
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning white light emission using single-component tetrachroic Dy3+ metallacrowns: the role of chromophoric building blocks†
Elvin V.
Salerno‡
a,
Svetlana V.
Eliseeva‡
 *b,
Stéphane
Petoud
*b and
Vincent L.
Pecoraro
*a
*b,
Stéphane
Petoud
*b and
Vincent L.
Pecoraro
*a
aDepartment of Chemistry, Willard H. Dow Laboratories, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: vlpec@umich.edu
bCentre de Biophysique Moléculaire, CNRS UPR 4301, Université d’Orléans, F-45071 Orléans Cedex 2, France. E-mail: svetlana.eliseeva@cnrs-orleans.fr; stephane.petoud@inserm.fr
First published on 8th May 2024
Abstract
White light production is of major importance for ambient lighting and technological displays. White light can be obtained by several types of materials and their combinations, but single component emitters remain rare and desirable towards thinner devices that are, therefore, easier to control and that require fewer manufacturing steps. We have designed a series of dysprosium(III)-based luminescent metallacrowns (MCs) to achieve this goal. The synthesized MCs possess three main structural types LnGa4(L′)4(L′′)4 (type A), Ln2Ga8(L′)8(L′′′)4 (type B) and LnGa8(L′)8(OH)4 (type C) (H3L′, HL′′ and H2L′′′ derivatives of salicylhydroxamic, benzoic and isophthalic acids, respectively). The advantage of these MCs is that, within each structural type, the nature of the organic building blocks does not affect the symmetry around Dy3+. By detailed studies of the photophysical properties of these Dy3+-based MCs, we have demonstrated that CIE coordinates can be tuned from warm to neutral to cold white by (i) defining the symmetry about Dy3+, and (ii) choosing appropriate chromophoric building blocks. These organic building blocks, without altering the coordination geometry around Dy3+, influence the total emission profile through changing the probability of different energy transfer processes including the 3T1 ← Dy3+* energy back transfer and/or by generating ligand-centered fluorescence in the blue range. This work opens new perspectives for the creation of white light emitting devices using single component tetrachroic molecular compounds.
Introduction
White light production is important in a modern society and has applications in ambient lighting and/or screen backlighting in liquid crystalline displays. Presently, light emitting diodes (LEDs) are becoming the state of the art in light production as they can be highly efficient and tunable.To obtain white light emission (WLE), multiple colours need to be combined on a single device since LEDs are usually monochromatic emitters.1 Hence, dichroic, trichroic and tetrachroic approaches have been used that combine blue/yellow, red/green/blue (RGB) or RGB/cyano emitters, respectively.2 The use of multiple colour components implies a significant difference in comparison to broadband emitters, such as the sun. Generally, the broader the emission profile, and the more colour components, the better is the colour rendition.3
The CIE (Commission Internationale de l'Eclairage) colour space is commonly used to standardize how the average human eye will perceive the colour of an object.4 The CIE chromaticity coordinates are defined by three numbers (x, y, z). The chromaticity of an object can be plotted in a two-dimensional CIE diagram, since it implies a normalization condition: z = 1 − x − y. The white light region lies in the centre of the diagram, with the neutral white light having coordinates (0.33, 0.33). White light can also be described by its Correlated Colour Temperature (CCT), where its quality is defined by the blackbody equivalent temperature. Modern white light emitting devices include sources such as fluorescent lamps,5 high-pressure sodium–mercury lamps,6 solid state light emitting diodes,7 organic light emitting diodes (OLEDs).8,9
Single component white light emitters are desirable because they allow for thinner materials with greater manufacturing reproducibility and enhanced stability over time.10,11 Several lanthanide(III) ions (Ln3+) emitting in the visible range, namely, Sm3+, Eu3+, Tb3+, Dy3+, Tm3+, have the potential to emit compatible lights. Typically, the creation of individual materials that display WLE involves various Ln3+ ions, each with different properties, either independently or in combination with the light emitted by organic chromophoric ligands or an inorganic matrix.
Several compounds possessing WLE capabilities have been created with a combination of different visible-emitting Ln3+. The variation of the concentrations of the individual Ln3+ allows one to tune the corresponding relative emission intensities and, thus, to control the CIE coordinates. The pertinence of this strategy has been demonstrated for molecular materials,12,13 Ln3+-based polymers,14–21 metal–organic frameworks (MOFs),22–26 inorganic materials,27–29 and hybrid organic/inorganic30–33 materials.
Alternatively, to produce WLE, the combination of emission signals arising from a single visible-emitting Ln3+ and organic ligands or inorganic matrix can be used.34,35 The validity of this approach has been demonstrated for molecular Eu3+ compounds,36 as well as for Dy3+ compounds. Some of these Dy3+ compounds were shown to be tunable by ligand modifications,37 as well as by excitation wavelength38 or by the change of the experimental temperatures.37 The appearance of the Ln3+ emission is dependent on the environment around Ln3+ that is controlled by the ligand field induced by the structure of this ligand. It will control the splitting of the different transitions as well as their relative emission intensities and, therefore, the CIE coordinates.39–42
Single component Dy3+ is a special lanthanide(III) ion for such application as its possesses two main groups of sharp emission bands in the green and in the red spectral domains that allow for the development of single-component WLE with the addition of a blue emission component arising from the same compound.46–55 Several inorganic compounds have shown an ability to tune the CIE coordinates by controlling the concentration of the Dy3+ dopant. These examples include Dy3+-doped chloroborosilicate glasses,56 barium silicate,57 zinc–aluminum–sodium–phosphate,58 or lithium–zinc borosilicate59 glasses.
The most studied transitions in Dy3+ compounds with respect to WLE46 are bands located in the visible range, in particular, 4F9/2 → 6H15/2 (blue), 4F9/2 → 6H13/2 (yellow), minimally 4F9/2 → 6H11/2 (red), and rarely 4F9/2 → 6H9/2 (red-brown) transitions. The emission intensity ratio I(4F9/2 → 6H13/2)/I(4F9/2 → 6H15/2) is sometimes used as a measurement of the effects of ligand field on the emission spectrum of Dy3+ (the “yellow/blue” or Y/B ratio). Modification of the Y/B ratio has been associated with Dy3+-ligand covalency,40,41 as well as with asymmetry of the coordination environment.58,60–63 Higher level of covalency and coordination sphere asymmetry apparently leads to a higher Y/B ratio, i.e. change of the Dy3+ emission profile and tuning of WLE properties.
The coordination environment around Ln3+ impacts its emission profile and, in turn, CIE coordinates. Ln3+-doped extended solids, amorphous inorganic compounds, such as ceramics, glasses, sol-gels or cementitious materials do not possess a single well-defined structure about each dopant.7 In crystalline materials, the dopants are dispersed statistically at allowed positions within the lattice, while the introduction of the dopant itself will induce the distortion of the structure of the lattice.64–67 Thus, in such materials, the correlation between the emission spectra of the doped Ln3+ with the symmetry about them is limited, as changes will be averaged by the environments around each dopant.61
Single Ln3+ molecular compounds provide the advantage of a unique and well-defined coordination environment around the lanthanide, especially when highly rigid. They have been studied for WLE in the past. For example, a single molecular white light emitting Eu3+ complex has been previously examined.36 This complex was formed with a 1,8-naphthalimide functionalized tetraazacyclododecane ligand. In this complex, the 1,8-naphthalimide chromophoric ligand generates a blue/purple fluorescent component, as well as a green component from a naphthalimide aggregation associated excimer state. This ligand also acts as a sensitizer for the generation of the red emission from Eu3+. Through their combination, these different components lead to the generation of WLE.
Dy3+-based molecular compounds have also shown a strong potential as single-component white light emitters. Thus, by combining Dy3+-centred electronic transitions with those located on the blue-emitting ligand, WLE was observed in a [Dy3+(TETP)(NO3)3]·4H2O MOF material [TETP = 1,1′,1′′-((2,4,6-triethyl-benzene-1,3,5-triyl)tris(methylene))tris(pyridin-4(1H)-one))]. The CIE coordinates of this MOF could be tuned to (0.33, 0.35) by varying the excitation wavelength.38
In a series of molecular binuclear Dy3+ complexes, [Dy2(L)2(NO3)2(solvent)2]·x(solvent) [L = 2,2′-[[(2-pyridinyl-methyl)imino]di(methylene)]bis(4 R-phenol), where R = CH3, Cl, and CH3O; solvent = methanol or DMF], the WLE could be tuned by varying both the substituents of the ligand and the experimental temperature.37 This thermal behaviour might be explained by the interactions of the Dy3+ with solvent vibrational modes and/or changes in energy transfer from the ligand. On the other hand, the influence of the substituents of the ligand on the spectra of Dy3+ was not fully elucidated but the modification of the CIE coordinates may be attributed to changes of the relative intensities of Dy3+- vs. ligand-centred emissions.
Molecular compounds are more suited probes of the effects of the ligand field on Dy3+ emission profile as they provide a defined environment about the coordinated ion. In a recent communication, we have demonstrated that, within a series of monomeric and dimeric Dy3+/Ga3+ metallacrowns (MCs) with a common [12-MCGaN(Shi)-4] motif, the Dy3+ emission profile can be tuned by modifying the symmetry about the Dy3+ ion.70 The changes of the relative intensities of different Dy3+ emission bands combined with the minimal ligand contribution led to tunable WLE arising from these MCs. Here, we expand this series of MCs with WLE properties by synthesizing eleven new Dy3+ MCs formed with different combinations of building blocks using substituted salicylhydroxamic, benzoic and isophthalic acids (Fig. 1) while preserving three main structural types: LnGa4(L′)4(L′′)4 (type A), Ln2Ga8(L′)8(L′′′)4 (type B) and LnGa8(L′)8(OH)4 (type C) (Fig. 1, H3L′ = salicylhydroxamic acid (H3shi), 5-methylsalicylhydroxamic acid (H3mshi), 5-methoxysalicylhydroxamic acid (H3moshi), 5-chlorosalicylhydroxamic acid (H3Cl-shi), 5-bromosalicylhydroxamic acid (H3Br-shi), 5-iodosalicylhydroxamic acid (H3I-shi); HL′′ = benzoic acid (Hbz), 2-methylbenzoic acid (Hmbz), 2-fluorobenzoic acid (HF-bz); H2L′′′ = isophthalic acid (H2iph), 5-iodoisophthalic acid (H2I-iph)). This approach allows the investigation of the influence of these modifications to the electronic structure of the ligand scaffold may have on the total emission profile of Dy3+ MCs and on the corresponding WLE properties reflected by the CIE coordinates. Moreover, to determine how such modifications affect the energy positions of the singlet and triplet states within this series of MCs, the corresponding Gd3+ analogues were synthesized and studied.
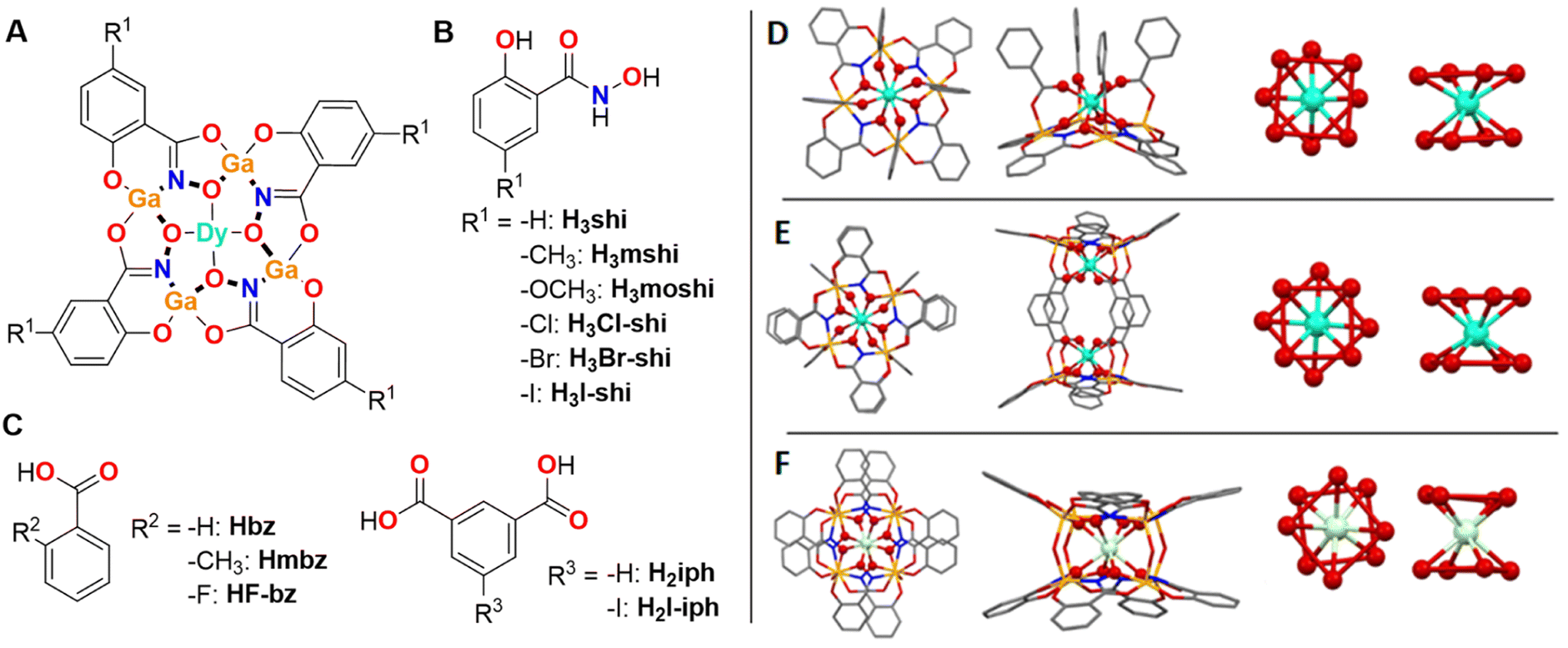 | ||
| Fig. 1 (A) Schematic presentation of a Dy3+[12-MCGaN(R1-shi)-4] MC core, where Ga3+ and R1-substituted H3shi (B) template the formation of the MC, while Dy3+ is linked to the central cavity by the derivatives of benzoic or isophthalic acids (C). (D–F) Top-down (left) and side-on (left-centre) views of the three Ga3+/Dy3+ MCs geometries and the corresponding primary coordination spheres about Dy3+ (centre-left and right views, the red lines between oxygen atoms are guides for the eyes to suggest O4 planes coming from the MC ring and otherwise). (D) [DyGa4(shi)4(bz)4] representing the LnM4(L′)4(L′′)4 structure (type A).43 (E) [Dy2Ga8(shi)8(iph)4] corresponding to the Ln2M8(L′)8(L′′′)4 structure (type B).44 (F) [DyGa8(shi)8(OH)4] corresponding to the LnM8(L′)8(OH)4 structure (type C).45 The structure was solved for isostructural [NdGa8(shi)8(OH)4]Na. Solvents of crystallization, counter ions, and hydrogen atoms are omitted for clarity. Central Ln3+ and coordinating oxygen atoms are bolded for highlight effect. Colour code: Ga, orange; Ln, teal; O, red; N, blue; C, grey. | ||
Experimental section
Ln3+/Ga3+ MCs were synthesized using the deprotonated forms of the ligands given in Fig. 1. All MC synthesis reactions were carried out aerobically under ambient conditions via single-pot self-assembly reactions. Elemental analyses were performed by Atlantic Microlabs Inc. ESI-MS spectra were collected with an Agilent 6230 TOF HPLC-MS mass spectrometer in negative ion mode (−350 V) on samples dissolved in methanol at a concentration of 2 mg mL−1. 1H NMR spectra were collected on a Varian MR400 NMR in deuterated DMSO at a concentration of 4 mg mL−1. Single crystal unit cell parameters were obtained by mounting samples on a Rigaku AFC10K Saturn 944+ CCD-based X-ray diffractometer equipped with a low temperature device and Micromax-007HF Cu-target micro-focus rotating anode (λ = 1.54187 Å) operating at 1.2 kW power (40 kV, 30 mA). The X-ray intensities were measured at 85(1) K with the detector placed at a distance of 42.00 mm from the crystal. Detailed synthetic procedures and characterizations are provided in ESI.† Photophysical characterization was carried out as described in ESI.†Results
Synthesis and characterization
Ln3+/M3+ MCs possess the remarkable property to form structurally closely similar compounds when the ligands used for the assembly possess identical binding motifs. Furthermore, by modifying the binding motif, the coordination geometry can also be tightly controlled and modified. Thus, independently of the nature of the Ln3+ (Ln3+ = Pr3+–Yb3+) and for a variety of M3+ metals located in the ring (M3+ = Al3+, Ga3+, Mn3+, Fe3+), the use of the salicylhydroximate ‘O–C–C–CO–NHOH’ motif in combination with carboxylic or hydroxy ligands has been shown to form similar monomeric LnM4(L′)4(L′′)4 (Fig. 1D),43,73–76 dimeric Ln2M8(L′)8(L′′′)4 (Fig. 1E)44,71,77–79 or LnM8(L′)8(OH)4 (Fig. 1F)45 structural topologies, respectively.Novel functionalized salicylhydroxamic acids were synthesized in this work by converting the appropriate carboxylic acids into the corresponding methyl esters followed by an exchange reaction with an excess of hydroxylamine. The MCs were synthesized by a self-assembly reaction between stoichiometric amounts of derivatives of salicylhydroxamic, benzoic or isophthalic acids with Ga3+ and Ln3+ nitrates in DMF or methanol in the presence of a base. MCs were obtained as pure crystalline products by the slow evaporation of the solvent. The composition of the MCs was confirmed by mass spectrometry and elemental analysis (ESI†). Each novel MC was screened for unit cell parameters by single crystal X-ray diffraction to confirm the crystallinity of the sample and to establish the crystallographic similarities for different Dy3+ and Gd3+ analogues. Several novel Dy3+ and Gd3+ MCs were synthesized using this approach. For the sake of comparison, the spectroscopic properties of these compounds are analysed and presented together with those obtained from several Dy3+ and Gd3+ MCs which we previously described.43–45,68,69
In total, eight of these complexes adopt the LnGa4(L′)4(L′′)4 topology (Type A), five adopt the Ln2Ga8(L′)8(L′′′)4 topology (Type B), and two adopt the LnGa8(L′)8(OH)4 topology (Type C). Most of these MCs crystallize with Na+ as counterions, while two possess a PyH+ (pyridinium) counterion and one include NH4+ counterions. Throughout this article, the counter-cations are Na+ or 2 Na+ unless stated otherwise. Each compound herein presented also co-crystallizes with a variable number of solvent molecules (DMF or methanol, and water). The details about the synthesis and the characterization of all studied MCs are given in the ESI.†
Photophysical properties
The photophysical properties of Dy3+ MCs were measured in the solid state at room temperature. Each of the complexes showed a characteristic Dy3+ emission profile under ligand excitation at 340 nm (Fig. 2). These profiles include three emission bands, two, being the most intense, correspond to the 4F9/2 → 6H15/2 (∼480 nm, blue) and 4F9/2 → 6H13/2 (∼575 nm, yellow) transitions, and one, being significantly less intense, is due to the 4F9/2 → 6H11/2 (∼660 nm, red) transition. A broad band located in the blue is attributed to the emission arising from the electronic structure of the chromophoric building blocks that constitute the different MCs. Important differences in intensities of these blue bands in comparison to the intensities of the Dy3+ transitions can be observed in the emission spectra (Fig. 2).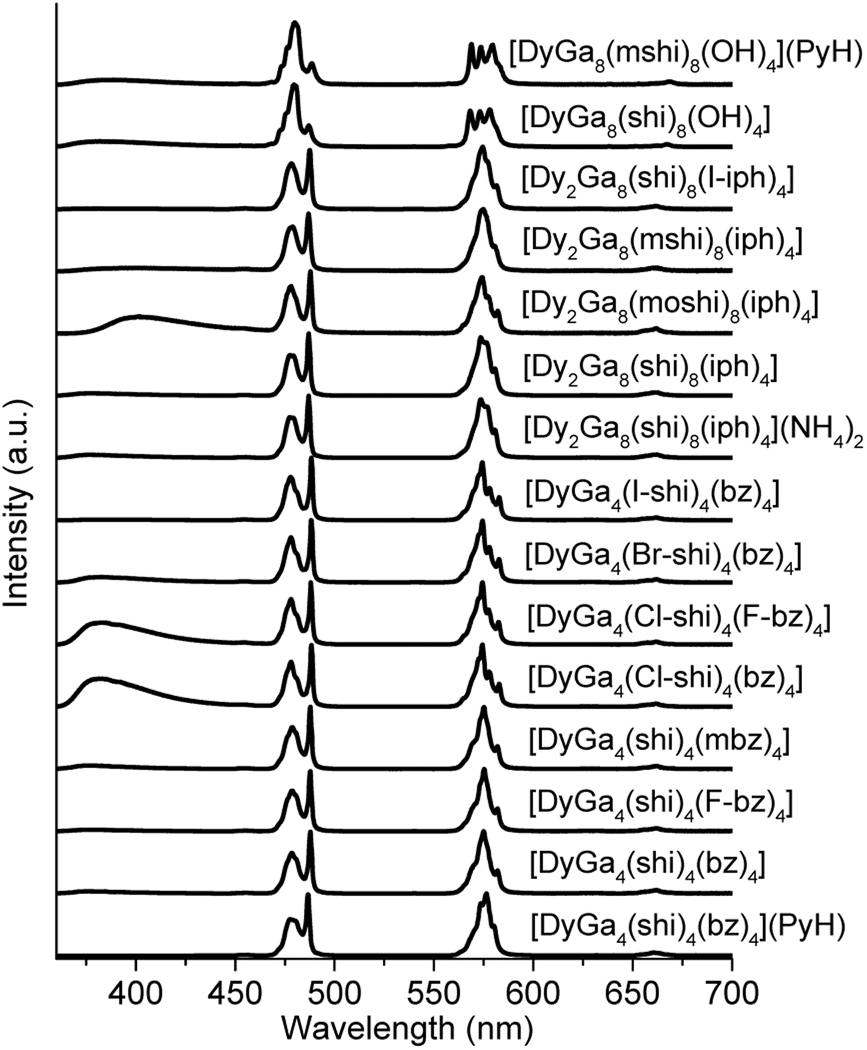 | ||
| Fig. 2 Corrected and normalized emission spectra of the Dy3+ MCs in the solid state upon ligand-centred excitation at 340 nm at room temperature. Counter-cation is either Na+ or 2 Na+ unless stated otherwise. | ||
Absolute quantum yields of the Dy3+-centred (QLDy) and ligand-centred (QLL) transitions were determined upon excitation in the range 330–350 nm. The total quantum yield value (QLtotal) was calculated using the formula: QLtotal = QLDy + QLL. The values of QLDy vary from 0.128(4)% to 8.3(3)% (Table 1), while those of QLL change from 0.0043(1)% to 0.607(3)%. The experimental decay of the luminescence of monomeric DyGa4(L′)4(L′′)4 are best described by a monoexponential function while those of LnGa8(L′)8(OH)4 and most of Ln2Ga8(L′)8(L′′′)4 exhibits a biexponential behaviour. The observed luminescence lifetimes of the Dy3+ emission vary from 4.1(1) to 61.1(7) μs. The excitation spectra are given in Fig. S1 (ESI).†
| Metallacrownb | Type | τ obs (μs) | Q L Dy , (%) | Q L L , (%) | Q L L/QLtotalg |
|---|---|---|---|---|---|
a Data for powder solid state samples of MCs. 2σ values are given between parenthesis. Estimated experimental errors: τobs, ±2%; QLDy, ±10%; QLL, ±10%.
b Counter-cation is Na+ or 2 Na+ unless otherwise specified.
c Under excitation at 355 nm. If a biexponential decay was observed, population parameters 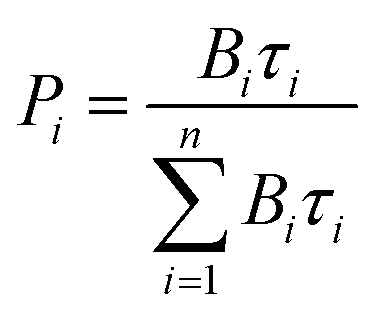 in % are given after the colon.
d Under excitation at 340 nm.
e Quantum yield of Dy3+-centred transitions.
f Quantum yield of ligand-centred transitions.
g
Q
L
total = QLDy + QLL. in % are given after the colon.
d Under excitation at 340 nm.
e Quantum yield of Dy3+-centred transitions.
f Quantum yield of ligand-centred transitions.
g
Q
L
total = QLDy + QLL.
|
|||||
| [DyGa4(shi)4(bz)4](PyH)43,68 | A | 21.2(2) | 1.23(2) | 0.0043(1) | 0.0035 |
| [DyGa4(shi)4(bz)4]68 | A | 50.9(6) | 8.3(3) | 0.54(2) | 0.060 |
| [DyGa4(shi)4(F-bz)4] | A | 61.1(7) | 4.26(2) | 0.314(1) | 0.069 |
| [DyGa4(shi)4(mbz)4] | A | 42.7(2) | 4.45(3) | 0.607(3) | 0.120 |
| [DyGa4(Cl-shi)4(bz)4] | A | 8.6(2) | 0.39(2) | 0.41(1) | 0.513 |
| [DyGa4(Cl-shi)4(F-bz)4] | A | 11.0(1) | 0.785(9) | 0.688(6) | 0.467 |
| [DyGa4(Br-shi)4(bz)4] | A | 10.5(2) | 0.86(2) | 0.174(3) | 0.168 |
| [DyGa4(I-shi)4(bz)4] | A | 11.3(2) | 0.779(9) | 0.0346(3) | 0.0425 |
| [Dy2Ga8(shi)8(iph)4](NH4)2 (ref. 44 and 68) | B | 15.0(1) | 0.85(1) | 0.086(1) | 0.092 |
| [Dy2Ga8(shi)8(iph)4] | B | 18(1): 16(4)%; 10.0(7): 74(4)% | 1.46(4) | 0.117(3) | 0.074 |
| [Dy2Ga8(moshi)8(iph)4]69 | B | 2.43(1): 91.8(1)%; 0.51(1): 8.2(1)% | 0.128(4) | 0.106(7) | 0.455 |
| [Dy2Ga8(mshi)8(iph)4] | B | 17.0(4): 61(4)%; 8.2(3): 39(4)% | 0.837(5) | 0.260(8) | 0.237 |
| [Dy2Ga8(shi)8(I-iph)4] | B | 40.7(1): 79.9(2)%; 12.3(3): 20.1(2)% | 1.2(1) | 0.052(4) | 0.043 |
| [DyGa8(shi)8(OH)4]45,68 | C | 37.7(8): 55.4(7)%; 4.1(1): 44.6(7)% | 1.42(8) | 0.34(1) | 0.193 |
| [DyGa8(mshi)8(OH)4](PyH) | C | 19.4(4): 81(1)%; 5.8(2): 19(1)% | 1.11(1) | 0.299(4) | 0.212 |
The diffuse reflectance spectra of Dy3+ and Gd3+ MCs were measured in the solid state at room temperature (Fig. S2, ESI†). The lowest excited singlet state (S1) energy levels were estimated by considering the red edge of these spectra (Table 2) or from the intersection of the emission spectrum of the ligand with the diffuse reflectance spectrum of the corresponding MC (Fig. S4, ESI†). The shapes of these two types of spectra match well with one another (Fig. S8, ESI†), and are summarized in Table 2. Energy values vary from 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 870 to 28860 cm−1. The corresponding measurements were also taken for the organic building blocks used to assemble MCs (Fig. S3, S5 and Table S2, ESI†).
870 to 28860 cm−1. The corresponding measurements were also taken for the organic building blocks used to assemble MCs (Fig. S3, S5 and Table S2, ESI†).
| Metallacrownb | Type | 3T1c | S1d | ΔE(3T1−4F9/2)e,c | ΔE(S1 − 3T1) |
|---|---|---|---|---|---|
|
a Data for powder solid state samples of MCs. 3T1, S1, and ΔE values are given in cm−1. The energy values of 3T1 and S1 determined using an alternative method are provided in Table S2.
b Counter-cation is Na+ or 2 Na+ unless otherwise specified.
c Determined from the Gaussian fitting of the 0–0 phonon line of the phosphorescence spectra of Gd3+ MCs measured at 77 K (Fig. S6, ESI).
d Determined from the red edge (10% of the maximum) of the solid-state diffuse reflectance spectra (Fig. S2, ESI).
e Energy of the emissive 4F9/2 level of Dy3+ is 21050 cm−1.72
f Gd3+ analogue could not be synthesized, so 3T1 is not determined.
g Determined for Dy3+ analogues.
|
|||||
| [LnGa4(shi)4(bz)4](PyH)43 | A | 22170 |
28290 |
1120 | 6120 |
| [LnGa4(shi)4(bz)4] | A | 22680 |
28735 |
1630 | 6055 |
| [LnGa4(shi)4(F-bz)4] | A | —f | 28695g |
— | — |
| [LnGa4(shi)4(mbz)4] | A | —f | 28860g |
— | — |
| [LnGa4(Cl-shi)4(bz)4] | A | 21620 |
27820 |
570 | 6200 |
| [LnGa4(Cl-shi)4(F-bz)4] | A | —f | 27470g |
— | — |
| [LnGa4(Br-shi)4(bz)4] | A | 21890 |
27700 |
840 | 5810 |
| [LnGa4(I-shi)4(bz)4] | A | —f | 27625g |
— | — |
| [Ln2Ga8(shi)8(iph)4](NH4)2(ref. 44) | B | 21980 |
28650 |
930 | 6670 |
| [Ln2Ga8(shi)8(iph)4]71 | B | 22385 |
28650 |
1335 | 6265 |
| [Ln2Ga8(moshi)8(iph)4]69 | B | 21640 |
25870 |
590 | 4230 |
| [Ln2Ga8(mshi)8(iph)4]69 | B | 21570 |
27780 |
520 | 6210 |
| [Ln2Ga8(shi)8(I-iph)4] | B | 22130 |
28170 |
1080 | 6040 |
| [LnGa8(shi)8(OH)4]45 | C | 22620 |
27815 |
1570 | 5195 |
| [LnGa8(mshi)8(OH)4](PyH) | C | 21945 |
27210 |
895 | 5265 |
The phosphorescence spectra were collected at 77 K for all the synthesized Gd3+ MCs in the solid state. The blue side of the observed phosphorescence spectrum was deconvoluted into several Gaussian curves. The highest-energy band was taken as the 0–0 phonon phosphorescence emission and considered as the energy of the lowest ligand triplet state (3T1). These values vary from 21570 to 22680 cm−1. Another estimate of the 3T1 energy level was also calculated as the centre of gravity of the phosphorescence spectrum. These values are presented in Table S2 (ESI†) and have a similar trend as the ones obtained from the 0–0 triplet state energy estimation (Fig. S8, ESI†). When sufficient emission signals could be obtained, the phosphorescence spectra of the free ligands were acquired in the solid state and analysed (Fig. S7 and Table S2, ESI†). The ligands singlet and triplet states are presented relatively to the relevant states of Dy3+ in Fig. 3.
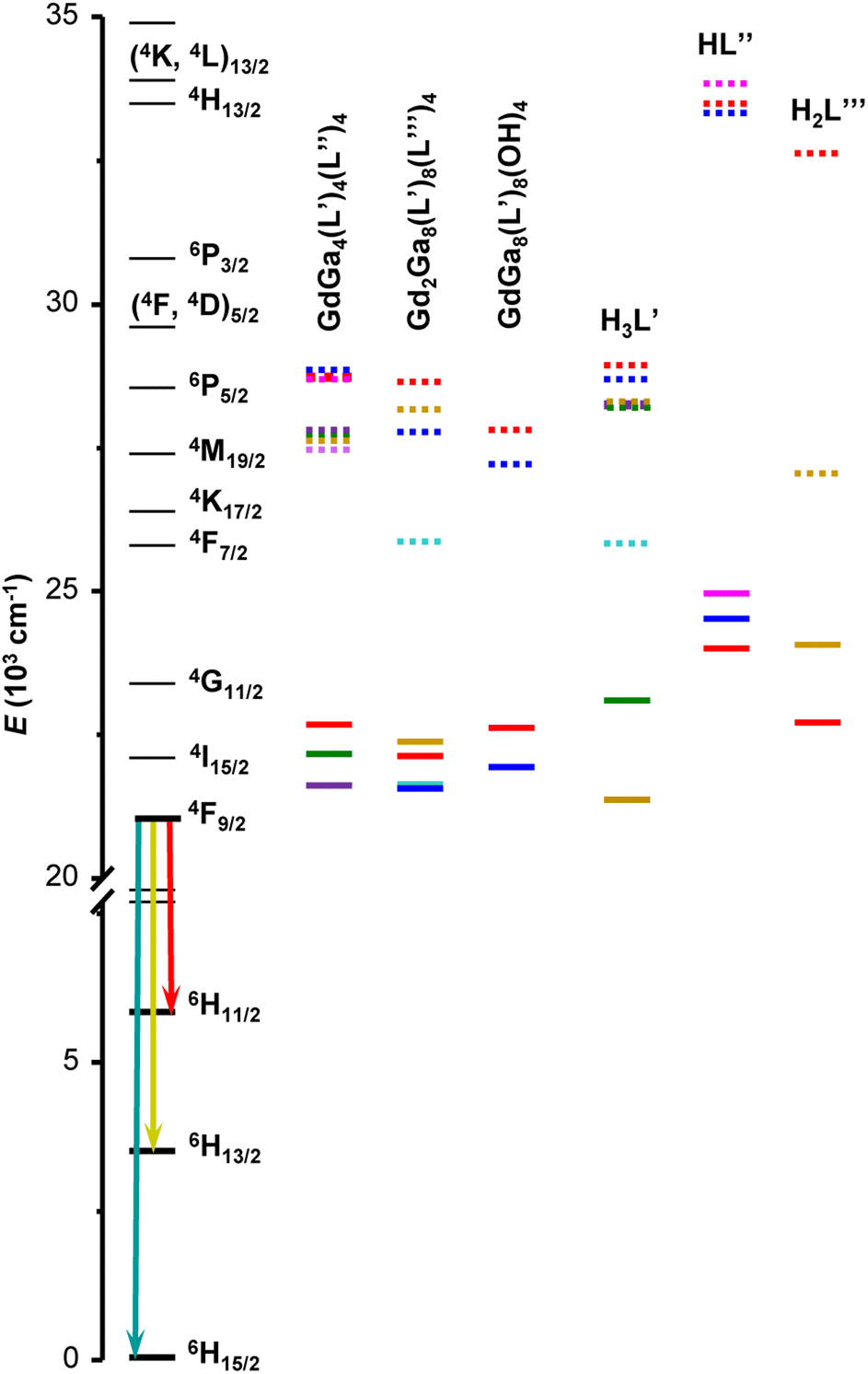 | ||
| Fig. 3 Diagram of Dy3+ energy levels (black traces), the lowest singlet (S1, dotted traces) and triplet (3T1, solid traces) states determined for Gd3+ MCs as well as for the organic building blocks used to assemble MCs. The list of energy levels is provided in Tables 2 and S2, ESI.† Chemical structures of the ligands and crystal structures of MCs are given in Fig. 1. | ||
Colorimetric properties. The colorimetric properties of the Dy3+ MCs were calculated from the corresponding visible emission spectra in the range of 370–700 nm (Fig. 2 and ESI†).80 Each MC showed CIE coordinates consistent with WLE, with x coordinates ranging from 0.29 to 0.35 and y coordinates ranging from 0.28 to 0.39 as compared to the neutral WLE coordinates of x, y = (0.33, 0.33) (Table 3). These results demonstrate the possibility to tune the WLE of Dy3+ MCs within the studied series based principally on the nature of the ligands and, to a lower extent, to the symmetry around the Dy3+ (Fig. 4). The CIE diagram is presented separately for each MC structure type in Fig. S11 (ESI).†
| MC | Type | Range: 370–700 nm | Range: 460–700 nm | |||||
|---|---|---|---|---|---|---|---|---|
| x | y | CCT (K) | x | y | CCT (K) | Y/B | ||
| a Calculated using corresponding emission spectra, Fig. 4. | ||||||||
| [DyGa4(shi)4(bz)4](PyH)43,68 | A | 0.35 | 0.39 | 4888 | 0.35 | 0.39 | 4891 | 1.16 |
| [DyGa4(shi)4(bz)4]68 | A | 0.35 | 0.39 | 4896 | 0.36 | 0.40 | 4821 | 1.19 |
| [DyGa4(shi)4(F-bz)4] | A | 0.35 | 0.39 | 4954 | 0.35 | 0.40 | 4882 | 1.13 |
| [DyGa4(shi)4(mbz)4] | A | 0.35 | 0.39 | 5029 | 0.35 | 0.40 | 4912 | 1.14 |
| [DyGa4(Cl-shi)4(bz)4] | A | 0.32 | 0.33 | 6291 | 0.35 | 0.40 | 4926 | 1.16 |
| [DyGa4(Cl-shi)4(F-bz)4] | A | 0.32 | 0.33 | 6289 | 0.35 | 0.39 | 5084 | 1.12 |
| [DyGa4(Br-shi)4(bz)4] | A | 0.34 | 0.38 | 5201 | 0.35 | 0.40 | 4961 | 1.15 |
| [DyGa4(I-shi)4(bz)4] | A | 0.34 | 0.39 | 5099 | 0.35 | 0.39 | 5035 | 1.11 |
| [Dy2Ga8(shi)8(iph)4](NH4)2 (ref. 44 and 68) | B | 0.35 | 0.39 | 4882 | 0.35 | 0.40 | 4818 | 1.22 |
| [Dy2Ga8(shi)8(iph)4] | B | 0.35 | 0.39 | 4859 | 0.36 | 0.40 | 4767 | 1.23 |
| [Dy2Ga8(moshi)8(iph)4]69 | B | 0.29 | 0.28 | 9117 | 0.34 | 0.39 | 5315 | 1.13 |
| [Dy2Ga8(mshi)8(iph)4] | B | 0.33 | 0.36 | 5546 | 0.35 | 0.40 | 4976 | 1.19 |
| [Dy2Ga8(shi)8(I-iph)4] | B | 0.35 | 0.39 | 4950 | 0.35 | 0.40 | 4895 | 1.17 |
| [DyGa8(shi)8(OH)4]45,68 | C | 0.31 | 0.33 | 6677 | 0.32 | 0.36 | 5920 | 0.89 |
| [DyGa8(mshi)8(OH)4](PyH) | C | 0.32 | 0.34 | 6034 | 0.33 | 0.37 | 5556 | 0.88 |
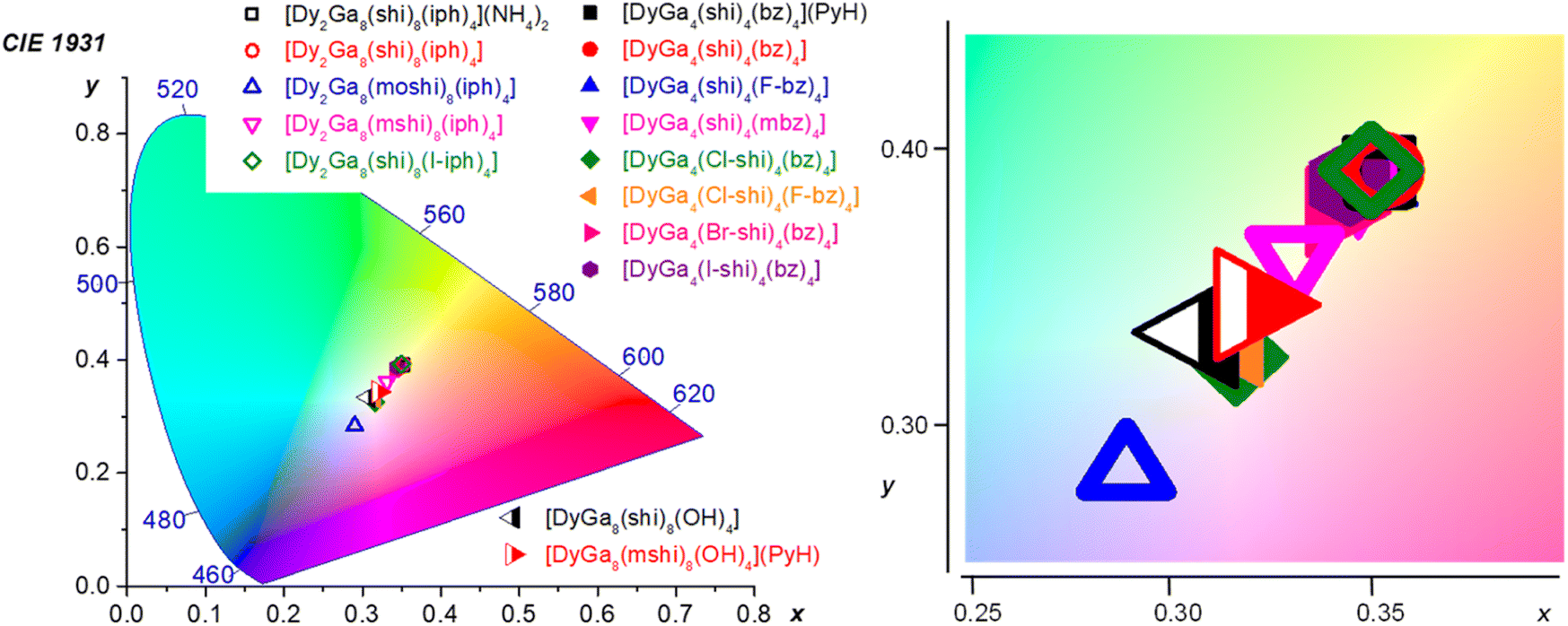 | ||
| Fig. 4 CIE 1931 diagram depicting the chromaticity coordinates (calculated for the 370–700 nm range) for all of the Dy3+ MCs presented herein. The CIE coordinates are specified in Table 3 and presented for each structure type separately in Fig. S11, ESI.† | ||
For the type A structures, each of the Dy3+ MCs possesses similar coordinates (0.34–0.35, 0.38–0.39). However, two of the compounds, DyGa4(Cl-shi)4(bz)4 and DyGa4(Cl-shi)4(F-bz)4 have disparate CIE coordinates (0.32, 0.33). Similarly, in the case of the type B structures, each Dy3+ MC has similar CIE coordinates (0.33–0.35, 0.36–0.39) except for the Dy2Ga8(moshi)8(iph)4 which has disparate coordinates (0.29, 0.28). For the type C structures, each of the two compounds has similar coordinates (0.31–0.32, 0.33–0.34).
Emission spectra of Dy3+ MCs, beside the emission signals due to the 4F9/2 → 6HJ (J = 15/2, 13/2, 11/2) Dy3+ transitions, exhibit broad ligand-centred emission bands possessing different intensities in the UV-blue range (Fig. 2). To estimate if ligand-centred emission signals affect CIE coordinates, they were re-calculated for the wavelength range comprised between 460 and 700 nm that covers exclusively the Dy3+-centred transitions (Table 3). The values obtained are much more similar for each MC structure type, i.e. (0.35–0.36, 0.39–0.40) for type A, (0.34–0.36, 0.39–0.40) for type B, and (0.32–0.33, 0.36–0.37) for type C, suggesting that the Dy3+ emission is influenced more by the symmetry of the coordination sphere rather than by the nature of the coordinating ligand. This is further confirmed by the small variations of the relative integral intensities of different Dy3+ transitions observed in the emission spectra, in particular the ratio of the yellow (4F9/2 → 6H13/2) to the blue (4F9/2 → 6H15/2) transitions, within each MC structural type (Tables 3 and S1†).
Discussion
Fifteen Dy3+ MCs formed with varied organic building blocks that belong to three different molecular topologies (Fig. 1) were synthesized and characterized. Their photophysical properties in the solid state, i.e. diffuse reflectance, excitation and emission spectra, observed luminescence lifetimes, ligand- and Dy3+-centred quantum yields were acquired and analysed. CIE 1931 and CCT have been calculated. Several Gd3+ analogues were also synthesized to provide complementary information about the ligand-centred electronic properties. This large array of compounds allows a fairly comprehensive exploration of whether the introduction of different substituents to the MC constituting ligands in a series of Dy3+/Ga3+ MCs adopting three different molecular topologies can influence the emission profiles toward the creation of tunable molecular-based WLE diodes.Among different visible-emitting Ln3+-based materials, Dy3+ ones are unique in their ability to produce WLE.46–55 Indeed, a typical emission spectrum of Dy3+ in the visible range contains three fundamental components: a blue, a yellow and a red one arising from the 4F9/2 → 6H15/2 (∼480 nm), 4F9/2 → 6H13/2 (∼575 nm), and 4F9/2 → 6H11/2 (∼660 nm) electronic transitions, respectively. Therefore, the WLE of the material can be tuned by modifying the Dy3+ emission profile and the relative intensities of each of these transitions. These parameters, in turn, can be controlled by the nature of the surrounding environment (in this case organic ligands) and the coordination symmetry about Dy3+. In addition, organic ligands may exhibit emission in the visible range and contribute to the CIE coordinates. Indeed, the generally accepted sensitization mechanism of Ln3+ in coordination compounds is assumed to be occurring through an energy transfer from the 3T1 (mainly) and/or S1 states of organic ligands, i.e. ‘antenna effect’. If this energy transfer is not complete, emission spectra may exhibit residual broad bands arising from the electronic structure of the ligand along with Ln3+-centred transitions. Therefore, the analysis of the effect of the nature of organic ligands on the emission spectra of Dy3+-based compounds is key to rationalize and further tune the WLE properties.
If one considers the ligand-centred photophysical properties in the studied series of MCs, more pronounced changes of the diffuse reflectance spectra (Fig. S3, ESI†), emission profiles (Fig. S4, ESI†) and energy positions of 3T1 and/or S1 states (Table 2 and Fig. 3) are observed upon introduction of different substituents to the salicylhydroximate ligands, H3L′. For example, 3T1 and S1 are lowered by 1060 and 915 cm−1 for [GdGa4(Cl-shi)4(bz)4] or by 745 and 2780 cm−1 for [Gd2Ga8(moshi)8(iph)4] compared to [GdGa4(shi)4(bz)4] or [Gd2Ga8(shi)8(iph)4], respectively. Accordingly, the maxima of the ligand-centred emission bands are red-shifted by 17 nm (from 360 nm for [GdGa4(shi)4(bz)4] to 377 nm for [GdGa4(Cl-shi)4(bz)4]) or 39 nm (from 364 nm for [Gd2Ga8(shi)8(iph)4] to 403 nm for [Gd2Ga8(moshi)8(iph)4]). If one compares the diffuse reflectance spectra of the LnGa4(L′)4(L′′)4, Ln2Ga8(L′)8(L′′′)4 and LnGa8(L′)8(OH)4 MCs (Ln = Gd3+ or Dy3+) with those of the corresponding constituent organic building blocks (Fig. S9, ESI†), the positions of the red edges of the spectra and, in turn, the S1 levels (Fig. 3 and Table S2, ESI†), are mainly determined by the nature of the ring salicylhydroximate ligands, H3L′. On the other hand, a similar comparison of the ligand-centred emission profiles (Fig. S10, ESI†) is more challenging. Generally, the maxima of the emission bands of the Gd3+ MCs are red-shifted by 20–40 nm compared to those of the ring H3L′ ligands. However, in the case of [GdGa4(Br-shi)4(bz)4] and [Gd2Ga8(moshi)8(iph)4], a blue shift of the emission maxima by 40 or 52 nm is observed, respectively, to these of H3Br-shi or H3moshi.
The emission spectra of all Dy3+ MCs exhibit three main bands in the visible range originating from the 4F9/2 → 6HJ (J = 15/2, 13/2, 11/2) electronic transitions along with the broad ligand-centred emission < 460 nm (Fig. 2 and Table 1). The relative intensity of the latter depends on the nature of the constituting organic ligands and is the most pronounced for [DyGa4(Cl-shi)4(bz)4], [DyGa4(Cl-shi)4(F-bz)4] and [Dy2Ga8(moshi)8(iph)4] compared to [DyGa4(shi)4(bz)4] or [Dy2Ga8(shi)8(iph)4]. On the other hand, relative integral intensities (Table S1, ESI†) and the ligand field splitting of the different Dy3+ transitions are determined by the symmetry about Dy3+. Moreover, relative integral intensities are similar for Dy3+ MCs of type A and B being in line with the analogous coordination environments around Dy3+ in both structures (Fig. 1). In contrast, significant differences are observed when comparing the emission spectra of types A and B versus type C, which has a more distinct Dy3+-coordination environment. All these variations, in turn, impact both the WLE properties and CIE coordinates (vide infra).
When it comes to quantitative characteristics, i.e. Dy3+-centred quantum yield QLDy and observed luminescence lifetimes (Table 1), more pronounced changes are again observed upon introduction of different substituents to the salicylhydroximate ligands. More specifically, for the halogenated analogues of Dy3+ MCs of type A the values of τobs are shorter by 4.5–5.9 times, while QLDy are lower by 9.7–21 times compared to those of the [DyGa4(shi)4(bz)4] MC. If one considers Dy3+ MCs of type B, the introduction of a –OCH3 substituent on the salicylhydroximate ligand leads to a 7.4-times shortening of the longest component of τobs and 11.4-times lowering of the QLDy relative to the [Dy2Ga8(shi)8(iph)4]. On the other hand, an introduction of iodine on the isophthalate ligand in [Dy2Ga8(shi)8(I-iph)4] results in a 2.2-times lengthening of the longest component of τobs (up to 40.7 μs) and an insignificant variation of the value of QLDy. A similar value of τobs (37.7 μs) is observed for [DyGa8(shi)8(OH)4]. These observations correlate well in general with the positions of the 3T1 relative to the 4F9/2 level of Dy3+: the smaller the difference between these two levels, the lower is the value of the QLDy because of the higher probability of back 3T1 ← Dy3+* energy transfer. We can also mention that the longest values of τobs are observed for MCs with a 3T1 state located at least 1000 cm−1 above the Dy3+ 4F9/2 accepting level and the S1 state not being in resonance with any upper lying Dy3+ states. However, among the Dy3+ MCs possessing very similar energy positions of the 3T1, e.g. [DyGa4(Cl-shi)4(bz)4], [Dy2Ga8(moshi)8(iph)4] and [Dy2Ga8(mshi)8(iph)4] (Table 2), the values of QLDy vary from 0.128 to 0.837% with the lowest one being for [Dy2Ga8(moshi)8(iph)4]. This observation can be explained by the lower energy position of the S1 state in this MC and its participation to energy transfer processes, including enhanced back transfer S1 ← 3T1 ← Dy3+*. We observed a similar situation for the analogues Tb3+ MCs.69 Enhanced back energy transfer processes also contribute to the relative enhancement of the ligand-centred emission and contribute to the CIE coordinates (vide infra).
WLE properties, CIE coordinates and CCT, of a material are usually determined in the visible range from 370 to 700 nm, where the human eye is sensitive. In the case of the Dy3+ MCs studied here, this range includes the broad ligand-centred bands (360–460 nm) and the three Dy3+ transitions, 4F9/2 → 6H15/2, 4F9/2 → 6H13/2 and 4F9/2 → 6H11/2 (460–700 nm). CIE coordinates (x, y) lie in the range (0.32–0.35, 0.33–0.39), (0.29–0.35, 0.28–0.39) and (0.31–0.32, 0.33–0.34) for Dy3+ MCs of type A, B and C, respectively (Table 3). Each of these coordinates correspond to a type of white appearing light, ranging from blue (or cool) to neutral (0.33, 0.33) and yellow (or warm) white (Fig. 4). This range represents an attractive tunability of the WLE in these series of Dy3+ MCs, controlled by the nature of the constituting organic ligands. The closest to the neutral white light CIE coordinates are observed for [DyGa4(Cl-shi)4(bz)4], [DyGa4(Cl-shi)4(F-bz)4] and Dy3+ MCs of type C.
Since the emission spectra of Dy3+ MCs originate from both the Dy3+ and the constituting ligands, another set of (x, y) was calculated considering only Dy3+-centred transitions in the range of 460–700 nm (Table 3) in order to understand which parameters and to what extent they contribute to the tunability of CIE coordinates. The obtained values indicate a significantly lower level of tunability: similar for MCs of types A and B while distinct for those of type C, and vary only within 0.02 units for both x and y for a specific MC topology. These results are in line with the previous point that the Dy3+ emission profile, i.e. the relative integral intensities of the 4F9/2 → 6HJ (J = 15/2, 13/2, 11/2) transitions and their ligand field splitting, is determined by the MC topology and the symmetry about Dy3+, and is less influenced by the nature of the constituent organic ligands. On the other hand, the latter has a significant impact on the general emission profile and contributes significantly to the tuning of the WLE properties within a series of Dy3+ MCs of a specific structure type.
Here, we have demonstrated that, among MCs of types A and B, the greatest divergence from average CIE coordinates, ca. (0.35, 0.39), is observed for [DyGa4(Cl-shi)4(bz)4] (0.32, 0.33), [DyGa4(Cl-shi)4(F-bz)4] (0.32, 0.33), [Dy2Ga8(mshi)8(iph)4] (0.33, 0.36), and [Dy2Ga8(moshi)8(iph)4] (0.29, 0.28). The origin of the disparities using these building blocks must come from the higher contribution of the ligand-centred bands to the total emission profile because of the similarity of the Dy3+-centred transitions for these types of MCs. Indeed, the ratio QLL/QLtotal is the largest for the selected Dy3+ MCs and it increases in the row [Dy2Ga8(mshi)8(iph)4] (0.237) ≪ [Dy2Ga8(moshi)8(iph)4] (0.435) < [DyGa4(Cl-shi)4(F-bz)4] (0.467) < [DyGa4(Cl-shi)4(bz)4] (0.513) (Table 1). In general, these results correlate well with the positions of the 3T1 states of the ligands relatively to the 4F9/2 level of Dy3+ and higher probability of the back 3T1 ← Dy3+* energy transfer that has been discussed above. However, one may notice that the most significant impact on CIE coordinates is observed for the [Dy2Ga8(moshi)8(iph)4] MC, that does not possess the highest QLL/QLtotal ratio. On the other hand, [Dy2Ga8(moshi)8(iph)4] exhibits the most red-shifted ligand-centred emission among the studied MCs (Fig. 2), that is located in the blue range of the spectrum and, in turn, has a larger contribution to the CIE coordinates. Indeed, a higher energy (ultraviolet) fluorescence is less visible to the human eye and has an insignificant impact on the tuning of WLE properties of the materials (Fig. S12, ESI†). Therefore, the CIE coordinates of Dy3+ MCs of a particular topology can be adjusted by the appropriate choice of the building blocks that will influence the total emission profile by changing the probability of different energy transfer processes including the 3T1 ← Dy3+* process and/or by exhibiting fluorescence in the blue range. In this way, the warm white emission of the majority of Dy3+ MCs of types A and B can be turned into neutral white in [DyGa4(Cl-shi)4(bz)4] (0.32,0.33), and [DyGa4(Cl-shi)4(F-bz)4] (0.32, 0.33), for example. To an extent, this can be predicted before the MC is synthesized, because the S1 energy of the ligand should generally inversely correlate with the wavelength of the emission, i.e. ligands with lower S1 will exhibit emission at longer wavelengths (Fig. S5, ESI†). However, as mentioned before, the direct correlation of the ligand-centred emission in the MC with the one of the constituent organic ligands is not straightforward. Several different parameters have to be considered. Moreover, in the solid state, additional parameters that will impact the ligand-centred emission need to be taken into account: these include the effect of the presence of the co-crystallized solvent molecules, the intramolecular interactions and interactions due to the packing of the molecules. One should also consider the ratio of the ligand fluorescence to phosphorescence, as a higher proportion of the latter will lead to a more redshifted total ligand-centred emission.
Conclusions
We have previously shown that Dy3+/Ga3+ metallacrowns possessing three different molecular topologies were capable of tuning CIE coordinates through the modification of the Dy3+ first coordination sphere symmetry. In this work, we subject these previously characterized systems to electronic perturbations via ligand functionalization to decipher the role of the different organic building blocks on the emission properties of these molecules.First, we observed that the Dy3+-centred transitions are essentially unperturbed by altering the electron donor or acceptor properties of the organic building blocks. This insensitivity of the Dy3+ emission profile to electronic perturbations is a consequence of the rigidity of this family of MCs meaning that one may modify the donor/acceptor properties of the organic ligand of the molecule while retaining the original lanthanide(III) spectral profile. Second, we have demonstrated that CIE coordinates can be extensively tuned from warm to neutral to cool white by first defining the symmetry about Dy3+ and then choosing appropriate organic ligands that will influence the total emission profile through changing the probability of different energy transfer processes including 3T1 ← Dy3+* energy back transfer and/or by generating ligand-centred fluorescence in the blue range. In particular, the overall WLE profile of the Dy3+/Ga3+ MCs can be changed by altering the relative proportion of blue ligand emissions. This behaviour is due to the more visible appearance of the lower energy ligand states' fluorescence (opposed to less-visible higher energy UV emissions), and also the effect that electronic perturbations on the ligands has on the proportion of Dy3+- vs. ligand-centred transitions, (QLL/QLtotal). Thus, by specifying an individual coordination geometry for the lanthanide(III) in the MC structural motif, one obtains an essentially invariant Dy3+ emission that can be fine-tuned to the desired CIE coordinates by controlling the electronic properties of organic building blocks. We have shown that this approach allows the creation of unique tunable WLE using single component tetrachroic, molecular chromophores.
Data availability
All data are available in the ESI.†Author contributions
E. V. S. – investigation, data curation, formal analysis, writing – original draft; S. V. E. – conceptualization, investigation, data curation, formal analysis, funding acquisition, writing – review & editing; S. P. – conceptualization, funding acquisition, supervision, writing – review & editing; V. L. P. – conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the National Science Foundation under grants CHE-1664964 and DGE-1256260, La Ligue Contre le Cancer du Grand Ouest (comités du Loiret, du Loir-et-Cher, d’Eure-et-Loir et de la Sarthe), the Réseau ‘Molécules Marines, Métabolisme & Cancer’ du Cancéropôle Grand Ouest and La Région Centre. V. L. P. thanks Le Studium Loire Valley Institute for Advanced Studies, Orléans & Tours, France as a partial support for a mobility in France. S. P. acknowledges support from Institut National de la Santé et de la Recherche Médicale (INSERM).Notes and references
- J. Cho, J. H. Park, J. K. Kim and E. F. Schubert, White Light-Emitting Diodes: History, Progress, and Future, Laser Photonics Rev., 2017, 11(2), 1600147, DOI:10.1002/lpor.201600147
.
- S. SeethaLekshmi, A. R. Ramya, M. L. P. Reddy and S. Varughese, Lanthanide Complex-Derived White-Light Emitting Solids: A Survey on Design Strategies, J. Photochem. Photobiol. C: Photochem. Rev., 2017, 33, 109–131, DOI:10.1016/j.jphotochemrev.2017.11.001
- W. A. Thornton, Luminosity and Color- Rendering Capability of White Light, J. Opt. Soc. Am. A, 1971, 61(9), 1155–1163, DOI:10.1364/josa.61.001155
-
A. K. R. Choudhury, Using Instruments to Quantify Colour, In Principles of Colour and Appearance Measurement, Woodhead Publishing, 2014, pp. 270–317, DOI:10.1533/9780857099242.270
-
D. Hofmann and E. Rasch, Chapter 7. Fluorescent Lamps, In Revolution in Lamps, ed. R. Kane and H. Sell, River Publishers, 2001, DOI:10.1201/9781003150985
-
T. Erdem and H. V. Demir, Common White Light Sources, In Color Science and Photometry for Lighting with LEDs and Semiconductor Nanocrystals, Springer, Singapore, 2019, pp. 27–34, DOI:10.1007/978-981-13-5886-9
-
B. D. Fahlman, Materials Chemistry, Springer, Dordrecht, 2007, DOI:10.1007/978-1-4020-6120-2
- H.-W. Chen, J.-H. Lee, B.-Y. Lin, S. Chen and S.-T. Wu, Liquid Crystal Display and Organic Light-Emitting Diode Display: Present Status and Future Perspectives, Light: Sci. Appl., 2018, 7(3), 17168, DOI:10.1038/lsa.2017.168
- N. Thejo Kalyani and S. J. Dhoble, Organic Light Emitting Diodes: Energy Saving Lighting Technology - A Review, Renewable Sustainable Energy Rev., 2012, 16(5), 2696–2723, DOI:10.1016/j.rser.2012.02.021
- S. K. Behera, R. Kainda, S. Basu and Y. S. Chaudhary, Single Organic Molecular Systems for White Light Emission and Their Classification with Associated Emission Mechanism, Appl. Mater. Today, 2022, 27, 101407, DOI:10.1016/j.apmt.2022.101407
- T. Yuan, F. Yuan, X. Li, Y. Li, L. Fan and S. Yang, Fluorescence-Phosphorescence Dual Emissive Carbon Nitride Quantum Dots Show 25% White Emission Efficiency Enabling Single-Component WLEDs, Chem. Sci., 2019, 10(42), 9801–9806, 10.1039/c9sc03492g
- O. Kotova, S. Comby, C. Lincheneau and T. Gunnlaugsson, White-Light Emission from Discrete Heterometallic Lanthanide-Directed Self-Assembled Complexes in Solution, Chem. Sci., 2017, 8(5), 3419–3426, 10.1039/c7sc00739f
- H. Zhang, X. Shan, L. Zhou, P. Lin, R. Li, E. Ma, X. Guo and S. Du, Full-Colour Fluorescent Materials Based on Mixed-Lanthanide(III) Metal-Organic Complexes with High-Efficiency White Light Emission, J. Mater. Chem. C, 2013, 1(5), 888–891, 10.1039/c2tc00414c
- Q. Li, J. Qian, J. Zhou, L. Du and Q. Zhao, Highly Chemically and Thermally Stable Lanthanide Coordination Polymers for Luminescent Probes and White Light Emitting Diodes, CrystEngComm, 2020, 22(15), 2667–2674, 10.1039/D0CE00228C
- W. Feng, G. Fu, H. Zheng, H. Zheng, S. Yuan and X. Lü, Single-Component White-Light-Emitting Eu3+-Metallopolymer for near-Ultraviolet White Light-Emitting Diode (n-UV-WLED), J. Lumin., 2021, 233, 117897, DOI:10.1016/j.jlumin.2021.117897
- R. Huo, X. Li and D. Ma, Lanthanide Coordination Frameworks Constructed from 1,3-Benzenedicarboxylate, Oxalate and 1,10-Phenanthroline: Crystal Structure, Multicolor Luminescence and High-Efficiency White Light Emission, CrystEngComm, 2015, 17(20), 3838–3844, 10.1039/C5CE00548E
- H. Zhang, R. Fan, Y. Dong, W. Chen, X. Du, P. Wang and Y. Yang, Assembly of One-, Two-, and Three-Dimensional Ln(III) Complexes Constructed from a Novel Asymmetric Tricarboxylic Acid: Synthesis, Structure, Photoluminescence and Tunable White-Light Emission, CrystEngComm, 2016, 18(20), 3711–3724, 10.1039/C6CE00483K
- P. A. Demakov, A. A. Ryadun, P. V. Dorovatovskii, V. A. Lazarenko, D. G. Samsonenko, K. A. Brylev, V. P. Fedin and D. N. Dybtsev, Intense Multi-Colored Luminescence in a Series of Rare-Earth Metal–Organic Frameworks with Aliphatic Linkers, Dalton Trans., 2021, 50(34), 11899–11908, 10.1039/D1DT00872B
- O. Guillou, C. Daiguebonne, G. Calvez and K. A. Bernot, Long Journey in Lanthanide Chemistry: From Fundamental Crystallogenesis Studies to Commercial Anticounterfeiting Taggants, Acc. Chem. Res., 2016, 49(5), 844–856, DOI:10.1021/acs.accounts.6b00058
- C. Blais, G. Calvez, Y. Suffren, C. Daiguebonne, C. Paranthoen, E. Bazin, S. Freslon, K. Bernot and O. Guillou, Luminance and Brightness: Application to Lanthanide-Based Coordination Polymers, Inorg. Chem., 2022, 61(48), 19588–19596, DOI:10.1021/acs.inorgchem.2c03500
- S. Freslon, Y. Luo, C. Daiguebonne, G. Calvez, K. Bernot and O. Guillou, Brightness and Color Tuning in a Series of Lanthanide-Based Coordination Polymers with Benzene-1,2,4,5-Tetracarboxylic Acid as a Ligand, Inorg. Chem., 2016, 55(2), 794–802, DOI:10.1021/acs.inorgchem.5b02242
- J. Liu, W. Sun and Z. Liu, White-Light Emitting Materials with Tunable Luminescence Based on Steady Eu(III) Doping of Tb(III) Metal-Organic Frameworks, RSC Adv., 2016, 6(31), 25689–25694, 10.1039/c6ra01931e
- V. Trannoy, I. N'Dala-Louika, J. Lhoste, T. Devic and H. Serier-Brault, Lanthanide Isophthalate Metal-Organic Frameworks: Crystal Structure, Thermal Behavior, and White Luminescence, Eur. J. Inorg. Chem., 2021, 2021(4), 398–404, DOI:10.1002/ejic.202000906
- L.-D. Zhao, B.-B. Ma, X.-S. Gao, L. Zhai and X.-M. Ren, Metal–Organic Framework Solid Solutions of Rare Earth Ions Tb3+, Eu3+ and Y3+ with Pyridine-2, 4, 6-Tricarboxylate Ligand Emitting High Quantum Yield White Light, J. Solid State Chem., 2022, 305, 122654, DOI:10.1016/j.jssc.2021.122654
- D. Ma, X. Li and R. Huo, A High-Efficiency White Light-Emitting Lanthanide–Organic Framework Assembled from 4,4′-Oxybis(Benzoic Acid), 1,10-Phenanthroline and Oxalate, J. Mater. Chem. C, 2014, 2(43), 9073–9076, 10.1039/C4TC01409J
- Y.-F. Li, D. Wang, Z. Liao, Y. Kang, W.-H. Ding, X.-J. Zheng and L.-P. Jin, Luminescence Tuning of the Dy–Zn Metal–Organic Framework and Its Application in the Detection of Fe(III) Ions, J. Mater. Chem. C, 2016, 4(19), 4211–4217, 10.1039/C6TC00832A
- L. Wu, Y. Zhang, M. Gui, P. Lu, L. Zhao, S. Tian, Y. Kong and J. Xu, Luminescence and Energy Transfer of a Color Tunable Phosphor: Dy3+-, Tm3+-, and Eu3+-Coactivated KSr4(BO3)3 for Warm White UV LEDs, J. Mater. Chem., 2012, 22(13), 6463–6470, 10.1039/c2jm15506k
- Y. Zhang, W. Gong, J. Yu, Y. Lin and G. Ning, Tunable White-Light Emission via Energy Transfer in Single-Phase LiGd(WO4)2:Re3+ (Re = Tm, Tb, Dy, Eu) Phosphors for UV-Excited WLEDs, RSC Adv., 2015, 5(117), 96272–96280, 10.1039/c5ra19345a
- Z. Fu, W. Xia, Q. Li, X. Cui and W. Li, Highly Uniform NaLa(MoO4)2:Ln3+ (Ln = Eu, Dy) Microspheres: Template-Free Hydrothermal Synthesis, Growing Mechanism, and Luminescent Properties, CrystEngComm, 2012, 14(14), 4618–4624, 10.1039/c2ce06682c
- B. W. Ennis, S. Muzzioli, B. L. Reid, D. M. D'Alessio, S. Stagni, D. H. Brown, M. I. Ogden and M. Massi, Recyclable Calix[4]Arene-Lanthanoid Luminescent Hybrid Materials with Color-Tuning and Color-Switching Properties, Dalton Trans., 2013, 42(19), 6894–6901, 10.1039/c3dt33059a
- Z. Sun, F. Bai, H. Wu, D. M. Boye and H. Fan, Monodisperse Fluorescent Organic/Inorganic Composite Nanoparticles: Tuning Full Color Spectrum, Chem. Mater., 2012, 24(17), 3415–3419, DOI:10.1021/cm3016287
- A. S. Kuznetsov, A. Nikitin, V. K. Tikhomirov, M. V. Shestakov and V. V. Moshchalkov, Ultraviolet-Driven White Light Generation from Oxyfluoride Glass Co-Doped with Tm3+-Tb3+-Eu3+, Appl. Phys. Lett., 2013, 102(16), 161916, DOI:10.1063/1.4803448
- M. Fang, A. N. C. Neto, L. Fu, R. A. S. Ferreira, V. DeZeaBermudez and L. D. Carlos, A Hybrid Materials Approach for Fabricating Efficient WLEDs Based on Di-Ureasils Doped with Carbon Dots and a Europium Complex, Adv. Mater. Technol., 2022, 7(3), 2100727, DOI:10.1002/admt.202100727
- D. Mara, F. Artizzu, J. Goura, M. Jayendran, B. Bokić, B. Kolaric, T. Verbiest and R. Van Deun, Molecular Dysprosium Complexes for White-Light and near-Infrared Emission Controlled by the Coordination Environment, J. Lumin., 2022, 243, 118646, DOI:10.1016/j.jlumin.2021.118646
- Q. Shi, J. Liu, J. Wang, X. Yang, X. Zhang, S. Li, P. Sun, J. Chen, B. Li and X. Lü, Color-Tunable White-Light of Binary Tris-β-Diketonate-(Dy3+, Gd3+) Complexes' Blend under Single Wavelength Excitation, Inorg. Chem. Commun., 2020, 113, 107814, DOI:10.1016/j.inoche.2020.107814
- A. H. Shelton, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Controllable Three-Component Luminescence from a 1,8-Naphthalimide/Eu(III) Complex: White Light Emission from a Single Molecule, Chem. Comm., 2012, 48(22), 2749–2751, 10.1039/c2cc17182a
- J. Manzur, R. C. De Santana, L. J. Q. Maia, A. Vega and E. Spodine, Tuning White Light Emission in Dinuclear Phenoxo Bridged DyIII Complexes, Inorg. Chem., 2019, 58(15), 10012–10018, DOI:10.1021/acs.inorgchem.9b01153
- Q. Y. Yang, K. Wu, J. J. Jiang, C. W. Hsu, M. Pan, J. M. Lehn and C. Y. Su, Pure White-Light and Yellow-to-Blue Emission Tuning in Single Crystals of Dy(III) Metal–Organic Frameworks, Chem. Comm., 2014, 50(57), 7702–7704, 10.1039/c4cc01763c
- C. Görller-Walrand and K. Binnemans, Chapter 167 Spectral Intensities of F-f Transitions, Handbook on the Physics and Chemistry of Rare Earths, 1998, 101–264, DOI:10.1016/S0168-1273(98)25006-9
- Q. Su, Influence of Environment on the Luminescence of Rare Earths, J. Lumin., 1988, 40–41, 113–114, DOI:10.1016/0022-2313(88)90113-5
- Q. Su, Z. Pei, L. Chi, H. Zhang, Z. Zhang and F. Zou, The Yellow-to-Blue Intensity Ratio (Y/B) of Dy3+ Emission, J. Alloys Compd., 1993, 192(1–2), 25–27, DOI:10.1016/0925-8388(93)90174-L
- D. Parker, E. A. Suturina, I. Kuprov and N. F. Chilton, How the Ligand Field in Lanthanide Coordination Complexes Determines Magnetic Susceptibility Anisotropy, Paramagnetic NMR Shift, and Relaxation Behavior, Acc. Chem. Res., 2020, 53(8), 1520–1534, DOI:10.1021/acs.accounts.0c00275
- C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, T. N. Nguyen, J. W. Kampf, S. Petoud and V. L. Pecoraro, Ga3+/Ln3+ Metallacrowns: A Promising Family of Highly Luminescent Lanthanide Complexes That Covers Visible and Near-Infrared Domains, J. Am. Chem. Soc., 2016, 138(15), 5100–5109, DOI:10.1021/jacs.6b00984
- T. N. Nguyen, C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, J. W. Kampf, I. Martinić, S. Petoud and V. L. Pecoraro, One-Step Assembly of Visible and Near-Infrared Emitting Metallacrown Dimers Using a Bifunctional Linker, Chem.–A Euro. J., 2018, 24(5), 1031–1035, DOI:10.1002/chem.201703911
- E. V. Salerno, S. V. Eliseeva, B. L. Schneider, J. W. Kampf, S. S. S. Petoud and V. L. V. Pecoraro, Near-Infrared, and Dual-Range Luminescence Spanning the 4f Series Sensitized by a Gallium(III)/Lanthanide(III) Metallacrown Structure, J. Phys. Chem. A, 2020, 124(50), 10550–10564, DOI:10.1021/acs.jpca.0c08819
- R. Shrivastava, J. Kaur and V. Dubey, White Light Emission by Dy3+ Doped Phosphor Matrices: A Short Review, J. Fluoresc., 2016, 26(1), 105–111, DOI:10.1007/s10895-015-1689-8
- J. Li, W. Wang, B. Liu, G. Duan and Z. Liu, Enhanced Dy3+ White Emission via Energy Transfer in Spherical (Lu,Gd)3Al5O12 Garnet Phosphors, Sci. Rep., 2020, 10(1), 2–10, DOI:10.1038/s41598-020-59232-8
- K. Reenabati Devi, S. Dorendrajit Singh and T. David Singh, Photoluminescence Properties of White Light Emitting La2O3:Dy3+ Nanocrystals, Indian J. Phys., 2018, 92(6), 725–730, DOI:10.1007/s12648-018-1159-7
- S. Sharma, N. Brahme, D. P. Bisen and P. Dewangan, Cool White Light Emission from Dy3+ Activated Alkaline Alumino Silicate Phosphors, Opt. Express, 2018, 26(22), 29495, DOI:10.1364/oe.26.029495
- B. V. Ratnam, M. Jayasimhadri, K. Jang, H. Sueb Lee, S. S. Yi and J. H. Jeong, White Light Emission from NaCaPO4:Dy3+ Phosphor for Ultraviolet-Based White Light-Emitting Diodes, J. Am. Ceram. Soc., 2010, 93(11), 3857–3861, DOI:10.1111/j.1551-2916.2010.03963.x
- Y. Dai, S. Yang, Y. Shan, C. G. Duan, H. Peng, F. Yang and Q. Zhao, Single-Composition White Light Emission from Dy3+ Doped Sr2CaWO6, Materials, 2019, 12(3), 431, DOI:10.3390/ma12030431
- S. Liu, B. Deng, J. Chen, H. Liu, C. S. Zhou and R. Yu, A Novel UV Pumped White-Emitting Phosphor La3SbO7:Dy3+ for White Light-Emitting Diodes, IOP Conf. Ser.: Earth Environ. Sci., 2019, 295(3), 4–7, DOI:10.1088/1755-1315/295/3/032035
- P. Narwal, M. S. Dahiya, A. Agarwal, A. Hooda and S. Khasa, White Light Emission in Dy3+ Doped NaCl−BaO Bismuth Borate Glasses, J. Lumin., 2019, 209(May 2019), 121–128, DOI:10.1016/j.jlumin.2019.01.042
- J. Kuang, Y. Liu and J. Zhang, White-Light-Emitting Long-Lasting Phosphorescence in Dy3+-Doped SrSiO3, J. Solid State Chem., 2006, 179(1), 266–269, DOI:10.1016/j.jssc.2005.10.025
- G. Che, C. Liu, Q. Wang and Z. Xu, White-Light-Emission Afterglow Phosphor CaZnGe2O6: Dy3+, Chem. Lett., 2008, 37(2), 136–137, DOI:10.1246/cl.2008.136
- N. Shasmal and B. Karmakar, White Light-Emitting Dy3+ -Doped Transparent Chloroborosilicate Glass: Synthesis and Optical Properties, J. Asian Ceram. Soc., 2019, 7(1), 42–52, DOI:10.1080/21870764.2018.1555883
- L. Mishra, A. Sharma, A. K. Vishwakarma, K. Jha, M. Jayasimhadri, B. V. Ratnam, K. Jang, A. S. Rao and R. K. Sinha, J. Lumin., 2016, 169, 121–127, DOI:10.1016/j.jlumin.2015.08.063
- K. Brahmachary, D. Rajesh and Y. C. Ratnakaram, Luminescence Properties of Dy3+ Doped Zinc-Aluminum-Sodium-Phosphate (ZANP) Glasses for White Light Applications, Optik, 2015, 126(23), 4050–4055, DOI:10.1016/j.ijleo.2015.08.001
- N. Jaidass, C. Krishna Moorthi, A. Mohan Babu and M. Reddi Babu, Luminescence Properties of Dy3+ Doped Lithium Zinc Borosilicate Glasses for Photonic Applications, Heliyon, 2018, 4(3), e00555, DOI:10.1016/j.heliyon.2018.e00555
- X. Li, L. Guan, M. Sun, H. Liu, Z. Yang, Q. Guo and G. Fu, Luminescent Properties of Dy3+ Doped SrMoO4 Phosphor, J. Lumin., 2011, 131(5), 1022–1025, DOI:10.1016/j.jlumin.2011.01.015
- P. Sailaja, S. Mahamuda, R. A. Talewar, K. Swapna and A. S. Rao, Spectroscopic Investigations of Dysprosium Ions Doped Oxy Chloro Boro Tellurite Glasses for Visible Photonic Device Applications, J. Alloys Compd., 2019, 789, 744–754, DOI:10.1016/j.jallcom.2019.03.148
- N. Luewarasirikul, H. J. Kim, P. Meejitpaisan and J. Kaewkhao, White Light Emission of Dysprosium Doped Lanthanum Calcium Phosphate Oxide and Oxyfluoride Glasses, Opt. Mater., 2017, 66, 559–566, DOI:10.1016/j.optmat.2017.02.049
- U. Fawad, H. J. Kim, S. Khan, M. Khan and L. Ali, Photoluminescent Properties of White-Light-Emitting Li6Y(BO3)3:Dy3+ Phosphor, Solid State Sci., 2016, 62(8), 1–5, DOI:10.1016/j.solidstatesciences.2016.08.008
- Y. Chen, J. Xu, S. Xie, Z. Tan, R. Nie, Z. Guan, Q. Wang and J. Zhu, Ion Doping Effects on the Lattice Distortion and Interlayer Mismatch of Aurivillius-Type Bismuth Titanate Compounds, Materials, 2018, 11(5), 821, DOI:10.3390/ma11050821
- F. Ma, D. Jiang, Z. Zhang, X. Tian, Q. Wu, J. Wang, X. Qian, Y. Liu and L. Su, Tailoring the Local Lattice Distortion of Nd3+ by Codoping of Y3+ through First Principles Calculation for Tuning the Spectroscopic Properties, Opt. Mater. Express, 2019, 9(11), 4256, DOI:10.1364/ome.9.004256
- K. R. Limmer, M. R. Neupane, R. E. Brennan, T. L. Chantawansri and S. Sinnott, Rare-Earth Dopant Effects on the Structural, Energetic, and Magnetic Properties of Alumina from First Principles, J. Am. Ceram. Soc., 2016, 99(12), 4007–4012, DOI:10.1111/jace.14445
- K. Leśniak, Crystal Fields and Local Lattice Distortions in Some Tetragonal Symmetry Centers in Fluorite Crystals Doped with Trivalent Rare-Earth Ions, J. Chem. Phys., 1991, 94(5), 3919–3927, DOI:10.1063/1.460668
- S. V. Eliseeva, E. V. Salerno, B. A. Lopez Bermudez, S. Petoud and V. L. Pecoraro, Dy3+ White Light Emission Can Be Finely Controlled by Tuning the First Coordination Sphere of Ga3+/Dy3+ Metallacrown Complexes, J. Am. Chem. Soc., 2020, 142(38), 16173–16176, DOI:10.1021/jacs.0c07198
- E. V. Salerno, A. N. Carneiro Neto, S. V. Eliseeva, M. A. Hernández-Rodríguez, J. C. Lutter, T. Lathion, J. W. Kampf, S. Petoud, L. D. Carlos and V. L. Pecoraro, J. Am. Chem. Soc., 2022, 144(40), 18259–18271, DOI:10.1021/jacs.2c04821
- E. V. Salerno, J. Zeler, S. V. Eliseeva, M. A. Hernández-Rodríguez, A. N. Carneiro Neto, S. Petoud, V. L. Pecoraro and L. D. Carlos, [Ga3+8Sm3+2, Ga3+8Tb3+2] metallacrowns are highly promising ratiometric luminescent molecular nanothermometers operating at physiologically relevant temperatures, Chem.–A Euro. J., 2020, 26(61), 13792–13796, DOI:10.1002/chem.202003239
- J. C. Lutter, S. V. Eliseeva, G. Collet, I. Martinić, J. W. Kampf, B. L. Schneider, A. Carichner, J. Sobilo, S. Lerondel, S. Petoud and V. L. Pecoraro, Iodinated Metallacrowns: Toward Combined Bimodal Near-Infrared and X-Ray Contrast Imaging Agents, Chem.–A Euro. J., 2020, 26(6), 1274–1277, DOI:10.1002/chem.201905241
-
J.-C. Bünzli and S. V. Eliseeva, Basics of Lanthanide Photophysics, In Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, Springer-Verlag Berlin Heidelberg, 2011, pp. 1–46, DOI:10.1007/978-3-642-21023-5
- T. T. Boron, J. C. Lutter, C. I. Daly, C. Y. Chow, A. H. Davis, A. Nimthong-Roldán, M. Zeller, J. W. Kampf, C. M. Zaleski and V. L. Pecoraro, The Nature of the Bridging Anion Controls the Single-Molecule Magnetic Properties of DyX4M 12-Metallacrown-4 Complexes, Inorg. Chem., 2016, 55(20), 10597–10607, DOI:10.1021/acs.inorgchem.6b01832
- M. R. Azar, T. T. Boron, J. C. Lutter, C. I. Daly, K. A. Zegalia, R. Nimthong, G. M. Ferrence, M. Zeller, J. W. Kampf, V. L. Pecoraro and C. M. Zaleski, Controllable Formation of Heterotrimetallic Coordination Compounds: Systematically Incorporating Lanthanide and Alkali Metal Ions into the Manganese 12-Metallacrown-4 Framework, Inorg. Chem., 2014, 53(3), 1729–1742, DOI:10.1021/ic402865p
- T. Lou, H. Yang, S. Zeng, D. Li and J. Dou, A New Family of Heterometallic LnIII [12-MCFeIIIn(Shi)-4] Complexes: Syntheses, Structures and Magnetic Properties, Crystals, 2018, 8(5), 229, DOI:10.3390/cryst8050229
-
F. Shao, Engineering Magnetic Anisotropy In Mononuclear Cobalt(II) Complexes And Lanthanide-Based Metallacrowns, PhD Thesis, Université Paris Saclay, 2017
- J. R. Travis, A. M. Smihosky, A. C. Kauffman, S. E. Ramstrom, A. J. Lewis, S. G. Nagy, R. E. Rheam, M. Zeller and C. M. Zaleski, Syntheses and Crystal Structures of Two Classes of Aluminum-Lanthanide-Sodium Heterotrimetallic 12-Metallacrown-4 Compounds: Individual Molecules and Dimers of Metallacrowns, J. Chem. Crystallogr., 2021, 51, 372–393, DOI:10.1007/s10870-020-00861-2
- J. C. Lutter, B. A. Lopez Bermudez, T. N. Nguyen, J. W. Kampf and V. L. Pecoraro, Functionalization of Luminescent Lanthanide-Gallium Metallacrowns Using Copper-Catalyzed Alkyne-Azide Cycloaddition and Thiol-Maleimide Michael Addition, J. Inorg. Biochem., 2019, 192(March 2019), 119–125, DOI:10.1016/j.jinorgbio.2018.12.011
- J. Wang, G. Lu, Y. Liu, S. G. Wu, G. Z. Huang, J. L. Liu and M. L. Tong, Building Block and Directional Bonding Approaches for the Synthesis of {DyMn4}n (n = 2, 3) Metallacrown Assemblies, Cryst. Growth Des., 2019, 19(3), 1896–1902, DOI:10.1021/acs.cgd.8b01879
-
A. K. R. Choudhury, Chapter 7: Using Instruments to Quantify Colour, In Principles of Colour and Appearance Measurement, 2014, pp. 270–317, DOI:10.1533/9780857099242.270
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed of the synthesis and characterization of MCs, additional information about photophysical studies, details about the determination of the singlet, triplet states, CIE 1931 coordinates and CCT. See DOI: https://doi.org/10.1039/d4sc00389f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |