
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Network topology diversification of porous organic salts†
Hiroi
Sei‡
a,
Kouki
Oka‡
 ab,
Yuta
Hori
c,
Yasuteru
Shigeta
c and
Norimitsu
Tohnai
*a
ab,
Yuta
Hori
c,
Yasuteru
Shigeta
c and
Norimitsu
Tohnai
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: tohnai@chem.eng.osaka-u.ac.jp
bInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
cCenter for Computational Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
First published on 1st May 2024
Abstract
Hydrogen-bonded organic frameworks (HOFs) are porous organic materials constructed via hydrogen bonds. HOFs have solubility in specific high-polar organic solvents. Therefore, HOFs can be returned to their components and can be reconstructed, which indicates their high recyclability. Network topologies, which are the frameworks of porous structures, control the pore sizes and shapes of HOFs. Therefore, they strongly affect the functions of porous materials. However, hydrogen bonds are usually weak interactions, and the design of the intended network topology in HOFs from their components has been challenging. Porous organic salts (POSs) are an important class of HOFs, are hierarchically constructed via strong charge-assisted hydrogen bonds between sulfonic acids and amines, and therefore are expected to have high designability of the porous structure. However, the network topology of POSs has been limited to only dia-topology. Here, we combined tetrasulfonic acid with the adamantane core (4,4′,4′′,4′′′-(adamantane-1,3,5,7-tetrayl)tetrabenzenesulfonic acid; AdPS) and triphenylmethylamines with modified substituents in para-positions of benzene rings (TPMA-X, X = F, methyl (Me), Cl, Br, I). We changed the steric hindrance between the adamantane and substituents (X) in TPMA-X and obtained not only the common dia-topology for POSs but also rare sod-topology, and lon- and uni-topologies that are formed for the first time in HOFs. Changing template molecules under preparation helped in successfully isolating the porous structures of AdPS/TPMA-Me with dia-, lon-, and sod-topologies which exhibited different gas adsorption properties. Therefore, for the first time, we demonstrated that the steric design of HOF components facilitated the formation, diversification, and control of the network topologies and functions of HOFs.
Introduction
Organic porous materials are composed of organic molecules and can be facilely functionalized via the molecular design of their components.1 Moreover, they are metal-free and composed of abundant elements (C, N, O, S, etc.) on earth, and are therefore expected to have high environmental acceptability.1c,2 Representative organic porous materials, covalent organic frameworks (COFs) constructed via covalent bonds, and hydrogen-bonded organic frameworks (HOFs) constructed via hydrogen bonds, are well-known. They have been aggressively investigated based on the design of nanospaces toward various functions and applications such as selective adsorption and separation of gas molecules,3 stimulated response to specific ions and molecules,4 ion conduction,5 and catalytic reactions.6 Among them, HOFs can be prepared under mild conditions such as recrystallization, and continuous bond formation and dissociation allow them to eliminate energetically unfavorable defects and to construct thermodynamically stable ordered structures with high crystallinity.7 Single-crystal X-ray diffraction analysis enables us to reveal the detailed crystal structure, such as the framework of the porous structures (network topology), the interpenetration of the frameworks, and the bond lengths and angles, and therefore, to investigate a correlation between their porous structures and functions. In addition, HOFs are soluble in specific high-polar solvents, return to their components facilely, and then are reproduced via recrystallization, which exhibits their high recyclability.8In general, network topologies control the pore sizes and shapes of the porous structures, and strongly affect their functions.9 Therefore, diversification and control of network topology are significantly important in the investigation of porous materials. The conventional studies of HOFs have aimed to form various network topologies by changing the combination between supramolecular synthons1a,10 which are the common patterns of hydrogen bonds in crystal structures, and tectons10b,11 which are the building blocks. However, the conventional HOFs are constructed by weak hydrogen bonds and form unintended bonds12 including the involvement of solvent molecules that are different from supramolecular synthons, which causes the construction of unexpected structures. Therefore, it is difficult to predict network topologies from supramolecular synthons and tectons.8 Some previous studies have reported that the change of the solvent condition in recrystallization enables HOFs to form porous structures with different network topologies.13 Furthermore, polymorphic and pseudopolymorphic porous structures of HOFs are often constructed even under the same solvent conditions in recrystallization.14 Therefore, isolating and selectively obtaining a porous structure with only a specific network topology has been difficult.
Among hydrogen bonds, the hydrogen bonds between strong acids such as sulfonic acids and bases such as amines, namely charge-assisted hydrogen bonds,15 have higher ionic character and binding energy than those of common hydrogen bonds. As shown in Fig. 1, we have reported that various sulfonic acids and bulky triphenylamine (TPMA) hierarchically construct porous organic salts (POSs).16 Four sulfo groups and four amino groups of TPMA form a tetrahedral [4 + 4] supramolecular cluster (Fig. 1 upper portion), and the clusters were connected to form diamondoid topology (dia-topology) (Fig. 1 right portion). Then, the frameworks were interpenetrated to construct a porous structure (Fig. 1 lower portion). Bulky trityl groups shielded sulfo groups and amino groups in the cluster from solvent molecules and prevented the involvement of solvent molecules in the charge-assisted hydrogen bonds (Fig. S1†),17 which enabled POSs surely to construct the porous structures composed of tetrahedral moieties. Recently, by using TPMA-X, where substituents (X) were introduced in the para-position of the benzene rings of TPMA, we successfully formed various environments in porous structures with dia-topology by distortion of the framework and change of the style of interpenetration.16d However, porous structures of POSs, i.e., types of network topologies, have been limited to the dia-topology.
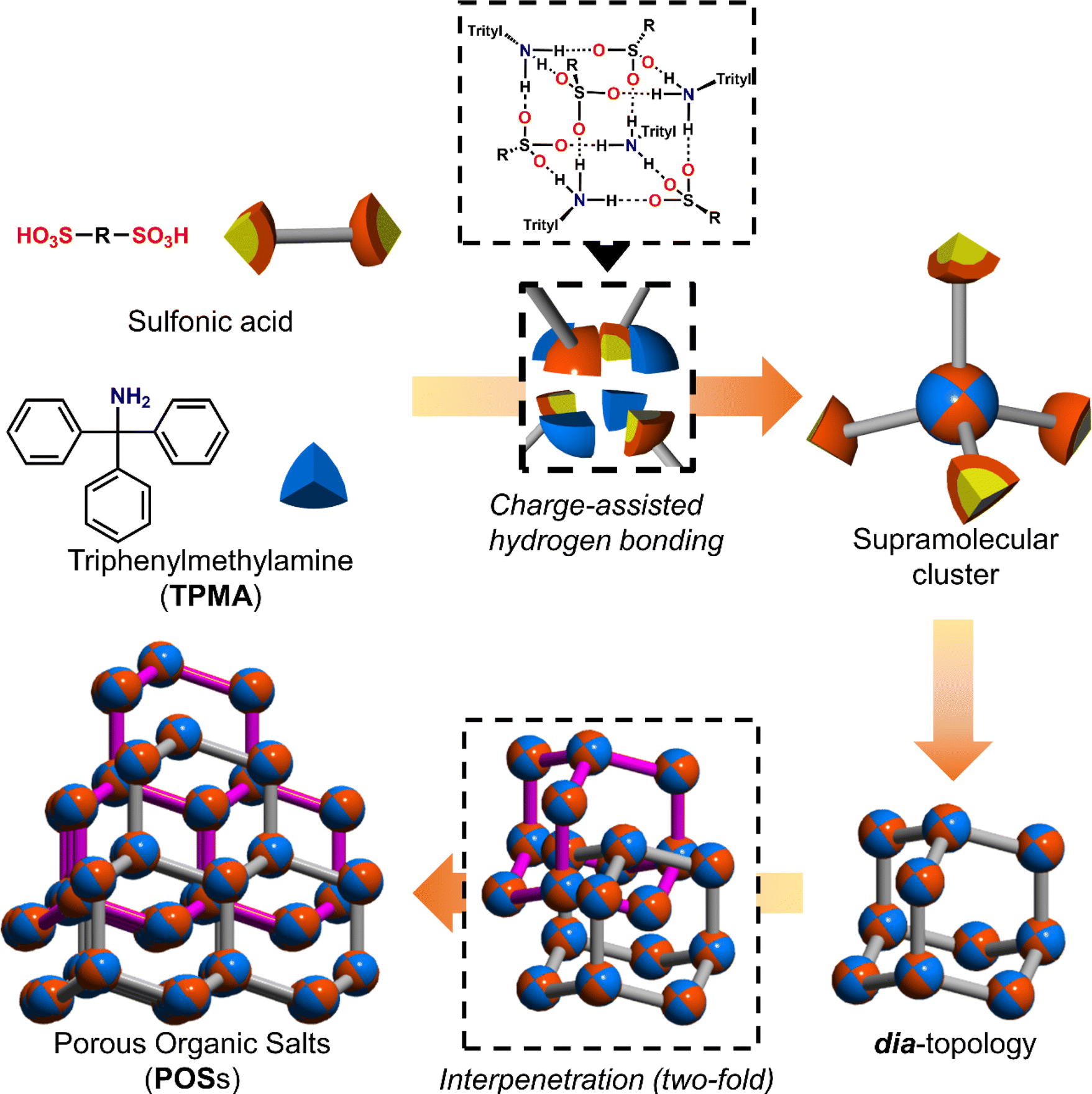 | ||
| Fig. 1 Schematic representation of the hierarchical construction of POSs with sulfonic acids and TPMA. | ||
Here, we aimed to control the network topology of HOFs toward diversification and functionalization of their porous structures. To form the different network topologies from the default network topology (dia-topology) in POSs, we focused on the conformation between connected two tetrahedral moieties (Fig. 2a), which are the core of tetrasulfonic acid (Fig. 2a blue tetrahedron, and Fig. 2b blue tetrahedron) and the supramolecular cluster (Fig. 2a orange tetrahedron, and Fig. 2b orange tetrahedron). We designed the POSs by combining tetrahedral-shaped tetrasulfonic acids with bulky adamantane (4,4′,4′′,4′′′-(adamantane-1,3,5,7-tetrayl) tetrabenzenesulfonic acid; AdPS) and TPMA-X (X = F, methyl (Me), Cl, Br, I) shown in Scheme 1, which provided the steric hindrance between adamantane (blue tetrahedron in Fig. 2b) and the substituents (X, purple ball in Fig. 2b) (as shown in Fig. 2b left portion), and we controlled the degree of steric hindrance by changing X. We changed the degree of steric hindrance and formed not only the common dia-topology in POSs but also the rare network topology (sod-topology) and lon- and uni-topologies that are formed for the first time in HOFs. In the case of AdPS/TPMA-F and AdPS/TPMA-Me with a medium degree of steric hindrance, multiple network topologies were formed depending on the template molecules in recrystallization, and we successfully isolated each porous structure with only a specific network topology. While the three types of AdPS/TPMA-Me with different network topologies (dia-, lon-, and sod-topologies) were composed of the same components, they exhibited significantly different gas adsorption properties depending on their network topologies. AdPS/TPMA-Me with dia-topology exhibited CO2, H2, and O2 adsorption and AdPS/TPMA-Me with lon- and sod-topologies exhibited selective CO2 adsorption.
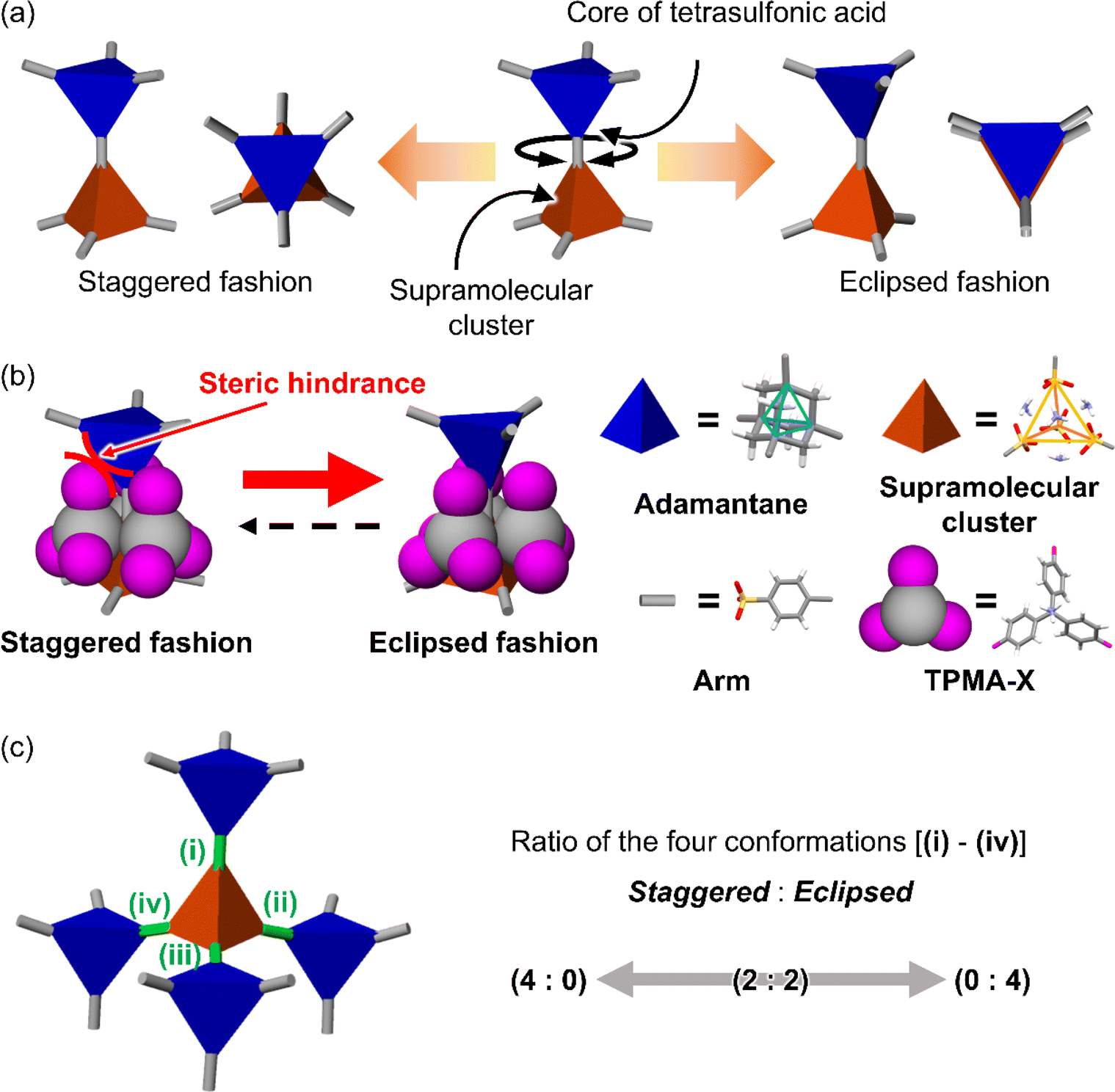 | ||
| Fig. 2 (a) Two connection fashions between the core of tetrasulfonic acid and the supramolecular cluster. (b) Conformation styles between the tetrahedron of the adamantane core (blue) of AdPS and the tetrahedron of the supramolecular cluster (orange), and the strategy to form eclipsed conformation by steric hindrance between the substituent X (pink) at TPMA-X and the adamantane core. (c) Schematic representations of the four conformations (green connection) between the four adamantane cores (blue) and a supramolecular cluster (orange) leading to the determination of the network topology of the porous structure. | ||
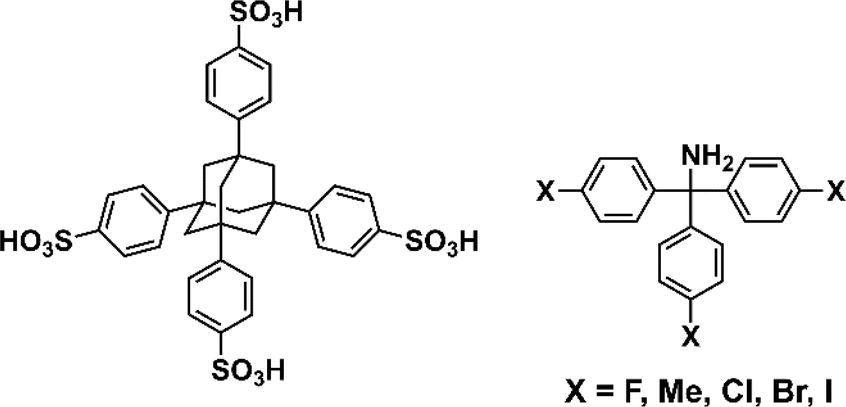 | ||
| Scheme 1 Chemical structures of porous organic salts. | ||
Results and discussion
POSs are composed of tetrahedral components (e.g., core of tetrasulfonic acid (Fig. 2a blue tetrahedron) and a supramolecular cluster (Fig. 2a orange tetrahedron)). The style of conformation between two tetrahedral components (Fig. 2a blue and orange tetrahedra) includes staggered fashion and eclipsed fashion. Structures composed of tetrahedral components have the conformation with staggered fashion18 to form highly symmetrical19 and stable dia-topology. In the case of POSs with tetrahedral-structured tetrasulfonic acid and TPMA (Fig. S2a†), two tetrahedra of the core of tetrasulfonic acid (Fig. S2b† blue tetrahedron) and the tetrahedral supramolecular cluster (Fig. S2b† orange tetrahedron) were linked in staggered fashion (Fig. S2b†) to form dia-topology.16d Therefore, to form the different network topologies from the default network topology (dia-topology), we aimed to change the style of conformation between the core of tetrasulfonic acid (Fig. 2b blue tetrahedron) and the supramolecular cluster (Fig. 2b orange tetrahedron) from staggered fashion to eclipsed fashion (Fig. 2b).In the conformation with staggered fashion (Fig. 2a left portion), the three arms (Fig. 2a gray sticks on the blue tetrahedron) from a core of tetrasulfonic acid (Fig. 2a blue tetrahedron) and the three arms (Fig. 2a gray sticks on the orange tetrahedron) from a supramolecular cluster (Fig. 2a orange tetrahedron) were arranged in staggered fashion. Herein, POSs had two components such as sulfonic acids and amines. Trityl groups of TPMA (Fig. 2b gray ball and purple balls) were located on the faces of the tetrahedron of the supramolecular cluster (Fig. 2b orange tetrahedron). Furthermore, in one connection moiety between the core of tetrasulfonic acid and the supramolecular cluster (Fig. 2b left portion), three trityl groups (Fig. 2b gray ball and purple balls) existed between the core of tetrasulfonic acid (Fig. 2b blue tetrahedron) and the supramolecular cluster (Fig. 2b orange tetrahedron). Therefore, we hypothesized that the occurrence of steric hindrances between tetrasulfonic acids (Fig. 2b blue tetrahedron) and trityl groups (Fig. 2b gray ball and purple balls) allowed the tetrasulfonic acid and supramolecular cluster to form the conformation with eclipsed fashion (Fig. 2b center portion).
To provide the steric hindrance between tetrasulfonic acids and trityl groups (Fig. 2b left portion), we prepared the organic salts from AdPS and TPMA-X where substituents were introduced into TPMA (Scheme 1). Furthermore, one supramolecular cluster was connected to four tetrasulfonic acids (Fig. 2c left portion). Therefore, the whole network topology was determined by the style (staggered or eclipsed) and the ratio of the four conformations. Thus, to control the style and the ratio of the conformations and to form various network topologies, we used halogens and the methyl group (X = F, methyl (Me), Cl, Br, I) with different bulkiness (Scheme 1) as the substituents (X) of TPMA-X (Fig. 2b gray ball with three purple balls) and changed the degree of steric hindrance between tetrasulfonic acids (Fig. 2b blue tetrahedron) and trityl groups (Fig. 2b gray ball and purple balls) systematically.
In the organic salt of AdPS and TPMA with no substituents (AdPS/TPMA), the bulkiness of only adamantane was not enough to provide steric hindrance; therefore, only dia-topology was formed (Fig. 3a Network topology). Furthermore, the frameworks with dia-topology were interpenetrated to construct the non-porous structure (Fig. 3a Structure).
 | ||
| Fig. 3 (a) Schematic representation of the resultant network topology, interpenetration, and the structure of AdPS/TPMA. Schematic representations of the resultant network topology, porous structure, and void structure of AdPS/TPMA-F with (b) dia-topology, (c) lon-topology, or (d) uni-topology, and (e) AdPS/TPMA-Cl with sod-topology. | ||
On the other hand, the organic salt of AdPS and TPMA-F where fluorine (F) was introduced into TPMA (AdPS/TPMA-F) formed dia-topology without interpenetration by the bulkiness of the substituents (Fig. 3b Network topology). The porous structure of dia-topology possessed the cage-like void (Fig. 3b Void) which was derived from the central void in the basic structure of dia-topology composed of four AdPS (Fig. 3b Network topology), and the narrow bottleneck to connect the voids (Fig. 3b Void). The maximum and minimum pore sizes (Table S1†) calculated with the Poreblazer v4.0 (ref. 20) program that was commonly used in previous investigations,21 were 8.92 Å and 3.61 Å respectively. Furthermore, the use of different template molecules with different electron densities and molecular structures (see the ESI;† Preparation of the single-crystal of AdPS/TPMA-F), enabled us to form AdPS/TPMA-F with lon-topology (Fig. 3c Network topology) and uni-topology (Fig. 3d Network topology), respectively.
lon -topology is the framework of hexagonal diamond (Lonsdaleite),22 which is an allotrope of diamond. Therefore, to form lon-topology, tetrahedral components are necessary, which is the same as the strategy for dia-topology formation;19 therefore, lon-topology could not be designed in HOFs. The porous structure of AdPS/TPMA-F with lon-topology possessed the columnar void (Fig. 3c Void) which was derived from the hexagonal structure in lon-topology, and the columnar voids were connected to form a three-dimensional void (Fig. 3c Void). The maximum and minimum pore sizes (Table S1†) calculated with the Poreblazer v4.0 (ref. 20) program were 8.37 Å and 4.96 Å respectively.
AdPS/TPMA-F with uni-topology had a helical framework and a chiral structure with a three-fold helix (Fig. 3d Network topology), where three AdPS existed per turn of the helix (Fig. S3†). The porous structure possessed a triangular columnar void (Fig. 3d Void) which was derived from the three-fold helical structure. The maximum and minimum pore sizes (Table S1†) calculated with the Poreblazer v4.0 (ref. 20) program were 7.45 Å and 4.84 Å respectively. Chiral HOFs have been often constructed using chiral building blocks as a component, i.e., the chirality of the building blocks has a direct effect.23 It should be noted that even though the helical structure of AdPS/TPMA-F with uni-topology was constructed from achiral building blocks, using R- or S-form of the chiral template molecules (e.g., carvone) in recrystallization enabled to us induce the corresponding chiralities of the helix, respectively. The R-form of carvone was used to form the left-handed helical structure (Fig. S3a and b†) and the S-form of carvone was used to form the right-handed helical structure (Fig. S3c and d†).
Next, the organic salt of AdPS and TPMA-Cl whose substituents (X) were bulkier than that of TPMA-F (AdPS/TPMA-Cl), formed sod-topology which was the same network topology as sodalite (Fig. 3e Network topology). Even by changing the template molecules, AdPS/TPMA-Cl formed only sod-topology. The maximum and minimum pore sizes (Table S2†) calculated with the Poreblazer v4.0 (ref. 20) program were 15.7 Å and 5.88 Å respectively. The basic structure of the sod-topology of AdPS/TPMA-Cl was composed of twelve AdPS (Fig. 3e Network topology) and formed a larger void (Fig. 3e Void) than those of the other network topologies. Generally, bulkier substituents decrease the pore size.16d,24 Although the maximum pore size of AdPS/TPMA-F with dia-topology, lon-topology, and uni-topology was 8.92 Å, 8.37 Å, and 7.45 Å respectively (Table S1†), the maximum pore size of AdPS/TPMA-Cl with sod-topology was 15.7 Å (Table S2†) because of the formation of sod-topology and large voids.
Powder X-ray diffraction (PXRD) patterns of AdPS/TPMA-X were measured (Fig. S4–S6†). Characteristic PXRD peaks (2θ = 6.22° (AdPS/TPMA-F with dia-topology), 5.76°, 6.06°, and 6.50° (AdPS/TPMA-F with lon-topology), 3.74°, and 6.36° (AdPS/TPMA-F with uni-topology), 6.08° (AdPS/TPMA-Me with dia-topology), 5.66°, 6.08°, and 6.48° (AdPS/TPMA-Me with lon-topology), 3.90° (AdPS/TPMA-Me with sod-topology), 4.08° (AdPS/TPMA-Cl with sod-topology), 4.04° (AdPS/TPMA-Br with sod-topology), 4.12° (AdPS/TPMA-I with sod-topology)) in the low angle region of immediately following formation via crystallization were identical to those of simulated patterns from the crystal structures, respectively. The previous investigations25 of PXRD patterns of porous materials indicated that differences in the higher angle region (2θ > 10°) were attributed to the existence of molecules in the pore, and identification of characteristic peaks in the lower angle region meant that structures were the same. Therefore, in our case, the structures formed via crystallization would be identical to their crystal structures, which indicated that AdPS/TPMA-X with different network topologies was isolated respectively. These obtained crystals were soluble in methanol and decomposable into their components, which indicated the high chemical recyclability of AdPS/TPMA-X. We summarized the obtained network topologies in order of the bulkiness of substituents (X) in Table 1. The organic salts that formed dia-topology were limited up to AdPS/TPMA-Me (Table 1). To investigate the reason for the topology change, we focused on the crystal structure of AdPS/TPMA-Me with dia-topology (Fig. 4a). As shown in Fig. 4b, in the structure of dia-topology, the methyl group of TPMA-Me was located close to the methylene moiety of adamantane, and the distance between the hydrogen atom of the methyl group and the hydrogen atom of adamantane was 2.4 Å (Fig. 4b). This distance was equal to the sum of the van der Waals radius of two hydrogen atoms (1.2 Å),26 which indicated the contact of the methyl group and the methylene moiety of adamantane. Therefore, when the substituent was bulkier than the methyl group, the conformation with staggered fashion could not be formed by steric hindrances, and the conformation was considered to change from staggered fashion to eclipsed fashion.
| AdPS/TPMA-X | Radius of X (Å)27 | Topology | |||
|---|---|---|---|---|---|
| a van der Waals radius parallel to the group axis. | |||||
| AdPS/TPMA | 1.20 | dia (two-fold) | lon | uni | sod |
| AdPS/TPMA-F | 1.47 | dia | lon | uni | sod |
| AdPS/TPMA-Me | 1.58a | dia | lon | uni | sod |
| AdPS/TPMA-Cl | 1.75 | dia | lon | uni | sod |
| AdPS/TPMA-Br | 1.85 | dia | lon | uni | sod |
| AdPS/TPMA-I | 1.98 | dia | lon | uni | sod |
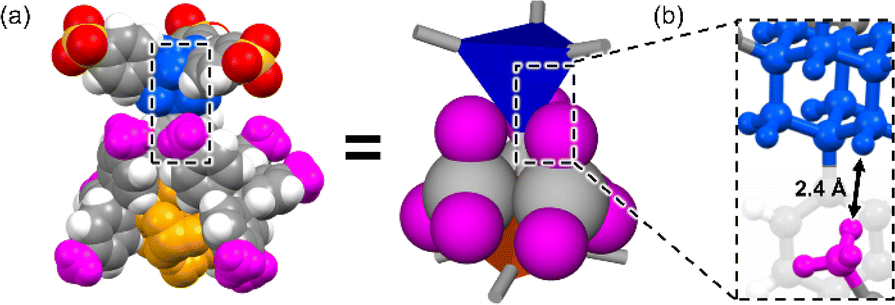 | ||
| Fig. 4 (a) Corey–Pauling–Koltun (CPK) molecular model and schematic representation of staggered conformation between the adamantane core (blue) and supramolecular cluster (orange) of AdPS/TPMA-Me with dia-topology. (b) Distance between the hydrogen of adamantane of AdPS and hydrogen of the methyl group (pink) of TPMA-Me. | ||
As shown in Fig. 2c left portion, one supramolecular cluster had four conformations, and the bulkier substituents changed the conformation from staggered fashion to eclipsed fashion. From the conformation between the core of tetrasulfonic acid (Fig. 5 blue tetrahedron) and the supramolecular cluster (Fig. 5 orange tetrahedron) based on the crystal structures (Fig. 5 left portion of first column) of the obtained four-type network topologies, the ratios of the conformations (staggered fashion versus eclipsed fashion) of each network topology were summarized in Fig. 5. When the styles of conformations were all staggered fashion, dia-topology was formed (Fig. 5dia-topology). However, when the style of one conformation was eclipsed fashion and that of other conformations was staggered fashion in the four conformations, lon-topology was formed (Fig. 5lon-topology). Then, when the style of two conformations was staggered fashion and that of other conformations was eclipsed fashion in the four conformations, uni-topology was formed (Fig. 5uni-topology), and when the styles of conformations were all eclipsed fashion, sod-topology was formed (Fig. 5sod-topology). Therefore, as the substituents were bulkier, the formation possibility of network topology increased in the order of dia-topology < lon-topology < uni-topology < sod-topology. In fact, as shown in Table 1, AdPS/TPMA with the lowest degree of steric hindrance formed only dia-topology where the style of all conformations was staggered fashion. AdPS/TPMA-F and AdPS/TPMA-Me had a higher degree of steric hindrance than AdPS/TPMA and formed not only dia-topology but also lon- and uni-topologies where the style of one or two conformations among the four was eclipsed fashion. Furthermore, AdPS/TPMA-Cl, AdPS/TPMA-Br, and AdPS/TPMA-I had the bulkier substituents than the methyl group, provided a high degree of steric hindrance, and formed only sod-topology where the style of all conformations was eclipsed fashion. In addition, to remove the difference of the condition in recrystallization, PXRD measurements of AdPS/TPMA-X (X= F, Me, Cl, Br, I), which was prepared under the same conditions (template molecule: 1-methylnaphthalene, solvent: methanol, temperature: 25 °C), were performed. AdPS/TPMA-X (X= Me, Cl, Br, I) had sod-topology but AdPS/TPMA-F had lon-topology (Fig. S7†), which supported that the network topologies with potential for formation were determined not by recrystallization conditions but by the degree of steric hindrance.
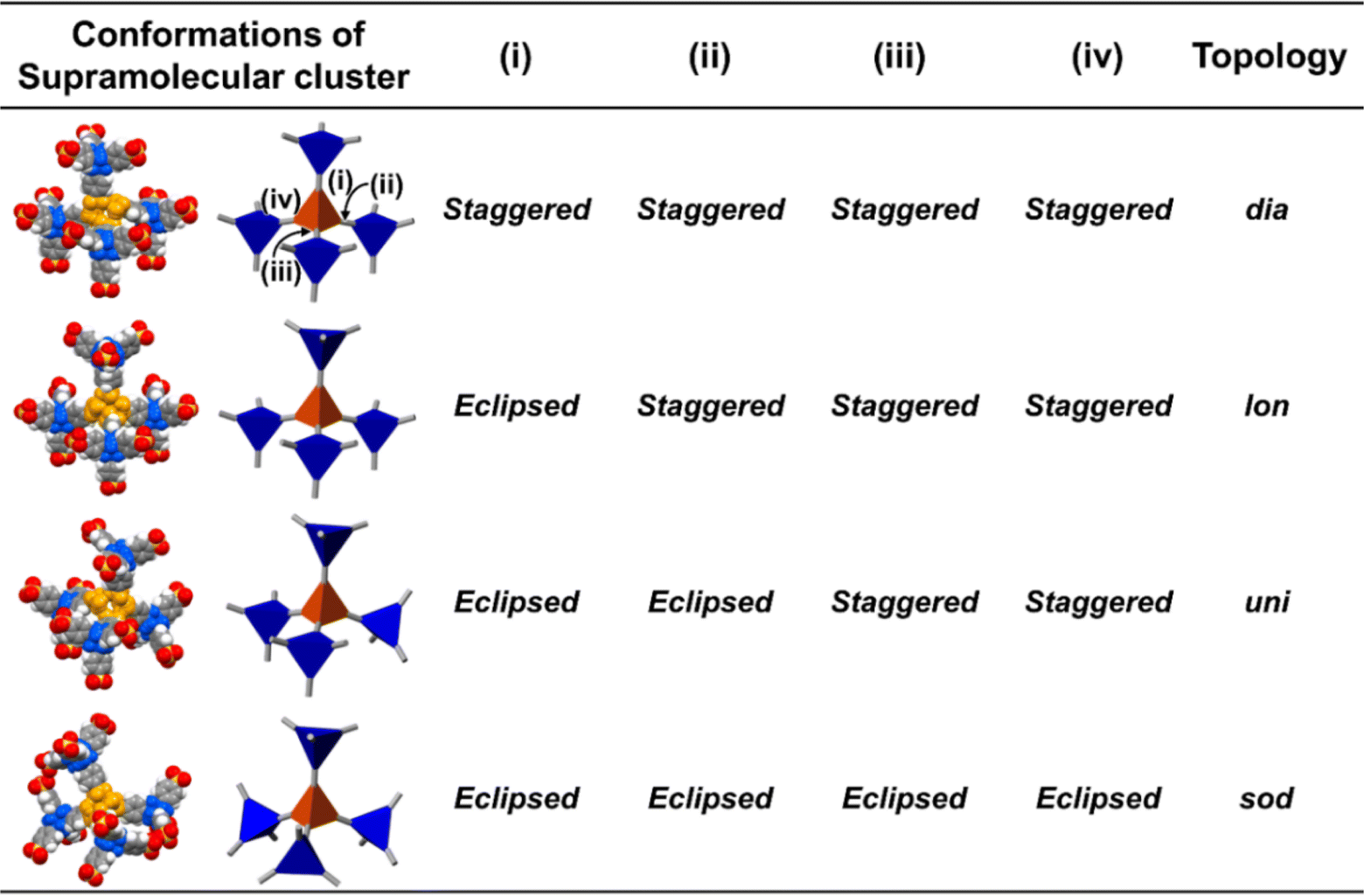 | ||
| Fig. 5 Corey–Pauling–Koltun (CPK) molecular model and schematic representations of the four conformations between the four adamantane cores (blue) and a supramolecular cluster (orange) in dia-topology, lon-topology, uni-topology, and sod-topology. | ||
According to Table 1, AdPS/TPMA-F and AdPS/TPMA-Me had a medium degree of steric hindrance, which enabled them to form multiple network topologies depending on the template molecules. For example, in the case of AdPS/TPMA-Me, when the template molecule was altered from (−)-β-pinene without aromatic rings to benzonitrile with an aromatic ring, the resultant network topology was changed from dia-topology to sod-topology (see the ESI;† Preparation of the single-crystal of AdPS/TPMA-Me). This was presumably because the template molecules which had strong interaction (e.g., π–π interaction) with the benzene rings in TPMA-X, predominately located close to TPMA-X, which increased the degree of steric hindrance surrounding a supramolecular cluster.
The difference in network topology is known to change the pore sizes of porous structures and even the gas adsorption properties.9c,e,28 To investigate the pure effect of the different network topologies on their gas adsorption properties, we used AdPS/TPMA-Me which had methyl groups with few interactions for gases and significantly different three-type network topologies (dia-, lon-, and sod-topologies). The porous structures of AdPS/TPMA-Me with dia-topology (Fig. S8a†) possessed the cage-like void (Fig. S8d†), and the maximum and minimum pore sizes (Table S3†) calculated with the Poreblazer v4.0 (ref. 20) program were 6.25 Å and 3.66 Å respectively. The porous structures of AdPS/TPMA-Me with lon-topology (Fig. S8b†) possessed the columnar void (Fig. S8e†), and the maximum and minimum pore sizes (Table S3†) calculated with the Poreblazer v4.0 (ref. 20) program were 7.06 Å and 5.65 Å respectively. The porous structures of AdPS/TPMA-Me with sod-topology (Fig. S8c†) possessed the cage-like void (Fig. S8f†), and the maximum and minimum pore sizes (Table S3†) calculated with the Poreblazer v4.0 (ref. 20) program were 15.6 Å and 4.57 Å respectively.
To perform gas adsorption measurements, we activated the porous structures of AdPS/TPMA-Me with dia-, lon-, and sod-topologies. AdPS/TPMA-Me with dia-topology was successfully activated by drying at 80 °C. However, in the case of AdPS/TPMA-Me with lon-topology and sod-topology, the template molecules were not completely removed by drying. Therefore, they were activated using supercritical CO2 fluid.
FT-IR measurements of the activated AdPS/TPMA-Me with dia-, lon-, and sod-topologies did not show NH stretching mode (3361 and 3296 cm−1) of TPMA-Me (Fig. S9†). In addition, S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the activated AdPS/TPMA-Me with dia-, lon-, and sod-topologies (1163, 1124, 1034, and 1007 cm−1) was blue shifted from that of AdPS (1123, 1032, 1003 cm−1), which indicated that protons of sulfo groups were captured by hydrogen bond acceptors. Therefore, these results indicated that all sulfo groups and amino groups formed charge-assisted hydrogen bonds (Fig. S9†). Furthermore, elemental analyses revealed that AdPS and TPMA-Me were in the ratio of 1 to 4 (see the ESI;† Element analyses of AdPS/TPMA-Me with dia, lon, and sod-topologies). These results indicated that supramolecular clusters were not damaged and amorphous impurities were not formed during activation.
O stretching mode of the activated AdPS/TPMA-Me with dia-, lon-, and sod-topologies (1163, 1124, 1034, and 1007 cm−1) was blue shifted from that of AdPS (1123, 1032, 1003 cm−1), which indicated that protons of sulfo groups were captured by hydrogen bond acceptors. Therefore, these results indicated that all sulfo groups and amino groups formed charge-assisted hydrogen bonds (Fig. S9†). Furthermore, elemental analyses revealed that AdPS and TPMA-Me were in the ratio of 1 to 4 (see the ESI;† Element analyses of AdPS/TPMA-Me with dia, lon, and sod-topologies). These results indicated that supramolecular clusters were not damaged and amorphous impurities were not formed during activation.
PXRD measurements of activated AdPS/TPMA-Me with dia-, lon-, and sod-topologies were also performed (Fig. S5†). In PXRD patterns after activation (Fig. S5a and c†), porous structures with dia-topology and sod-topology were not changed by activation. However, in the case of the porous structure with lon-topology, the wide-angle shift of the diffraction peaks by activation was observed (Fig. S5b†). The diffraction peak at 5.7° of the porous structure with lon-topology immediately after recrystallization corresponded to the (100) and (001) planes, and the diffraction peak at 6.5° corresponded to the (011) and (110) planes (Fig. S5b†). In the porous structure after activation, the former peak shifted to 6.4° and the latter shifted to 7.2°.
The peak shapes of AdPS/TPMA-Me with lon-topology before and after activation (Fig. S5b†) were similar; FT-IR spectra (Fig. S9†) indicated that the supramolecular clusters connecting AdPS and TPMA-Me were not damaged, and peak locations of FT-IR spectra before and after activation (Fig. S10†) did not have much difference; therefore, the porous structure would shrink by 1.4–1.8 Å along three directions while maintaining lon-topology.
To investigate the retainment of the network topology in detail, we performed variable-temperature (VT) PXRD (Fig. S11a†). VT-PXRD of the porous structure with lon-topology showed the wide-angle shifts of the diffraction peaks in the range of 70 °C to 119 °C, and these shifts had the same trend as those of lon-topology after activation. In addition to thermogravimetric analysis (TGA) results (Fig. S11c†), these indicated structural transition following the removal of template molecules. Furthermore, from 119 °C, the low-angle shifts of diffraction peaks occurred, and the peak returned in the original peak location of the porous structure with lon-topology at 194 °C (Fig. S11b†). From these results it can be inferred that the removal of template molecules induced the structure with lon-topology to shrink, but the network topology was maintained up to 194 °C and also after activation.
We performed CO2, N2, O2, and H2 gas adsorption measurements of the activated porous materials at 195 K, 77 K, 77 K, and 77 K, respectively. Adsorption isotherms of AdPS/TPMA-Me with different network topologies are shown in Fig. 6a–c, respectively. All porous structures of AdPS/TPMA-Me adsorbed more CO2 than the other gas, and this adsorption trend was also observed in the adsorption isotherms of CO2, N2, and O2 at 195 K (Fig. S12†), which exhibited a higher affinity for CO2. This was presumably due to the following three reasons: (1) the smaller kinetic diameter of CO2 (3.30 Å (ref. 29)) than those of N2 (3.80 Å (ref. 29)) and O2 (3.46 Å (ref. 29)), (2) the quadrupole–quadrupole interaction30 between CO2 and benzene rings30 with a large quadrupole moment, and (3) the interaction31 between CO2 and polar moieties of the porous structure that was composed of sulfonic acid and amines.
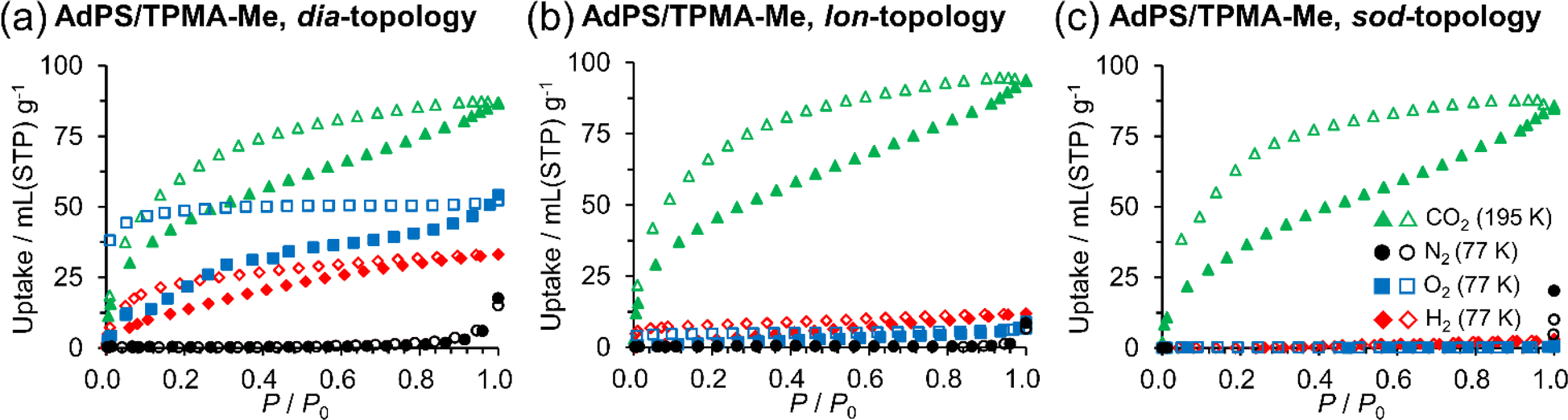 | ||
| Fig. 6 Gas adsorption isotherms of dia-topology (a), lon-topology (b), and sod-topology (c) of AdPS/TPMA-Me: CO2 (195 K), N2 (77 K), O2 (77 K), H2 (77 K). Filled symbols: adsorption process, open symbols: desorption process. P denotes the pressure at adsorption and P0 denotes the atmospheric pressure. | ||
To confirm this hypothesis (3), density functional theory (DFT) calculation of a supramolecular cluster in AdPS/TPMA-Me with dia-topology and a CO2 molecule was performed (Fig. S13†). As shown in Fig. 7a, the electrostatic potential map of the plane composed of a sulfur atom (S) and an oxygen atom (O) in a sulfo group, and a nitrogen atom (N) in an amino group showed that oxygen atoms of sulfo groups were negatively charged and amino groups were positively charged, which indicated that the polar moieties existed in the supramolecular clusters. Furthermore, as shown in Fig. 7b, the electrostatic potential map of the plane composed of a carbon atom (C) and an oxygen atom (O1) in the CO2, and an oxygen atom (O2) in the sulfo group showed that O2 was negatively charged and C was positively charged, and the distance between O2 and C was 2.97 Å. These results indicated that oxygen atoms in sulfo groups interacted with CO2. The calculated adsorption energy (−45.98 kJ mol−1) located in a strong physisorption range (30–50 kJ mol−1),32 which supported that AdPS/TPMA-Me with polar moieties had a higher affinity for CO2.
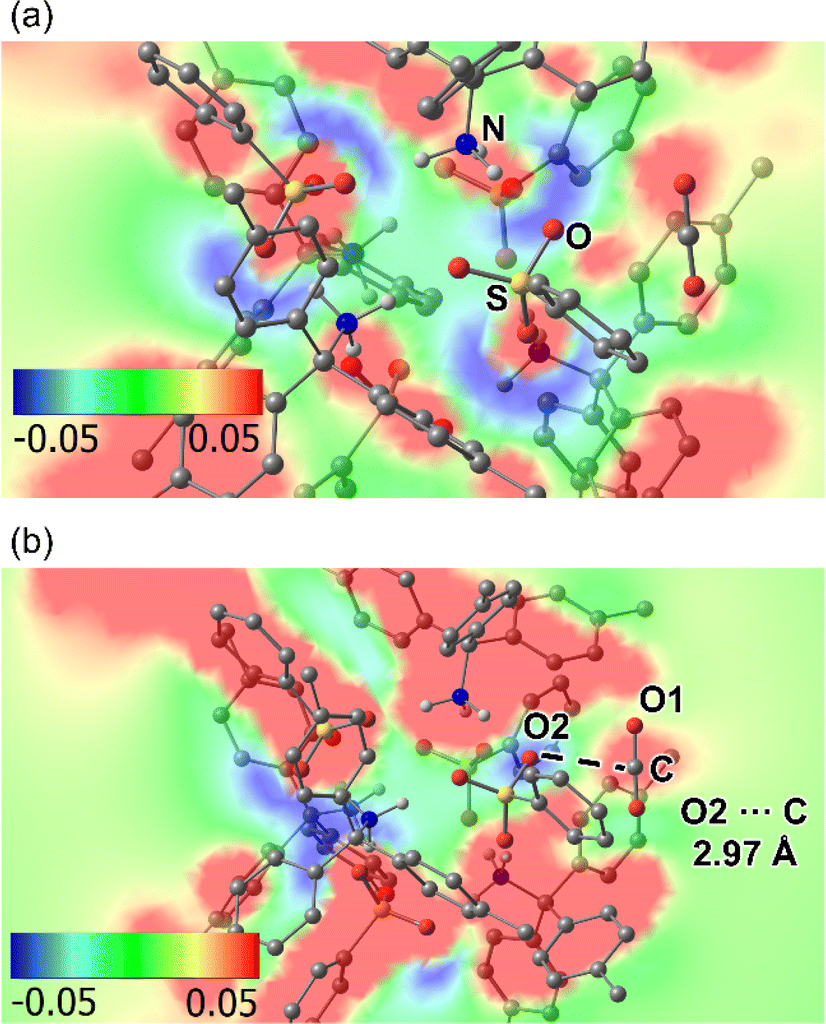 | ||
| Fig. 7 The electrostatic potential map of the supramolecular cluster in AdPS/TPMA-Me with dia-topology and a CO2: (a) the plane composed of S, O, and N. (b) The plane composed of C, O1, and O2. | ||
As depicted in Fig. 6b and c, the porous structures with lon-topology and sod-topology selectively adsorbed CO2, and their CO2 isotherms showed hysteresises. This should be attributed to strong interaction between CO2 and the polar moieties on their pore surfaces. The previous investigations31b,33 indicated that pore surfaces with polarity cannot provide sufficient energy of adsorption for non-polar gas molecules, and the porous materials with polarity did not adsorb them. In our case, supramolecular clusters with the polar moiety existed in the pore surfaces of AdPS/TPMA-Me with not only dia-topology (Fig. 7) but also lon-topology and sod-topology; therefore, based on the effect of polarity of the supramolecular cluster, these porous materials did not adsorb N2, O2, and H2. In order to investigate the influence of the substituents (X) on gas adsorption properties, the gas adsorption measurement of AdPS/TPMA-F with lon-topology was performed. In the case of the same lon-topology, despite the difference of substituents (X), AdPS/TPMA-F with lon-topology showed similar gas adsorption properties (Fig. S14†) to that of AdPS/TPMA-Me with lon-topology. The CO2 isotherm of AdPS/TPMA-F with lon-topology had larger hysteresis than that of AdPS/TPMA-Me with lon-topology (Fig. 6b and S14†), which indicated that the high electronegativity of fluorine atoms34 increased the interaction between CO2 and the substituent (X = F) on the pore surface.
As depicted in Fig. 6a, AdPS/TPMA-Me with dia-topology adsorbed not only CO2 but also H2 and O2. Furthermore, hystereses were observed in isotherms of CO2, H2, and O2, which suggested that they were attributed to not only the interaction between CO2 and the polar moieties but also the narrow bottleneck. Despite the effect of the polar pore, the porous structure with dia-topology adsorbed non-polar H2 and O2. This might be because the size of the bottleneck (3.66 Å (Table S3†)) was similar to that of O2 (3.46 Å (ref. 29)) and H2 (2.89 Å (ref. 29)), which induced the van der Waals forces with gas molecules to get stronger.24,26a,35 Therefore, the van der Waals forces overcome the effect of the polar pore, and the porous structure with dia-topology also adsorbed O2 and H2. While O2 and H2 were adsorbed at 77 K, N2 was not adsorbed (Fig. 6a). This was presumably because N2 (3.80 Å (ref. 29)) with a larger kinetic diameter than the size of the bottleneck (3.66 Å (Table S3†)) did not pass through the bottleneck.
Finally, Kr adsorption measurements of AdPS/TPMA-Me with dia-, lon-, and sod-topologies at 77 K were conducted to calculate the Brunauer–Emmett–Teller (BET) surface areas and pore volumes (Fig. S15†). The surface area analysis with Kr has higher sensitivity than those with N2 and Ar.36 However, AdPS/TPMA-Me with dia-, lon-, and sod-topologies did not adsorb Kr (3.66 Å (ref. 29)) presumably due to the narrow bottleneck (3.66 Å (Table S3†) in dia-topology) and the effect of the polar pore. In previous investigation,37 BET surface area of HOFs with no adsorption for N2 was calculated based on the CO2 isotherm; therefore, in the case of AdPS/TPMA-Me, BET surface areas and pore volumes were calculated based on the CO2 isotherms to be 228 m2 g−1 and 0.236 cm3 g−1 in the porous structure with dia-topology, 228 m2 g−1 and 0.255 cm3 g−1 in the porous structure with lon-topology, and 194 m2 g−1 and 0.231 cm3 g−1 in the porous structure with sod-topology respectively (Table S4†).
We revealed that despite the same components, the difference in network topologies enables a change in gas adsorption properties dramatically.
Conclusions
We focused on the steric hindrance between tetrasulfonic acid with a bulky adamantane core (AdPS) and TPMA-X. By changing the substituents (X) in TPMA-X, we changed the style of conformation between two tetrahedra (adamantane and supramolecular cluster) from staggered fashion to eclipsed fashion. Therefore, the four-type network topologies (dia-, lon-, uni-, and sod-topologies) were successfully formed and isolated. lon- and uni-topologies were obtained for the first time in HOFs. AdPS/TPMA-Me had a medium degree of steric hindrance, which enabled the formation of three-type network topologies (dia-, lon-, and sod-topologies); therefore, the change of template molecules used in recrystallization helped in successfully isolating these network topologies facilely. Further analyses of the relationship between network topologies, substituents, and recrystallization conditions will be performed in our future work. Despite the same components, AdPS/TPMA-Me with different network topologies exhibited significantly different gas adsorption properties. Based on these gas adsorption properties, AdPS/TPMA-Me with dia-, lon-, and sod-topologies is expected for applications such as the generation of high-purity oxygen and the separation of CO2 from air. In our future work, we will investigate the relationship between substituents, network topologies, and gas adsorption properties. We showed that changing the conformation style of components determined the network topologies that are difficult to form or have not been formed in HOFs and achieved the proof-of-concept for the formation, diversification, and control of the network topologies and concomitant functions of HOFs. Furthermore, we revealed the relationship between lon- and uni-topologies and the conformation style and ratio. Therefore, this investigation leads to structural diversification and functionalization of HOFs, COFs, and metal–organic frameworks.Data availability
Details of materials, instruments, experimental procedures, crystallographic, and DFT calculations are given in the ESI† for this manuscript.Author contributions
Hiroi Sei: conceptualization, methodology, formal analysis, data curation, investigation, and writing – original draft. Kouki Oka: conceptualization, methodology, formal analysis, data curation, project administration, validation, supervision, and writing – original draft. Yuta Hori: software, formal analysis, and writing – review & editing. Yasuteru Shigeta: software, formal analysis, and writing – review & editing. Norimitsu Tohnai: conceptualization, methodology, data curation, project administration, validation, supervision, writing – review & editing, funding acquisition, and resources.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the Grants-in-Aid for Scientific Research (20H02548, and 22K14732) from MEXT, Japan. K. O. also acknowledges the support from LNest Grant from Nipponham, FUSO Innovative Technology Fund, Masuyakinen Basic Research Foundation, and the Shorai Foundation for Science and Technology. H. S. acknowledges the support from JST, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2125. H. S. thanks the Materials Science Research Unit, Osaka University and Honors Program for Graduate Schools in Science, Engineering and Informatics, and the Super Hierarchical Materials Science program in Osaka University.Notes and references
- (a) L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Mater. Adv., 2022, 3, 3680–3708 RSC; (b) R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC; (c) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC; (d) S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- (a) S. Bhunia, K. A. Deo and A. K. Gaharwar, Adv. Funct. Mater., 2020, 30, 2002046 CrossRef CAS; (b) J. D. Wuest, Nat. Commun., 2020, 11, 4652 CrossRef CAS PubMed.
- (a) D. B. Shinde, M. Ostwal, X. Wang, A. M. Hengne, Y. Liu, G. Sheng, K.-W. Huang and Z. Lai, CrystEngComm, 2018, 20, 7621–7625 RSC; (b) P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. O'Keeffe, Y. Han and B. Chen, Angew. Chem., Int. Ed., 2015, 54, 574–577 CrossRef CAS PubMed; (c) W. Yang, J. Wang, H. Wang, Z. Bao, J. C.-G. Zhao and B. Chen, Cryst. Growth Des., 2017, 17, 6132–6137 CrossRef CAS; (d) Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed; (e) D. D. Zhou, Y. T. Xu, R. B. Lin, Z. W. Mo, W. X. Zhang and J. P. Zhang, ChemComm, 2016, 52, 4991–4994 RSC; (f) H. Wang, H. Wu, J. Kan, G. Chang, Z. Yao, B. Li, W. Zhou, S. Xiang, J. Cong-Gui Zhao and B. Chen, J. Mater. Chem. A, 2017, 5, 8292–8296 RSC.
- (a) S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed; (b) S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed; (c) I. Hisaki, Y. Suzuki, E. Gomez, Q. Ji, N. Tohnai, T. Nakamura and A. Douhal, J. Am. Chem. Soc., 2019, 141, 2111–2121 CrossRef CAS PubMed; (d) Z. Sun, Y. Li, L. Chen, X. Jing and Z. Xie, Cryst. Growth Des., 2015, 15, 542–545 CrossRef CAS.
- (a) H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed; (b) A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed.
- (a) M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed; (b) Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed; (c) B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- (a) Y. Byun, D. Jo, D. N. Shin and S. B. Hong, ACS Catal., 2014, 4, 1764–1776 CrossRef CAS; (b) I. Matito-Martos, A. Martin-Calvo, J. J. Gutierrez-Sevillano, M. Haranczyk, M. Doblare, J. B. Parra, C. O. Ania and S. Calero, Phys. Chem. Chem. Phys., 2014, 16, 19884–19893 RSC; (c) L. Fan, L. Yue, W. Sun, X. Wang, P. Zhou, Y. Zhang and Y. He, ACS Appl. Mater. Interfaces, 2021, 13, 40788–40797 CrossRef CAS PubMed; (d) J. Lyu, X. Zhang, K. I. Otake, X. Wang, P. Li, Z. Li, Z. Chen, Y. Zhang, M. C. Wasson, Y. Yang, P. Bai, X. Guo, T. Islamoglu and O. K. Farha, Chem. Sci., 2019, 10, 1186–1192 RSC; (e) F. Chen, D. Bai, Y. Wang, M. He, X. Gao and Y. He, Dalton Trans., 2018, 47, 716–725 RSC.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed., 1995, 34, 2311–2327 CrossRef CAS; (b) M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2018, 19, 1426–1453 CrossRef; (c) Y.-F. Han, Y.-X. Yuan and H.-B. Wang, Molecules, 2017, 22 Search PubMed.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737–2738 CrossRef CAS.
- (a) K. Kobayashi, A. Sato, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 3035–3045 CrossRef CAS PubMed; (b) Y. Zhou, L. Kan, J. F. Eubank, G. Li, L. Zhang and Y. Liu, Chem. Sci., 2019, 10, 6565–6571 RSC.
- (a) J. Gao, Y. Cai, X. Qian, P. Liu, H. Wu, W. Zhou, D. X. Liu, L. Li, R. B. Lin and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 20400–20406 CrossRef CAS PubMed; (b) Y. Shi, Y. Ding, W. Tao and P. Wei, ACS Appl. Mater. Interfaces, 2022, 14, 36071–36078 CrossRef CAS PubMed.
- (a) E. M. L. Lippitt, C. Ennis, S. C. Moratti and L. R. Hanton, Cryst. Growth Des., 2020, 20, 7805–7821 CrossRef CAS; (b) I. Hisaki, H. Toda, H. Sato, N. Tohnai and H. Sakurai, Angew. Chem., Int. Ed., 2017, 56, 15294–15298 CrossRef CAS PubMed.
- (a) M. D. Ward, ChemComm, 2005, 5838–5842, 10.1039/b513077h; (b) R. Akai, K. Oka, S. Dekura, H. Mori and N. Tohnai, Bull. Chem. Soc. Jpn., 2022, 95, 1178–1182 CrossRef CAS.
- (a) A. Yamamoto, S. Uehara, T. Hamada, M. Miyata, I. Hisaki and N. Tohnai, Cryst. Growth Des., 2012, 12, 4600–4606 CrossRef CAS; (b) A. Yamamoto, T. Hirukawa, I. Hisaki, M. Miyata and N. Tohnai, Tetrahedron Lett., 2013, 54, 1268–1273 CrossRef CAS; (c) A. Yamamoto, T. Hasegawa, T. Hamada, T. Hirukawa, I. Hisaki, M. Miyata and N. Tohnai, Chem.–Eur. J., 2013, 19, 3006–3016 CrossRef CAS PubMed; (d) T. Ami, K. Oka, K. Tsuchiya and N. Tohnai, Angew. Chem., Int. Ed., 2022, 61, e202202597 CrossRef CAS PubMed.
- N. Tohnai, Y. Mizobe, M. Doi, S.-i. Sukata, T. Hinoue, T. Yuge, I. Hisaki, Y. Matsukawa and M. Miyata, Angew. Chem., Int. Ed., 2007, 119, 2270–2273 CrossRef.
- N. Alsadun, G. Mouchaham, V. Guillerm, J. Czaban-Jozwiak, A. Shkurenko, H. Jiang, P. M. Bhatt, P. Parvatkar and M. Eddaoudi, J. Am. Chem. Soc., 2020, 142, 20547–20553 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- L. Sarkisov, R. Bueno-Perez, M. Sutharson and D. Fairen-Jimenez, Chem. Mater., 2020, 32, 9849–9867 CrossRef CAS.
- (a) M.-M. Fu, C. Liu and G.-Y. Dong, CrystEngComm, 2021, 23, 7397–7405 RSC; (b) F. Steinke, T. Otto, S. Ito, S. Wöhlbrandt and N. Stock, Eur. J. Inorg. Chem., 2022, 2022, e202200562 CrossRef CAS.
- F. P. Bundy and J. S. Kasper, J. Chem. Phys., 1967, 46, 3437–3446 CrossRef CAS.
- Y. Zhou, B. Liu, X. Sun, J. Li, G. Li, Q. Huo and Y. Liu, Cryst. Growth Des., 2017, 17, 6653–6659 CrossRef CAS.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- (a) Q. L. Guan, Y. H. Xing, J. Liu, C. Han, C. Y. Hou and F. Y. Bai, J. Phys. Chem. C, 2019, 123, 23287–23296 CrossRef CAS; (b) I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, J. Am. Chem. Soc., 2016, 138, 6617–6628 CrossRef CAS PubMed; (c) L. Peng, J. Sun, J. Huang, C. Song, Q. Wang, L. Wang, H. Yan, M. Ji, D. Wei, Y. Liu and D. Wei, Chem. Mater., 2022, 34, 2886–2895 CrossRef CAS.
- (a) J. L. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS PubMed; (b) R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- M. Charton, J. Am. Chem. Soc., 1968, 91, 615–618 CrossRef.
- X. Gong, H. Noh, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 6146–6151 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- W. Steele, Chem. Rev., 1993, 93, 2355–2378 CrossRef CAS.
- (a) A. K. Chaudhari, S. Mukherjee, S. S. Nagarkar, B. Joarder and S. K. Ghosh, CrystEngComm, 2013, 15, 9465–9471 RSC; (b) P. Kanoo, A. C. Ghosh, S. T. Cyriac and T. K. Maji, Chem.–Eur. J., 2012, 18, 237–244 CrossRef CAS PubMed; (c) S. Yu, G. L. Xing, L. H. Chen, T. Ben and B. L. Su, Adv. Mater., 2020, 32, e2003270 CrossRef PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Angew. Chem., Int. Ed., 2012, 51, 1826–1829 CrossRef CAS PubMed.
- A. Pal, S. Chand, D. G. Madden, D. Franz, L. Ritter, B. Space, T. Curtin, S. Chand Pal and M. C. Das, ACS Appl. Mater. Interfaces, 2020, 12, 41177–41184 CrossRef CAS PubMed.
- A. Comotti, F. Castiglioni, S. Bracco, J. Perego, A. Pedrini, M. Negroni and P. Sozzani, Chem. Commun., 2019, 55, 8999–9002 RSC.
- (a) A. Rehman and S.-J. Park, Chem. Eng. J., 2019, 362, 731–742 CrossRef CAS; (b) S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688–1700 CrossRef CAS PubMed.
- M. Thommes and K. A. Cychosz, Adsorption, 2014, 20, 233–250 CrossRef CAS.
- I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Chem.–Eur. J., 2017, 23, 11611–11619 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2190716, 2190957, 2191243, 2209699, 2209700, 2209701, 2209702, 2209745, 2209746, 2210216 and 2238308. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01218f |
| ‡ H. S. and K. O. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |