ ions in the transition to Zn3WN4via a tetrahedral (1) to octahedral (2) to tetrahedral (3) sequence, while the orange dashed arrow shows an alternative migration direction. Side views of three layers of (c) Li6WN4 and (d) Zn3WN4 stacked along the [201] and [001] directions, respectively. The shading of the [WN4] units in (c) indicates depth. Li and Zn are omitted for clarity; they are shown in Fig. S13.† | ||
The synthesis reported here is distinct from literature on prior metathesis syntheses of nitrides in that the ions undergoing exchange have different formal charges. All prior reports on nitride metathesis reactions have exchanged ions of the same charge (e.g., displacing Na+ with Cu+ in ATaN2, or Ca2+ with Mg2+ in A2Si5N8; where A represents the exchangeable cations).68,71,72 Here, we replace a monovalent cation (Li+) with a divalent cation (Zn2+). While such exchange has been conducted in oxides86,87,94,95 and sulfides (e.g., 2 NaCrS2 + MgCl2 → MgCr2S4 + 2 NaCl),96 to the best of our knowledge this is the first report of such an exchange in nitrides. The resulting decrease in the cation:anion ratio (from 7:4 to 4:4) means that a truly topotactic replacement is unlikely to occur. However, the transformation appears to be near-topotactic.
Generalizability to other materials
There are numerous Zn–M–N phases that have been demonstrated to be synthesizable via thin film sputtering but that have not yet been made in bulk. In addition to Zn3WN4,23,89 sputtering has been used to synthesize fully nitridized transition metal ternaries: ZnTiN2, ZnZrN2, Zn2VN3, Zn2NbN3, Zn2TaN3, and Zn3MoN4.23,24,47,48,52 Although computational predictions for these thin film materials find that cation-ordered structures are the thermodynamic ground state (Fig. 5f), these sputtered films tend to form in cation-disordered structure variants (Fig. 5e).23,24 This disorder tends to decrease the bandgap of the material by creating localized electronic states.29,92 Bulk syntheses could advance the development of these new semiconductors by studying the effect of structure (e.g., ordering) on optoelectronic properties of these new materials.The synthesis of Zn3WN4 from Li6WN4 and ZnX2 suggests a promising strategy for future materials discovery of cation-ordered heterovalent ternary nitrides via metathesis from lithium-based ternary nitride precursors. Lithium-based ternary nitrides are the most well-studied subset of ternary nitrides,23 suggesting that many Li–M–N phases exist that could be used to synthesize additional A–M–N phases via exchange with AXn (where A and M are metals and X is a halide). Following our results here, X should be selected to minimize reaction energy and thus minimize the risk of decomposing the target phase via gaseous N2 loss. To demonstrate this point, we also synthesized Zn3MoN4 from Li6MoN4 and ZnBr2 (Fig. S4†). Zn3MoN4 was the main product, but some decomposition products were also observed, indicating that additional reaction optimization is needed. Unlike in Zn3WN4, the ZnBr2-based reaction was not sufficiently low-energy to fully avoid this decomposition for Zn3MoN4. While we did not synthesize phase-pure Zn3MoN4 here, further reaction engineering, like adding NH4Cl to manage heat flow,62–65 may be able to produce phase-pure Zn3MoN4. As we found that the reaction onset temperature is correlated with AX2 melting point, phases with high-melting temperature precursors may be difficult to synthesize below the decomposition point of the targeted ternary. Therefore, future work should consider ways to decouple the reaction onset from the AX2 melting point. In sum, this work shows how Li–M–N phases can be promising precursors for accelerating the discovery of new ternary nitrides.
Conclusions
Here, we report the bulk synthesis of cation-ordered Zn3WN4, through metathesis reactions beginning from a Li-based ternary nitride precursor: Li6WN4 + ZnX2 → Zn3WN4 + 6 LiX (X = Br, Cl, F). These reactions proceed directly (i.e., without nitride intermediates), as measured by in situ synchrotron powder X-ray diffraction and differential scanning calorimetry. The reaction onset temperature correlates with the melting point of the ZnX2 precursor, allowing ZnCl2- and ZnBr2-based reactions to proceed at ≤300 °C. The more exothermic reactions lead to greater degrees of Zn3WN4 decomposition, meaning that the least exothermic reaction (with ZnBr2) is the most favorable for synthesis. The high resolution synchrotron powder X-ray diffraction data are consistent with cation-ordered Zn3WN4 (Pmn21). This finding is distinct from prior thin film syntheses, which yielded cation-disordered P63mc Zn3WN4. Diffuse reflectance spectroscopy shows that Zn3WN4 powders exhibit absorption onsets near 2.5 eV and 4.0 eV, consistent with a small degree of cation disorder in the mostly long-range ordered Pmn21 phase. Preliminary work targeting Zn3MoN4 from Li6MoN4 and ZnBr2 suggests this synthesis approach may readily extend to other systems. These findings indicate that Li–M–N compounds may serve as precursors for synthesizing numerous other ternary nitrides.Data availability
Density functional theory calculations can be found at the NREL MatDB (https://materials.nrel.gov/). The ESI† contains additional data on experimental conditions, additional PXRD patterns, in situ variable temperature SPXRD measurements, DSC measurements, compositional characterization, structural models, and magnetic susceptibility measurements.Author contributions
Christopher L. Rom: conceptualization, investigation, formal analysis, visualization, writing – original draft preparation, writing – reviewing and editing Shaun O'Donnell: investigation Kayla Huang: investigation Ryan A. Klein: investigation, formal analysis Morgan J. Kramer: investigation, formal analysis Rebecca W. Smaha: investigation, writing – reviewing and editing Andriy Zakutayev: conceptualization, funding acquisition, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored at the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by DOE Basic Energy Sciences Early Career Award “Kinetic Synthesis of Metastable Nitrides.” This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. R. A. K. gratefully acknowledges support from the U.S. DOE Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office (HFTO). R. W. S. acknowledges support from the Director's Fellowship within NREL's Laboratory Directed Research and Development program, and K. H. acknowledges support from the DOE Science Undergraduate Laboratory Internships (SULI) program. The authors thank Dr Robert Bell and Dr Noemi Leick for technical support with DSC measurements, Dr Fred Baddour for support with the laboratory PXRD measurements, Dr Wenqian Xu for support with the in situ SPXRD measurements, Dr Daniyal Kiani and Dr Dan Ruddy for support with the diffuse reflectance measurements, and Dr Stephan Lany for calculating the absorption coefficient of Pmn21 Zn3WN4. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government.References
- G. Tammann, Fr. Westerhold, B. Garre, E. Kordes, H. Kalsing and Z. Für, Z. Anorg. Allg. Chem., 1925, 149, 21–98 CrossRef CAS.
- D. Astruc, New J. Chem., 2005, 29, 42–56 RSC.
- C. P. Casey, J. Chem. Educ., 2006, 83, 192–195 CrossRef CAS.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- R. R. Schrock, Adv. Synth. Catal., 2007, 349, 41–53 CrossRef CAS.
- B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Sci., 2014, 5, 2489–2497 RSC.
- C. Deraedt, M. d'Halluin and D. Astruc, Eur. J. Inorg. Chem., 2013, 2013, 4881–4908 CrossRef CAS.
- M. Cui and G. Jia, J. Am. Chem. Soc., 2022, 144, 12546–12566 CrossRef CAS PubMed.
- J.-M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS PubMed.
- A. M. Belenguer, T. Friščić, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696–700 RSC.
- C. K. Brozek and M. Dincă, Chem. Sci., 2012, 3, 2110–2113 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- L. Zou, D. Feng, T.-F. Liu, Y.-P. Chen, S. Yuan, K. Wang, X. Wang, S. Fordham and H.-C. Zhou, Chem. Sci., 2016, 7, 1063–1069 RSC.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158–165 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2015, 7, 30–38 RSC.
- R. H. Grubbs and W. Tumas, Science, 1989, 243, 907–915 CrossRef CAS PubMed.
- L. Pettazzoni, M. Ximenis, F. Leonelli, G. Vozzolo, E. Bodo, F. Elizalde and H. Sardon, Chem. Sci., 2024, 15, 2359–2364 RSC.
- I. P. Parkin and A. Kafizas, Exothermic Metathesis Reactions, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 471–490 Search PubMed.
- A. Wustrow and J. R. Neilson, Metathesis Routes to Materials, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, Oxford, 3rd edn, 2023, pp. 24–39 Search PubMed.
- J. R. Neilson, M. J. McDermott and K. A. Persson, J. Mater. Res., 2023, 38, 2885–2893 CrossRef CAS.
- A. L. Greenaway, C. L. Melamed, M. B. Tellekamp, R. Woods-Robinson, E. S. Toberer, J. R. Neilson and A. C. Tamboli, Annu. Rev. Mater. Res., 2021, 51, 591–618 CrossRef CAS.
- W. Sun, C. J. Bartel, E. Arca, S. R. Bauers, B. Matthews, B. Orvañanos, B.-R. Chen, M. F. Toney, L. T. Schelhas, W. Tumas, J. Tate, A. Zakutayev, S. Lany, A. M. Holder and G. Ceder, Nat. Mater., 2019, 18, 732–739 CrossRef CAS PubMed.
- A. Zakutayev, S. R. Bauers and S. Lany, Chem. Mater., 2022, 34, 1418–1438 CrossRef CAS.
- A. Zakutayev, J. Mater. Chem. A, 2016, 4, 6742–6754 RSC.
- Bond Dissociation Energies in Diatomic Molecules, in CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 104th edn, 2023 Search PubMed.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- E. W. Blanton, K. He, J. Shan and K. Kash, J. Cryst. Growth, 2017, 461, 38–45 CrossRef CAS.
- R. R. Schnepf, J. J. Cordell, M. B. Tellekamp, C. L. Melamed, A. L. Greenaway, A. Mis, G. L. Brennecka, S. Christensen, G. J. Tucker, E. S. Toberer, S. Lany and A. C. Tamboli, ACS Energy Lett., 2020, 5, 2027–2041 CrossRef CAS.
- I. S. Khan, K. N. Heinselman and A. Zakutayev, JPhys Energy, 2020, 2, 032007 CrossRef CAS.
- A. D. Martinez, A. N. Fioretti, E. S. Toberer and A. C. Tamboli, J. Mater. Chem. A, 2017, 5, 11418–11435 RSC.
- F. Kawamura, N. Yamada, M. Imai and T. Taniguchi, Cryst. Res. Technol., 2016, 51, 220–224 CrossRef CAS.
- J. Häusler, S. Schimmel, P. Wellmann and W. Schnick, Chem.–Eur. J., 2017, 23, 12275–12282 CrossRef PubMed.
- R. Juza and F. Hund, Naturwissenschaften, 1946, 33, 121–122 CrossRef CAS.
- M. Y. Chern and F. J. DiSalvo, J. Solid State Chem., 1990, 88, 528–533 CrossRef CAS.
- H. Yamane and F. J. DiSalvo, J. Solid State Chem., 1995, 119, 375–379 CrossRef CAS.
- W. L. Larson, H. P. Maruska and D. A. Stevenson, J. Electrochem. Soc., 1974, 121, 1673 CrossRef CAS.
- Q.-H. Zhang, J. Wang, C.-W. Yeh, W.-C. Ke, R.-S. Liu, J.-K. Tang, M.-B. Xie, H.-B. Liang and Q. Su, Acta Mater., 2010, 58, 6728–6735 CrossRef CAS.
- M. Wintenberger, M. Maunaye and Y. Laurent, Mater. Res. Bull., 1973, 8, 1049–1053 CrossRef CAS.
- O. E. O. Zeman, F. O. von Rohr, L. Neudert and W. Schnick, Z. Anorg. Allg. Chem., 2020, 646, 228–233 CrossRef CAS.
- Y. Sun, Y. Guo, Y. Tsujimoto, J. Yang, B. Shen, W. Yi, Y. Matsushita, C. Wang, X. Wang, J. Li, C. I. Sathish and K. Yamaura, Inorg. Chem., 2013, 52, 800–806 CrossRef CAS PubMed.
- S. Deng, Y. Sun, H. Wu, Z. Shi, L. Wang, J. Yan, K. Shi, P. Hu, X. Diao, Q. Huang, C. Sürgers and C. Wang, Scr. Mater., 2018, 146, 18–21 CrossRef CAS.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman, P. Eklund, P. O. Å. Persson, S. Du, Z. Chai, Z. Huang and Q. Huang, J. Am. Chem. Soc., 2019, 141, 4730–4737 CrossRef CAS PubMed.
- O. Beckmann, H. Boller, H. Nowotny and F. Benesovsky, Monatsh. Chem., 1969, 100, 1465–1470 CrossRef CAS.
- W. Jeitschko, H. Nowotny and F. Benesovsky, Monatsh. Chem. Verw. Teile Anderer Wiss., 1964, 95, 1212–1218 CrossRef CAS.
- W. Wang, X. Kan, X. Liu, Z. Cheng, C. Liu, M. Shezad, Y. Yang, Q. Lv and K. Mehmood Ur Rehman, Eur. Phys. J. Plus, 2020, 135, 505 CrossRef CAS.
- A. L. Greenaway, S. Ke, T. Culman, K. R. Talley, J. S. Mangum, K. N. Heinselman, R. S. Kingsbury, R. W. Smaha, M. K. Gish, E. M. Miller, K. A. Persson, J. M. Gregoire, S. R. Bauers, J. B. Neaton, A. C. Tamboli and A. Zakutayev, J. Am. Chem. Soc., 2022, 144, 13673–13687 CrossRef CAS PubMed.
- R. Woods-Robinson, V. Stevanović, S. Lany, K. N. Heinselman, M. K. Horton, K. A. Persson and A. Zakutayev, Phys. Rev. Mater., 2022, 6, 043804 CrossRef CAS.
- D. Chu, Y. Huang, C. Xie, E. Tikhonov, I. Kruglov, G. Li, S. Pan and Z. Yang, Angew. Chem., 2023, 135, e202300581 CrossRef.
- Physical Constants of Inorganic Compounds, in CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 104th edn, 2023 Search PubMed.
- G. Paniconi, Z. Stoeva, R. I. Smith, P. C. Dippo, B. L. Gallagher and D. H. Gregory, J. Solid State Chem., 2008, 181, 158–165 CrossRef CAS.
- E. Arca, S. Lany, J. D. Perkins, C. Bartel, J. Mangum, W. Sun, A. Holder, G. Ceder, B. Gorman, G. Teeter, W. Tumas and A. Zakutayev, J. Am. Chem. Soc., 2018, 140, 4293–4301 CrossRef CAS PubMed.
- E. Horvath-Bordon, R. Riedel, A. Zerr, P. F. McMillan, G. Auffermann, Y. Prots, W. Bronger, R. Kniep and P. Kroll, Chem. Soc. Rev., 2006, 35, 987–1014 RSC.
- A. J. Martinolich, J. A. Kurzman and J. R. Neilson, J. Am. Chem. Soc., 2016, 138, 11031–11037 CrossRef CAS PubMed.
- E. G. Gillan and R. B. Kaner, Inorg. Chem., 1994, 33, 5693–5700 CrossRef CAS.
- L. Rao and R. B. Kaner, Inorg. Chem., 1994, 33, 3210–3211 CrossRef CAS.
- E. G. Gillan and R. B. Kaner, Chem. Mater., 1996, 8, 333–343 CrossRef CAS.
- C. H. Wallace, T. K. Reynolds and R. B. Kaner, Chem. Mater., 1999, 11, 2299–2301 CrossRef CAS.
- J. L. O'Loughlin, C. H. Wallace, M. S. Knox and R. B. Kaner, Inorg. Chem., 2001, 40, 2240–2245 CrossRef PubMed.
- J. B. Wiley and R. B. Kaner, Science, 1992, 255, 1093–1097 CrossRef CAS PubMed.
- C. H. Wallace, S.-H. Kim, G. A. Rose, L. Rao, J. R. Heath, M. Nicol and R. B. Kaner, Appl. Phys. Lett., 1998, 72, 596–598 CrossRef CAS.
- R. A. Janes, M. A. Low and R. B. Kaner, Inorg. Chem., 2003, 42, 2714–2719 CrossRef CAS PubMed.
- R. A. Janes, M. Aldissi and R. B. Kaner, Chem. Mater., 2003, 15, 4431–4435 CrossRef CAS.
- A. J. Anderson, R. G. Blair, S. M. Hick and R. B. Kaner, J. Mater. Chem., 2006, 16, 1318–1322 RSC.
- R. W. Cumberland, R. G. Blair, C. H. Wallace, T. K. Reynolds and R. B. Kaner, J. Phys. Chem. B, 2001, 105, 11922–11927 CrossRef CAS.
- C. L. Rom, M. J. Fallon, A. Wustrow, A. L. Prieto and J. R. Neilson, Chem. Mater., 2021, 33, 5345–5354 CrossRef CAS.
- P. K. Todd, M. J. Fallon, J. R. Neilson and A. Zakutayev, ACS Mater. Lett., 2021, 3, 1677–1683 CrossRef CAS PubMed.
- P. Bielec and W. Schnick, Angew. Chem., Int. Ed., 2017, 56, 4810–4813 CrossRef CAS PubMed.
- P. Bielec, O. Janka, T. Block, R. Pöttgen and W. Schnick, Angew. Chem., Int. Ed., 2018, 57, 2409–2412 CrossRef CAS PubMed.
- C. L. Rom, A. Novick, M. J. McDermott, A. A. Yakovenko, J. R. Gallawa, G. T. Tran, D. C. Asebiah, E. N. Storck, B. C. McBride, R. C. Miller, A. L. Prieto, K. A. Persson, E. Toberer, V. Stevanović, A. Zakutayev and J. R. Neilson, J. Am. Chem. Soc., 2024, 146, 4001–4012 CrossRef CAS PubMed.
- U. Zachwieja and H. Jacobs, Eur. J. Solid State Inorg. Chem., 1991, 28, 1055–1062 CAS.
- M. Yang, A. Zakutayev, J. Vidal, X. Zhang, D. S. Ginley and F. J. DiSalvo, Energy Environ. Sci., 2013, 6, 2994–2999 RSC.
- A. Miura, M. Lowe, B. M. Leonard, C. V. Subban, Y. Masubuchi, S. Kikkawa, R. Dronskowski, R. G. Hennig, H. D. Abruña and F. J. DiSalvo, J. Solid State Chem., 2011, 184, 7–11 CrossRef CAS.
- A. Stein, S. W. Keller and T. E. Mallouk, Science, 1993, 259, 1558–1564 CrossRef CAS PubMed.
- C. Chieh and M. A. White, Z. Kristallogr., 1984, 166, 189–197 CAS.
- W. X. Yuan, J. W. Hu, Y. T. Song, W. J. Wang and Y. P. Xu, Powder Diffr., 2005, 20, 18–21 CrossRef CAS.
- A. Pfitzner, J. K. Crockcroft, I. Solinas and H. D. Lutz, Z. Anorg. Allg. Chem., 1993, 619, 993–998 CrossRef CAS.
- J. N. Law, S. Pandey, P. Gorai and P. C. St. John, JACS Au, 2023, 3, 113–123 CrossRef CAS PubMed.
- C. Martin, A. Durif-Varambon and J.-C. Joubert, Bull. Soc. Fr. Mineral. Cristallogr., 1965, 88, 141 CAS.
- S. J. Shaw and G. S. Perry, Thermochim. Acta, 1989, 155, 87–96 CrossRef CAS.
- H. Yokokawa, J. Phase Equil., 1999, 20, 258–287 CrossRef CAS.
- P. K. Todd, M. J. McDermott, C. L. Rom, A. A. Corrao, J. J. Denney, S. S. Dwaraknath, P. G. Khalifah, K. A. Persson and J. R. Neilson, J. Am. Chem. Soc., 2021, 143, 15185–15194 CrossRef CAS PubMed.
- A. Jain, G. Hautier, S. P. Ong, C. J. Moore, C. C. Fischer, K. A. Persson and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045115 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- A. Wustrow, G. Huang, M. J. McDermott, D. O'Nolan, C.-H. Liu, G. T. Tran, B. C. McBride, S. S. Dwaraknath, K. W. Chapman, S. J. L. Billinge, K. A. Persson, K. Thornton and J. R. Neilson, Chem. Mater., 2021, 33, 3692–3701 CrossRef CAS.
- A. Wustrow, M. J. McDermott, D. O'Nolan, C.-H. Liu, G. T. Tran, B. C. McBride, S. M. Vornholt, C. Feng, S. S. Dwaraknath, K. W. Chapman, S. J. L. Billinge, W. Sun, K. A. Persson and J. R. Neilson, Chem. Mater., 2022, 34, 4694–4702 CrossRef CAS.
- P. K. Todd, A. M. M. Smith and J. R. Neilson, Inorg. Chem., 2019, 58, 15166–15174 CrossRef CAS PubMed.
- P. K. Todd and J. R. Neilson, J. Am. Chem. Soc., 2019, 141, 1191–1195 CrossRef CAS PubMed.
- B. E. Matthews, PhD dissertation, Oregon State University, 2018.
- S. Lany, J. Phys. Condens. Matter, 2015, 27, 283203 CrossRef PubMed.
- S. Lany, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 085112 CrossRef.
- J. J. Cordell, G. J. Tucker, A. Tamboli and S. Lany, APL Mater., 2022, 10, 011112 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- T. K. Mandal and J. Gopalakrishnan, J. Mater. Chem., 2004, 14, 1273–1280 RSC.
- T. Sivakumar and J. Gopalakrishnan, Chem. Mater., 2002, 14, 3984–3989 CrossRef CAS.
- A. Miura, H. Ito, C. J. Bartel, W. Sun, N. Carolina Rosero-Navarro, K. Tadanaga, H. Nakata, K. Maeda and G. Ceder, Mater. Horiz., 2020, 7, 1310–1316 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2308271 and 2308272. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00322e |
| This journal is © The Royal Society of Chemistry 2024 |