
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient mRNA display protocol yields potent bicyclic peptide inhibitors for FGFR3c: outperforming linear and monocyclic formats in affinity and stability†
Camille
Villequey
 *a,
Silvana S.
Zurmühl‡
a,
Christian N.
Cramer‡
a,
Bhaskar
Bhusan
b,
Birgitte
Andersen
c,
Qianshen
Ren
d,
Haimo
Liu
d,
Xinping
Qu
d,
Yang
Yang
d,
Jia
Pan
d,
Qiujia
Chen
d and
Martin
Münzel
a
*a,
Silvana S.
Zurmühl‡
a,
Christian N.
Cramer‡
a,
Bhaskar
Bhusan
b,
Birgitte
Andersen
c,
Qianshen
Ren
d,
Haimo
Liu
d,
Xinping
Qu
d,
Yang
Yang
d,
Jia
Pan
d,
Qiujia
Chen
d and
Martin
Münzel
a
aGlobal Research Technologies, Novo Nordisk A/S, Novo Nordisk Park, 2760 Måløv, Denmark. E-mail: cmvq@novonordisk.com
bDepartment of Chemistry, Oxford University, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, UK
cGlobal Drug Discovery, Novo Nordisk A/S, Novo Nordisk Park, 2760 Måløv, Denmark
dNovo Nordisk Research Center China, Novo Nordisk A/S, Shengmingyuan West Ring Rd, Changping District, Beijing, China
First published on 18th March 2024
Abstract
Macrocyclization has positioned itself as a powerful method for engineering potent peptide drug candidates. Introducing one or multiple cyclizations is a common strategy to improve properties such as affinity, bioavailability and proteolytic stability. Consequently, methodologies to create large libraries of polycyclic peptides by phage or mRNA display have emerged, allowing the rapid identification of binders to virtually any target. Yet, within those libraries, the performance of linear vs. mono- or bicyclic peptides has rarely been studied. Indeed, a key parameter to perform such a comparison is to use a display protocol and cyclization chemistry that enables the formation of all 3 formats in equal quality and diversity. Here, we developed a simple, efficient and fast mRNA display protocol which meets these criteria and can be used to generate highly diverse libraries of thioether cyclized polycyclic peptides. As a proof of concept, we selected peptides against fibroblast growth factor receptor 3c (FGFR3c) and compared the different formats regarding affinity, specificity, and human plasma stability. The peptides with the best KD's and stability were identified among bicyclic peptide hits, further strengthening the body of evidence pointing at the superiority of this class of molecules and providing functional and selective inhibitors of FGFR3c.
Introduction
Introducing cyclisations into a peptide structure limits its conformational flexibility which in turn can improve multiple aspects of its therapeutic properties. First, structural constraints limit the entropic penalty upon target binding, which translates into higher affinities.1 Second, the ring-shaped structure makes it more difficult for proteases to access the peptide backbone and degrade it into smaller fragments, which can significantly impact the plasma stability of peptide drugs as well as their resistance to digestive enzymes when taken orally.2,3 Finally, peptide macrocyclization has furthermore demonstrated advantages for passive diffusion across cell membranes which facilitates the access of peptide drugs to potential intracellular targets and enhances their ability to cross stomach or gut epithelia.4 Considering all the aforementioned benefits, it is not a surprise that cyclization is a preferred tool for the maturation of peptides into potent drug candidates.5 Besides, multiple macrocyclic linkages in a given molecule are expected to yield even better results.6–8 One recent example is the clinical candidate MK-0616, a PCSK9 inhibitor developed by Merck currently evaluated in phase II for the treatment of elevated cholesterol levels in patients with renal impairment. In the hit-to-lead campaign, the addition of two stabilizing cyclisations into the monocyclic hit compound synergistically increased the affinity more than 500 fold, while maintaining favourable properties for oral bioavailability and plasma stability.9However, introducing cyclisations in the correct geometry after hit identification is often a complex and time-consuming task for medicinal chemists and much time and effort can be saved if large libraries of tightly knotted peptides are screened right at the beginning of a drug discovery campaign. This has previously been achieved through powerful in vitro display techniques such as mRNA or phage display in which large collections of linear peptides were cyclized either via oxidation of one or several pairs of cysteines,7,10 bridging two or more side chains with small reactive crosslinkers,9,11–16 incorporation of non-natural amino acids with new reactivities17,18 or the use of several orthogonal cyclization reactions together.19 Despite the range of possibilities, the majority of the peptides generated via those methods are monocyclic, possibly due to simpler procedures for library generation and more convenient synthetic routes. In addition, very few studies have focused on whether naïve libraries of polycyclic peptides are truly delivering better hits to develop into drugs than their monocyclic counterparts. In theory, genetically encoded libraries offer the opportunity to perform such an evaluation by directly screening binders to a given target for the three formats, namely linear, mono- and bicyclic in parallel. In practice, however, small variations in the library quality and diversity can greatly affect the output of the Darwinian selection systems, and therefore make any comparison difficult or irrelevant. This means that, in order to compete fairly, the chemical steps to generate the different formats must be equally efficient and should not interfere with the binder selection procedures.
Here we report an efficient and robust protocol to generate mRNA display libraries of mono- and bicyclic peptides allowing this type of comparative study. We decided to use cysteine bridging with thiol-reactive crosslinkers as they offer good ring stability in different redox environments and enable a multitude of geometries.20–22 Besides, their use in phage display selections is well-documented and has enabled the identification of numerous high-affinity bicyclic peptides with clinical relevance.13,23–25 Several procedures in which those linkers are used in connection with mRNA display have been published but the protocols, when disclosed, are often tedious, requiring multiple purifications of the chemically modified library as well as up to 10 rounds of selection. Additionally, the identified binders were inferior to compounds reported elsewhere with other peptide display methodologies.26–28 Our first goal was thus to prove that the mRNA display protocol could accommodate such peptide cyclization procedures and afford a similar success rate as observed with other protocols. After validation of the system, we aimed at conducting a comparative study, using a linear library devoid of cysteines, a monocyclic library where a pair of cysteines was cyclized with m-dibromoxylene (DBX) and a bicyclic library where 3 cysteines could be cyclized with 1,3,5-tris(bromomethyl)benzene (TBMB) (Fig. 1). Specifically, we aimed at generating ligands for the extra cellular domain of fibroblast growth factor receptor 3c (FGFR3c), which is a tyrosine kinase and an important target both in cancer and metabolic disease.29 In total, four FGF receptor families are encoded in the human genome, but 48 splice variants exist at the proteome level.30 These receptors are bound by in total 21 individual fibroblast growth factor (FGF) ligands which exhibit overlapping FGFR selectivity.31 In nature, no subfamily selective ligands are found, hence selective binders of individual FGFRs would be valuable research tools to decipher their exact biological role.
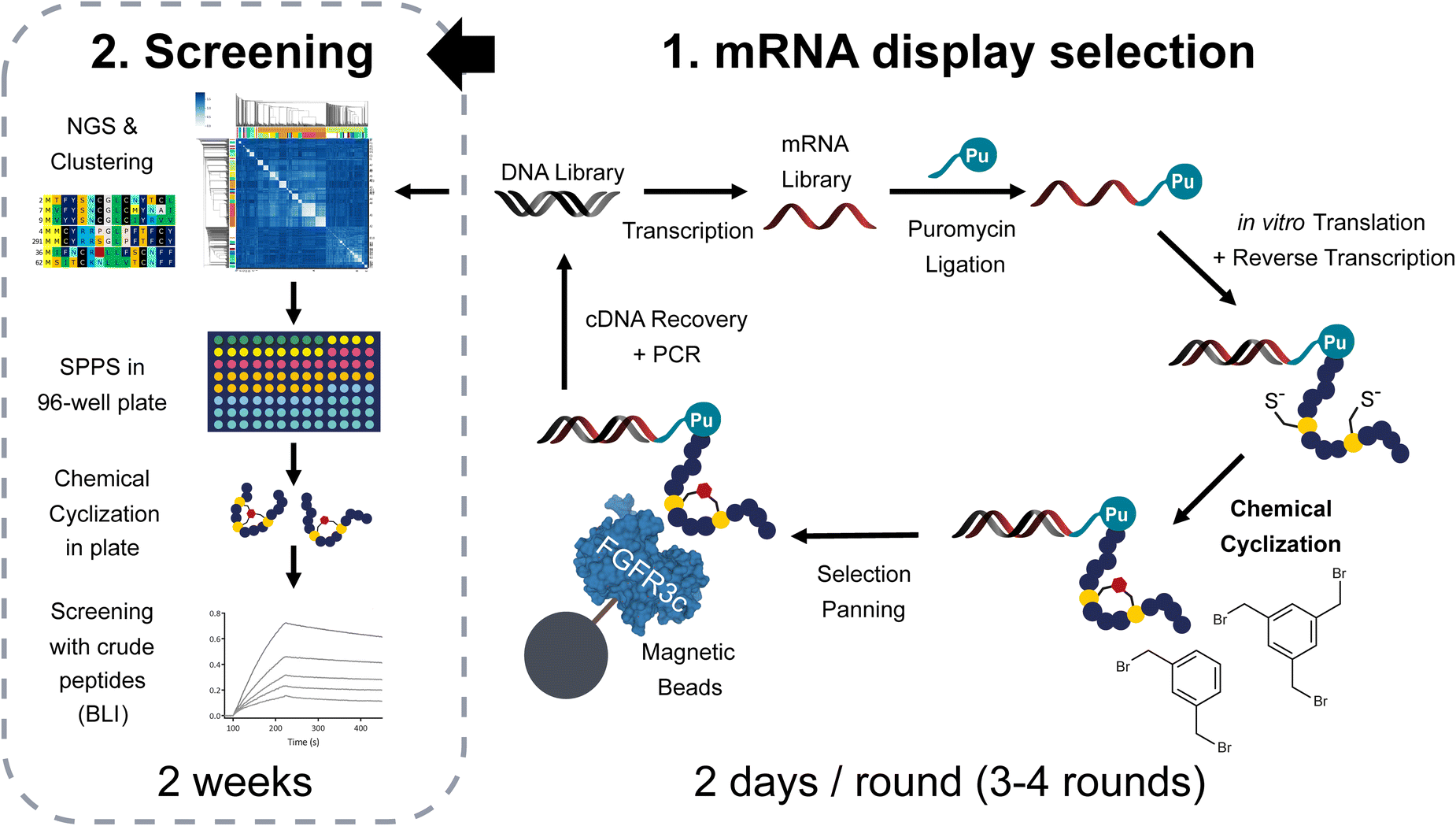 | ||
| Fig. 1 Methodology applied in this report to generate and screen polycyclic libraries of peptides via mRNA display. The procedure starts with a DNA-encoded library of peptides which undergoes transcription, puromycin attachment via T4 ligation and translation in the PURE system. The peptides are maintained in a reduced state by the presence of DTT. After reverse transcription and heat precipitation of heat unstable proteins at 60 °C, the mixture is subjected to 200 μM (if not specified otherwise) of TBMB or DBX cyclization in 60 mM NH4HCO3 buffer pH8. The cyclized library is panned against biotinylated FGFR3c immobilized on streptavidin beads and the cDNA is later recovered by heat denaturation at 95 °C to either continue with the next round or decode the results using next generation sequencing. The obtained sequences are analysed using our bioinformatic pipelines and sequence clustering tools. Representative sequences from the different families of peptides are synthesized in plate format for subsequent hit identification using high throughput biolayer interferometry. | ||
Results
Reaction of a thiol-reactive scaffold with an in vitro translated peptide
To be applicable to mRNA display procedures, the cyclization chemistry we chose needs to be (1) near quantitative under the conditions of the protocol, (2) must not lead to major unwanted by-products and (3) may not affect the nucleic acid moiety allowing to decode the selection results. With these criteria in mind, we decided to investigate the reactivity of TBMB with an in vitro translated peptide similar to PK15 (amino acid sequence: fMet-ACSDRFRNCPADEALC-(GS)3), a bicyclic molecule previously identified by phage display.12 The PURE system was chosen as cell-free translation media, as it allows strict control of the translation machinery components thereby limiting possible side reactions. Moreover, proteins and ribosomes, that could potentially react with TBMB, were heat-precipitated for 15 min at 60 °C, pelleted and removed from the reaction mixture immediately after completion of the translation step without the need for further purification. We hypothesized that the 8 mM DTT contained in the RNase inhibitor added to the translation (160 μM in the translation mix, about 3 equivalents of the quantity of ribosomes) was sufficient to keep the cysteines of PK15 reduced over the course of the experiment. The translation was then followed by a reverse transcription to mimic the conditions of an actual mRNA display selection. Finally, different concentrations of TBMB (50, 150 and 300 μM) were applied to the reaction mixture, which was further diluted in ammonium bicarbonate buffer at pH 8. The reaction was incubated for 30 min at 30 °C and the product formation was subsequently analysed by LC-MS (ESI Fig. S1a†). We observed close to 100% conversion to the bicyclic peptide when 300 μM TBMB was added to the reaction (ESI Fig. S1b and c†). At this concentration, no traces of the linear peptide were detected on our instrument, with no indication of partial reaction nor hydrolysis of the bromo-ethyl group. To validate the third and last condition, i.e. the absence of reaction between TBMB and the nucleic acids, we first verified that the reverse transcribed mRNA in our experiment could still be used as a template for PCR amplification. An agarose gel of the PCR products showed that no concentration of TBMB affected the yield or the size of the isolated PCR amplicon (ESI Fig. S1d†). This likely indicates that no chemical modification of the cDNA has led to interference with the polymerase activity. In addition, LC-MS showed no modification of the MS signal of 20 μM of mRNA coding for PK15 when incubated for 2 h with 2 mM TBMB at 30 °C in 80 mM NH4HCO3 pH8, 20% DMSO v/v (ESI Fig. S1e†).Enrichment of bicyclic peptide binders in mRNA display selection
Although no apparent difficulties were identified when cyclizing a single in vitro translated peptide, the performance of the thiol-alkylation chemistry in the context of a selection still needed to be assessed. Indeed, a mRNA display library contains trillions of different peptide sequences that may not cyclize as efficiently as PK15 when exposed to TBMB. Moreover, while the concentration of TBMB in our first experiment was within the same range as the one of the overexpressed peptide (around 100 μM), we now expect it to be in large excess (by several orders of magnitude) in the mRNA display protocol, which may affect both the selectivity and speed of the reaction. In principle, if our method to generate TBMB-cyclized libraries of peptides is sufficiently robust, it should yield results of equal quality to those reported with disulfide-cyclized peptides. To validate this, we designed an experiment in which mRNA display libraries of peptides encoded by XmCXnCXo (where m + n + o = 12, m, o ≥ 0 and n > 0, and X positions are encoded by NNK codons and can give any of the 20 canonical amino acids including cysteine) were either disulfide cyclized using previously published procedures or cyclized with various concentrations of TBMB (40, 200 or 1000 μM) using the workflow depicted in Fig. 1. The different libraries were panned against FGFR3c. The enrichment of binders specific to the target was measured by qPCR quantification of the cDNA recovered after each round of selection (Fig. 2a). The percentage of cDNA/peptide captured on magnetic beads coated with FGFR3c increased similarly to around 1% over input for all concentrations of TBMB and the control disulfide-cyclized library. There was no indication of major non-specific recovery on non-coated beads for any of the libraries. After four rounds of selection, the DNA was sequenced using Illumina Miseq, and the reads were clustered into groups of peptides sharing similar consensus sequences and cysteine topologies32 (Fig. 2b). Both library types yielded a highly diverse, yet distinct, set of clusters. Indeed, while the disulfide-cyclized libraries gave mostly hits with an even number of cysteines, the clusters identified for the TBMB-cyclized libraries often comprised peptides with 3 cysteines, which are required for cyclization with the crosslinker. This suggests that nearly no peptide remained disulfide-cyclized in the libraries after the chemical reaction step with TBMB. In addition, roughly the same clusters were identified for all 3 concentrations of TBMB, indicating that this parameter had little impact on the selection output.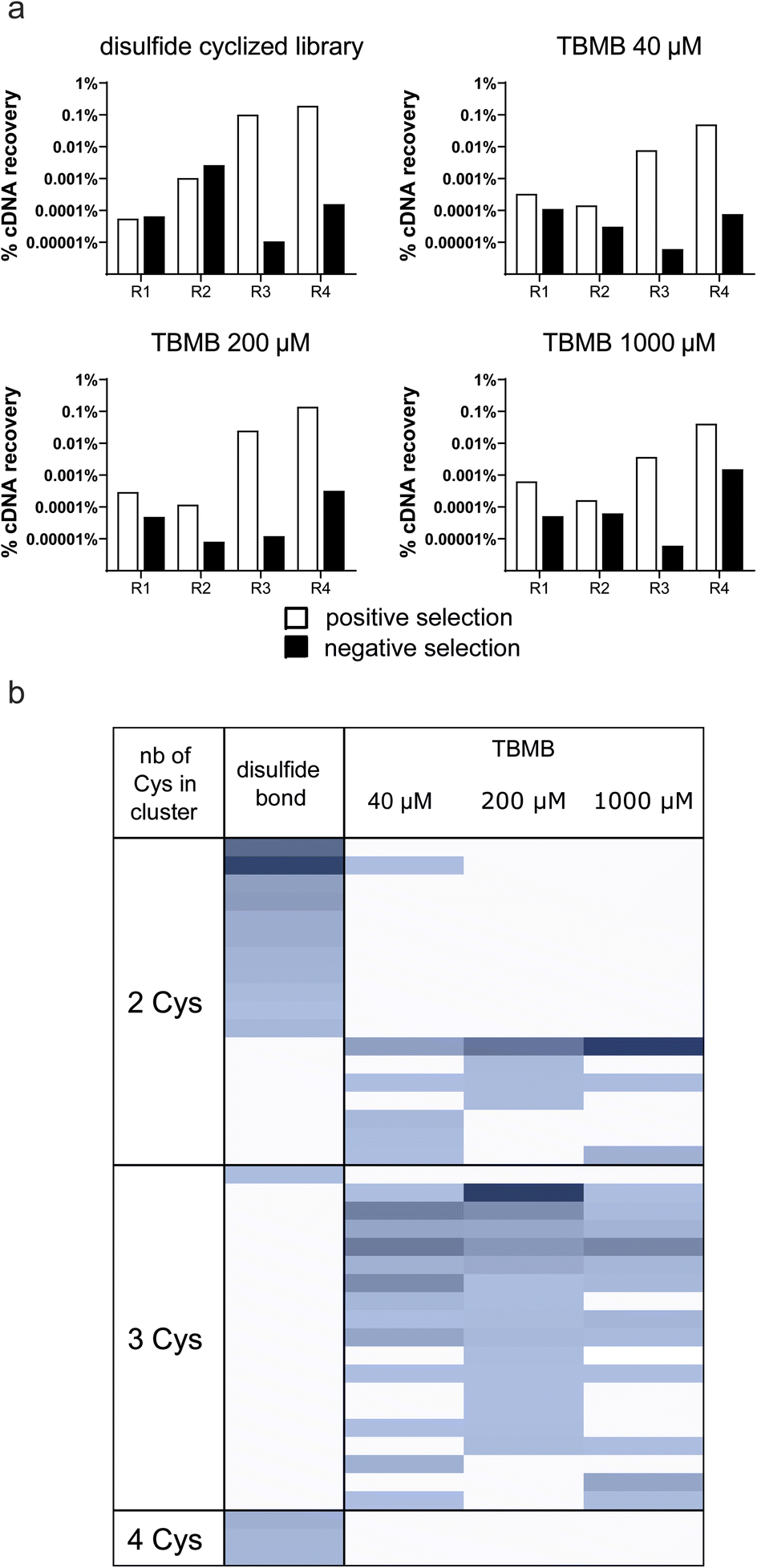 | ||
| Fig. 2 Enrichment of bicyclic peptides in mRNA display selections (a) cDNA recovered after each round of panning for the different libraries. The white bars indicate cDNA recovered on streptavidin beads coated with the biotinylated ECD of FGFR3c and the black bars indicate cDNA recovery on streptavidin beads alone. (b) Clusters obtained for the different screened conditions. Each line in the heatmap corresponds to one particular cluster, or peptide family, identified in the selection while the columns indicate the different conditions. The color intensity is proportional to the abundance/frequency of this cluster in the dataset. The different clusters are organized by a number of cysteines. | ||
Enrichment of linear, monocyclic, and bicyclic peptides against FGFR3c
Libraries of either linear, monocyclic, or bicyclic peptides were panned against FGFR3c for direct comparison of the three formats. The linear libraries consisted of 14 amino acid long sequences encoded by trinucleotide building blocks coding for all canonical amino acids but cysteine and methionine (the latter is prone to oxidation, often not compatible with drug development). The monocyclic and bicyclic libraries were based on the same NNK-based XmCXnCXo format described earlier, cyclized with either DBX or TBMB, respectively. Four rounds of selections were conducted, followed by next generation sequencing and sequence clustering (Fig. 1). A similar sequence consensus was observed across the different datasets, for example, the presence of the motif TL/F in many of the hits, which was also identified by another group in a phage display selection of peptides against FGFR3c.33 Based on the clustering, we selected around 20 sequences for each format (Fig. 3) for high throughput solid phase peptide synthesis in a 96-well format. Each peptide was synthesized with an N-terminal acylated methionine to mimic fMet present in the display selection as well as a C-terminal GSGSGS-DYKDDDDK-NH2 FLAG tag to ensure good solubility in buffer (previous work by our lab showed no or little impact of the tag on binding affinity32). Single enriched sequences were made in duplicate to avoid false positive/negative results in case of failed syntheses. After synthesis, peptides were cleaved under reducing conditions and about 50 nmoles of peptides were cyclized with the different crosslinkers directly in a 96-well plate format. To do so, the peptides were first incubated for 15 min at room temperature in 750 μL of 60 mM NH4HCO3-buffer (pH 8) containing 300 μM TCEP (approx. 5 eq.) and 100 μL of DMSO. Next 6 eq. of the chemical linkers in DMSO were added and reacted with the peptide for 1 h, followed by quenching with 10 eq. of L-cysteine. | ||
| Fig. 3 Linear, mono- and bicyclic peptides identified after selection against FGFR3c. The conserved amino acids in each cluster are highlighted with color. For each peptide, we display the abundance in the NGS dataset as well as the RU and estimated KD for the single concentration BLI measurement. The compounds that were further selected for in-depth characterization are numbered. (*) The full sequence usually contains a C-terminal GSGSGS-DYKDDDDK-NH2 tag; (**) the KD value is only indicative and should not be considered a precise number. | ||
We also included an additional plate duplicate as a control, in which the peptides were oxidized (20% DMSO in HEPES buffer, pH 7.4, overnight),32 instead of being cyclized with thiol-bridging reagents. The formation and purity of the macrocycles were assessed by UPLC-MS followed by quantification in UPLC-CAD. We could observe the desired product for the majority of the compounds, with an average crude purity of 30–50% which was sufficient for the initial screening steps as described earlier32 (ESI Table ‘Summary HTP synthesis.xlsx’†). In order to identify peptide hits for further detailed follow-up, the peptides plates were then screened for binding to the target using single concentration biolayer interferometry (BLI) using biotinylated FGFR3c extra-cellular domain (ECD) immobilized on streptavidin biosensors (peptide concentrations ranging from 200 to 1000 nM, same FGFR3c construct as used during the mRNA display selection). Response units and estimated KD's are reported in Fig. 3 for all synthesized compounds and were used for the selection of hits for further characterization. A total of 40 binders were identified for all 3 formats, but as depicted in Fig. 3, a preliminary analysis at this stage revealed that the estimated KD's appeared better for the mono- and bicyclic peptides than for the linear ones. To our satisfaction, only five of the tested monocyclic peptides also showed binding in the DMSO treated plate, indicating that they may have been in a disulfide-cyclized form during selection, while the rest of the hits exclusively bound when cyclized with DBX (ESI Fig. S2†).
Characterization of linear, mono- and bicyclic peptides for affinity and selectivity
For each format, we selected the five to six structurally diverse compounds that appeared to have the best affinities, in their cyclized (chemical linker) but not oxidized (disulfide) form, for further characterization. These peptides were either purified directly from the plate if present in sufficient quantity or newly synthesized by SPPS (ESI Table S2 and Fig. S3†). While most compounds were obtained efficiently, monocyclic peptide M6 could not be purified successfully, and further characterization of this compound was not completed. Precise binding affinities were measured using multiple concentration BLI (Table 1 and ESI Fig. S4†). For some peptides, such as B4, we observed binding curves that may indicate multiple binding equilibrium and we could not accurately determine the KD. Two of the sixteen tested peptides (L5 and M4) did not show binding after purification, indicating that they may have been false positive hits in the primary screen. We observed that the best affinities were obtained with the bicyclic peptides, which was the only class delivering compounds with single digit nanomolar affinities (compounds B3 and B5). As a comparison, four out of the five tested linear peptides exhibited binding affinities in the triple digit nM range (or were not binding), with only one, L4, having a KD = 39 nM. Finally, the four binding mono-cyclic peptides showed affinities in the mid-nM range, which nicely indicates an improvement compared to the linear ones but not to the extent of what was reached with bicyclization.| Target | FGFR3c | FGFR1c | FGFR4c | |
|---|---|---|---|---|
| Peptide | Geometry | K D (nM) | K D (nM) | K D (nM) |
| a Due to the presence of multiple binding equilibria the KD could not be calculated (see ESI Fig. S4). | ||||
| L1 | Linear | 350.0 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
2390.0 |
| L2 | 140.0 | >10000 |
>10000 |
|
| L3 | 640.0 | >10000 |
>10000 |
|
| L4 | 39.4 | >10000 |
56.1 | |
| L5 | >10000 |
>10000 |
>10000 |
|
| M1 | Monocyclic | 90.7 | >10000 |
>10000 |
| M2 | 13.4 | >10000 |
>10000 |
|
| M3 | 100.0 | >10000 |
>10000 |
|
| M4 | >10000 |
>10000 |
>10000 |
|
| M5 | 340.0 | >10000 |
>10000 |
|
| B1 | Bicyclic | 13.3 | 4150.0 | 91.7 |
| B2 | 6.7 | >10000 |
1310.0 | |
| B3 | 76.1 | >10000 |
>10000 |
|
| B4 | 7.5 | 840.0 | NDa | |
| B5 | 33.2 | >10000 |
>10000 |
|
To assess the selectivity of our peptides, we then measured the affinity to two additional FGF receptors, FGFR1c and FGFR4c. This family of receptors is particularly suited for this exercise as all members share high sequence identity and structural similarity, particularly in the domains D2 and D3 of the ECD (around 74% and 76% identity, respectively).34 Five of the sixteen characterized peptides displayed some measurable binding to at least one other FGF receptor, among which 3 were bicyclic and 2 were linear. Thus, unlike for binding affinity, increased peptide rigidity did not coincide with increased selectivity for the target. Nevertheless, except for the linear compound L4 and the bicycle B1, the difference in affinity was one or more orders of magnitude. In particular, the best linear binder (L4) also displayed the highest degree of poly-specificity to another FGFR, as the reported KD of 56 nM to FGFR4c, was nearly on par with the affinity for original target FGFR3c. To verify that the peptides could compete with the natural ligands FGF21 (for FGFR3c and FGFR1c) and FGF19 (for FGFR4c), we tested the five bicyclic peptides in an alphascreen binding assay in the presence of the co-receptor beta-klotho (ESI Fig. S5†). With this experiment, we confirmed the good affinity and receptor selectivity of B2, B3 and B5, as observed in BLI. In contrast, peptide B4 for which we could not precisely determine the KD to FGFR4 also showed strong binding to this receptor.
Plasma stability of the linear, mono- and bicyclic peptides
To evaluate the impact of cyclization on plasma proteolytic stability, we incubated 4 linear, 4 monocyclic and 4 bicyclic compounds in human plasma and quantified the remaining amount of intact peptide over the course of 5 hours (Fig. 4a). Most peptides showed some conversion over time, which arose either from proteolytic degradation and/or oxidation of the N-terminal acetyl-methionine. However, 3 peptides, linear L3 and two monocyclic M1 and M3 showed faster degradation, at a rate up to that of native GLP-1 which was included as a control. To determine the mechanism behind the fast degradation of the two monocyclic peptides M1 and M3, we performed metabolite identification and found an arginine in both peptides to be the hotspot for proteolytic cleavage followed by sequential degradation of exposed single amino acids until the cysteine residue involved in cyclization (ESI Fig. S6†). This is in alignment with previous observations that arginine together with lysine are residues highly sensitive to serine proteases abundant in human plasma.35 Interestingly, the monocyclic M2 peptide which belongs to the same sequence cluster but does not contain an arginine displayed a slower degradation over time. In a larger metabolite identification effort, we then investigated whether all basic residues were hotspots for proteolytic degradation independent of the position in the monocyclic and bicyclic peptides. We found that arginine residues located outside the macrocyclic structure constituted the primary source of metabolism as observed for the M1, M3, B1 and B4 peptides. In contrast, an arginine or lysine residue located in the macrocycle of M2 and B5 remained fully stable. In fact, we did not detect any proteolytic degradation inside of the macrocyclic structures in any of the peptides and all metabolism was restricted to amino acids outside of the rings. As a result of this, the bicyclic B5 peptide, in which nearly all amino acids are located within the macrocyclic rings, showed full stability in human plasma over the course of 24 h (Fig. 4b and S6†).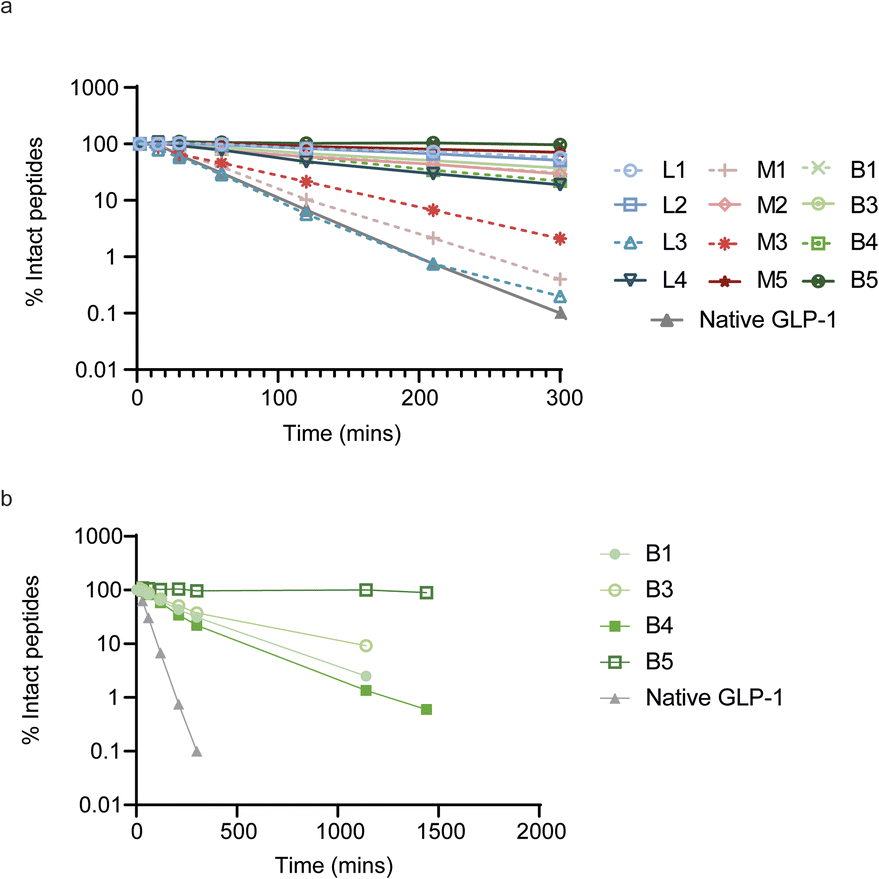 | ||
| Fig. 4 Stability of linear, mono- and bicyclic peptides after in vitro incubation in human plasma. (a) Percentage of intact peptide after up to 300 min incubations in human plasma. (b) Stability profile over the course of 24 h for the bicyclic peptides. | ||
Peptide B5 is a functional and selective antagonist of FGFR3c
In a final experiment we decided to assess whether the good binding affinities and selectivity of our bicyclic peptides would result in functional inhibition of FGFR3c signalling in a BaF3 cell line, an immortalized murine bone marrow-derived pro-B-cell cell line having no endogenous FGF receptor expression, stably transfected with the human beta klotho receptor and either FGFR1c, FGFR3c or FGFR4. The FGFR-induced signalling was monitored via quantitative detection of phospho-ERK 1/2 in cell lysate. Only two peptides, B1 and B5, showed inhibition of pERK activity at nanomolar concentrations with B5 being the only FGF receptor selective peptide (Fig. 5). Together with its strong proteolytic stability, B5 is a promising tool compound to study FGFR3c biology and would serve as the most promising molecule to develop therapeutic candidates.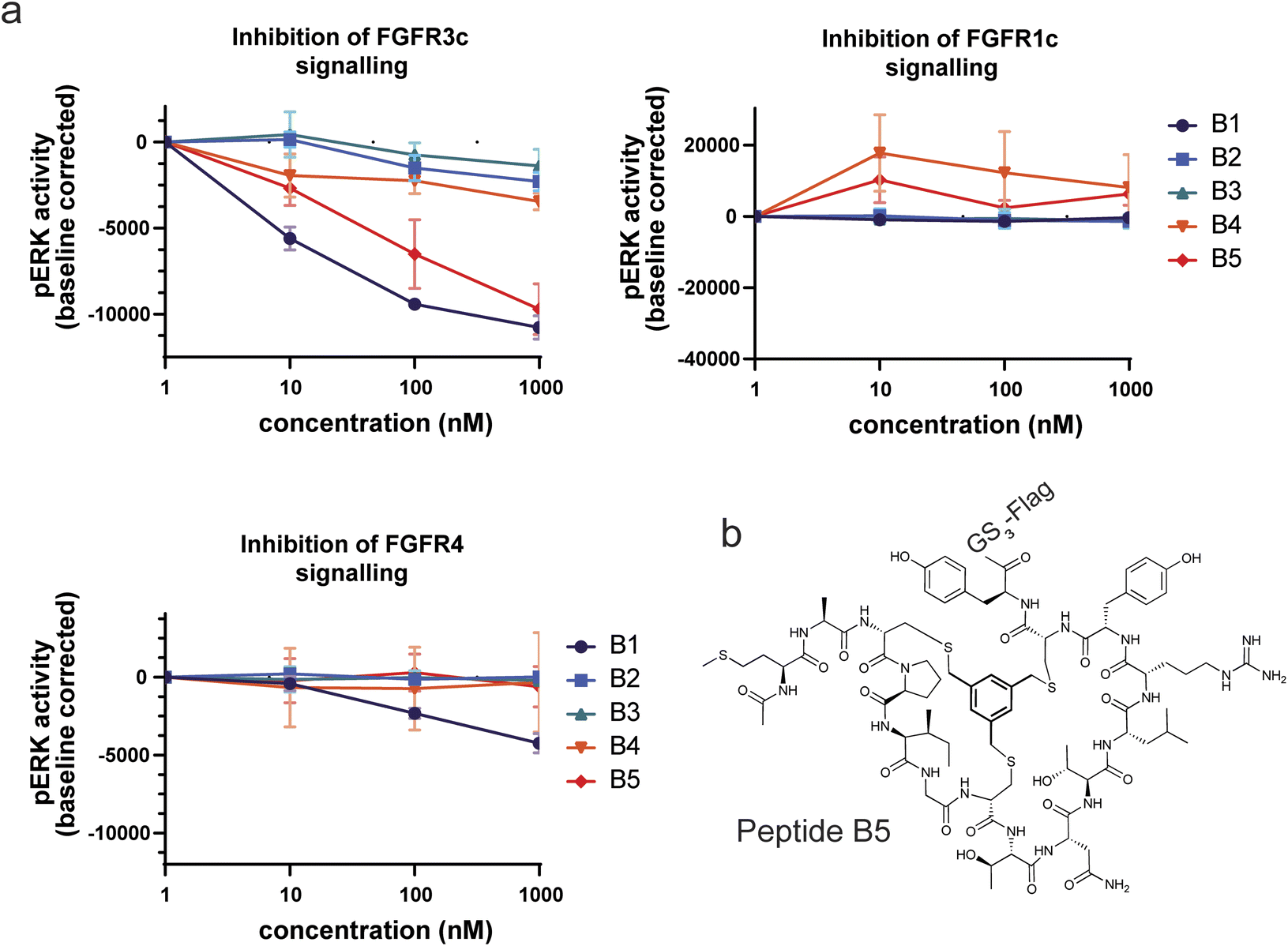 | ||
| Fig. 5 Inhibition of FGF receptor signalling in stably transfected BAF3 cells. (a) The 5 bicyclic peptides were tested for inhibition at 1, 10, 100 and 1000 nM. The assay was done in triplicate. (b) Structure of B5, the most potent and selective bicyclic peptide. | ||
Discussion
Here, we outlined a simple method to efficiently generate high quality mRNA display libraries of mono- and bicyclic peptides cyclized via thiol-reactive crosslinkers. Through careful evaluation of the different parts of the display protocol, we could show that the added chemical steps required for cyclization had no negative impact on the selection procedures and our ability to rapidly identify high affinity peptide binders in sufficient numbers. This was ensured by keeping the procedures simple with no extra purification required prior to selection panning (with the exception of a desalting step), which is often a source of diversity loss. Each round of selection can be completed in two days (about 4 rounds are needed for sufficient binder enrichment) and can be combined with high throughput next generation sequencing analysis, synthesis and screening reported by our laboratory earlier.32 Applying this technique, we could evaluate the performance of linear, mono- and bicyclic peptide libraries in their ability to deliver strong and specific binders to FGFR3c with good plasma stability. As anticipated from previous studies and theoretical considerations, bicyclic peptides outperformed the two other formats in terms of affinity, delivering the only binders with single-digit nM affinity despite sharing the same sequence motif TL/F as other, less potent, linear or monocyclic peptides. This means that, although those peptides likely interact with the same epitope on the target and engage in similar interactions, the additional constraints in their structure are driving substantial affinity gain. We also confirmed the benefit of cyclization for protecting peptides against proteolytic degradation. While none of the residues located inside the macrocyclic rings of the compounds studied underwent proteolytic cleavage, the residues outside of the rings remained exposed. This emphasizes the importance of getting as much as possible of the amino acid sequence into a rigid macrocyclic structure to fully benefit from this particular resistance, which is more easily achieved with bicyclic formats. Our last comparison point was target selectivity, using binding to other structurally related FGFRs as criteria. Here, we could not identify any apparent correlation between the peptide formats and results. Although it has been shown that cyclization could tune the selectivity of RGD peptides to particular integrin receptors by favoring a more constrained geometry,36 specificity englobes several notions and is governed by various complex molecular properties. Indeed, one can distinguish non-specificity, which can be driven by sequence features such as sticky hydrophobic or charged patches, from poly-specificity where similarity of epitopes is an essential parameter. Consequently, it is difficult to compare the peptides in our study, for which the amino acid content and distribution vary greatly, and which may bind different epitopes on FGFR3c.The outcome of our study seems to be in line with empirical evidence as well as results obtained with rational engineering of peptides. To the best of our knowledge, only one similar study has been reported before.19 Here, researchers directly compared mRNA display libraries of linear, mono- and bi-cyclic peptides, generated by a combination of copper-mediated azide–alkyne cycloaddition and cysteine bisalkylation with DBX, for binding to streptavidin. In contrast to the present work and to the current knowledge in the field, no added benefits of mono- nor bicyclization were reported regarding binding affinity. However, the reported affinities are in the mid-micromolar range, well above reported KD's for the same target in the literature,37–40 hinting at issues with the cyclization and/or display procedures. This further underlines the importance of using robust library generation protocols when performing such comparative studies.
Conclusions
To summarize, this work emphasizes the superiority of polycyclic peptide libraries for the identification of hits with more promising affinity and proteolytic stability, ensuring a faster path towards a mature drug candidate. Specifically, we identified peptide B5 which shows potent inhibition of FGFR3c signalling, good receptor selectivity and full plasma stability over the course of 24 h. While only TBMB and DBX were used as cyclization agents in this work, we believe that this method is compatible with the wide range of structurally diverse thiol-reactive crosslinkers, which could give access to a plethora of peptide formats with potentially improved pharmacological properties.13Author contributions
C. V. and M. M. conceived the idea. C. V. established the mRNA display procedures. C. V. and S. S. Z. performed the mRNA display selections and analysed the next generation sequencing data. C. V. and S. S. Z. synthesized and purified the peptides. S. S. Z. performed the biolayer interferometry experiments. C. N. C. designed, performed, and analysed the plasma stability and metabolite identification experiments. B. B. and B. A. designed, performed, and analysed the FGF receptor inhibition experiments. Q. R. designed the protein constructs. Q. X., H. L., J. P., Q. C. and Y. Y. expressed and purified the recombinant FGF receptors. C. V., S. S. Z., C. N. C and M. M. wrote the manuscript. All the authors approved the final version of the manuscript.Conflicts of interest
All authors apart from B. B. are or were employees and shareholders of Novo Nordisk A/S.Acknowledgements
The authors would like to thank Johnny Madsen, David Hansen, Kirsten V. Haugegaard, Kira Meyhoff-Madsen, Vladimir Bariak (Apigenex), Pavla Kovarova (Apigenex), and Aleš Halama (Apigenex) for excellent technical assistance.References
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- L. Gentilucci, R. De Marco and L. Cerisoli, Curr. Pharm. Des., 2010, 16, 3185–3203 CrossRef CAS PubMed.
- A. F. B. Rader, M. Weinmuller, F. Reichart, A. Schumacher-Klinger, S. Merzbach, C. Gilon, A. Hoffman and H. Kessler, Angew Chem. Int. Ed. Engl., 2018, 57, 14414–14438 CrossRef PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- V. Baeriswyl and C. Heinis, ChemMedChem, 2013, 8, 377–384 CrossRef CAS PubMed.
- S. Chen, I. Rentero Rebollo, S. A. Buth, J. Morales-Sanfrutos, J. Touati, P. G. Leiman and C. Heinis, J. Am. Chem. Soc., 2013, 135, 6562–6569 CrossRef CAS PubMed.
- C. A. Rhodes and D. Pei, Chemistry, 2017, 23, 12690–12703 CrossRef CAS PubMed.
- S. E. Iskandar and A. A. Bowers, ACS Med. Chem. Lett., 2022, 13, 1379–1383 CrossRef CAS PubMed.
- S. Hansen, Y. Zhang, S. Hwang, A. Nabhan, W. Li, J. Fuhrmann, Y. Kschonsak, L. Zhou, A. H. Nile, X. Gao, R. Piskol, E. M. F. de Sousa, F. J. de Sauvage and R. N. Hannoush, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207327119 CrossRef CAS PubMed.
- J. T. Hampton, T. J. Lalonde, J. M. Tharp, Y. Kurra, Y. R. Alugubelli, C. M. Roundy, G. L. Hamer, S. Xu and W. R. Liu, ACS Chem. Biol., 2022, 17, 2911–2922 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- S. W. Millward, T. T. Takahashi and R. W. Roberts, J. Am. Chem. Soc., 2005, 127, 14142–14143 CrossRef CAS PubMed.
- T. R. Oppewal, I. D. Jansen, J. Hekelaar and C. Mayer, J. Am. Chem. Soc., 2022, 144, 3644–3652 CrossRef CAS PubMed.
- J. Y. Wong, R. Mukherjee, J. Miao, O. Bilyk, V. Triana, M. Miskolzie, A. Henninot, J. J. Dwyer, S. Kharchenko, A. Iampolska, D. M. Volochnyuk, Y. S. Lin, L. M. Postovit and R. Derda, Chem. Sci., 2021, 12, 9694–9703 RSC.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120–129 CrossRef CAS PubMed.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed.
- D. E. Hacker, N. A. Abrigo, J. Hoinka, S. L. Richardson, T. M. Przytycka and M. C. T. Hartman, ACS Comb. Sci., 2020, 22, 306–310 CrossRef CAS PubMed.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew Chem. Int. Ed. Engl., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- L. E. Smeenk, N. Dailly, H. Hiemstra, J. H. van Maarseveen and P. Timmerman, Org. Lett., 2012, 14, 1194–1197 CrossRef CAS PubMed.
- Y. Wang and D. H. Chou, Angew Chem. Int. Ed. Engl., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- V. Baeriswyl, S. Calzavarini, S. Chen, A. Zorzi, L. Bologna, A. Angelillo-Scherrer and C. Heinis, ACS Chem. Biol., 2015, 10, 1861–1870 CrossRef CAS PubMed.
- K. Hurov, J. Lahdenranta, P. Upadhyaya, E. Haines, H. Cohen, E. Repash, D. Kanakia, J. Ma, J. Kristensson, F. You, C. Campbell, D. Witty, M. Kelly, S. Blakemore, P. Jeffrey, K. McDonnell, P. Brandish and N. Keen, J. ImmunoTher. Cancer, 2021, 9(11), e002883 CrossRef PubMed.
- G. E. Mudd, H. Scott, L. Chen, K. van Rietschoten, G. Ivanova-Berndt, K. Dzionek, A. Brown, S. Watcham, L. White, P. U. Park, P. Jeffrey, M. Rigby and P. Beswick, J. Med. Chem., 2022, 65, 14337–14347 CrossRef CAS PubMed.
- C. Alleyne, R. P. Amin, B. Bhatt, E. Bianchi, J. C. Blain, N. Boyer, D. Branca, M. W. Embrey, S. N. Ha, K. Jette, D. G. Johns, A. D. Kerekes, K. A. Koeplinger, D. LaPlaca, N. Li, B. Murphy, P. Orth, A. Ricardo, S. Salowe, K. Seyb, A. Shahripour, J. R. Stringer, Y. Sun, R. Tracy, C. Wu, Y. Xiong, H. Youm, H. J. Zokian and T. J. Tucker, J. Med. Chem., 2020, 63, 13796–13824 CrossRef CAS PubMed.
- D. E. Hacker, J. Hoinka, E. S. Iqbal, T. M. Przytycka and M. C. Hartman, ACS Chem. Biol., 2017, 12, 795–804 CrossRef CAS PubMed.
- Y. V. Schlippe, M. C. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef PubMed.
- M. Katoh, Nat. Rev. Clin. Oncol., 2019, 16, 105–122 CrossRef CAS PubMed.
- L. Duchesne, B. Tissot, T. R. Rudd, A. Dell and D. G. Fernig, J. Biol. Chem., 2006, 281, 27178–27189 CrossRef CAS PubMed.
- N. Itoh and D. M. Ornitz, Trends Genet., 2004, 20, 563–569 CrossRef CAS PubMed.
- B. Bhushan, D. Granata, C. S. Kaas, M. A. Kasimova, Q. Ren, C. N. Cramer, M. D. White, A. M. K. Hansen, C. Fledelius, G. Invernizzi, K. Deibler, O. D. Coleman, X. Zhao, X. Qu, H. Liu, S. S. Zurmuhl, J. T. Kodra, A. Kawamura and M. Munzel, Chem. Sci., 2022, 13, 3256–3262 RSC.
- M. Jin, Y. Yu, H. Qi, Y. Xie, N. Su, X. Wang, Q. Tan, F. Luo, Y. Zhu, Q. Wang, X. Du, C. J. Xian, P. Liu, H. Huang, Y. Shen, C. X. Deng, D. Chen and L. Chen, Hum. Mol. Genet., 2012, 21, 5443–5455 CrossRef CAS PubMed.
- J. Nishita, S. Ohta, S. B. Bleyl and G. C. Schoenwolf, Dev. Dyn., 2011, 240, 1537–1547 CrossRef CAS PubMed.
- R. Bottger, R. Hoffmann and D. Knappe, PLoS One, 2017, 12, e0178943 CrossRef PubMed.
- A. Roxin and G. Zheng, Future Med. Chem., 2012, 4, 1601–1618 CrossRef CAS PubMed.
- S. Bellotto, S. Chen, I. Rentero Rebollo, H. A. Wegner and C. Heinis, J. Am. Chem. Soc., 2014, 136, 5880–5883 CrossRef CAS PubMed.
- L. B. Giebel, R. T. Cass, D. L. Milligan, D. C. Young, R. Arze and C. R. Johnson, Biochemistry, 1995, 34, 15430–15435 CrossRef CAS PubMed.
- M. R. Jafari, L. Deng, P. I. Kitov, S. Ng, W. L. Matochko, K. F. Tjhung, A. Zeberoff, A. Elias, J. S. Klassen and R. Derda, ACS Chem. Biol., 2014, 9, 443–450 CrossRef CAS PubMed.
- T. Lamla and V. A. Erdmann, J. Mol. Biol., 2003, 329, 381–388 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04763f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |