DOI:
10.1039/D4SC00581C
(Edge Article)
Chem. Sci., 2024,
15, 6130-6140
Intramolecular chaperone-assisted dual-anchoring activation (ICDA): a suitable preorganization for electrophilic halocyclization†
Received
24th January 2024
, Accepted 20th March 2024
First published on 20th March 2024
Abstract
The halocyclization reaction represents one of the most common methodologies for the synthesis of heterocyclic molecules. Many efforts have been made to balance the relationship between structure, reactivity and selectivity, including the design of new electrophilic halogenation reagents and the utilization of activating strategies. However, discovering universal reagents or activating strategies for electrophilic halocyclization remains challenging due to the case-by-case practice for different substrates or different cyclization models. Here we report an intramolecular chaperone-assisted dual-anchoring activation (ICDA) model for electrophilic halocyclization, taking advantage of the non-covalent dual-anchoring orientation as the driving force. This protocol allows a practical, catalyst-free and rapid approach to access seven types of small-sized, medium-sized, and large-sized heterocyclic units and to realize polyene-like domino halocyclizations, as exemplified by nearly 90 examples, including a risk-reducing flow protocol for gram-scale synthesis. DFT studies verify the crucial role of ICDA in affording a suitable preorganization for transition state stabilization and X+ transfer acceleration. The utilization of the ICDA model allows a spatiotemporal adjustment to straightforwardly obtain fast, selective and high-yielding synthetic transformations.
Introduction
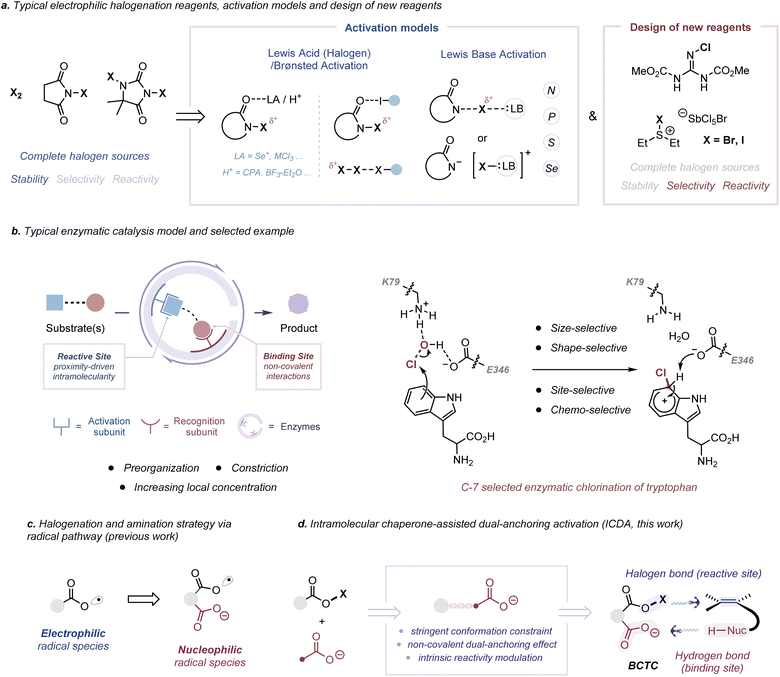
Halogen-induced intramolecular cyclization of unactivated alkenes has emerged as a powerful strategy to rapidly build up molecular complexity, due to the concomitant formation of a nascent heterocyclic moiety and halide functionalities.1 However, a harmonious relationship between structure, reactivity and selectivity for electrophilic halocyclization has proven to be a long-standing problem. For example, advancements in medium-sized and large-sized halocyclization,2 polyene-like domino halocyclization,3 asymmetric variants of electrophilic halocyclization4 and clear-cut reaction mechanism determination5 remain challenging (Fig. S1 to S3†) due to the case-by-case practice for different substrates or different cyclization models. To this end, a variety of electrophilic halogenation reagents, from molecular halogens (X2, X = Cl, Br, I) to stable and available reagents bearing the N–X bond, have been developed successively (Scheme 1a, left).6 Nevertheless, a catalyst or an extra additive was generally required to increase the electrophilic character of N-haloamides, which is typically realized by Lewis/Brønsted acid activation7 or Lewis base activation8 (Scheme 1a, middle, and Fig. S4a†). More recently, significant efforts have also been devoted to the elaboration of practical and highly reactive electrophilic halogenation reagents, including Et2SX·SbCl5X (X = Br or Cl),9 Palau’chlor10 and Py2IBF4,11 for direct halogenation (Scheme 1a, right, and Fig. S4b†). Nevertheless, the range of applications has been restricted by their incomplete halenium sources and stability liabilities from moisture, light and so on.12 Therefore, it is desired to develop powerful electrophilic halogenation strategies that can balance the relationship between structure, reactivity and selectivity.
 |
| | Scheme 1 Summary of halogenation reagents, enzymatic catalysis and our strategy design. | |
In the pursuit of fast, selective and high-yielding organic transformations, the proximity and orientation effects have been central chemical tenets in regulating many important biological processes.13 The recognition site of an enzyme usually makes use of a combination of multiple non-covalent interactions to bring the substrates in spatial proximity to the reaction site and increase the reaction rate (Scheme 1b, left, and Fig. S5a†). The proximity and orientation effects allow a maximally reactive conformation of substrates and further stabilize the transition state because of the specific enzyme–substrate preorganization (Fig. S5b and S6†).14 For an efficient and position-specific halogen installation, enzymatic halogenation reactions are generally predominated by an oxidative strategy in which an oxygen-based oxidant is utilized to oxidize halide ions (X−) into the corresponding hypohalite (−OX) intermediates (Fig. S4c†).15 In a polar pathway, these intermediates react as activated X+ equivalents with electron-rich substrates owing to the high electrophilicity of the oxygen atom (χP = 3.44).16 In regard to the regioselective C-7 chlorination of tryptophan by flavin-dependent halogenases, HOCl, for example, is generated by the reaction of flavin hydroperoxide and the chloride ion (Scheme 1b, right).17 Chlorination by free HOCl lacks both regiospecificity and substrate specificity. Before electrophilic chlorination, the K79 residue may hydrogen bond to free HOCl, thus positioning it proximal to tryptophan and increasing the electrophilicity of Cl+. During the chlorination, the carboxylate group of the proximal E346 residue is oriented to stabilize and deprotonate the resulting Wheland intermediate, leading to a C-7 selective chlorination product. Such a spatially confined activating step may not only enable the chlorination of less reactive substrates but also protect nearby tryptophan residues.
In our previous work, we have found that the combination of phthaloyl peroxide (PPO) and X− can generate Brønsted base Covalently Tethered Carbonyl hypohalites (BCTC) via an oxidative strategy.18 Taking advantage of the spatially confined non-covalent orientation and proximity effect, our previous studies have developed a tether-tunable distonic radical anion mediated approach, wherein the installation of an intramolecular chaperone-like carboxylate anion leads to a polarity inversion or umpolung strategy, for the direct generation of heteroatom-centered radicals from O–H or N–H bonds (Scheme 1c).18 Along these lines, except for the radical (1e−) process, we envisioned that BCTC would act as an in situ generated electrophilic reagent in a closed shell (2e−) process for halocyclization reactions. In the proposal, this protocol allows the utilization of a covalently integrated halogen bond donor and hydrogen bond acceptor for dual-anchoring orientation (Scheme 1d). At the molecular level, this Intramolecular Chaperone-assisted Dual-anchoring Activation (ICDA) model could afford an induced-fit conformational change in a tailored environment and create a specific reactive conformation, thus pre-paying for the entropy loss required for the formation of the transition state to accelerate the reaction.19
Results and discussion
Optimization of the reaction conditions
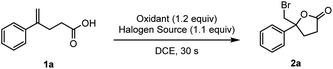
To validate our hypothesis, 4-phenyl-4-pentenoic acid 1a was selected as the model substituted alkene for optimization studies (Table 1, see more details in Tables S1 to S4†). The reaction rate of this turbo-charged bimolecular process is increased by orders of magnitude compared to the three-molecule process induced by benzoyl peroxide (BPO) (the reaction was finished in 30 s; see the video in the ESI†) via ICDA (Table 1, entries 1 to 4). Tetrabutylammonium bromide (TBAB) was proved to be the optimal halogen source (Table 1, entries 5 and 6, and Table S2†). Furthermore, bromolactonization of 1a was conducted under the irradiation of a blue LED (400 nm), and the yield of 2a-Br was decreased to 51% which might be due to the undesired radical side-reactions (Table 1, entry 7).18b
Table 1 Optimization studya
|
|
| Entry |
Oxidant |
Halogen source |
Yieldb of 2a [%] |
|
Reaction conditions: 1a (0.3 mmol, 1 equiv.), oxidant (0.36 mmol, 1.2 equiv.), Br source (0.33 mmol, 1.1 equiv.) in DCE (3 mL, 0.1 M) at room temperature, under an air atmosphere, 30 seconds.
Yields of isolated products after chromatographic purification.
Reaction time: 17 hours.
The reaction was performed with 400 nm LEDs (10 W). PPO = phthaloyl peroxide; BPO = benzoyl peroxide; MPO = malonoyl peroxide; LPO = lauroyl peroxide; TBAB = tetrabutylammonium bromide.
|
| 1 |
PPO |
TBAB |
>96 |
| 2 |
MPO |
TBAB |
90 |
| 3 |
BPO |
TBAB |
<5c (90%) |
| 4 |
LPO |
TBAB |
<5c (60%) |
| 5 |
PPO |
CsBr |
13c |
| 6 |
PPO |
NBS |
6c |
| 7 |
PPO |
TBAB |
51d |
|
Synthesis of five-membered and six-membered heterocyclic rings enhanced by ICDA
With the optimized conditions in hand, we assessed the scope of alkenoic acids to test the functional group tolerance of this protocol (Scheme 2). Generally, electron-donating, electron-neutral, and electron-withdrawing substituents on 4-aryl-4-enoic acids exhibited similar tolerance: methoxy, alkyl, aryl, halo, trifluoromethyl, nitryl, cyan, methoxycarbonyl, sulfonyl, and dimethylcarbamoyl were sufficiently compatible, and the corresponding γ-lactones (2b–2l) were obtained in 80 to 96% yields within two minutes. Different steric substituted groups were found to have negligible effects on the bromolactonization (2m–2p). Pyridyl (2q) and alkynyl (2r) were compatible with the oxidizing atmosphere. The bromolactonization of α,β-unsaturated enone 1s with a comparatively inert and sluggish π-bond was implemented successfully (2s, 84%).20 Our approach is also suitable for the synthesis of 3,3-disubstituted phthalide (2t) and halogenated α-exo-methylene-lactone (2u). The BCTC-induced bromolactonization of substrates bearing an endo-cyclic π-bond (1v–1z) proceeded smoothly with exclusive diastereoselectivities superior to the previous report.21 The bromolactonization of mono-substituted alkene 1aa definitely furnished the exo-lactone 2aa as a single diastereomer with 81% yield. The reaction of the (Z)-1,2-disubstituted alkene (1ab) proceeded with exclusive diastereoselectivity and regioselectivity. A mixture of δ-valerolactone (2ac) and γ-butyrolactone (2ac′) in a ratio of 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was obtained when (E)-5-phenylpent-4-enoic acid (1ac) was used as the substrate. Furthermore, our protocol is also competent for the synthesis of six-membered lactones (2ad–2ak) and E-bromo enol lactone (2al) in 40 to 96% yields via exo-cyclization.
:1 was obtained when (E)-5-phenylpent-4-enoic acid (1ac) was used as the substrate. Furthermore, our protocol is also competent for the synthesis of six-membered lactones (2ad–2ak) and E-bromo enol lactone (2al) in 40 to 96% yields via exo-cyclization.
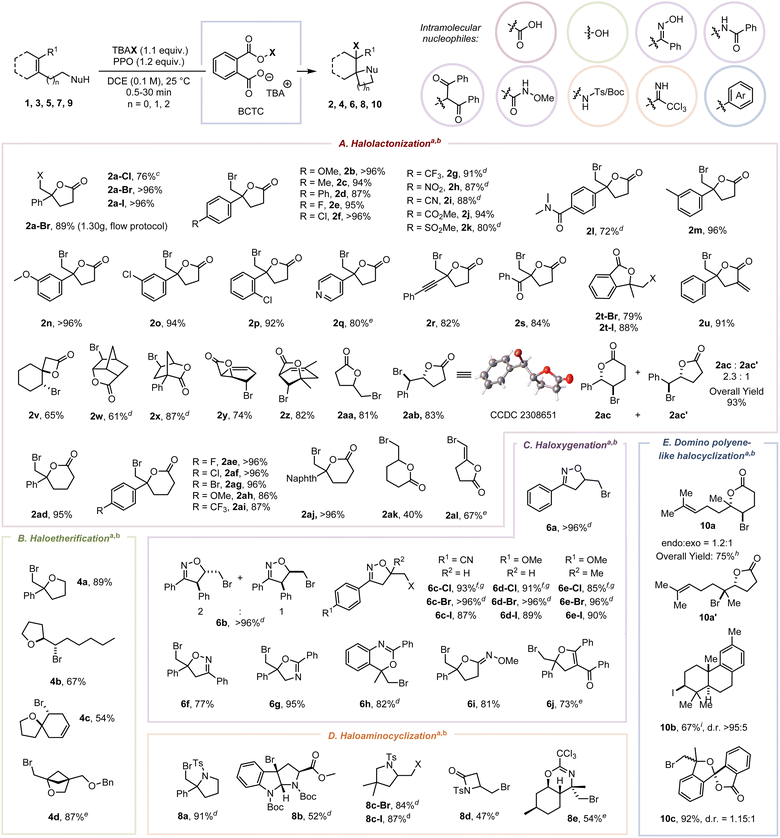 |
| | Scheme 2 Substrate scope for electrophilic halogenation of 1, 3, 5, 7, and 9 with BCTC. a Reaction conditions: olefin (0.3 mmol, 1 equiv.), PPO (0.36 mmol, 1.2 equiv.), TBAB (0.33 mmol, 1.1 equiv.) in DCE (3 mL, 0.1 M) at room temperature, under an air atmosphere, 30 seconds. b Yields of isolated products after chromatographic purification. c TBACl (2.2 equiv.) and PPO (2.2 equiv.) were used. d Reacted for 2 minutes instead of 30 seconds. e Reacted for 10 minutes instead of 30 seconds. f Reacted for 30 minutes instead of 30 seconds. g TBACl (3.0 equiv.) and PPO (3.0 equiv.) were used. h Reaction conditions: 9a (0.1 mmol, 1 equiv.), HFIP (0.5 mmol, 5.0 equiv.), PPO (0.11 mmol, 1.1 equiv.), TBAB (0.11 mmol, 1.1 equiv.) in DCE (3.3 mL, 0.033 M) at −30 °C, under an air atmosphere, 5 minutes. i Reaction conditions: 9b (0.1 mmol, 1 equiv.), HFIP (1.5 mmol, 15.0 equiv.), PPO (0.11 mmol, 1.1 equiv.), TBAI (0.11 mmol, 1.1 equiv.) in DCE (5 mL, 0.02 M) at 0 °C, under an air atmosphere, 5 minutes. Ts = tosyl; Boc = t-butyloxy carbonyl; Tr = triphenyl; Bn = benzyl; Naphth = naphthyl; TBACl = tetrabutylammonium chloride, TBAI = tetrabutylammonium iodide, DCE = 1,2-dichloroethane. | |
This protocol was also suitable for the synthesis of substituted tetrahydrofurans (4a–4c) in 54–89% yields. A transannular bromoetherification of 3d allowed for the high-efficiency synthesis of 2-oxabicyclo[2.1.1]hexane, which is regarded as a saturated benzene mimetic and an analogue of bicyclo[1.1.1]pentane (BCP).22 Additionally, multiple substituted β,γ-unsaturated allylic ketoximes participated smoothly in the chloro-, bromo- and iodooxygenation (6a–6f). The vicinal brominated iminolactones (6g–6i) were obtained in 81–95% yields via 5- or 6-exo O-attacked cyclization. These results were consistent with the hard/soft acid–base theory (HSAB), where a carbocation as a hard acid was inclined to be attacked by a hard nucleophile with higher electronegativity.23 The bromoxygenation of a 1,3-dicarbonyl compound, which contains an acidic proton and an olefinic side chain at an appropriate distance, allows the synthesis of highly functionalized cyclic ether 6j. The successful construction of pyrrolidines (8a–8c) and N-sulfonate ester (8d) reveals the foundational synthetic competence of N-attacked cyclization. Notably, the bromocyclization of chiral alkene imidate 7e afforded the exclusive diastereoselective formation of 8e in 54% yield, the structure of which was determined by 1H–1H COSY.
ICDA enhanced domino polyene-like halocyclization and gram-scale flow protocol
Due to the dual-anchoring orientation, the Br+ was transferred to the π-bond proximal to the carboxylic acid instead of the distal π-bond, resulting in the formation of mixed monocyclization products 10a and 10a′ in 75% total yield. Conversely, without a strong hydrogen bond donor, the iodiranium-induced polyene cyclization furnished the corresponding bicyclic product 10b in 67% yield when an electron-rich aryl served as the intramolecular nucleophile. Moreover, a spiroketal framework (10c, 1.15:1 d.r.) was constructed in 92% yield via halocyclization and spiroketalization.4e Recognizing the potential safety concerns associated with high-energy peroxide-containing compounds, we implemented the flow protocol to produce PPO through a packed bed reactor (Fig. S7 to S9†).24 By combining with batch reaction, this approach allows a safer electrophilic bromolactonization to offer 2a-Br (1.30 g, 89% yield) by minimizing the accumulation of PPO and obviating the necessity for isolating and recrystallizing substantial quantities of PPO. Furthermore, a preliminary investigation into the enantioselective bromocyclization of 1a and 7a offered enantiomeric excesses (ee) of 25% and 36% with yields of 80% and 74%, respectively, when employing C11 as the chiral catalyst (Tables S6 and S7†).
Construction of medium-sized and large-sized rings and synthesis of a difluoromethylene unit containing compounds enhanced by ICDA
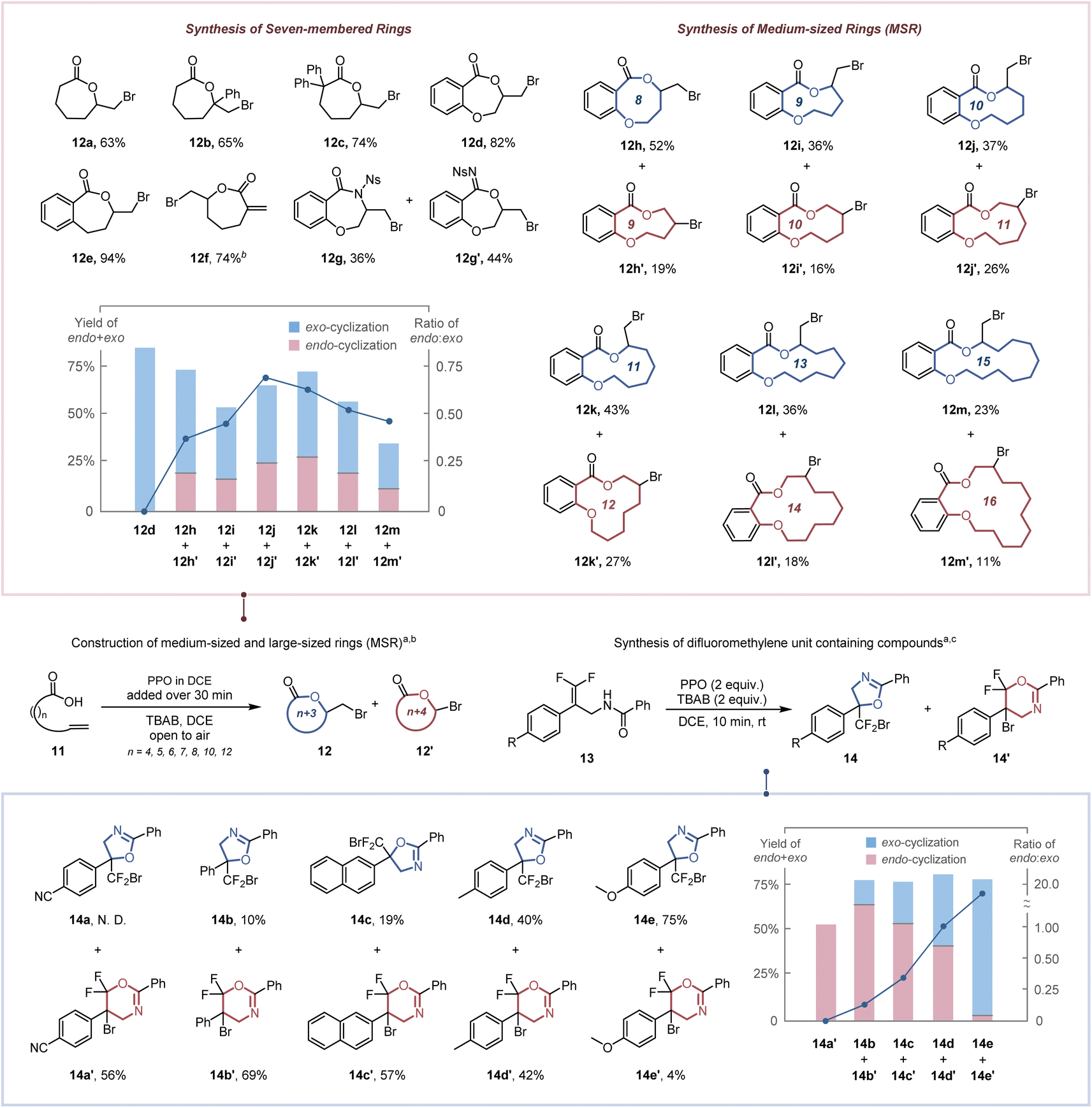
As a consequence of enthalpic (e.g., transannular interactions, torsional and bond strains) and entropic influences,25 the construction of seven-membered rings, medium-sized rings (MSR, typically eight-to-eleven membered rings) and large-sized rings is more challenging. Therefore, the high dilution and slow addition methodologies were utilized to reduce the competitive intermolecular addition (Table S5†).26 The seven-membered exo-lactones (12a–12f) were obtained specifically in 63–92% yields (Scheme 3a). The inferior result obtained by replacing PPO with BPO demonstrates the importance of ICDA in the spatial adjustment of the reactive conformation for constructing medium-sized and large-sized rings (Table S5,† entry 6). Notably, our protocol has a clear advantage over previous reports in synthesizing seven-membered lactone 12d (82% vs. 48%2b and 42%2c). The synthesis of seven-membered α-exo-methylene-lactone 12f, which is critical for the anti-inflammatory exhibition of several natural and bioactive molecules,27 was accomplished in 74% yield. A competition of O-attacked and N-attacked cyclization was observed when 11g was employed as a starting material, resulting in the generation of both O-cyclization product 12g and N-cyclization product 12g′ in 36% and 44% yields, respectively. This may be due to that altering the substituent group from –OMe (electron-donating group) to –Ns (electron-withdrawing group) significantly increases the acidity of the relevant N–H bonds (the pKa value in MeCN reduced from 19.08 to 16.05),28 which accelerated the corresponding deprotonation process and the generation of the N-attacked cyclization product. For the construction of medium-sized and large-sized rings, both endo- and exo-cyclization products (12h–12m′) were observed in 34–71% total yields. The increase in the ratio of endo lactone with chain length ring sizes of 8–10 implies that the steric hindrance has a weaker effect in an endo approach than in an exo approach. Nevertheless, the transannular interaction29 (H/H repulsions for methylene groups across the ring) was decreased for constructing larger rings while an increase of the exo lactone ratio was observed for chain lengths 11, 13 and 15. Our protocol was further applied to access a tetrasubstituted difluoromethylene unit (Scheme 3b), which confers increased lipophilicity, oxidative stability and modulated bioavailability to pharmaceutical molecules.30 Although it has been demonstrated that increasing the number of fluorine atoms on the π-bond could decrease its reactivity toward electrophilic reagents,31 the desired difluoromethyl-containing oxazolines (14a–14e) could also be obtained via exo-cyclization or endo-cyclization in 56–82% total yields within 10 min.
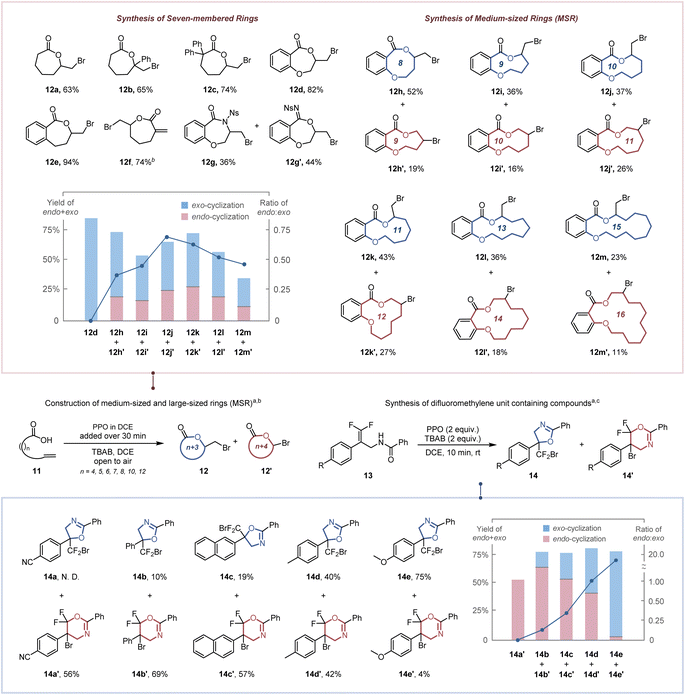 |
| | Scheme 3 Construction of medium-sized and large-sized rings and synthesis of difluoromethylene unit containing compounds. a Yields of isolated products after chromatographic purification. b Reaction conditions: olefin (0.3 mmol, 1 equiv.), PPO (0.60 mmol, 2.0 equiv.), TBAB (0.60 mmol, 2.0 equiv.) in DCE (15 mL, 0.02 M) at room temperature, under an air atmosphere, 30 minutes. c Reaction conditions: olefin (0.3 mmol, 1 equiv.), PPO (0.6 mmol, 2.0 equiv.), TBAB (0.6 mmol, 2.0 equiv.) in DCE (1 mL, 0.1 M) at room temperature, under an air atmosphere, 10 minutes. Ns = nosyl. | |
Mechanism study
2H KIE values were measured to investigate the reactive intermediate by considering two factors: (1) the adjacent C–H bonds of the carbenium ion center are expected to stabilize the carbon cation via the hyperconjugation effect; (2) the formation of the β-halo-carbenium ion should be slower in the labeled substrates 1a-D2 and 1b-D2 than in their parents (1a and 1b).32 The following 2H KIE measurements for 1a-D2 and 1b-D2 were 1.10 and 1.20 (Scheme 4a), respectively. These values showed a β-secondary isotope effect and supported the presence of a β-halo-carbenium intermediate.5a To discriminate the face selectivity of nucleophilic attack during the reaction process, the bromolactonization of deuterated substrate 1a-D was investigated (Scheme 4b).33 As a consequence, the bromolactonization of 1a-D under standard conditions led to 2a-D with a significant predominance of an anti stereopreference (anti:syn = 94.2:5.8). The influence of the intramolecular nucleophile on the reaction rate of bromocyclization has been evaluated by intermolecular competition experiments.341a, 3a, 5f, 5g, 5i and 7a were subjected to competitive bromocyclization with others, respectively, and the resulting competition ratios are illustrated in Scheme 4c. According to the previous reports,5a,35 we assumed that the differences in reaction rates were related to the nucleophilicity of the attacking group. For intramolecular reactions, knowledge of Baldwin's rules and effective molarity (EM) is required, that is, a correction term to adjust the prediction of the correlation equation to these processes.36 Nevertheless, in light of the circumstance where the intramolecular reactions mentioned above undergo the same 5-exo-trig cyclization and involve the similar benzylic carbocation acceptors, we attempted to ignore the effect of the correction term36 and utilized the nucleophilicity of the attacking groups to qualitatively evaluate the relative rate. The nucleophilicity parameters (N) of six molecules were predicted with the aid of the rSPOC model developed by Luo et al.,37 and the reaction rates of 1a, 5g, 5i, and 7a show a positive correlation with the N values of their equivalents (Fig. S23,† top). The different ratios of 2aversus4a in this work and a previous study,5a as well as the mismatches between the N values of 3a and 5f and the reaction rates (Scheme 4c, bottom), indicate that nucleophilicity is not the only factor influencing the reaction rate (Section S9.3). Next, the competition experiments of chlorolactonization, bromolactonization and iodolactonization showed that the iodolactonization was the fastest, while the chlorocyclization was the slowest (Scheme 4d, and Section S3), which may be due to the original difference in Lewis acidity of XB donors (I > Br > Cl).38
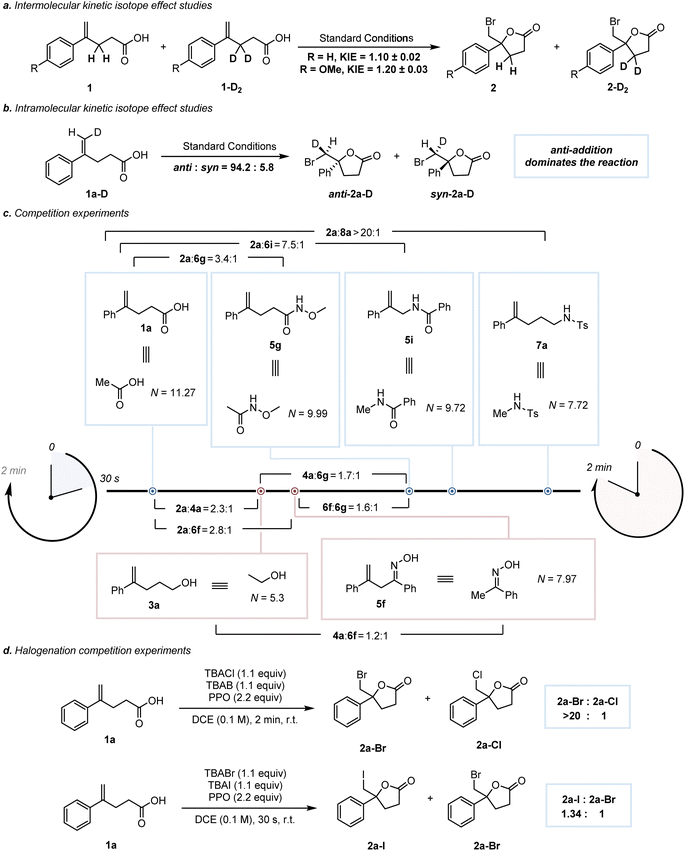 |
| | Scheme 4 KIE experiments and competition experiments. | |
Density functional theory (DFT) calculations
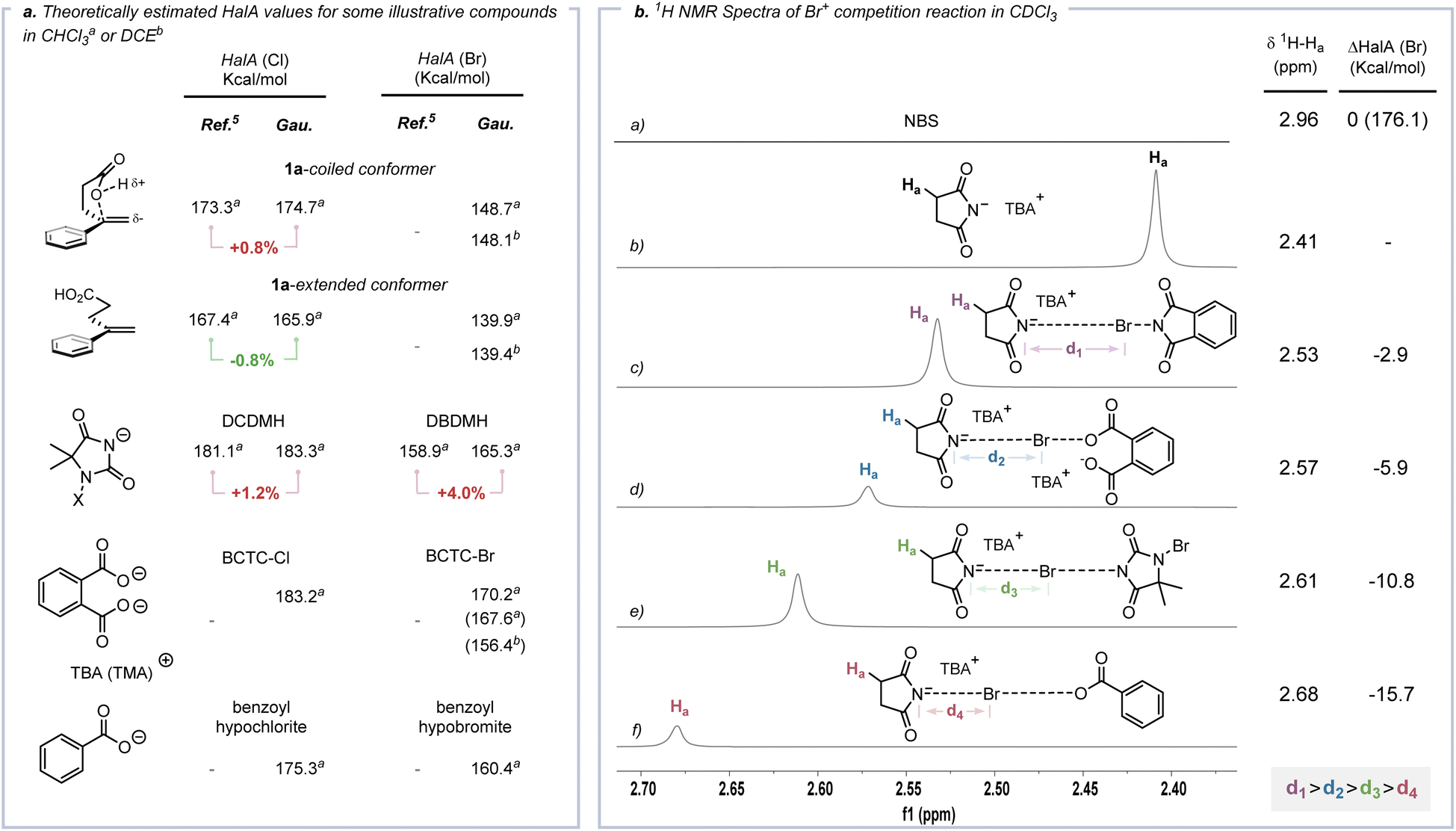
Cation affinity evaluation has been widely applied under thermodynamically controlled conditions. For example, proton affinity (PA) evaluation is important for rationalizing the biological functions of nucleic bases and designing hyperstrong neutral organic bases.39 Analogously, Borhan et al. introduced halenium affinity (HalA) to quantificationally describe the bond strengths of various Lewis bases to X+.40 Recently, HalA evaluation has been employed to interpret, estimate and predict electrophilic halogenation reactions (Fig. S26†). Therefore, density functional theory (DFT) calculations (see the ESI† for details) were performed to evaluate the HalA scales of 1a, anions of BCTC-X, benzoyl hypohalite (BPO-X) and commonly employed X+ sources (Scheme 5a and Fig. S27a,† X = Cl and Br). The HalA value of the anion of BCTC-Br (170.2 kcal mol−1) is higher than those of BPO-Br (160.4 kcal mol−1) and DBDMH (165.3 kcal mol−1) in CHCl3. That is, the introduction of an intramolecular chaperone-like carboxylate anion slightly increases the O–Br bond strength of BCTC-Br. According to the definition of the HalA parameter, the anion with a higher HalA value has a greater ability to capture a Br+, thus showing a stronger tendency to form the corresponding electrophilic bromination reagent. From this point of view, the intermolecular competition reactions of tetra-n-butylammonium succinimidate and electrophilic halogenation reagents were employed to visualize the Br+ transfer. As shown in Scheme 5b, the chemical shift of Ha decreased from 2.41 ppm to 2.53, 2.57, 2.61 and 2.68 ppm when tetra-n-butylammonium succinimidate was mixed 1:1 with N-bromophthalimide (NBP), BCTC-Br, 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) and BPO-Br, respectively. Namely, the capacity to capture Br+ attenuated sequentially from NBP to BPO-Br, which accords with the HalA(Br) of the anion of NBS versus other anions (ΔHalA(Br) = −2.9, −5.9, −10.8 and −15.7 kcal mol−1, Scheme 5b). The NPA charges of BPO-Br and BCTC-Br on the Br atom were calculated at the same level of theory and further proved the lower electrophilicity of the Br nucleus in BCTC-Br (Fig. S27c†). Comparing the HalA(Br) value of unactivated 1a (139.4 kcal mol−1) or nucleophile assisted alkene activation (NAAA)5a enabled 1a (148.1 kcal mol−1) with the anion of BCTC-Br (154.7 kcal mol−1) in DCE, one would predict that simple Br+ transfer to 1a would be untenably endothermic (ΔHalA(Br) = −17.0 kcal mol−1). In computational studies of the potential pathway, the ICDA affects the elongation of the O–Br bond and the conformation of the olefin, thus resulting in an effective ΔHalA(Br) (17.0 kcal mol−1) relative to the unperturbed compounds (Scheme 6b). That is, the suitable preorganization overcame the thermodynamic disadvantage of X+ transfer. The reaction process depends not only on the nature of 1a and BCTC, but also on the specific reactive conformation created in a precisely tailored environment.
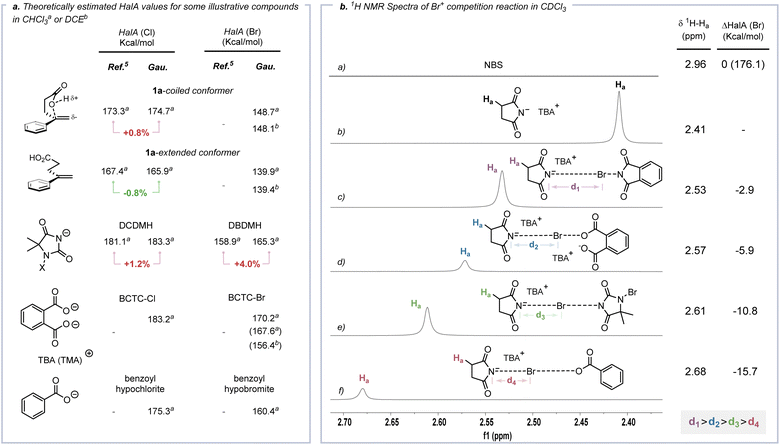 |
| | Scheme 5
HalA valuation. | |
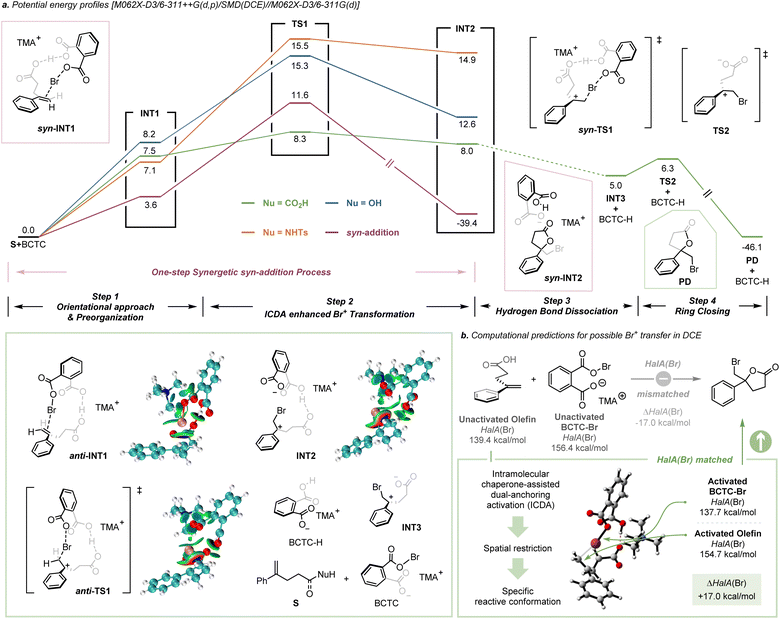 |
| | Scheme 6 Potential energy profiles and possible X+ transfer via HalA prediction. | |
DFT calculations were carried out to provide further insight into the mechanism of the ICDA enhanced electrophilic halocyclization (see the ESI† for details). As illustrated in Scheme 6a, the whole reaction consists of four steps. Orientational approach & preorganization: BCTC-Br approached the olefin via hydrogen bond interaction and the bromenium species was positioned in proximity to the π-bond. This preorganization process requires a certain amount of energy, which is less than 10 kcal mol−1. ICDA enhanced Br+ transfer: a cooperative activation of hydrogen bond and halogen bond interaction provided strict conformational control in a precisely tailored environment. Br+ transfer takes place with a relatively small barrier (0.8 kcal mol−1), which might be due to the preorganization process having pre-paid the entropy loss of the transition state. The rate-determining step is S+BCTC-Br → TS1. During the transfer of Br+, it is obvious the halogen bond between the π-bond and Br+ becomes stronger (Table S10†). In our calculations, three different substrates were chosen to study the effect of intramolecular nucleophiles. It is clear that for different nucleophilic groups, the reaction barrier changes, that is, for –CO2H it is the lowest, while for −NHTs it is the highest. Since the 1H NMR experiments ruled out the possibility of hydrogen transfer as the initiation step (Fig. S28†), the hydrogen bond dissociation and ring closure take place successively after Br+ transformation, with the O atom attacking the carbon cation from the opposite side. Furthermore, we found that the one-step syn-addition is thermodynamically unfavorable since its barrier is higher than that of the anti-addition process in our case, which is consistent with the intramolecular KIE experiment.
Conclusions
In conclusion, we have reported here a practical, versatile, catalyst-free and rapid electrophilic halocyclization that can afford structurally diversified valuable heterocyclic molecules in 30 s to 30 min. The synthetic ability can be categorized as follows: (a) synthesis of five-membered and six-membered substituted lactones, cyclic ethers, iminolactones and pyrrolidines, (b) domino polyene-like halocyclization, (c) a risk-reducing flow protocol for gram-scale synthesis, (d) construction of medium-sized and large-sized rings, (e) synthesis of difluoromethylene unit containing compounds, and (f) preliminary exploration of the catalytic asymmetric version. As shown in Fig. S30,† the ICDA enhanced halocyclization was demonstrated to be a powerful and superior protocol for the fast and high-yielding construction of seven types of heterocyclic units. DFT calculations verify the importance of suitable preorganization and the crucial role of the ICDA model in affording an induced-fit conformational change, stabilizing the transition state and accelerating X+ transfer in the rate-determining step. The utilization of the ICDA model may open up straightforward access to fast, selective, and high-yielding synthetic transformations. Further development of new synthetic strategies is still underway in our laboratory.
Data availability
All data associated with this article are available from ESI.†
Author contributions
L. S. designed and directed the project. X. Y. performed the experiments and developed the reactions. X. Y. explored the substrate scope. X. Y. and H. G. developed the synthesis of substrates. H. G. and J. Y. helped in collecting some of the experimental data. X. Y. prepared the ESI.† J. Z. directed the DFT calculations. J. Z. and X. Y. performed DFT calculations and drafted the DFT sections. L. S. and X. Y. wrote the paper. All authors discussed the results and commented on the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the financial support from the National Natural Science Foundation of China (22271069, 21871067), the Guangdong Basic and Applied Basic Research Foundation (2023A1515012457, 2023A1515011332, 2021A1515010190), the State Key Laboratory of Urban Water Resources and Environment (Harbin Institute of Technology) (2022TS36), the Fundamental Research Funds for the Central Universities (HIT.OCET.2021035), Shenzhen Science and Technology Program (GXWD20231130100539001) and the Open Project Program of State Key Laboratory of Elemento-Organic Chemistry (202009). Computer time made available by the National Supercomputing Center of China in Shenzhen (Shenzhen Cloud Computing Center) is gratefully acknowledged. We deeply thank Prof. Herbert Mayr (Ludwig-Maximilians-Universität München) for providing suggestions for the intensive investigation of this research, Prof. Min Wang (Hangzhou Normal University) for the assistance with the measurement of High-Resolution Mass Spectrometry, and Dr Denghu Chang for the assistance and advice on the experimental operation.
Notes and references
-
(a) X.-W. Liang, C. Zheng and S.-L. You, Chem.–Eur. J., 2016, 22, 11918–11933 CrossRef CAS PubMed;
(b) S. A. Snyder, D. S. Treitler and A. P. Brucks, Aldrichimica Acta, 2011, 44, 27–40 CAS;
(c) S. E. Denmark and M. T. Burk, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20655–20660 CrossRef CAS PubMed;
(d) C. Qi, G. Force, V. Gandon and D. Leboeuf, Angew. Chem., Int. Ed., 2021, 60, 946–953 CrossRef CAS PubMed;
(e) Q. Yin and S.-L. You, Org. Lett., 2012, 14, 3526–3529 CrossRef CAS PubMed;
(f) K. D. Ashtekar, H. Gholami, M. Moemeni, A. Chakraborty, L. Kiiskila, X. Ding, E. Toma, C. Rahn and B. Borhan, Angew. Chem., Int. Ed., 2022, 61, e202115173 CrossRef CAS PubMed;
(g) M. Sofiadis, D. Xu, A. J. Rodriguez, B. Nissl, S. Clementson, N. N. Petersen and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 21760–21765 CrossRef CAS PubMed.
-
(a) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, J. Org. Chem., 2016, 81, 545–552 CrossRef CAS PubMed;
(b) Y. A. Cheng, T. Chen, C. K. Tan, J. J. Heng and Y.-Y. Yeung, J. Am. Chem. Soc., 2012, 134, 16492–16495 CrossRef CAS PubMed;
(c) A. Verma, S. Jana, C. D. Prasad, A. Yadav and S. Kumar, Chem. Commun., 2016, 52, 4179–4182 RSC.
-
(a) A. M. Arnold, A. Pöthig, M. Drees and T. Gulder, J. Am. Chem. Soc., 2018, 140, 4344–4353 CrossRef CAS PubMed;
(b) S. A. Snyder, D. S. Treitler and A. P. Brucks, J. Am. Chem. Soc., 2010, 132, 14303–14314 CrossRef CAS PubMed;
(c) R. C. Samanta and H. Yamamoto, J. Am. Chem. Soc., 2017, 139, 1460–1463 CrossRef CAS PubMed.
-
(a) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS PubMed;
(b) J. Yan, Z. Zhou, Q. He, G. Chen, H. Wei and W. Xie, Org. Chem. Front., 2022, 9, 499–516 RSC;
(c) Y. Lu, H. Nakatsuji, Y. Okumura, L. Yao and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 6039–6043 CrossRef CAS PubMed;
(d) Y. M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928–12931 CrossRef CAS PubMed;
(e) T. Zheng, X. Wang, W.-H. Ng, Y.-L. S. Tse and Y.-Y. Yeung, Nat. Catal., 2020, 3, 993–1001 CrossRef CAS;
(f) K. Yamashita, R. Hirokawa, M. Ichikawa, T. Hisanaga, Y. Nagao, R. Takita, K. Watanabe, Y. Kawato and Y. Hamashima, J. Am. Chem. Soc., 2022, 144, 3913–3924 CrossRef CAS PubMed;
(g) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS PubMed;
(h) M. T. Knowe, M. W. Danneman, S. Sun, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2018, 140, 1998–2001 CrossRef CAS PubMed;
(i) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332–7335 CrossRef CAS PubMed.
-
(a) K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2016, 138, 8114–8119 CrossRef CAS PubMed;
(b) R. Van Lommel, J. Bock, C. G. Daniliuc, U. Hennecke and F. De Proft, Chem. Sci., 2021, 12, 7746–7757 RSC.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7024 CrossRef CAS PubMed.
-
(a) W. Wang, X. Li, X. Yang, L. Ai, Z. Gong, N. Jiao and S. Song, Nat. Commun., 2021, 12, 3873 CrossRef CAS PubMed;
(b) S. D. Vaidya, S. T. Toenjes, N. Yamamoto, S. M. Maddox and J. L. Gustafson, J. Am. Chem. Soc., 2020, 142, 2198–2203 CrossRef CAS PubMed;
(c) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068–6071 CrossRef CAS PubMed;
(d) X. He, X. Wang, Y.-L. S. Tse, Z. Ke and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2018, 57, 12869–12873 CrossRef CAS PubMed;
(e) F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 CrossRef CAS PubMed;
(f) P. Zhou, L. Lin, L. Chen, X. Zhong, X. Liu and X. Feng, J. Am. Chem. Soc., 2017, 139, 13414–13419 CrossRef CAS PubMed;
(g) Y.-C. Chan and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2018, 57, 3483–3487 CrossRef CAS PubMed.
-
(a) A. Jaganathan, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14806–14813 CrossRef CAS PubMed;
(b) X. Xiong and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2016, 55, 16101–16105 CrossRef CAS PubMed;
(c) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627–5630 CrossRef CAS PubMed;
(d) Y. Nishii, M. Ikeda, Y. Hayashi, S. Kawauchi and M. Miura, J. Am. Chem. Soc., 2020, 142, 1621–1629 CrossRef CAS PubMed;
(e) Z. Tao, K. A. Robb, K. Zhao and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 3569–3573 CrossRef CAS PubMed;
(f) A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Nat. Chem., 2015, 7, 146–152 CrossRef CAS PubMed;
(g) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900–903 CrossRef CAS PubMed.
-
(a) S. A. Snyder, A. Gollner and M. I. Chiriac, Nature, 2011, 474, 461–466 CrossRef CAS PubMed;
(b) S. A. Snyder, Z.-Y. Tang and R. Gupta, J. Am. Chem. Soc., 2009, 131, 5744–5745 CrossRef CAS PubMed.
- R. A. Rodriguez, C.-M. Pan, Y. Yabe, Y. Kawamata, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 6908–6911 CrossRef CAS PubMed.
- J. Barluenga, M. Trincado, E. Rubio and J. M. González, J. Am. Chem. Soc., 2004, 126, 3416–3417 CrossRef CAS PubMed.
- S. A. Snyder and D. S. Treitler, Angew. Chem., Int. Ed., 2009, 48, 7899–7903 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps and A. Volbeda, Chem. Rev., 2022, 122, 12110–12131 CrossRef CAS PubMed.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 CrossRef CAS PubMed.
- J. K. Nagle, J. Am. Chem. Soc., 1990, 112, 4741–4747 CrossRef CAS.
- C. Dong, S. Flecks, S. Unversucht, C. Haupt, K.-H. van Pée and J. H. Naismith, Science, 2005, 309, 2216–2219 CrossRef CAS PubMed.
-
(a) R. Zhao, K. Fu, Y. Fang, J. Zhou and L. Shi, Angew. Chem., Int. Ed., 2020, 59, 20682–20690 CrossRef CAS PubMed;
(b) R. Zhao, Y. Yao, D. Zhu, D. Chang, Y. Liu and L. Shi, Org. Lett., 2018, 20, 1228–1231 CrossRef CAS PubMed.
- N. London, D. Movshovitz-Attias and O. Schueler-Furman, Structure, 2010, 18, 188–199 CrossRef CAS PubMed.
- X. Jiang, S. Liu, S. Yang, M. Jing, L. Xu, P. Yu, Y. Wang and Y.-Y. Yeung, Org. Lett., 2018, 20, 3259–3262 CrossRef CAS PubMed.
- T. Chen, T. J. Y. Foo and Y.-Y. Yeung, ACS Catal., 2015, 5, 4751–4755 CrossRef CAS.
- V. V. Levterov, Y. Panasyuk, V. O. Pivnytska and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 7161–7167 CrossRef CAS PubMed.
- H. Mayr, M. Breugst and A. R. Ofial, Angew. Chem., Int. Ed., 2011, 50, 6470–6505 CrossRef CAS PubMed.
- A. M. Eliasen, R. P. Thedford, K. R. Claussen, C. Yuan and D. A. Siegel, Org. Lett., 2014, 16, 3628–3631 CrossRef CAS PubMed.
- G. A. Molander, Acc. Chem. Res., 1998, 31, 603–609 CrossRef CAS.
- W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680–4683 CrossRef CAS PubMed.
- T.-C. Tsai, H.-Y. Chen, J.-H. Sheu, M.-Y. Chiang, Z.-H. Wen, C.-F. Dai and J.-H. Su, J. Agric. Food Chem., 2015, 63, 7211–7218 CrossRef CAS PubMed.
- Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2020, 59, 19282–19291 CrossRef CAS PubMed.
- M.-C. Roux, R. Paugam and G. Rousseau, J. Org. Chem., 2001, 66, 4304–4310 CrossRef CAS PubMed.
-
(a) E. Miller, S. Kim, K. Gibson, J. S. Derrick and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 8946–8952 CrossRef CAS PubMed;
(b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed;
(c) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed;
(d) A. P. Combs, J. Med. Chem., 2010, 53, 2333–2344 CrossRef CAS PubMed;
(e) Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949–3952 CrossRef CAS PubMed;
(f) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- D. F. Shellhamer, J. L. Allen, R. D. Allen, D. C. Gleason, C. O'Neil Schlosser, B. J. Powers, J. W. Probst, M. C. Rhodes, A. J. Ryan, P. K. Titterington, G. G. Vaughan and V. L. Heasley, J. Org. Chem., 2003, 68, 3932–3937 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- R. Yousefi, K. D. Ashtekar, D. C. Whitehead, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14524–14527 CrossRef CAS PubMed.
- R. Huisgen, Angew. Chem., Int. Ed., 1970, 9, 751–762 CrossRef CAS.
- H. Mayr and A. R. Ofial, Pure Appl. Chem., 2005, 77, 1807–1827 CrossRef CAS.
-
(a) L. Shi, M. Horn, S. Kobayashi and H. Mayr, Chem.–Eur. J., 2009, 15, 8533–8541 CrossRef CAS PubMed;
(b) J. E. Baldwin, J. Chem. Soc. Chem. Commun., 1976, 734–736 RSC;
(c) J. E. Baldwin, J. Cutting, W. Dupont, L. Kruse, L. Silberman and R. C. Thomas, J. Chem. Soc. Chem. Commun., 1976, 736–738 RSC;
(d) J. E. Baldwin, R. C. Thomas, L. I. Kruse and L. Silberman, J. Org. Chem., 1977, 42, 3846–3852 CrossRef CAS;
(e) C. D. Johnson, Acc. Chem. Res., 1993, 26, 476–482 CrossRef CAS;
(f) R. Cacciapaglia, S. Di Stefano and L. Mandolini, Acc. Chem. Res., 2004, 37, 113–122 CrossRef CAS PubMed;
(g) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS.
- Y. Liu, Q. Yang, J. Cheng, L. Zhang, S. Luo and J.-P. Cheng, ChemPhysChem, 2023, 24, e202300162 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
-
(a) Z. B. Maksić, B. Kovačević and R. Vianello, Chem. Rev., 2012, 112, 5240–5270 CrossRef PubMed;
(b) K. Vazdar, D. Margetić, B. Kovačević, J. Sundermeyer, I. Leito and U. Jahn, Acc. Chem. Res., 2021, 54, 3108–3123 CrossRef CAS PubMed.
- K. D. Ashtekar, N. S. Marzijarani, A. Jaganathan, D. Holmes, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2014, 136, 13355–13362 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Haowei
Gao
a,
Jiale
Yan
a,
Jia
Zhou
a,
Haowei
Gao
a,
Jiale
Yan
a,
Jia
Zhou