
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceG-quadruplex-guided cisplatin triggers multiple pathways in targeted chemotherapy and immunotherapy†
Tian-Zhu
Ma
a,
Liu-Yi
Liu
 a,
You-Liang
Zeng
a,
Ke
Ding
c,
Hang
Zhang
a,
Wenting
Liu
*a,
Qian
Cao
a,
Wei
Xia
a,
Xushen
Xiong
*c,
Chao
Wu
*b and
Zong-Wan
Mao
*a
a,
You-Liang
Zeng
a,
Ke
Ding
c,
Hang
Zhang
a,
Wenting
Liu
*a,
Qian
Cao
a,
Wei
Xia
a,
Xushen
Xiong
*c,
Chao
Wu
*b and
Zong-Wan
Mao
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, IGCME, GBRCE for Functional Molecular Engineering, Sun Yat-Sen University, Guangzhou 510275, P. R. China. E-mail: liuwenting@mail.sysu.edu.cn; cesmzw@mail.sysu.edu.cn
bDepartment of Neurology, The First Affiliated Hospital, Guangdong Provincial Key Laboratory of Diagnosis and Treatment of Major Neurological Diseases, National Key Clinical Department and Key Discipline of Neurology, Sun Yat-sen University, Guangzhou 510080, P. R. China. E-mail: wuch35@mail.sysu.edu.cn
cThe Second Affiliated Hospital & Liangzhu Laboratory, Zhejiang University School of Medicine, Hangzhou 311121, P. R. China. E-mail: xiongxs@zju.edu.cn
First published on 15th May 2024
Abstract
G-quadruplexes (G4s) are atypical nucleic acid structures involved in basic human biological processes and are regulated by small molecules. To date, pyridostatin and its derivatives [e.g., PyPDS (4-(2-aminoethoxy)-N2,N6-bis(4-(2-(pyrrolidin-1-yl) ethoxy) quinolin-2-yl) pyridine-2,6-dicarboxamide)] are the most widely used G4-binding small molecules and considered to have the best G4 specificity, which provides a new option for the development of cisplatin-binding DNA. By combining PyPDS with cisplatin and its analogs, we synthesize three platinum complexes, named PyPDSplatins. We found that cisplatin with PyPDS (CP) exhibits stronger specificity for covalent binding to G4 domains even in the presence of large amounts of dsDNA compared with PyPDS either extracellularly or intracellularly. Multiomics analysis reveals that CP can effectively regulate G4 functions, directly damage G4 structures, activate multiple antitumor signaling pathways, including the typical cGAS-STING pathway and AIM2-ASC pathway, trigger a strong immune response and lead to potent antitumor effects. These findings reflect that cisplatin-conjugated specific G4 targeting groups have antitumor mechanisms different from those of classic cisplatin and provide new strategies for the antitumor immunity of metals.
Introduction
Nucleic acids are the most important genetic material of life. As a special and stable nucleic acid secondary structure, G-quadruplexes (G4s) play a vital role in DNA replication, transcription, and genome maintenance.1 G4 structures are stacked by G-tetrads formed by Hoogsteen hydrogen bonding of guanine bases in G-rich DNA or RNA sequences.2–5 G4 structures are widely distributed in the human genome, and more than 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 G4s with different topologies have been identified by G4 sequencing technology (G4-seq).6 Since nuclear expression levels of G4 structures may be higher in tumor cells than in normal cells,4,7 overexpression of protooncogenes with G4 folding is widely found in most types of cancers, such as c-MYC and VEGF, which play crucial roles in tumor cell growth, proliferation, migration and angiogenesis.8,9 Recent studies have shown that G4 damage can trigger genome instability, eventually leading to immune gene activation and related processes in cancer cells because cytoplasmic DNA/RNA can act as an alarm molecule to activate innate immune gene pathways in tumor cells. These cellular processes have the potential to harness the innate immune system for cancer immunotherapy.10,11 Therefore, G4 receives great attention as a potentially effective pharmacological target for cancer therapy and provides a great opportunity for drug design towards a new cancer strategy.
000 G4s with different topologies have been identified by G4 sequencing technology (G4-seq).6 Since nuclear expression levels of G4 structures may be higher in tumor cells than in normal cells,4,7 overexpression of protooncogenes with G4 folding is widely found in most types of cancers, such as c-MYC and VEGF, which play crucial roles in tumor cell growth, proliferation, migration and angiogenesis.8,9 Recent studies have shown that G4 damage can trigger genome instability, eventually leading to immune gene activation and related processes in cancer cells because cytoplasmic DNA/RNA can act as an alarm molecule to activate innate immune gene pathways in tumor cells. These cellular processes have the potential to harness the innate immune system for cancer immunotherapy.10,11 Therefore, G4 receives great attention as a potentially effective pharmacological target for cancer therapy and provides a great opportunity for drug design towards a new cancer strategy.
Many organic small molecules have been discovered or designed to recognize and target G4 structures,12–16 such as pyridostatin (PDS) and its analogs,17 phen-DC3 (ref. 18) and BRACO19.19 Metal complexes have been reported to achieve G4 recognition and targeting.20,21 Among all ligands, PDS and its analogs [e.g., PyPDS (4-(2-aminoethoxy)-N2,N6-bis(4-(2-(pyrrolidin-1-yl) ethoxy) quinolin-2-yl) pyridine-2,6-dicarboxamide)] have attracted great attention due to their high specificity and affinity for G4 targeting, which were recently elucidated by us through solution structure analysis of the PDS/PyPDS-G4 complexes.22 The structures indicated that the aromatic scaffold of the ligands adaptively matched with the G-tetrad via π–π stacking for specific structure recognition, and the flexible aliphatic amine side chains interacted with G-tetrad or phosphate backbones via hydrogen bonding and electrostatic interactions to further increase affinity. To date, PDS/PyPDS, as a G4-targeting module, has been widely used in the selective recognition of G4s, cell imaging, proteomics and functional regulation23–33 and provides guidance for the design of antitumor drugs. PyPDS targets G4 structures that can inhibit both DNA and RNA polymerase activity, regulate telomerase function, activate DNA damage response and repress gene expression.27,34,35 In addition, PDS was found to stimulate the cGAS-STING pathway at low doses.36,37 However, the antitumor activity of PDS has not been well explored and appears to be modest.35 Although PDS as a targeting ligand has been applied in various aspects of research, few reports have described the biological response or corresponding antitumor immune response induced by PDS as a G4 targeting module in combination with drugs, especially in combination with clinical cisplatin. Obviously, the G4-guided module enabling traditional chemotherapy drugs has the potential to improve the genome-wide targeting of drugs to improve antitumor efficacy, but there is still a lack of research.
Nucleic acid-binding platinum drugs have achieved great success in clinical cancer therapy. The most representative cisplatin is listed as an essential drug by the World Health Organization and is currently used in chemotherapy regimens in 50% of tumor patients and in combination with other therapies such as immunization.38 Cisplatin itself has particular activity in certain tumors, such as ovarian and BRCA1/2 sensitive breast cancers.39,40 However, the nontargeting of platinum drugs limits their further development, and it needs to be addressed urgently.41 Various platinum complexes targeting biomolecules have been extensively reported, which also include some organic molecules that act as G4 binders coupled to platinum complexes to achieve interactions with G4s, leading to telomere loss and tumor cell death.41–44 However, the targeting of these G4 binders was very limited. Therefore, it is necessary to explore PDS/PyPDS as a G4 targeting module to increase the genome-wide targeting of platinum drugs and enhance the antitumor immune effect.
Based on our previous studies of PDS/PyPDS on G4 interactions and antitumor platinum complexes, we designed and prepared three PyPDS derivatives, cis-[Pt(NH3)2(PyPDS)Cl]NO3 (CP), trans-[Pt(NH3)2(PyPDS)Cl]NO3 (TP) and [Pt(dien)(PyPDS)](NO3)2 (DP), named PyPDSplatins. After various chemical biology experiments and multiomics analyses, we found that PyPDSplatins have a highly enhanced specificity for G4 binding and antitumor effects, especially CP (Scheme 1). CP entered the nucleus, covalently bound to genomic G4s specifically, and strongly inhibited cancer-regulated gene expression, represented by c-MYC and VEGF, thereby inhibiting tumor growth. On the other hand, CP caused G4 DNA damage. The damaged nucleic acid spills into the cytoplasm, finally activating the cGAS-STING and AIM2-ASC dual signaling pathways, resulting in pyroptosis. Finally, we investigated the antitumor immunity of CPin vivo. CP inhibited tumor growth by inducing the release of proinflammatory cytokines such as cGAMP and IL-1β/IL-18, as well as increasing the infiltration of CD8+ and CD4+ T cells. At the same time, the maturation of dendritic cells (DCs) led to the activation of T cells, which effectively inhibited the growth of distal tumors. In summary, we designed G4-guided PyPDSplatins that induced antitumor immune effects by activating the cGAS-STING and AIM2-ASC dual signaling pathways, which provides a new strategy for designing antitumor drugs targeting G4.
 | ||
| Scheme 1 The PyPDSplatins can not only effectively inhibit the expressions of c-MYC and VEGF to inhibit tumor proliferation, but also damage G4 structures to activate the immune-related cGAS-STING pathway and AIM2-ASC-related pyroptosis, triggering a strong immune response and significantly enhancing the therapeutic effect. | ||
Results and discussion
Design and synthesis of PyPDSplatins
The design of PyPDSplatins was based on our recently reported NMR structures of PDS/PyPDS-G4 complexes.22PyPDS is an analog of PDS and has a similar G4 targeting effect. In contrast to PDS, the two pyrrole side chains of PyPDS replace the two amine group side chains, which retain both the electrostatic and hydrogen bonding roles of the amine group but avoid coordination with the metal ions. Therefore, we chose PyPDS rather than PDS as the targeting molecule for G4 in our study. We coupled the primary amine of PyPDS to cisplatin and its analogs to generate a series of G4-targeted platinum complexes, CP, TP and DP, which were named PyPDSplatins (Fig. 1a). These platinum complexes were systematically characterized (Fig. S1–S8†). In the designed CP and TP structures, the chloride ligand is cis and trans to the amino group of the organic ligand, respectively. The Pt–Cl bonds of CP and TP will hydrolyze and can covalently bind to the DNA domain theoretically. For CP and TP, thus, the Pt atom acts as G4 locking, while PyPDS acts as G4 navigation.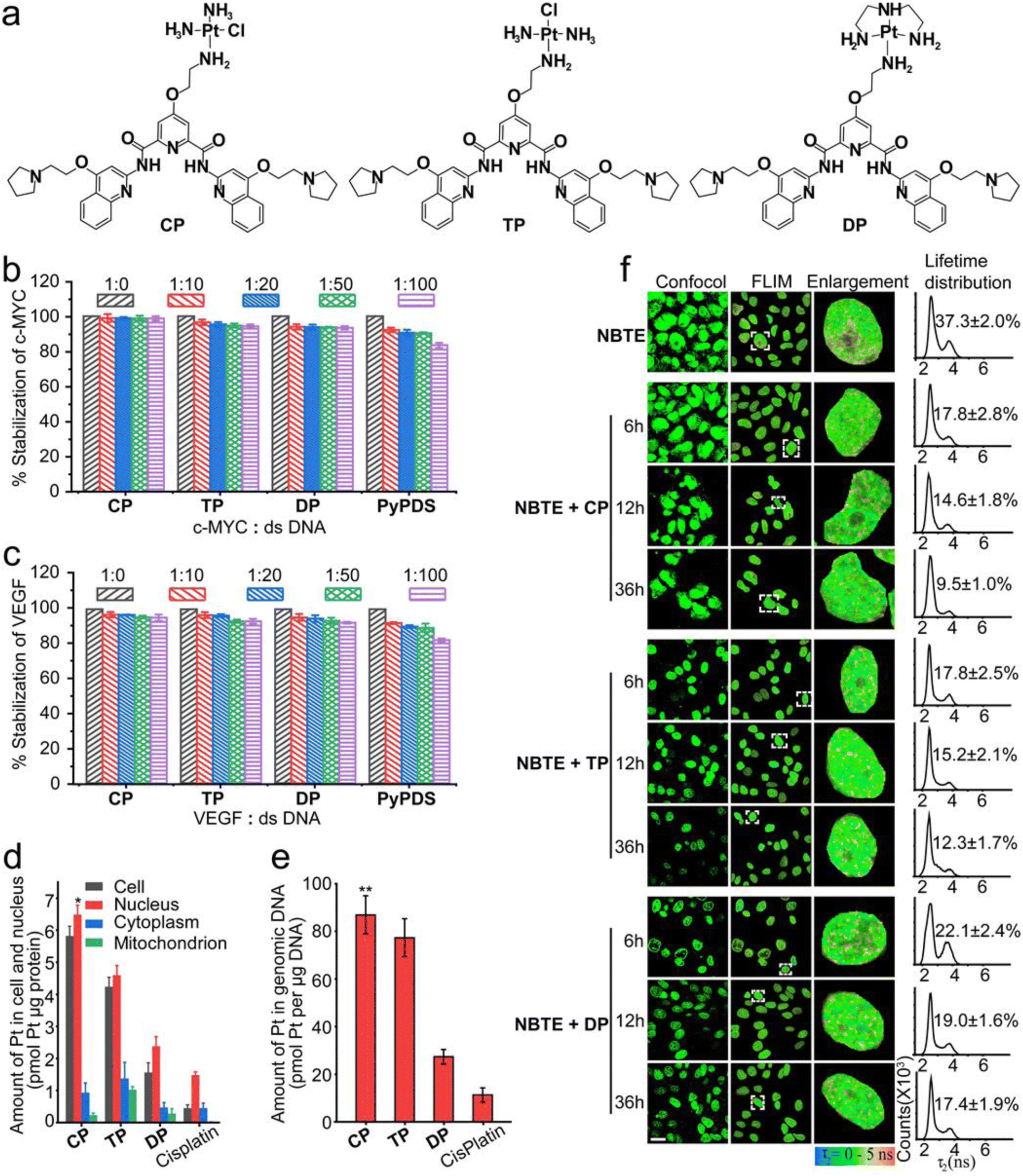 | ||
| Fig. 1 The structure of the PyPDSplatins, the FRET competition experiments and FLIM experiments of live cells incubated with the DNA probe (NBTE) while competing with PyPDSplatins. (a) Chemical structures of PyPDSplatins. (b and c) Thermal stabilization of c-MYC and VEGF with CP, TP, DP and PyPDS while competing with increasing ratios of dsDNA for binding to ligands. After mixing the compound and DNA, the samples were tested immediately. Different proportions of dsDNA (ds-12CT) were added to CP, TP, DP or PyPDS (1600 nM) with c-MYC, VEGF or MYT1L G4 DNA (400 nM) systems for competition. “dsDNA” means “ds-12CT DNA”. Errors are standard deviation (s.d.) with at least three biological replicates (n ≥ 3). (d and e) The amount of Pt in the cell and nucleus and the amount of Pt in the genomic DNA of MDA-MB-231 cells after treatment with different concentrations of CP, TP and DP for 24 h. The compound incubation concentrations were as follows: CP: 6 μM, TP: 6 μM, DP: 9 μM and PyPDS: 9 μM. Errors are s.d. (n ≥ 3). *p < 0.05, **p < 0.01. (f) FLIM experiments of live cells incubated with the DNA probe (NBTE, 20 μM, 24 h) while competing with CP, TP and DP (5 μM) for binding to G4s at 0 h, 6 h, 12 h and 36 h. The results showed that CP, TP and DP could target G4s in live cells. Errors are s.d. (n = 3). Scale bar = 20 μm. | ||
Selectivity and covalent binding to G4 structures
We selected c-MYC, VEGF and MYT1L G4s to study their interactions with PyPDSplatins. The c-MYC and VEGF proto-oncogenes have been proven to be crucial in the malignant progression of many human tumors and are potent immunosuppressive factors in anti-tumor immunity.45–47 MYT1L is a regulating gene in the nerve cell and plays a significant role in mental and neurodegenerative diseases. Studies have shown to identify G4 folding in these genes which can regulate their gene expressions.48 Moreover, we have previously investigated the NMR structure of PyPDS specifically binding to MYT1L G4, revealing design guidance for G4-targeted PyPDSplatins.22 Therefore, studying the bindings of PyPDSplatins to c-MYC, VEGF and MYT1L G4s provides insights into understanding G4-dependent gene regulation as a potential approach for the development of disease therapeutics.To evaluate the potential interactions of CP with c-MYC, VEGF and MYT1L G4s, molecular docking studies were conducted (Fig. S9 and Table S1–3†). In the binding of CP to all c-MYC, VEGF and MYT1L G4s, the aromatic scaffold of CP, which is composed of one central pyridine group, two amide bonds and two quinoline groups, were stacked on top of three bases of the G-tetrad via extensive π–π stacking interactions. Three side chains of one cisplatin group and two pyrrolidine groups extended into the grooves of the G4 structures with hydrogen bonding and electrostatic interactions. These interactions highlighted the potential for the covalent binding of the cisplatin group with the N7 of MYT1L G25 residue, the N7 of VEGF G20 residue, and the N7 of c-MYC G16 residue, showcasing the strong ability of CP to form stable covalent bonds with these G4 structures.
To further investigate the selectivity of three PyPDSplatins (CP, TP and DP) to c-MYC, VEGF and MYT1L G4s (Table S4†), we performed fluorescence resonance energy transfer (FRET) competition experiments. As shown by the FRET competition experiments (Fig. 1b–c and S10†), after PyPDSplatins were added to the fluorescently labeled G4 DNA solution plus 100 equivalents of ds-12CT DNA, the thermal stability of c-MYC, VEGF and MYT1L G4 DNA interacting with CP, TP and DP remained over 93.1 ± 0.3% when immediately tested, and 96.5 ± 0.2% with 48 h incubation time, respectively, which is much higher than the reported range of 60–70% of other platinum complexes.14 The results indicate that these three PyPDSplatins could selectively bind to G4 DNA and dsDNA displays a very limited effect on the binding of PyPDSplatins to G4 structures. The covalent binding of the three platinum complexes to the G4 structure was further investigated by ESI-MS spectrometry. The ESI-MS experiments showed mass peaks with a 1:1 ratio of complexes to three G4s (c-MYC G4, VEGF G4 and MYT1L G4), where CP had a high ion abundance for all G4 complexes and TP had a moderate ion abundance for partial G4 complexes (Fig. S11†). The ion abundance after DP binding to any G4 is not found. And the ESI-MS experiments showed that there was no corresponding ion abundance for the PyPDSplatins and dsDNA complexes. These results indicate that CP and TP are able to form covalent compounds with G4 structures but not duplex DNA, especially CP. The Pt(II) ion in CP and TP would readily react with water (hydrolyze) to lose the chloride ligand and would react with DNA to form a mono-DNA adduct. This kinetically inert adduct would involve a covalent bond between the Pt(II) ion and a nitrogen site on a base, usually guanine.49,50 Since the Pt(II) ion in DP is substitution inert in aqueous, the platinum part of the compound could only interact with anionic DNA via an electrostatic interaction.
We next conducted LC-MS experiments to identify the covalent bound nucleobases after incubation of CP and TP with c-MYC, VEGF and MYT1L G4s. As displayed in the LC-MS spectra, CP, TP reacted mainly with buffer because the total amount of hydrate and NH3 adduct products was the largest in the reaction mixture (Fig. 2), which is consistent with literature reports.24 The detected covalent conjugates of CP, TP and nucleobases were as follows: single adenine adduct with N1 and outer ring NH2, and single guanine adduct with N7 and N1 were minor products in the respective incubation systems of CP with c-MYC and MYT1L G4s (Fig. 2a and c). In the incubation system of CP with VEGF G4 and TP with c-MYC G4, the single guanine adducts with N7 and N1 were minor products (Fig. 2b and d). The covalent binding sites were confirmed by the analysis of the relative base composition of the mixtures before and after treatment with different doses of CP and TP (0.2–5 μM) in HPLC. Dose-dependent covalent binding of adenine and guanine was confirmed by incubation of c-MYC, VEGF, and MYT1L G4s with CP and TP, respectively (Fig. 2e–h). Among them, the incubation of TP with c-MYC G4 and CP with VEGF G4 showed only covalent binding to guanine, while the incubation of CP with c-MYC and VEGF G4s showed covalent binding to guanine and adenine. At the same time, the covalent binding rate of CP to guanine was faster than that to adenine in the system incubated separately with c-MYC and MYT1L G4s. Moreover, CP binds guanine more rapidly than TP, mainly because monofunctional cisplatin has less stereochemical restriction51 than monofunctional trans-platinum when covalently bound to bases, thus confirming the difference in structure-specific reactions. Therefore, as per the results of previous molecular docking (Fig. S9†), guanine in G4s is a common site for CP and TP to achieve covalent binding, which is also consistent with previous reports.24,52,53 It further provided strong evidence that PyPDSplatins affect the biological process of tumor cell-related enriched genes by covalently binding with guanine in G4, and achieving the purpose of anti-tumor.
 | ||
| Fig. 2 The covalent bound nucleobases of c-MYC, VEGF and MYT1L G4s upon CP and TP binding were determined by LC-MS and HPLC experiments. The LC-MS detection of covalent adducts of adenines and guanines in nuclease-digested c-MYC, VEGF and MYT1L G4s after incubation with (a–c) CP, (d) TP for 1 h, respectively. Relative base abundance was determined by HPCL after treatment of c-MYC, VEGF and MYT1L G4s with different doses of (e–g) CP, and (h) TP and nuclease, respectively. Errors are s.d. (n = 3). | ||
Cellular localization of G4 targeting in live cells and antiproliferative activities
To investigate the cellular distribution of these PyPDSplatins, we performed ICP-MS experiments and determined the cellular uptake and distribution of three PyPDSplatins (CP, TP and DP). The three PyPDSplatins were able to enter the cell efficiently, with the highest uptake of CP (Fig. 1d). At the same time, we also detected the content of the three PyPDSplatins in genomic DNA. CP-treated cells had the highest platinum content in genomic DNA, and the localization of CP at low and high concentrations was consistent with the above results (Fig. 1e and S12†), which was also related to the highest accumulation of CP in the nucleus (Fig. 1d).The cellular targeting of the PyPDSplatins was further investigated by fluorescence lifetime imaging microscopy (FLIM) in live cells. We used our recently reported fluorescence lifetime probe, NBTE (4,4′,4′′-(nitrilotris(benzene-4,1-diyl)) tris(1-ethylpyridin-1-ium) iodide),54,55 which can detect whether a certain ligand binds G4 in living cells through FLIM competitive experiments. In the FLIM, the ratio value of the DNA peak area to the total area counting from the lifetime distribution reflects the relative percentage of the photon counts contributed by different DNA. As we can see from the FLIM competitive results (Fig. 1f), the ratio of the G4 peak area decreased from 17.8% to 9.5%, 17.8% to 12.3% and 22. 1% to 17.4% after we added CP, TP and DP, respectively, implying that PyPDSplatins can partially replace NBTE and bind G4 DNAs. Therefore, the FLIM competitive results demonstrated that PyPDSplatins can target G4 structures more than other DNAs in living cells. The more potently the complex binds G4 covalently, the stronger the G4 targeting in living cells.
We investigated the antitumor activities (IC50 values) of PyPDSplatins, PyPDS and cisplatin on different cell lines, including cervical cancer cell (HeLa), breast cancer cell (MDA-MB-231 and Mcf-7), lung cancer cell (A549), ovarian cancer cell (SK-OV-3), BRCA1/2 sensitive breast cancer cell (HCC1937), cisplatin-resistant cell (A549R) and normal cell (Mcf-10A), by MTT assay and indicated in Table 1. The results showed that CP, TP, DP and PyPDS were more toxic to the tested cancer cells than cisplatin. Among them, the cytotoxicity of CP was comparable to TP but higher than that of DP. The cytotoxicity of CP, TP, DP and PyPDS against cisplatin resistant cell line (A549R) was similar to that of non-cisplatin-resistant cancer cell lines. The cytotoxic effect of cisplatin on ovarian cancer SK-OV-3 cell was similar to that of other non-sensitive tumor cells. Notably, CP and TP were more toxic to MDA-MB-231 cells compared to other cells, suggesting that these three PyPDSplatins may have different intracellular mechanisms of action from cisplatin (Table 1).
| CP | TP | DP | PyPDS | Cisplatin | |
|---|---|---|---|---|---|
| a Cells were incubated with the complexes for 48 h and measured by an MTT assay. | |||||
| Hela | 0.62 ± 0.1 | 1.42 ± 0.2 | 3.54 ± 0.2 | 1.25 ± 0.2 | 19.82 ± 1.5 |
| MDA-MB-231 | 0.63 ± 0.1 | 0.69 ± 0.1 | 4.91 ± 0.2 | 0.48 ± 0.1 | 31.35 ± 2.0 |
| Mcf-7 | 2.42 ± 0.3 | 2.23 ± 0.3 | 6.43 ± 0.4 | 1.80 ± 0.6 | 25.50 ± 3.0 |
| A549 | 1.42 ± 0.2 | 1.65 ± 0.3 | 8.98 ± 0.4 | 0.80 ± 0.1 | 26.02 ± 2.2 |
| HCC1937 | 2.06 ± 0.32 | 2.42 ± 0.34 | 6.54 ± 0.46 | 1.61 ± 0.31 | 4.32 ± 0.33 |
| SK-OV-3 | 2.88 ± 0.51 | 3.05 ± 0.08 | 7.28 ± 0.28 | 2.10 ± 0.30 | 32.30 ± 2.86 |
| A549R | 1.51 ± 0.1 | 1.62 ± 0.2 | 7.85 ± 0.4 | 1.61 ± 0.2 | 126.20 ± 2.2 |
| 4T1 | 1.65 ± 0.1 | 2.40 ± 0.1 | 3.35 ± 0.5 | 2.22 ± 0.1 | 14.70 ± 2.1 |
| Mcf-10A | 1.41 ± 0.2 | 2.64 ± 0.5 | 2.28 ± 0.3 | 2.10 ± 0.5 | 25.31 ± 1.5 |
G4-associated genomic and pathway enrichments of PyPDSplatins on the whole genome
We further investigated the genomic effects of PyPDSplatins at the cellular level. Based on whole-genome sequencing, we defined a deletion site containing a (GnNx)4 (n ≥ 2, n stands for any base) fragment as a G4-mediated deletion, named G4Del, compared with the control group. We systematically analyzed the genomic distribution and biological pathway enrichments of the PyPDSplatin targets associated with G4Del. Chromosomal distribution analysis revealed that G4 sites are strongly enriched in multiple chromosomes, including chr16, chr17, chr19, chr20, and chr21, and are depleted for most of the other chromosomes (Fig. 3a and S13a†). By analyzing the location of G4Del, we found that the deletion sites were mainly distributed in the pseudogene and promoter regions of the PyPDSplatin treatment groups (Fig. 3b), in line with the fact that gene promoter is the major genome region where the G4 structure exists.2 We next studied the biological pathways underlying PyPDSplatin-targeting genes by carrying out a gene ontology (GO) analysis for each group of the G4 deletion sites (Fig. S13b†). Intriguingly, we observed strong and specific enrichments of immune system processes and immune response for the genes associated with CP-bound G4 deletions, suggesting that CP might have effective antitumor immune activity. As a comparison, G4 deletions induced by other PyPDSplatins are preferentially enriched in the genes relevant to several other pathways, such as small molecule and anion binding for TP, biosynthetic processes for DP, and organelle organization and macromolecule metabolic process for PyPDS. Collectively, the PyPDSplatin-mediated deletions of G4 sites are involved in the regulation of fundamental biological processes. | ||
| Fig. 3 The effect of G4 deletion mediated by PyPDSplatins and PyPDS. (a) Distribution on chromosomes, (b) HOMER annotation, (c) distribution on genes, (d) schematic diagram of (G2Nx)4 G4 containing two G tetrads and (G≥3Nx)4 G4 containing three or more G tetrads, (e) histogram, (f and g) the mRNA transcription levels of MYC and VEGF were inhibited after CP, TP, DP and PyPDS treatment, as shown by RT-qPCR in the MDA-MB-231 cell lines. Errors are s.d. (n ≥ 3). ***p < 0.001. (h) Western blot analysis showing the impact of compounds on the expression of the indicated proteins. Cells were treated with compounds for 24 h. The un-cropped western blotting image is shown in Fig. S25.† | ||
To understand the potential biological effects caused by the PyPDSplatin-mediated deletion of G4, we assigned each G4 to its nearest gene given the overall proximal regulatory mode in the genome (Table S5†). We found that the majority of genes were associated with only one G4 deletion in CP, TP, DP and PyPDS treatments (Fig. 3c). For CP, the majority of 382 genes have one G4Del site, 40 genes have two G4Del sites, 6 genes have three G4Del sites and 2 genes have four G4Del sites. The quadruplex forming G-rich sequences (QGRS) in the significant enrichment genes in the CP group were also analyzed (Table S6†). Among these candidate target genes, the CDH4 gene, a potential biomarker related to breast cancer progression and metastasis,56 has the most QGRS of 6129 in full gene sequence and 17 QGRS in promoter sequence, suggesting that CP might have particular effects on breast cancer through G4-targeting. And the c-MYC and VEGF genes we studied here were also enriched in the immune response pathway. The QGRS in the full gene sequence of c-MYC and VEGF is 74 and 207 and in promoter sequence is 17 and 24 (Table S6†).
The number of genes associated with (G≥3Nx)4 G4s which contain three or more G-tetrads and (G2Nx)4 G4s which contain two G-tetrads was calculated (Fig. 3d). As shown in the figure, the number of G4Del genes in the PyPDSplatin-treated MDA-MB-231 cells were 715, 693 and 696, respectively (Fig. 3e). The number of shared G4Del genes was 312 (Fig. S14a†). Compared with the reported NDIplatins (NDI, naphthalene-1,4:5,8-bis(dicarboximide)),57 the deletion sites of both systems remained essentially consistent, but the number of total G4Del genes in PyPDSplatins increased by 26.2–56.1%, implying that the G4 targeting of PyPDS plays an important role. Moreover, the occupancy ratio of (G≥3Nx)4 G4Del in the total number of G4Del genes was 24.3–25.7% and 16.0–17.7% for PyPDSplatins and NDIplatins, respectively, indicating that PyPDSplatins have a stronger regulatory ability for higher order G4 structures.
In the CP group, 28 genes with the highest proportion of G4 deletions were selected by whole-genome sequencing (Table S7†), including 11 genes (39.3%) related to tumor growth inhibition, such as c-MYC and VEGF, and 7 genes (25%) related to antitumor immunity, such as MYO10, which induces the cGAS-STING pathway (Fig. S14b†).58 The results suggest that CP-induced G4 deletion may play a major role in the antitumor effect. To investigate the antitumor effect of G4 gene deletion caused by PyPDSplatins, we chose the c-MYC and VEGF genes as examples for qRT-PCR and western blot experiments. As shown in the results, CP can inhibit the transcription of the c-MYC and VEGF genes (Fig. 3f–h). This may be related to more G4 structures in cancer cells. These results indicate that CP is an effective inhibitor of c-MYC and VEGF, which may inhibit the mRNA transcriptional levels and then downregulate the protein expression of the c-MYC and VEGF genes through the binding and stabilization of the c-MYC and VEGF promoter G4 structures (Fig. 3f–h), finally repressing the proliferation of tumor cells and tumor angiogenesis to achieve the goal of inhibiting tumor growth.55,59
Effects of PyPDSplatins on gene transcription and G4 damage
To investigate the effects of these three PyPDSplatins on gene expression patterns, we performed RNA sequencing (RNA-Seq) with MDA-MB-231 cells (Fig. S15–S22†). Gene Ontology (GO) enrichment RNA-Seq analysis showed that PyPDSplatins mainly affected the regulation of transcription from RNA, protein metabolic process, endomembrane system, nucleolus, transcription factor activity, and RNA binding (Fig. S21†). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of RNA-seq revealed that PyPDSplatins mainly affected pathways in cancer, cytosolic DNA sensing, and immunity (Fig. S22†). Among them, CP has a prominent effect on immune-related pathways, including the interleukin-7 (IL-7) signaling pathway, tumor necrosis factor (TNF) signaling pathway, and nuclear factor-κb (NF-κB) signaling pathway. Since traditional enrichment analysis can only map to function, that is, to answer which functions these differential genes are related to but not the overall manifestation of a pathway, we need to conduct gene set enrichment analysis (GSEA).By RNA-seq studies, we listed the 20 top down-regulated genes and found their number of potential G4 structure formations (Table S8†). Among the genes down-regulated by CP, the CDH4 gene, TACC2 gene, c-MYC gene and VEGF gene, had the most significant effects. Studies have shown that TACC2 plays a cancer-promoting role in breast cancer by promoting cell proliferation activity. Residual TACC2-positive breast cancer cells after surgery still have a high potential for rapid growth and/or metastasis despite adjuvant therapy.60 Therefore, the significant down-regulation of the TACC2 gene mediated by CP can help to inhibit the proliferation and metastasis of breast cancer cells, and positively improve the prognosis of breast cancer after surgery. Meanwhile, c-MYC and VEGF are potent immunosuppressive factors in anti-tumor immunity. The formation of G4 structures in the promoter regions of c-MYC and VEGF can inhibit their gene transcription and expression in vivo.45–47 Therefore, the significant down-regulation of c-MYC and VEGF gene mediated by CP may help to inhibit tumor growth and achieve the purpose of anti-tumor. Among the TP-mediated down-regulated genes, the KANK1 gene had the most G4 content. Studies have shown that the KANK1 gene is positively correlated with the clinical treatment outcome of tumors and is involved in tumor growth and migration. Therefore, TP down-regulating the KANK1 gene is beneficial to anticancer therapy.61
Then we further studied the up-regulated genes. GSEA showed that G4 DNA binding and G4 DNA unwinding were significantly upregulated in the CP treatment group compared with the DNA damage response, dsDNA break repair, and DNA repair groups, indicating that CP selectively binds to G4 in cells to cause G4 damage (Fig. 4a–e). The G4-DNA and dsDNA damage induced by PyPDSplatins were further investigated using immunofluorescence co-localization analysis and western blot analysis, respectively. As displayed in Fig. 4f, the colocalization of γH2AX and BG4 (a specific antibody to G4 structures62) foci indicated that three PyPDSplatins can damage G4 structures, especially CP. The expression of phosphorylated histone H2AX (γH2AX) in Fig. 4g shows that three PyPDSplatins induce double-strand DNA damage, showing a similar trend. The ability of PyPDSplatins to induce significant DNA damage is associated with their DNA reactivity. DNA damage may cause abnormal leakage of nuclear DNA into the cytoplasm. Therefore, we used a HeLa cell line with mitochondrial DNA deletion (HeLa ρ0) for further confirmation. Upon treatment with PyPDSplatins, DNA release into the cytoplasm and chromatin shrinkage were clearly detected in cells (Fig. 4h). Our results verify that three PyPDSplatins cause genetic damage, leading to the release of ds DNA into the cytoplasm. Thus, PyPDSplatins might exert genome-wide effects by DNA damage, leading to activation of an immune response through possible STING signaling.36
 | ||
| Fig. 4 The effect of gene transcription and G4 damage mediated by PyPDSplatins and PyPDS. (a–e) GSEA reveals positive enrichment of genes altered in cells subjected to CP, TP, DP and PyPDS treatment. (f) Immunofluorescence study of γH2AX and BG4 antibodies causing nuclear G4 damage in MDA-MB-231 cells. White or yellow in the center of the magnification represents G4 damage, and cyan represents undamaged G4. The compound incubation concentrations were CP: 3 μM, TP: 3 μM, DP: 6 μM, PyPDS: 3 μM and cisplatin: 3 μM. Scale bar = 5 μm. (g) Western blot analysis showing the impact of compounds on the expression of the indicated proteins. Cells were treated with compounds for 24 h. The un-cropped western blotting image is shown in Fig. S26.† (h) Cytosolic dsDNA studies of HeLa ρ0 cells treated with compounds and PicoGreen staining. Scale bar = 20 μm. | ||
Pyroptosis and signaling pathways caused by PyPDSplatins
In addition to the DNA damage response and DNA repair, GSEA showed that pyroptosis was upregulated in PyPDSplatins (Fig. 5b). The epithelial–mesenchymal transition and the p53 pathway that mediate pyroptosis were also observed in the GSEA (Fig. S23†). Meanwhile, scanning electron microscopy showed that the cells treated with CP showed typical characteristics of pyroptosis, including cell membrane rupture and the formation of a large number of vesicles (Fig. 5a). We used several specific inhibitors to further confirm the mode of cell death induced by PyPDSplatins (Fig. S24†), including necrostatin-1 (Nec-1, necrosulfonamide); NSA (an inhibitor of programmed necrosis and pyroptosis) attenuated cell death. Moreover, western blotting showed that the expression level of GSDMD-N63,64 was significantly increased in cells subjected to CP/TP (Fig. 5g). These results indicate that CP and TP mainly induce pyroptosis.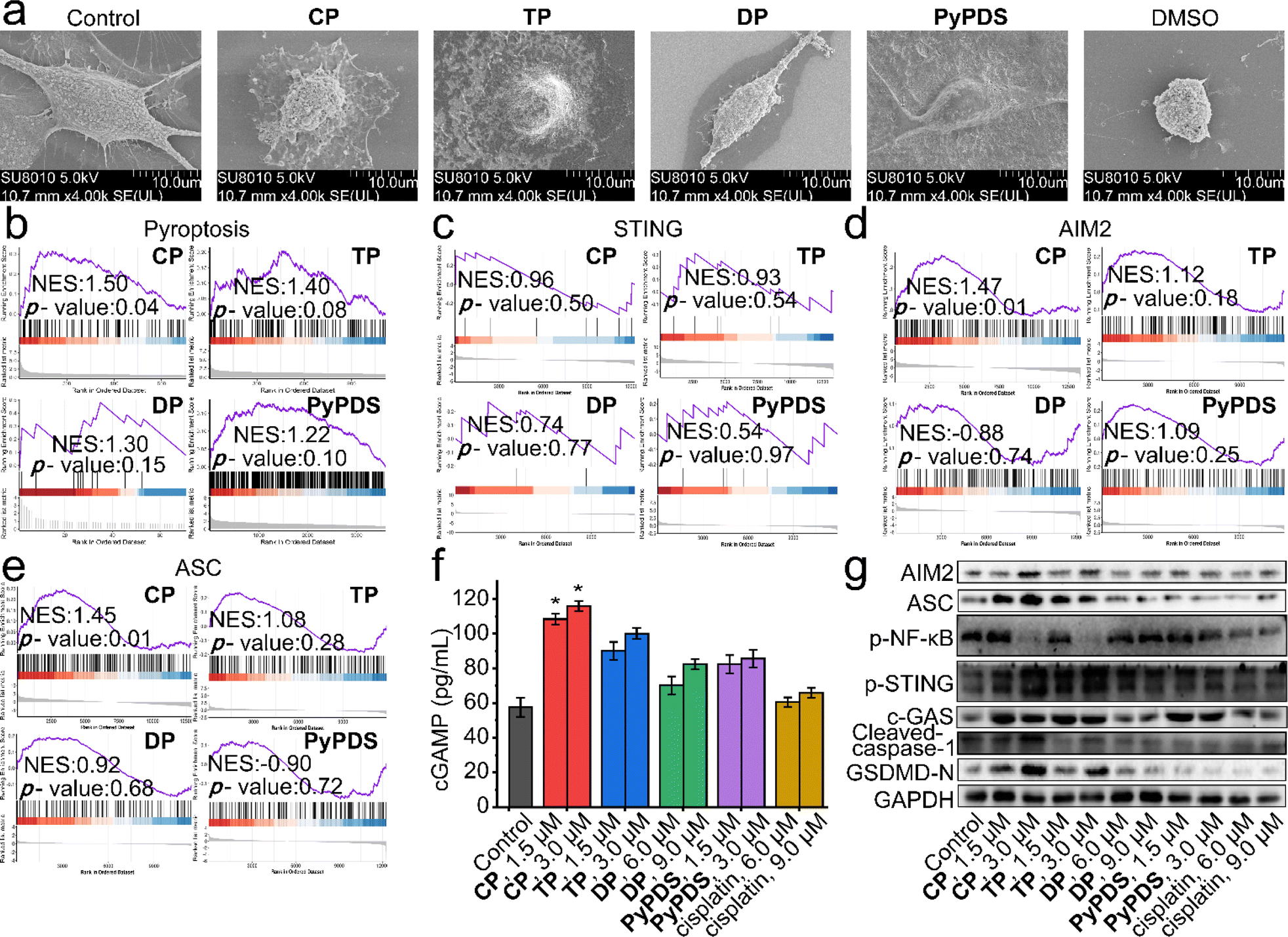 | ||
| Fig. 5 The AIM2-ASC and cGAS-STING signaling pathways activated by PyPDSplatins and PyPDS. (a) SEM images of dying MDA-MB-231 cells. DMSO, CP, TP, DP and PyPDS treatment resulted in apoptosis and pyroptosis, respectively. Imaged by scanning electron microscopy at 5.0 kV. (b–e) GSEA reveals positive enrichment of genes altered in cells subjected to CP, TP, DP and PyPDS treatment. (f) ELISA quantification of cGAMP in lysates of MDA-MB-231 cells subjected to compounds. Errors are s.d. (n ≥ 3). *p < 0.05. (g) Western blot analysis showing the impact of compounds on the expression of the indicated proteins. Cells were treated with compounds for 24 h. The un-cropped western blotting image is shown in Fig. S27.† | ||
The signaling pathways leading to pyroptosis were analyzed using GSEA, which showed that the cGAS-STING and AIM2-ASC signaling pathways play crucial roles in the antitumor immune response and antipathogen host defense. The activation of cytosolic DNA sensors cGAS and AIM2 promotes the production of the downstream cGAMP and the binding of AIM2 to ASC to form inflammasomes, respectively. STING activation leads to the production of immune factors such as the type I interferon signaling cascade (Fig. 5c), and next, caspase-1 precursors are cleaved to activated caspase-1, inducing the production of proinflammatory cytokines. The cGAMP levels of CP- and TP-treated cells were increased by approximately 1.8-fold and 1.5-fold, respectively, compared with those of control and cisplatin-treated cells (Fig. 5f). Thus, CP- and TP-treated cells showed increased expression of cGAS and phosphorylated STING (p-STING) and decreased expression of phosphorylated NF-κB (p-NF-κB) compared with control cells (Fig. 5g). Cisplatin had no significant effect on the expression of cGAMP, cGAS, p-STING or p-NF-κB. The results indicated that CP and TP can significantly activate cGAS-STING signaling pathways, being different from the reported NDIplatins.57
In addition, pyroptosis is critical to immune processes by promoting inflammation.65–69 Western blotting experiments showed that after incubation with CP and TP, the expression levels of AIM2, ASC, cleaved caspase-1, and GSDMD-N, a protein that oligomerizes to form pores in the plasma membrane and increases membrane permeability during pyroptosis, were significantly increased in the cells (Fig. 5d–g). The results indicated that CP and TP can significantly activate AIM2-ASC signaling pathways to produce proinflammatory cytokines and eventually lead to pyroptosis. The reason why this is different from the antitumor immune signaling pathway of NDIplatins is that the number of G4 deletions caused by PyPDSplatins is much higher than the number of G4 deletions caused by NDIplatins,57 mainly because CP and TP have higher binding ability and selectivity to G4s in vivo than NDIplatins. In turn, it causes damage to a wider range of G4-related genes, ultimately activating different antitumor immune-related pathways.
Immunogenic cell death caused by PyPDSplatins
CP and TP treatments can affect immune-related pathways and activate the cGAS-STING and AIM2-ASC pathways. Interestingly, the immune response was upregulated after treatment with PyPDSplatins, as shown by GSEA (Fig. 6a). These findings suggest that the synergistic behaviors between platinum complexes and DNA play important roles in the immune response and underscore a crucial need for further research on their potential use in cancer therapy. Therefore, we further evaluated their ability to induce immunogenic cell death (ICD) in vitro. ICD of tumor cells is triggered by the release of damp-associated molecular patterns (DAMPs), namely, calreticulin (CRT), high-mobility group protein B1 (HMGB1), adenosine triphosphate (ATP) and proinflammatory cytokines (i.e., interleukins IL-1β, IL-6, and IL-8). ICD inducers not only directly kill tumor cells but also enhance tumor immunogenicity and induce antitumor immune responses.70 | ||
| Fig. 6 Immunogenic cell death caused by PyPDSplatins and PyPDSin vitro. (a) GSEA reveals positive enrichment of genes altered in cells subjected to CP, TP, DP and PyPDS treatment. (b) Confocal images of HMGB1 and CRT in MDA-MB-231 cells treated with CP, TP, DP and PyPDS. Scale bar = 20 μm. (c) Flow cytometric analysis of HMGB1 and ecto-CRT in MDA-MB-231 cells treated with CP, TP, DP and PyPDS. (d) ATP levels in cell culture supernatants after compound treatment. Errors are s.d. (n ≥ 3). **p < 0.01, ***p < 0.001. | ||
Immunofluorescence imaging and flow cytometry experiments were used to detect the effect of ICD (Fig. 6b–c). The results showed that the fluorescence of HMGB1 in cells treated with CP and TP decreased significantly, suggesting that intracellular HMGB1 was secreted out of the cells. CRT showed fluorescence on the cell surface after CP and TP treatments, indicating that CRT was exposed to the cell surface. The results of flow cytometry showed that the intracellular fluorescence of HMGB1 decreased and CRT increased, respectively, in a concentration-dependent manner after CP and TP treatment. Moreover, the extracellular ATP concentration of CP-treated cells also increased most significantly in a concentration-dependent manner (Fig. 6d), indicating that CP most significantly caused ICD. In contrast, DP and PyPDS had a weaker effect on ICD.
Cancer immune response in vivo
To investigate their antitumor effects and potential to enhance immune responses in vivo, we used an immunocompetent mouse model carrying a homologous 4T1 tumor instead of human MDA-MB-231. A BALB/c mouse model bearing bilateral 4T1 tumors was established, with the primary tumor inoculated on the right side and the distant tumor inoculated on the left side (Fig. 7a). They were randomly assigned to 4 groups (n = 5) in which primary tumors received compound treatment (CP, cisplatin: 1.5 mg Pt kg; PyPDS: 5.9 mg kg−1),71 whereas distal tumors received no compound treatment. The compounds were administered every 3 days (on days 3, 6, 9, 12, 15) after intratumoral injection of a low dose (CP, cisplatin: 1.5 mg Pt kg; PyPDS: 5.9 mg kg−1). Cisplatin and PyPDS groups were set as the control group. The volumes of primary and distant tumors were measured every 2 days. On day 16, all mice were sacrificed, and tumors were removed. As shown in Fig. 7b–d, the inhibitory effect of CP was stronger than that of PyPDS and cisplatin on primary tumors. At the same time, the untreated distant tumor growth rate was also monitored, and the results showed that distant tumor growth was significantly inhibited in the CP group compared with the control group, and there was no regrowth at day 24 (Fig. S28a and b†). However, there was no significant inhibition in the cisplatin group and PyPDS group. At the end of treatment, tumor suppression rates were 68%, 51%, and 31% in the CP, PyPDS and cisplatin groups, respectively. This favorable abnormal effect indicates that CP not only has a good antitumor effect but also has the ability to activate antitumor immunity. Meanwhile, the body weight of mice in the CP group remained stable during the treatment (Fig. 7e), and there was no organ damage (Fig. S29†). This indicates that cells treated with G4-guided PyPDS cisplatin CP have a lower potential for systemic toxicity. | ||
| Fig. 7 Antitumor efficacy and antitumor immune response caused by CP, PyPDS and cisplatin in vivo. (a) In vivo therapeutic protocol using a bilateral model. (b) Representative photographs of mice at 16 days before sacrifice. (c) Volume changes of primary and distant tumors (n = 4). Errors are s.d. (n ≥ 3). (d) Photographs of the tumor collected at the end of the therapeutic period. (e) The weight of the mice during the treatment. Errors are s.d. (n ≥ 3). (f and g) Expression of CD3+ and CD4+, CD3+ and CD8+, and CD80+ and CD86+ T cells quantitatively detected by flow cytometry in distant tumors (n = 3). Errors are s.d. (n ≥ 3). (h) The secretion of cytokines (IL-6, IL-12p70, TNF-α, IFN-β) in serum on day 16. Errors are s.d. (n ≥ 3). *p < 0.05, **p < 0.01, ***p < 0.001. | ||
CD4+ T cells are also involved in the immune response and can assist cytotoxic T cells (CTLs). As shown in the results (Fig. 6f and g), in the CP-treated group, the percentage of CD3+CD4+ T cells was 46.6%, which was significantly higher than that in the PyPDS-treated group (18.9%), cisplatin-treated group (0.95%), and control group (0.77%) (Fig. 7f and g). In addition, the presence of CD8+ T cells in the tumor is considered to be a marker of the T-cell-mediated immune response and is strongly related to the prognosis of the tumor. CP treatment increased the proportion of CD3+CD8+ T cells to 47.2%, which was higher than that in the PyPDS (20.4%), cisplatin treatment (0.58%), and control groups (0.23%).
DCs are antigen-presenting cells (APCs). Mature DCs (CD86+CD80+ cells) can efficiently activate T cells and play an important role in the initiation, regulation and maintenance of the immune response. Compared with the control group (0.28%), the proportion of CD86+CD80+ cells increased to 46.3% in the CP treatment group, which was higher than that in the PyPDS (17.1%) and cisplatin treatment groups (0.76%) (Fig. 7f and g). The secretion of cytokines has a significant effect on the activation and regulation of the immune response. The proinflammatory cytokines interleukin-6 (IL-6), interleukin-12/P70 (IL-12/P70), tumor necrosis factor-α (TNF-α), and interferon IFN β were elevated in serum (Fig. 7h). These results suggest that CP can induce a robust immune response in vivo. The activation of cGAMP has an important effect on the cGAS-STING pathway, and cisplatin has the greatest effect on cGAMP levels compared with PyPDS in the CP-treated group. These results suggest that CP induces a strong cancer immune response in vivo by activating the cGAS-STING pathway (Fig. S28c–e†). At the same time, serum levels of IL-18 and IL-1β, inflammasomes associated with pyroptosis, were also increased, suggesting that pyroptosis occurred in the CP group (Fig. S29b†). These in vivo results indicate that CP could induce tumor pyroptosis and an adaptive immune response, which had a good antitumor immunotherapy effect.
Conclusion
After conjugation to PyPDS, the chemical biological properties and antitumor effects of PyPDSplatins were significantly improved. Compared with cisplatin, CP selectively targets G4 rather than dsDNA with a high cellular uptake, which probably benefits from the lipid solubility of PyPDS and protonation of amine groups.35 Increased targeting and uptake also promoted a significantly enhanced antitumor effect of PyPDSplatins, ∼10-fold higher than that of cisplatin. More importantly, CP has a different mechanism of action from cisplatin,72 which can regulate the G4 gene to inhibit tumor growth, especially by activating cell immunity. There are several signaling pathways that are different from cisplatin, for example, the cGAS-STING and AIM2-ASC signaling pathways that ultimately cause tumor pyroptosis, which is different from tumor cell apoptosis caused by cisplatin. The main reason is that after CP-induced G4 damage in breast cancer cells, the damaged G4 sprays into the cytoplasm and is recognized by cytoplasmic DNA receptors cGAS and AIM2 inflammasomes. Therefore, low-dose CP can activate the cGAS-STING and AIM2-ASC signaling pathways and kill tumor cells without light, making it more genetically safe than exogenous dsDNA. Compared with other targeted-modified cisplatin,20 both parts of PyPDSplatins act on the same nucleic acid, forming a resultant binding force, which can greatly improve the affinity of nucleic acid and antitumor efficiency. Other targeted-modified platinum complexes either act on different types of biomolecules or are taken up together and then dissociate and act on different targets separately, which is common in Pt(IV) prodrugs.73–75 Because PyPDS is able to lock G4 targeting by covalent binding of platinum and base, it greatly enhances the selectivity of CP for G4 nucleic acids, further reducing its toxicity and improving safety.Compared with our reported NDIplatins,57 PyPDSplatins bind extensive G4 structures with higher selectivity in vivo, implying that the G4 targeting of PyPDSplatins plays a more important role. After the study, we found that the number and sequence features of G4-associated deletion genes in PyPDSplatins and NDIplatins are significantly different. The number of G4-associated deletion genes in PyPDSplatins is 26.2–56.1% higher than that in NDIplatins, which causes damage to a wider range of genes. And the occupancy ratio of deletion higher-order G4 forming sequences in PyPDSplatins is 1.5 times that in NDIplatins, indicating that PyPDSplatins have a stronger regulatory ability for higher order G4 structures. Thus, the differences in binding selectivity, structure and number of G4 targeted by PyPDSplatins and NDIplatins ultimately activate different antitumor immune-related signaling pathways.
In summary, CP generated by PyPDS conjugated to cisplatin was found to display stronger specificity for G4 structures even in the presence of large amounts of dsDNA extracellularly or intracellularly. We found that CP can not only decrease the mRNA transcription level in a cluster of oncogenes involving G4 folding, such as c-MYC and VEGF, which then downregulates their protein expressions and finally inhibits tumor proliferation, but also damage G4 structures to activate the immune-related cGAS-STING pathway and AIM2-ASC-related pyroptosis, which causes immunogenic cell death and increases cytotoxic T cells in tumors, ultimately triggering a strong immune response and significantly enhancing the therapeutic effect. This is the first study to demonstrate that PyPDS binds covalently to platinum to achieve tumor growth inhibition. The PyPDS in the CP acts as G4 navigation, while the platinum unit acts as G4 locking through covalent binding of platinum and DNA. Therefore, the classical structure–activity relationship combination and the coexistence of covalent and noncovalent interactions can be fully considered in the design of anticancer drugs, which can improve the selectivity and efficacy of platinum drugs and reduce side effects. The combination of chemotherapy and immunotherapy with G4-guided cisplatin achieves more extensive and efficient antitumor therapy, which opens up a way for the study of traditional platinum drugs to enhance efficacy by coupling G4-guided modules.
Experimental methods
DNA sample preparation
The oligonucleotides were purchased from Sangon Biotechnology (China) and purified by HPLC. The DNA sequences used in this study are shown in Table S4.† The G-quadruplex (G4) structure was formed by heating at 95 °C for 5 min, followed by slow cooling in buffer to room temperature overnight. The DNA concentration was measured using a Nanodrop 200/200c (Thermo Science).Molecular docking
DNA preparation: the structure of the DNA–ligand complex was downloaded from RCSB Protein Data Bank (PDB code 6JJ0 for c-MYC G4 DNA, PDB code 6LNZ for VEGF G4 DNA, and PDB code 7X2Z for MYT1L G4 DNA). The original ligand was removed before docking. The docking calculations were performed with the AutoDock 4 suite of programs,76 using a ligand flexible docking approach that allows ligand flexibility. The Lamarckian genetic algorithm77 was chosen as the search protocol using the default parameters except for the number of GA runs (ga_run = 50) and the maximum number of energy evaluations (ga_num_evals = 2500000). The displayed images were rendered with PyMOL.78Fluorescence resonance energy transfer (FRET) melting assay
DNA was fluorescently labeled with FAM (6-carboxyfluorescein) at the 5′-end and TAMRA (6-carboxytetramethylrhodamine) at the 3′-end for FRET melting assays. The DNA sequences used are shown in Table S4.† Fluorescent DNA samples were prepared in 25 mM potassium phosphate and 70 mM KCl buffer (pH 7.4). For competition experiments, different proportions of dsDNA (ds-12CT) were added to CP, TP, DP or PyPDS (1600 nM) with c-MYC, VEGF or MYT1L G4 DNA (400 nM) systems for competition. The samples were incubated for 0 h or 48 h. Fluorescent melting curves were obtained using a Roche Light Cycler 2.0 real-time PCR instrument with an excitation wavelength of 470 nm and a detection wavelength of 530 nm. The data were read in the range from 37 to 99 °C once every 1 °C rise, and a constant temperature was maintained for 30 s before each reading.Mass spectrometry
c-MYC, VEGF, MYT1L G4 DNA and dsDNA were prepared with 40 mM ammonium acetate buffer (pH 7.4). The concentration of DNA was 30 μM. CP, TP, and DP were added. The mixture was incubated for 48 h at 37 °C and centrifuged with an AMICON ULTRA 0.5ML 3K 96PK, and the filtrate was collected. A German LTQ Orbitrap Elite mass spectrometer was used for negative spray ionization mass spectrometry.LC-MS analysis
All DNA oligonucleotides used in this study are shown in Table S4.† The oligonucleotides were annealed at a concentration of 400 nM, in 100 mM KCl, Tris.HCl buffer, pH 7.4, by heating at 94 °C for 10 min followed by slow cooling to rt at a controlled rate of 0.1 °C min−1. The annealed oligonucleotides were incubated with different doses of CP and TP in the same buffer at 37 °C for 1 h. Incubated DNA was nuclease digested by following a literature protocol,24 purified with Amicon Ultra 0.5 mL 3 kDa columns and analysed using an Agilent Technologies 1290 Infinity II 6545 Q-TOF LC/MS. The system was equipped with an Eclipse Agilent column EC-C18 3.0 × 150 mm. The column temperature was maintained at 45 °C. Eluting buffers were buffer A (500 mM ammonium acetate (Fisher) pH 5), buffer B (acetonitrile) and buffer C (H2O). Buffer A was held at 1% throughout the whole run and the gradient for the remaining buffers was 0 min – 5% B, 20 min – 50% B, 40 min – 80% B, 45 min – 95% B.Fluorescence lifetime imaging microscopy (FLIM)
FLIM imaging was performed using a Zeiss LSM 880 NLO multiphoton microscope equipped with a bh TCSPC FLIM system (Becker & Hickl GmbH, Berlin, Germany). Samples were two-photon excited using a femtosecond Ti:sapphire laser (Coherent Chamole), and emitted light was collected using a Zeiss BiG-2 GaAsP detector. The TCSPC module type is the SPC-150 module, and the image resolution is 512 × 512 pixels. The G4 probe NBTE (20 μM) was incubated with HeLa cells for 18 h at 37 °C. They were then further incubated with CP, TP or DP (5 μM) at 37 °C for 0 h, 6 h, 12 h or 36 h. Then, they were washed 3 times with ice-cold PBS buffer and detected with the FLIM system. The two-photon excitation wavelength of NBTE was 810 nm, and the emission was recorded from 545 nm to 590 nm. The acquisition time was sufficient, and the signal-to-noise ratio was good. Lifetime data were fitted to a three-exponential function for each pixel using SPC-Image software. For lifespan distributions, approximately 100 cells were analyzed in each sample. Images are representative of repeated experiments (n = 3). The percentage of G4 peaks was calculated by dividing the G4 peak area (3.0 to 5.0 ns) by the total area (0 to 5.0 ns).Platinum level in cells and genomic DNA
MDA-MB-231 cells were incubated for 24 h in a 10 cm dish. To observe the accumulation of platinum complexes in cells, after 24 h of treatment with the indicated concentrations of CP, TP, DP, and cisplatin, cells were harvested and washed three times with PBS. Nuclei were isolated using a nuclear protein extraction kit (C500009, Sangon Biotechnology, China) according to the manufacturer's instructions. Mitochondria were isolated using a mitochondrial protein extraction kit (20128ES50, YEASEN). The cells or nuclei were lysed with radioimmunoprecipitation (RIPA) buffer, and the protein concentration was determined by the bicinchoninic acid (BCA) method. For platinum levels in genomic DNA, genomic DNA was extracted using a GeneJET Genomic DNA Purification Kit (Thermo Fisher) after 24 h of CP, TP, DP and cisplatin treatment. The DNA concentration was determined by using a Nanodrop 200/200c (Thermo Science). The mixture of total cell, nuclear, mitochondrial, cytoplasmic or genomic DNA was digested in 400 μL 60% HNO3 solution at 60 °C for 1 h and diluted to 10 mL with Milli-Q water (containing 10 ppb platinum as an internal standard), and the platinum concentration was determined using an XSERIES 2 ICP-MS.Whole genome sequencing, transcriptome sequencing and data study
MDA-MB-231 cells were incubated in 10 cm dishes for 24 h and then treated with CP (3.0 μM), TP (3.0 μM), DP (9.0 μM) and PyPDS (3.0 μM) for 24 h. DNA was collected for whole-genome sequencing. Total RNA was extracted with the TRIzol reagent (Life Technologies, USA), and mRNA was purified with poly-T oligo-attached magnetic beads and used for RNA-Seq. Sequencing was performed by Biomarker Technologies.Whole-genome sequencing was performed according to standard procedures provided by Illumina. Raw reads were assessed for quality and filtered to obtain clean reads for downstream bioinformatics analysis. Sequencing reads were relocated to the human reference genome using BWA software. A deletion containing a fragment of (GnNx)4 (n≥2, n denotes any base) was defined as a G4-mediated gene deletion and named G4Del compared to the control group.
For RNA-seq, libraries were sequenced on an Illumina NovaSeq platform to produce 150 bp paired-end reads. For quality control, Q20, Q30, GC content, and sequence repeat levels of clean data were calculated, and high-quality clean data were used for downstream bioinformatics analysis. The human genome (Homo_sapiens. GRCh38_release95. genome.fa) was used as the reference genome sequence and mapped using HISA2T. Gene expression levels were estimated from fragments per kilobase of transcript per million fragments mapped. DESeq2 was used to analyze differentially expressed genes (DEGs) between the two groups. DEGs were assigned as genes with a corrected p value ≤ 0.01 and fold change ≥2 found by DESeq2.
Immunofluorescence (IF) staining
IF staining was performed as reported in the literature.60 Briefly, MDA-MB-231 cells were seeded into 96-well plates, incubated for 24 h, treated with the indicated concentrations of CP, TP, DP, PyPDS and cisplatin for 24 h and washed three times with precooled PBS buffer. Cells were then fixed with 4% paraformaldehyde and permeabilized in 0.5% Triton X-100 (1×PBS). Cells were then incubated with anti-G-tetraploid (BG4) antibody for 2 h at room temperature. Subsequently, the cells were incubated with anti-Flag antibody and anti-γH2AX primary antibody, rabbit monoclonal to HMGB1, and rabbit monoclonal to calreticulin (CRT) antibodies overnight at 4 °C. Next, the cells were washed three times with PBST and incubated with secondary antibodies DyLight 549-conjugated anti-mouse and DyLight 647-conjugated anti-rabbit (Multi Sciences, China) for 1 h at 37 °C. The cells were stained with DAPI or Hoechst 33342. Fluorescence was detected using a 63× oil immersion objective confocal microscope (LSM 710, Carl Zeiss, Germany).
Detection of cytosolic dsDNA in mitochondrial DNA-depleted HeLa cells (ρ0)
MtDNA was removed by continuous incubation of HeLa cells for 3 weeks in a medium containing EtBr (50 ng mL−1), sodium pyruvate (110 mg mL−1), uridine 50 (μg mL−1), and glutamine (4 mM) as described in the previous relevant literature. Cells were then rested in the presence of uridine for 2 weeks to obtain mtDNA-depleted ρ0 cells. Mitochondrial and nuclear gene expression was detected by real-time quantitative polymerase chain reaction (RT-qPCR) to confirm the loss of mtDNA. HeLa ρ0 cells were treated with CP, TP, DP and PyPDS for 24 h and then incubated with PicoGreen (1 μg mL−1, 15 min) to detect the release of ncDNA from the cells. Images were observed by confocal microscopy. PicoGreen: λex = 488 nm, λem = 520 ± 20 nm.HMGB1 and CRT levels detected by flow cytometry
MDA-MB-231 cells were seeded into 6 cm dishes and cultured for 24 h in complete DMEM. Cells were incubated with the corresponding concentrations of CP, TP, DP and PyPDS for 24 h. Cells were carefully harvested and then fixed, permeabilized, blocked, and incubated with Alexa Fluor® 488 rabbit monoclonal HMGB1 or Alexa Fluor® 647 rabbit monoclonal calreticulin solution overnight at 4 °C. They were then washed with PBST, resuspended in 500 μL of PBS solution, and analyzed by flow cytometry. The median of Alexa Fluor®-488 fluorescence (FITC channel detection)/Alexa Fluor®-647 fluorescence (APC channel detection) was used to determine the intracellular HMGB-1/CRT protein content of the analyzed population. FlowJo software was used to analyze the data from three independent experiments.Determination of extracellular ATP concentration
MDA-MB-231 cells were incubated in 6-well plates and grown in complete DMEM for 24 h at 37 °C in a cell incubator with 5% CO2. The cells were incubated with CP, TP, DP, PyPDS and cisplatin at the corresponding concentrations for 24 h, and the extracellular ATP concentration was detected by luciferase luminescence assay using an ATP assay kit.Real-time quantitative PCR (RT-qPCR)
RT-qPCR experiments were performed in the MDA-MB-231 cell line. The RNA was extracted with an EZ-press RNA Purification Kit, and the genome was removed and transcribed in one step with a Color All-in-one Reverse Transcription Kit (with DNase). qPCR was performed by adding the template ctDNA, primers and ddH2O to the color qPCR kit (premixed ROX1). Real-time quantitative PCR (RT-qPCR) was performed in triplicate using a LightCycler 480 system (Roche Diagnostics, USA). The following primers were used: c-MYC (forward primer: 5′-GCTGCTTAGACGCTGGATT-3′; reverse primer: 5′-TCCTCCTCGTCGCAGTAGA-3′), VEGF (forward: 5′-TGGGGGAGCGTGTCAGAAT-3′; reverse: 5′-CCGCTTTAATTGTGTGATTGGAC-3′) and GAPDH (forward primer: 5′-GGTGGTCTCCTCTGACTTCAACA-3′, reverse primer: 5′-GTTGCTGTAGCCAAATTCGTTGT-3′). Melting curves were used to detect and control the specificity of PCR products. The 2−ΔΔct method was used to calculate relative gene expression levels, where the amounts of MYC mRNA and VEGF mRNA were normalized to an endogenous reference (GAPDH) and ct represents the threshold cycle.Western blot
Western blotting was performed according to the manufacturer's protocol with slight modifications. Rabbit anti-GAPDH antibody (37 kDa) was used as a loading control. MDA-MB-231 cells were incubated with the indicated concentrations of CP, TP, DP, PyPDS and cisplatin at 37 °C for 24 h and then collected and treated with RIPA lysis buffer containing 100 μg mL−1 PMSF for 30 min on ice. The samples were then centrifuged at 10000×g for 15 min at 4 °C. The supernatant was added to loading buffer, heated at 100 °C for 10 min, and stored at −20 °C for further electrophoresis.
Antitumor capacity and the effects of different inhibitors on cell death induced by complexes
The antiproliferation activities of CP, TP, DP, PyPDS and cisplatin in vitro were detected by the methyl thiazol tetrazolium (MTT) assay. Cells were first seeded in 96-well microplates and incubated for 24 h at 37 °C in a cell incubator with 5% CO2. We then incubated the cells with the test concentration of the complex. The incubation was continued for 44 h, and then MTT solution (20 μL, 5 mg mL−1, dissolved in 1× PBS) was added to each well. After 4 h of incubation, the culture was removed, and 150 μL of DMSO solution was added to each well. The optical density of each well was measured with a microplate spectrophotometer at a wavelength of 595 nm. The percentage of cell viability was calculated using the following formula: (mean OD value of cells in the treatment group/mean OD value of cells in the control group) × 100%. Cells treated with solvent control (1% DMSO) were used as the control group. Data are presented as the mean ± standard deviation (n = 3). To evaluate the effect of different inhibitors on cell death induced by CP, TP, DP and PyPDS, inhibitors (Nsa: 10 μm; Nec-1: 100 μM) were incubated for 1 h and then coincubated with CP, TP, DP and PyPDS for 48 h. Each experiment was repeated at least three times to obtain the mean value.Scanning electron microscopy
MDA-MB-231 cells were incubated with DMSO and CP, TP, DP, PyPDS and cisplatin for 24 h, and then the cells were fixed with 4% electron microscope fixable solution at 4 °C overnight and washed three times with PBS. The samples were dehydrated using 30, 50, 70, 80, 90 and 100% ethanol, dried by the tert-butyl alcohol method, and imaged by scanning electron microscopy at 5.0 kV.In vivo antitumor efficacy and the abscopal effect
All of the animal experiments were approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University. The assigned Animal Ethics Approval number is 2022002845. All animal operations were in accord with the guidelines of the above institution and the approved experiment number is C2022-0501XS. SPF female BALB/c mice aged 4–5 weeks were purchased from Guangdong Medical Laboratory Animal Center and raised in Sun Yat-sen University Experimental Animal Center. 4T1 tumor cells were subcutaneously inoculated on the right side of female BALB/c mice, and 3 days later, 4T1 tumor cells (containing 2 × 106 cells) were subcutaneously inoculated on the left side. Dissolved in (100 μL of 1 × PBS) (Protocol S2). The right tumor was used as the primary tumor for incubation with complexes (CP, cisplatin: 1.5 mg Pt kg; PyPDS: 5.9 mg kg−1), and the left tumor was used as the distant tumor for detection of antitumor immunity. The tumor-bearing mice were randomly divided into four groups: the control group, CP group, PyPDS group and cisplatin group. The complexes were sequentially dissolved in 2% DMSO and 4% PEG 400, 4% Tween 80, and 92% PBS. The drug was administered every three days (on days 3, 6, 9, 12, 15). Mouse body weight and tumor volume were recorded every 2 days. The tumor volume (V) was calculated as V = W2 × L/2, where W was the tumor width and L was the tumor length. Mice were sacrificed on day 16 for further testing. Hematoxylin-eosin (H&E) staining was used to observe the changes in tumors and major organs (heart, liver, spleen, lung, kidney) in each group.In vivo antitumor immunity
To systematically study antitumor immunity in vivo, a series of immune cells were analyzed by flow cytometry after antibody staining. Mouse distant tumors (3 mice in each group) were harvested, and single-cell suspensions were made with 1 mg mL−1 collagenase IV and 0.1 mg mL−1 DNase I and incubated for 1 h at 37 °C. Blood samples were filtered through a 70 μm nylon cell filter, and red blood cells were removed with red blood cell lysis buffer. The obtained DCS and T lymphocytes were stained with anti-CD80-mouse-APC, anti-CD86-mouse-PE, anti-CD8a-mouse-APC, anti-CD4-mouse-PerCP and anti-CD3-mouse-FITC. Flow cytometry was used for identification. After the above treatments, mouse serum samples were isolated and stored in liquid nitrogen for analysis. ELISA kits were used to detect the secretion of serum proinflammatory cytokines (IL-12p70, TNF-α), interleukin (IL)-18, Il-1β, interferon (IFN)-β, and cGAMP.Statistical analysis
All experiments were repeated three times. The statistical significance in this study was determined by unpaired, two-tailed Student's t-test at *p < 0.05, **p < 0.01, and ***p < 0.001. Data are presented as the means ± standard deviations (SD).All materials were purchased from commercial sources unless otherwise stated. NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer (Germany). Chemical shift referencing was done based on the signal of TMS. ESI-MS spectra were recorded on a Thermo Finnigan LCQ DECA XP spectrometer (USA). The quoted m/z values represent the main peaks of the isotopic distribution.
Data availability
The sequencing data were deposited at the Genome Sequence Archive (GSA) with the code CRA016164. All the data are available from the corresponding author upon reasonable request.Author contributions
Tian-Zhu Ma: methodology, investigation, writing – original draft, writing – review & editing. Liu-Yi Liu: investigation. You-Liang Zeng: investigation. Ke Ding: investigation. Hang Zhang: investigation. Wenting Liu: conceptualization, methodology, writing – review & editing, funding acquisition, supervision. Qian Cao: supervision. Wei Xia: supervision. Xushen Xiong: supervision. Chao Wu: supervision, writing – review & editing. Zong-Wan Mao: conceptualization, writing – review & editing, funding acquisition, supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China [92353301, 22277151, 22293053, 22007103, 21837006, and 22293050], National Key Research and Development Program of China [2022YFB3804502], Natural Science Foundation of Guangdong Province [2024B1515020083], Guangzhou Science and Technology Plan Project [2023A04J1941] and Fundamental Research Funds for the Central Universities.Notes and references
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, The regulation and functions of DNA and RNA G-quadruplexes, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- J. Dong, M. P. O'Hagan and I. Willner, Switchable and dynamic G-quadruplexes and their applications, Chem. Soc. Rev., 2022, 51, 7631–7661 RSC.
- A. Guédin, J. Gros, P. Alberti and J.-L. Mergny, How long is too long? Effects of loop size on G-quadruplex stability, Nucleic Acids Res., 2010, 38, 7858–7868 CrossRef PubMed.
- R. Hansel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, G-quadruplex structures mark human regulatory chromatin, Nat. Genet., 2016, 48, 1267–1272 CrossRef CAS PubMed.
- J. T. Davis, G-quartets 40 years later: from 5'-GMP to molecular biology and supramolecular chemistry, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, High-throughput sequencing of DNA G-quadruplex structures in the human genome, Nat. Biotechnol., 2015, 33, 877–881 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. Miller, W. J. Howat and S. Balasubramanian, Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues, PLoS One, 2014, 9, e102711 CrossRef PubMed.
- R. S. Apte, D. S. Chen and N. Ferrara, VEGF in signaling and disease: beyond discovery and development, Cell, 2019, 176, 1248–1264 CrossRef CAS PubMed.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS PubMed.
- G. Miglietta, J. Marinello, M. Russo and G. Capranico, Ligands stimulating antitumour immunity as the next G-quadruplex challenge, Mol. Cancer, 2022, 21, 180 CrossRef PubMed.
- S. F. Bakhoum and L. C. Cantley, The multifaceted role of chromosomal instability in cancer and its microenvironment, Cell, 2018, 174, 1347–1360 CrossRef CAS PubMed.
- J. E. Reed, A. A. Arnal, S. Neidle and R. Vilar, Stabilization of G-quadruplex DNA and inhibition of telomerase activity by square-planar nickel (II) complexes, J. Am. Chem. Soc., 2006, 128, 5992–5993 CrossRef CAS PubMed.
- L. He, X. Chen, Z. Y. Meng, J. T. Wang, K. Y. Tian, T. H. Li and F. W. Shao, Octahedral ruthenium complexes selectively stabilize G-quadruplexes, Chem. Commun., 2016, 52, 8095–8098 RSC.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z. W. Mao, G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- L. He, Z. Y. Meng, Y. Q. Xie, X. Chen, T. H. Li and F. W. Shao, Aza-bridged bisphenanthrolinyl Pt (II) complexes: Efficient stabilization and topological selectivity on telomeric G-quadruplexes, J. Inorg. Biochem., 2017, 166, 135–140 CrossRef CAS PubMed.
- Z. Chen, K. W. Zheng, Y. H. Hao and Z. Tan, Reduced or diminished stabilization of the telomere G-quadruplex and inhibition of telomerase by small chemical ligands under molecular crowding condition, J. Am. Chem. Soc., 2009, 131, 10430–10438 CrossRef CAS PubMed.
- R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux, J. F. Riou and S. Balasubramanian, A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres, J. Am. Chem. Soc., 2008, 130, 15758–15759 CrossRef CAS PubMed.
- W. J. Chung, B. Heddi, F. Hamon, M. P. Teulade-Fichou and A. T. Phan, Solution structure of a G-quadruplex bound to the bisquinolinium compound Phen-DC (3), Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS PubMed.
- A. M. Burger, F. Dai, C. M. Schultes, A. P. Reszka, M. J. Moore, J. A. Double and S. Neidle, The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function, Cancer Res., 2005, 65, 1489–1496 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Interaction of metal complexes with G-quadruplex DNA, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- J. Wirmer-Bartoschek, L. E. Bendel, H. R. A. Jonker, J. T. Grün, F. Papi, C. Bazzicalupi, L. Messori, P. Gratteri and H. Schwalbe, Solution NMR structure of a ligand/hybrid-2-G-quadruplex complex reveals rearrangements that affect ligand binding, Angew. Chem., Int. Ed., 2017, 56, 7102–7106 CrossRef CAS PubMed.
- L. Y. Liu, T. Z. Ma, Y. L. Zeng, W. Liu and Z. W. Mao, Structural basis of pyridostatin and its derivatives specifically binding to G-quadruplexes, J. Am. Chem. Soc., 2022, 144, 11878–11887 CrossRef CAS PubMed.
- X. C. Chen, G. X. Tang, W. H. Luo, W. Shao, J. Dai, S. T. Zeng, Z. S. Huang, S. B. Chen and J. H. Tan, Monitoring and modulating mtDNA G-quadruplex dynamics reveal its close relationship to cell glycolysis, J. Am. Chem. Soc., 2021, 143, 20779–20791 CrossRef CAS PubMed.
- M. Di Antonio, K. I. E. McLuckie and S. Balasubramanian, Reprogramming the mechanism of action of chlorambucil by coupling to a G-quadruplex ligand, J. Am. Chem. Soc., 2014, 136, 5860–5863 CrossRef CAS PubMed.
- Y. Z. Hou, T. L. Gan, T. T. Fang, Y. Zhao, Q. Luo, X. K. Liu, L. Y. Qi, Y. Y. Zhang, F. F. Jia, J. J. Han, S. M. Li, S. J. Wang and F. Y. Wang, G-quadruplex inducer/stabilizer pyridostatin targets SUB1 to promote cytotoxicity of a transplatinum complex, Nucleic Acids Res., 2022, 50, 3070–3082 CrossRef CAS PubMed.
- P. Murat, M. V. Gormally, D. Sanders, M. Di Antonio and S. Balasubramanian, Light-mediated in cell downregulation of G-quadruplex-containing genes using a photo-caged ligand, Chem. Commun., 2013, 49, 8453–8455 RSC.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Small-molecule-induced DNA damage identifies alternative DNA structures in human genes, Nat. Chem. Biol., 2012, 8, 301–310 CrossRef CAS PubMed.
- K. Chen, Z. He, W. Xiong, C. J. Wang and X. Zhou, Enantioselective Diels–Alder reactions with left-handed G-quadruplex DNA-based catalysts, Chin. Chem. Lett., 2021, 32, 1701–1704 CrossRef CAS.
- M. Di Antonio, A. Ponjavic, A. Radzevicius, R. T. Ranasinghe, M. Catalano, X. Zhang, J. Shen, L. M. Needham, S. F. Lee, D. Klenerman and S. Balasubramanian, Single-molecule visualization of DNA G-quadruplex formation in live cells, Nat. Chem., 2020, 12, 832–837 CrossRef CAS PubMed.
- S. Jia, W. J. Wang, S. Qin, S. Xie, L. Zhan, Q. Wei, Z. Lu, X. Zhou, C. Chen, K. Chen, S. Yan, C. Tan, Z. W. Mao and X. Zhou, The development of an iridium (III) complex functionalized G-quadruplex probe for the stability of G-quadruplex and lifetime image in cytoplasm, Chin. Chem. Lett., 2023, 34, 107517 CrossRef CAS.
- F. Wu, C. Liu, Y. Chen, S. Yang, J. Xu, R. Huang, X. Wang, M. Li, W. Liu, W. Mao and X. Zhou, Visualization of G-quadruplexes in gel and in live cells by a near-infrared fluorescent probe, Sensor. Actuator. B Chem., 2016, 236, 268–275 CrossRef CAS.
- C. Zhao, G. Qin, J. Niu, Z. Wang, C. Wang, J. Ren and X. Qu, Targeting RNA G-quadruplex in SARS-CoV-2: a promising therapeutic target for COVID-19?, Angew. Chem., Int. Ed., 2020, 60, 432–438 CrossRef PubMed.
- Y. Dai, X. Teng and J. Li, Single-cell visualization of monogenic RNA G-quadruplex and occupied G-quadruplex ratio through a module-assembled multifunctional probes assay (MAMPA), Angew. Chem., Int. Ed., 2022, 61, e202111132 CrossRef CAS PubMed.
- D. Koirala, S. Dhakal, B. Ashbridge, Y. Sannohe, R. Rodriguez, H. Sugiyama, S. Balasubramanian and H. Mao, A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands, Nat. Chem., 2011, 3, 782–787 CrossRef CAS PubMed.
- S. Müller, D. A. Sanders, M. Di Antonio, S. Matsis, J. F. Riou, R. Rodriguez and S. Balasubramanian, Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells, Org. Biomol. Chem., 2012, 10, 6537–6546 RSC.
- F. J. Groelly, M. Porru, J. Zimmer, H. Benainous, Y. De Visser, A. A. Kosova, S. Di Vito, V. Serra, A. Ryan, C. Leonetti, A. Bruna, A. Biroccio and M. Tarsounas, Anti-tumoural activity of the G-quadruplex ligand pyridostatin against BRCA1/2-deficient tumours, EMBO Mol. Med., 2022, 14, e14501 CrossRef CAS PubMed.
- G. Miglietta, M. Russo, R. C. Duardo and G. Capranico, G-quadruplex binders as cytostatic modulators o innate immune genes in cancer cells, Nucleic Acids Res., 2021, 49, 6673–6686 CrossRef CAS PubMed.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Stimuli-responsive therapeutic metallodrugs, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- S. Bu, Q. Wang, Q. Zhang, J. Sun, B. He, C. Xiang, Z. Liu and D. Lai, Human endometrial mesenchymal stem cells exhibit intrinsic anti-tumor properties on human epithelial ovarian cancer cells, Sci. Rep., 2016, 6, 37019 CrossRef CAS PubMed.
- K. Xu, Y. Shi, X. Wang, Y. Chen, L. Tang and X. Guan, A novel BRCA1 germline mutation promotes triple-negative breast cancer cells progression and enhances sensitivity to DNA damage agents, Cancer Genet., 2019, 239, 26–32 CrossRef CAS PubMed.
- K. Peng, B. B. Liang, W. Liu and Z. W. Mao, What blocks more anticancer platinum complexes from experiment to clinic: major problems and potential strategies from drug design perspectives, Coord. Chem. Rev., 2021, 449, 214210 CrossRef CAS.
- A. M. Burger, J. A. Double and D. R. Newell, Inhibition of telomerase activity by cisplatin in human testicular cancer cells, Eur. J. Cancer, 1997, 33, 638–644 CrossRef CAS PubMed.
- T. Ishibashi and S. J. Lippard, Telomere loss in cells treated with cisplatin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4219–4223 CrossRef CAS PubMed.
- Y. Y. Ling, X. Y. Xia, L. Hao, W. J. Wang, H. Zhang, L. Y. Liu, W. Liu, Z. Y. Li, C. P. Tan and Z. W. Mao, Simultaneous photoactivation of cGAS-STING pathway and pyroptosis by platinum (II) triphenylamine complexes for cancer immunotherapy, Angew. Chem., Int. Ed., 2022, 61, e202210988 CrossRef CAS PubMed.
- S. M. Gaikwad, Z. Phyo, A. Q. Arteaga, S. Gorjifard, D. R. Calabrese, D. Connors, J. Huang, A. M. Michalowski, S. L. Zhang, Z. G. Liu, J. S. Schneekloth and B. A. Mock, A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma, Cancers, 2020, 12, 2952–2976 CrossRef CAS PubMed.
- K. M. Felsenstein, L. B. Saunders, J. K. Simmons, E. Leon, D. R. Calabrese, S. L. Zhang, A. Michalowski, P. Gareiss, B. A. Mock and J. S. Schneekloth, Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression, ACS Chem. Biol., 2016, 11, 139–148 CrossRef CAS PubMed.
- W. S. Lee, H. Yang, H. J. Chon and C. Kim, Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity, Exp. Mol. Med., 2020, 52, 1475–1485 CrossRef CAS PubMed.
- M. Mall, M. S. Kareta, S. Chanda, H. Ahlenius, N. Perotti, B. Zhou, S. D. Grieder, X. Ge, S. Drake, C. Euong Ang, B. M. Walker, T. Vierbuchen, D. R. Fuentes, P. Brennecke, K. R. Nitta, A. Jolma, L. M. Steinmetz, J. Taipale, T. C. Südhof and M. Wernig, Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates, Nature, 2017, 544, 245–249 CrossRef CAS PubMed.
- H. Huang, L. Zhu, B. R. Reid, G. P. Drobny and P. B. Hopkins, Solution structure of a cisplatin-induced DNA interstrand cross-link, Science, 1995, 270, 1842–1845 CrossRef CAS PubMed.
- S. E. Sherman, D. Gibson, A. H. Wang and S. J. Lippard, X-ray structure of the major adduct of the anticancer drug cisplatin with DNA: cis- Pt(NH3)2(d(pGpG)), Science, 1985, 230, 412–417 CrossRef CAS PubMed.
- A. Zákovská, O. Nováková, Z. Balcarová, U. Bierbach, N. Farrell and V. Brabec, DNA interactions of antitumor trans-[PtCl2(NH3)(quinoline)], Eur. J. Biochem., 1998, 254, 547–557 CrossRef PubMed.
- D. P. Bancroft, C. A. Lepre and S. J. Lippard, Platinum-195 NMR kinetic and mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to DNA, J. Am. Chem. Soc., 1990, 112, 6860–6871 CrossRef CAS.
- F. Paquet, M. Boudvillain, G. Lancelot and M. Leng, NMR solution structure of a DNA dodecamer containing a transplatin interstrand GN7-CN3 cross-link, Nucleic Acids Res., 1999, 27, 4261–4268 CrossRef CAS PubMed.
- L. Y. Liu, W. Liu, K. N. Wang, B. C. Zhu, X. Y. Xia, L. N. Ji and Z. W. Mao, Quantitative detection of G-quadruplex DNA in live cells based on photon counts and complex structure discrimination, Angew. Chem., Int. Ed., 2020, 59, 9719–9726 CrossRef CAS PubMed.
- B. C. Zhu, J. He, W. Liu, X. Y. Xia, L. Y. Liu, B. B. Liang, H. G. Yao, B. Liu, L. N. Ji and Z. W. Mao, Selectivity and targeting of G-quadruplex binders activated by adaptive binding and controlled by chemical kinetics, Angew. Chem., Int. Ed., 2021, 60, 15340–15343 CrossRef CAS PubMed.
- S. C. Ku, H. L. Liu, C. Y. Su, I. J. Yeh, M. C. Yen, G. Anuraga, H. D. K. Ta, C. C. Chiao, D. T. M. Xuan, F. B. Prayugo, W. J. Wang and C. Y. Wang, Comprehensive analysis of prognostic significance of cadherin (CDH) gene family in breast cancer, Aging, 2022, 14, 8498–8567 CrossRef CAS PubMed.
- L. Y. Liu, T. Z. Ma, Y. L. Zeng, W. Liu, H. Zhang and Z. W. Mao, Organic-platinum hybrids for covalent binding of G-quadruplexes: structural basis and application to cancer immunotherapy, Angew. Chem., Int. Ed., 2023, 12, e202305645 Search PubMed.
- F. M. Pozo, X. R. Geng, M. Miyagi, A. L. Amin, A. Y. Huang and Y. W. Zhang, MYO10 regulates genome stability and cancer inflammation through mediating mitosis, Cell Rep., 2023, 42, 112531 CrossRef PubMed.
- K. J. Magnet, M. S. Orr, J. L. Cleveland, C. Rodriguez-Galindo, H. Yang, C. Y. Yang, Y. M. Di, P. T. Jain and D. A. Gewirtz, Suppression of c-myc expression and c-Myc function in response to sustained DNA damage in MCF-7 breast tumor cells, Biochem. Pharmacol., 2001, 62, 593–602 CrossRef CAS PubMed.
- Y. Onodera, K. Takagi, Y. Miki, K. Takayama, Y. Shibahara, M. Watanabe, T. Ishida, S. Inoue, H. Sasano and T. Suzuki, TACC2 (transforming acidic coiled-coil protein 2) in breast carcinoma as a potent prognostic predictor associated with cell proliferation, Cancer Med., 2016, 5, 1973–1982 CrossRef CAS PubMed.
- A. Tadijan, I. Samaržija, J. D. Humphries, M. J. Humphries and A. Ambriović-Ristov, KANK family proteins in cancer, Int. J. Biochem. Cell Biol., 2021, 131, 105903 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Quantitative visualization of DNA G-quadruplex structures in human cells, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- X. X. Su, W. J. Wang, Q. Cao, H. Zhang, B. Liu, Y. Y. Ling, X. T. Zhou and Z. W. Mao, A carbonic anhydrase IX (CAIX)-anchored rhenium(I) photosensitizer evokes pyroptosis for enhanced anti-tumor immunity, Angew. Chem., Int. Ed., 2022, 61, e202115800 CrossRef CAS PubMed.
- Y. P. Wang, W. Q. Gao, X. Y. Shi, J. J. Ding, W. Liu, H. B. He, K. Wang and F. Shao, Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin, Nature, 2017, 547, 99–103 CrossRef CAS PubMed.
- T. Bürckstümmer, C. Baumann, S. Blüml, E. Dixit, G. Dürnberger, H. Jahn, M. Planyavsky, M. Bilban, J. Colinge, K. L. Bennett and G. Superti-Furga, An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome, Nat. Immunol., 2009, 10, 266–272 CrossRef PubMed.
- T. Fernandes-Alnemri, J. W. Yu, P. Datta, J. Wu and E. S. Alnemri, AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA, Nature, 2009, 458, 509–513 CrossRef CAS PubMed.
- V. Hornung, A. Ablasser, M. Charrel-Dennis, F. Bauernfeind, G. Horvath, D. R. Caffrey, E. Latz and K. A. Fitzgerald, AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC, Nature, 2009, 458, 514–518 CrossRef CAS PubMed.
- Z. Zhou, C. Li, T. Bao, X. Zhao, W. Xiong, C. Luo, G. Yin and J. Fan, Exosome-shuttled miR-672-5p from anti-inflammatory microglia repair traumatic spinal cord injury by inhibiting AIM2/ASC/Caspase-1 signaling pathway mediated neuronal pyroptosis, J. Neurotrauma, 2022, 39, 1057–1074 CrossRef PubMed.
- J. Ding, K. Wang, W. Liu, Y. She, Q. Sun, J. Shi, H. Sun, D. C. Wang and F. Shao, Pore-forming activity and structural autoinhibition of the gasdermin family, Nature, 2016, 535, 111–116 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Immunogenic cell death and DAMPs in cancer therapy, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- T. Yang, S. Zhang, H. Yuan, Y. Wang, L. Cai, H. Chen, X. Wang, D. Song, X. Wang, Z. Guo and X. Wang, Platinum-based tREM2 inhibitor suppresses tumors by remodeling the immunosuppressive microenvironment, Angew. Chem., Int. Ed., 2023, 62, e202213337 CrossRef CAS.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Platinum coordination compounds with potent anticancer activity, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- M. M. Gaidt, T. S. Ebert, D. Chauhan, K. Ramshorn, F. Pinci, S. Zuber, F. O'Duill, J. L. Schmid-Burgk, F. Hoss, R. Buhmann, G. Wittmann, E. Latz, M. Subklewe and V. Hornung, The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3, Cell, 2017, 171, 1110–1124 CrossRef CAS PubMed.
- A. Barbanente, V. Gandin, N. Ditaranto, C. Marzano, J. D. Hoeschele, G. P. Suranna, P. Papadia, G. Natile and N. Margiotta, A Pt (IV) prodrug of kiteplatin with the bone-targeting pyrophosphate ligand, Inorg. Chim. Acta, 2019, 494, 98–104 CrossRef CAS.
- Y. Dai, Y. Zhu, J. J. Cheng, J. Shen, H. Huang, M. M. Liu, Z. L. Chen and Y. Z. Liu, Nitric oxide-releasing platinum(iv) prodrug efficiently inhibits proliferation and metastasis of cancer cells, Chem. Commun., 2020, 56, 14051–14054 RSC.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart and R. K. Belew and A, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- W. L. Delano, The PyMOL Molecular Graphics System, DeLano Scientific, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00643g |
| This journal is © The Royal Society of Chemistry 2024 |