
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of O, S, and P in transition-metal compounds on the adsorption and catalytic ability of sulfur cathodes in lithium–sulfur batteries†
Meng
Du
a,
Jiakang
Shi
a,
Yuxiao
Shi
a,
Guangxun
Zhang
a,
Yan
Yan
a,
Pengbiao
Geng
c,
Ziqi
Tian
 b and
Huan
Pang
*a
b and
Huan
Pang
*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225009, P. R. China. E-mail: panghuan@yzu.edu.cn
bNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, 315201, P. R. China
cSchool of Materials Science and Engineering, Suzhou University of Science and Technology, Suzhou, 215009, P. R. China
First published on 28th May 2024
Abstract
Transition-metal compounds (TMCs) have recently become promising candidates as lithium–sulfur (Li–S) battery cathode materials because they have unique adsorption and catalytic properties. However, the relationship between the anionic species and performance has not been sufficiently revealed. Herein, using FeCoNiX (X = O, S, and P) compounds as examples, we systematically studied the effects of the anion composition of FeCoNiX compounds on the adsorption and catalytic abilities of sulfur cathodes in Li–S batteries. Adsorption tests and density functional theory calculations showed that the adsorption ability toward lithium polysulfides follows the order: FeCoNiP > FeCoNiO > FeCoNiS, while in situ ultraviolet-visible spectroscopy and cyclic voltammetry revealed that the catalytic ability for lithium polysulfide conversion follows the order: FeCoNiP > FeCoNiS > FeCoNiO. These results indicate that FeCoNiP is an excellent polysulfide immobilizer and catalyst that restricts shuttling and improves reaction kinetics. Electrochemical tests further demonstrated that the FeCoNiP cathode delivered superior cycling performance to FeCoNiO or FeCoNiS. In addition, the battery performance order is consistent with that of catalytic ability, which suggests that catalytic ability plays a key influencing role in batteries. This study provides new insight into the use of O-, S-, and P-doped TMCs as functional sulfur carriers.
Introduction
The increasing demand for portable electronic devices and electric vehicles has accelerated the exploration of advanced energy storage devices with high energy density.1,2 Among various energy-storage systems, the lithium–sulfur (Li–S) battery has become the focus of attention owing to its ultrahigh theoretical specific capacity (1672 mA h g−1) and prominent theoretical energy density (2600 W h kg−1).3,4 Moreover, sulfur as an active substance is earth-abundant, inexpensive and nontoxic. Despite these potential advantages, the commercialization of Li–S batteries still faces a multitude of obstacles, such as the poor conductivity of sulfur and its solid-state discharge products (Li2S/Li2S2), the shuttle effect caused by the dissolution and diffusion of lithium polysulfides (LiPS), and the slow kinetics of the LiPS redox reaction.5,6To address these issues, numerous strategies have been proposed to design and construct sulfur hosts. The most extensively used strategy is to employ carbon materials with a high surface area as the sulfur host for providing high electrical conductivity.7,8 Nevertheless, although a variety of carbon-based materials, including carbon fibers, carbon nanotubes, graphene, and carbon spheres, have been utilized as sulfur hosts in Li–S battery cathodes, the physical confinement of LiPS is finite owing to weak van der Waals interactions between polar LiPS and nonpolar carbons. Recently, polar materials that exhibit catalytic effects, such as transition-metal oxides, phosphides, and sulfides, have been investigated with the aim of inhibiting LiPS shuttling and improving the reaction kinetics for LiPS conversion.9,10 Strong polar–polar interactions between transition metal compound (TMC) hosts and LiPS can moderate the diffusion of intermediate polysulfides, while fast sulfur redox reactions can improve the use of sulfur.11,12 However, most of the earlier reported TMCs were applied in the form of irregular particles, with insufficient porosity; hence, they can only adsorb LiPS near the surface.13 Therefore, TMCs derived from metal–organic frameworks (MOFs) are promising materials for constructing high-performance sulfur cathodes because MOF-derived materials, to some extent, inherit the porosities and high surface areas of their MOF precursors, which is beneficial for exposing active sites and anchoring polysulfides.14–17 As special MOFs, Prussian blue analogues (PBAs) have been demonstrated to be outstanding precursors for preparing porous nanostructured TMCs.18 Notably, the obtained transition metal phosphides usually exhibit better conductivity in comparison with their oxide and sulfide counterparts; however, the adsorption strengths and catalytic abilities of these polar hosts toward polysulfides remain unclear (Fig. 1).
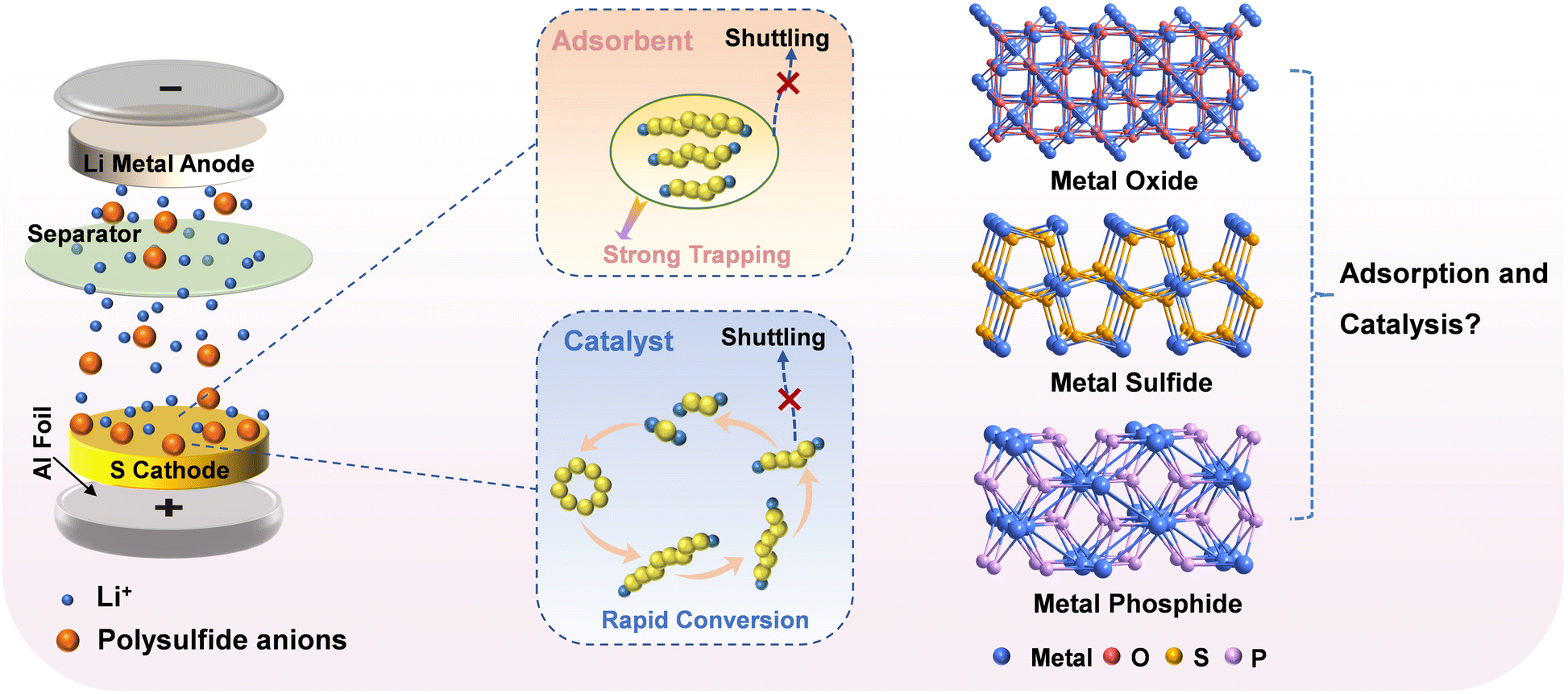 | ||
| Fig. 1 The importance of adsorbents and catalysts in sulfur cathodes, and the influence of metal oxides, sulfides and phosphides on adsorption and catalytic ability. | ||
Herein, we aimed to study the effects of O, S, and P in TMCs on the adsorption and catalytic ability of sulfur cathodes in Li–S batteries. Specifically, we reported the controllable synthesis of FeCoNi-oxide (FeCoNiO), FeCoNi-sulfide (FeCoNiS), and FeCoNi-phosphide (FeCoNiP) as sulfur host materials, using FeCoNi-PBA nanocubes as precursors. Adsorption tests and density functional theory (DFT) calculations showed that the ability to capture LiPS followed the order: FeCoNiP > FeCoNiO > FeCoNiS, which indicated that FeCoNiP was capable of acting as a polysulfide adsorbent that greatly confined LiPS shuttling. In situ ultraviolet-visible (UV-vis) spectroscopy and cyclic voltammetry (CV) revealed that the ability to catalyze the conversion of LiPS followed the order: FeCoNiP > FeCoNiS > FeCoNiO, which indicated that FeCoNiP can realize the rapid kinetics of the LiPS redox reaction. Electrochemical tests showed that, compared with FeCoNiO and FeCoNiS, the FeCoNiP cathode achieved better cyclic capability with a stable capacity of 525.8 mA h g−1 after 200 cycles at 0.5C. In addition, the order of battery performance and catalytic ability was consistent, indicating that the catalytic ability was more dominant than the adsorption ability. This study provides an opportunity to construct an ideal sulfur host that exhibits dual “adsorption–catalysis” behavior.
Results and discussion
FeCoNiX (X = O, S, P) was synthesized in a controlled manner using FeCoNi-PBA as the precursor. The formation processes are schematically presented in Fig. 2a and described in detail in the Experimental section of the ESI.† The materials obtained after calcination for 1, 2, and 3 h are referred to as FeCoNiX-1h, FeCoNiX-2h, and FeCoNiX-3h, respectively. The material that did not thermally decompose completely is referred to as FeCoNiX@FeCoNi-PBA. FeCoNi-PBA nanocubes were first prepared using a simple coprecipitation process. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) revealed that the FeCoNi-PBA particles were cubic in morphology with smooth surfaces and solid in nature (Fig. S1a and b†). The cubes are approximately 200 nm in size. Several PBA derivatives were subsequently synthesized by pyrolyzing FeCoNi-PBA. Herein, we discussed the influence of calcination time on the morphologies of the obtained materials. FeCoNiO-2h was formed by pyrolyzing FeCoNi-PBA at 350 °C in air for 2 h. SEM and TEM images displayed that FeCoNiO-2h retained the original cubic morphology of FeCoNi-PBA and possessed coarse surfaces (Fig. 2b). FeCoNiO-1h exhibited a relatively low surface roughness, while FeCoNiO-3h exhibited a partially collapsed structure (Fig. S2a and b†). FeCoNiS-2h was obtained by annealing the FeCoNi-PBA precursor with sublimed sulfur in a N2 atmosphere at 350 °C for 2 h. Morphological characterization showed that FeCoNiS-2h had a cubic structure and rich particles on its surface (Fig. 2f). FeCoNiS-1h had a similar structure to FeCoNiS-2h and partially broken particles were observed on the surfaces of FeCoNiS-3h (Fig. S3a and b†). Moreover, FeCoNi-PBA nanocubes were phosphatized at 350 °C for 2 h under N2, during which the PH3 gas released from the NaH2PO2 reacted with them to generate FeCoNiP-2h products. Fig. 2j shows that FeCoNiP-2h had slightly truncated structures at the eight corners of its cubes and cracked surfaces. The morphology of FeCoNiP-1h was similar to that of FeCoNiP-2h; however, FeCoNiP-3h exhibited a partially broken structure (Fig. S4a and b†). Three types of composites were obtained by controlling the calcination temperature and the amount of doping sources. The morphology of FeCoNiX@FeCoNi-PBA was consistent with that of FeCoNiX-2h (Fig. S5a–c†). | ||
| Fig. 2 (a) Schematic diagram of the preparation process for FeCoNiO-S, FeCoNiS-S, and FeCoNiP-S. (b) SEM and TEM images of FeCoNiO-2h. (c) Magnified TEM image and SAED pattern, (d) HRTEM image, and (e) elemental mapping images of FeCoNiO-2h-S. (f) SEM and TEM images of FeCoNiS-2h. (g) Magnified TEM image and SAED pattern, (h) HRTEM image, and (i) elemental mapping images of FeCoNiS-2h-S. (j) SEM and TEM images of FeCoNiP-2h. (k) Magnified TEM image and SAED pattern, (l) HRTEM image, and (m) elemental mapping images of FeCoNiP-2h-S. | ||
X-ray diffraction (XRD) analysis was performed to investigate the crystalline structures of FeCoNi-PBA, FeCoNiX@FeCoNi-PBA, and FeCoNiX. The diffraction peaks of FeCoNi-PBA ascribed to FeNi-PBA (JCPDS No. 51-1897) and FeCo-PBA (JCPDS No. 46-0907) can be identified (Fig. S6†). Among the various FeCoNiX-2h samples, the characteristic peaks of FeCoNiO-2h were well indexed to Fe3O4 (JCPDS No. 75-0449), Co3O4 (JCPDS No. 74-2120), and NiO (JCPDS No. 75-0197) (Fig. S7a†). The diffraction peaks observed for FeCoNiS-2h were well assigned to the standard patterns of FeS2 (JCPDS No. 79-0617), CoS2 (JCPDS No. 89-3056), and NiS2 (JCPDS No. 80-0377) (Fig. S8a†). The XRD pattern of FeCoNiP-2h indicated that FeP (JCPDS No. 89-2746), CoP (JCPDS No. 29-0497), and Ni2P (JCPDS No. 89-4864) were its main phases (Fig. 3a). The characteristic peaks of FeCoNiX-1h and FeCoNiX-3h were consistent with those of FeCoNiX-2h (Fig. S7–S9†). The main peaks observed for FeCoNiO at 36.1° and 44.3°, FeCoNiS at 32.4° and 55.3°, and FeCoNiP at 48.3° and 56.6° gradually intensified with increasing calcination time, consistent with enhanced degrees of oxidation, sulfidation, and phosphatization. Furthermore, the characteristic peaks of FeCoNiX@FeCoNi-PBA were well indexed to FeCoNiX and FeCoNi-PBA (Fig. S10†). The chemical structures of PBA and its derivatives were studied using Fourier transform infrared (FTIR) spectroscopy. The peaks observed at 2165 and 2092 cm−1 for FeCoNi-PBA were attributable to the characteristic stretching vibrations of the CN group (Fig. 3b).19 The FeCoNiX@FeCoNi-PBA hybrids exhibited weaker characteristic CN peaks following calcination, which indicated that some CN remained in the untransformed PBA frameworks (Fig. S11†).20 Moreover, the characteristic CN peak was not observed in the spectrum of FeCoNiX, which indicated that the PBA materials had fully decomposed (Fig. S12†).
 | ||
| Fig. 3 (a) XRD patterns of FeCoNiP-2h and FeCoNiP-2h-S. (b) FTIR spectroscopy of FeCoNi-PBA, FeCoNiO-2h, FeCoNiS-2h, and FeCoNiP-2h. (c) N2 adsorption–desorption isotherms and pore size distribution of FeCoNiP-2h and FeCoNiP-2h-S. (d) Raman spectrum of FeCoNiO-2h-S, FeCoNiS-2h-S, and FeCoNiP-2h-S. High-resolution XPS spectra of (e) Fe 2p, (f) Co 2p, (g) Ni 2p, (h) P 2p of FeCoNiP-2h and FeCoNiP-2h-S. (i) S 2p XPS spectra of FeCoNiO-2h-S, FeCoNiS-2h-S, and FeCoNiP-2h-S. | ||
Sulfur molecules were introduced into the FeCoNi-PBA, FeCoNiX@FeCoNi-PBA, and FeCoNiX samples via a liquid diffusion process.21 After sulfur loading, the FeCoNi-PBA-S, FeCoNiX@FeCoNi-PBA-S, FeCoNiX-1h-S, FeCoNiX-2h-S, and FeCoNiX-3h-S composites maintained their original nanostructures (Fig. S1–S5 and S13†). By controlling the feeding ratio, the sulfur content was governed to be 70 wt%, as confirmed by elemental analysis data (Table S1†).22 The FeCoNiX-2h-S samples were subjected to high-resolution TEM (HRTEM) observation. The enlarged TEM images show that many nanoparticles were produced within these materials during calcination, with their selected area electron diffraction (SAED) patterns confirming their good polycrystalline feature (Fig. 2c,g and k). The pattern of FeCoNiO-2h-S revealed three spacings of 0.251, 0.243, and 0.208 nm that originated from the (311) plane of Fe3O4, the (311) plane of Co3O4, and the (200) plane of NiO, respectively (Fig. 2d). Three obvious fringes observed for FeCoNiS-2h-S were consistent with spacings of 0.272, 0.247, and 0.227 nm that corresponded to the (200) facet of FeS2, the (210) facet of CoS2, and the (211) plane of NiS2 crystals, respectively (Fig. 2h). Fig. 2l shows that FeCoNiP-2h-S was well crystallized with explicit lattice spacings of 0.196, 0.283, and 0.221 nm that belonged to the (112) facet of FeP, the (011) facet of CoP, and the (111) facet of Ni2P, respectively. The high-angle annular dark field scanning TEM (HAADF-STEM) and elemental mapping images demonstrated that Fe, Co, Ni, O, P, and S were uniformly distributed in the FeCoNiX-2h-S samples (Fig. 2e, i and m).
Raman analysis was employed to further corroborate the success of sulfur loading. The peak at 2168 cm−1 of the FeCoNi-PBA precursor corresponded to the vibration of the CN group23 and disappeared after calcination, which meant that this precursor had been decomposed (Fig. S14a and S15a†). The Raman spectrum of FeCoNiS and FeCoNiP showed weak G- and D-bands (∼1580 and ∼1350 cm−1, respectively), which corresponded to the typical E2g vibration mode of sp2 carbon.24 This showed that a small amount of carbon derived from the cyano group existed in the FeCoNiS and FeCoNiP samples. After sulfur loading, three main peaks observed at 154, 219, and 471 cm−1 belonged to S8 molecules (Fig. 3d and S14–S15†).25 The XRD patterns of all sulfur-impregnated products also exhibited obvious S8 signals (Fig. S6–S10†). Additionally, compared to pristine FeCoNiX-2h, the N2 absorption quantity and pore volume of FeCoNiX-2h-S decreased significantly, indicating that S8 was well embedded in the FeCoNiX-2h pores (Fig. 3c and S16–S17†).26 The pore-size distributions suggested that the FeCoNiX-2h samples contained both micropores and mesopores in their structures, which contributed to anchoring sulfur and polysulfides.
The FeCoNiX-2h and FeCoNiX-2h-S samples were subjected to X-ray photoelectron spectroscopy (XPS) to clarify their elemental compositions and chemical bonding. Fig. 3e–h show the bonding states of M (M = Fe/Co/Ni) and P in FeCoNiP-2h and FeCoNiP-2h-S. The Fe, Co, and Ni 2p XPS spectra of FeCoNiP-2h showed that metal ions primarily exist in +2 and +3 valence states (Fig. 3e–g).27 These three spectra showed peaks at 707.1/720.2, 778.5/793.8, and 853.5/870.4 eV that were characteristic of Fe–P, Co–P, and Ni–P bonds, respectively.28,29 Sulfur loading did not appear to affect the valence states of these metals. Moreover, each M–P peak was more intense after sulfur loading, which indicated the presence of M–S bonds and confirmed that electrons were transferred between the metal atoms in the surface oxidation layer of the FeCoNiP-2h nanoparticles and the S atoms.30 The P 2p spectrum of FeCoNiP-2h showed a pair of P 2p3/2 and P 2p1/2 peaks at 129.4 and 130.3 eV, respectively, corresponding to the formation of P–Fe/Co/Ni (Fig. 3h).31 The peak at 133.9 eV reflected the P–O bonds produced by the oxidation of the sample in air. The P 2p spectrum in FeCoNiP-2h-S showed that the component of P–Fe/Co/Ni increased, which may be induced by sulfur molecules. The M and O XPS spectra of FeCoNiO-2h and FeCoNiO-2h-S are shown in Fig. S18,† which revealed that Fe, Co and Ni also existed in bivalent and trivalent forms. The O 1s spectrum at 529.8 eV corresponded to oxygen atoms bound to metals.32 After the sulfur loading process, the peaks appeared at 707.5/720.2 eV, 778.7/793.4 eV, and 853.6/870.9 eV, which can be assigned to Fe–S, Co–S, and Ni–S bonds, respectively.33 M–S bonds are formed through good interactions between the transition metal atoms on the FeCoNiO-2h surface and the sulfur atoms. Fig. S19† shows the bonding states of M and S in FeCoNiS-2h and FeCoNiS-2h-S; the cations in FeCoNiS had valences that were consistent with those in FeCoNiP and FeCoNiO. After sulfur loading, the intensity of M–S components did not show a significant increase trend, indicating that the interaction between FeCoNiS-2h and sulfur molecules was relatively poor. To further illustrate the interaction between the three FeCoNiX-2h samples and sulfur, the S 2p XPS spectra of FeCoNiO-2h-S, FeCoNiS-2h-S and FeCoNiP-2h-S were analyzed (Fig. 3i). The peaks at ∼163.6 and ∼164.8 eV were assigned to S 2p3/2 and S 2p1/2, respectively,34 and the peak at ∼169.2 eV indicated the generation of sulfate species. In addition, the S 2p spectra of FeCoNiP-2h-S exhibited a clearer S–M signal, confirming that FeCoNiP-2h and sulfur are strongly chemically bonded.
Coin cells were assembled to estimate the battery performance using metal oxides, sulfides and phosphides as sulfur host materials. FeCoNiO-2h-S, FeCoNiS-2h-S and FeCoNiP-2h-S were subjected to CV in the 1.7–2.7 V voltage range at 0.1 mV s−1 to investigate the reversibility of the sulfur redox reaction (Fig. S20†). The FeCoNiP-2h-S cathode exhibited a peak at 2.31 V that corresponded to the conversion of S8 into high-order LiPS and a peak at 2.05 V that corresponded to the conversion of high-order LiPS into Li2S2/Li2S (Fig. S20c†).35 The anodic peak at 2.42 V corresponded to the oxidation process of Li2S2/Li2S to S8. The cathodic peaks overlapped well over the next two cycles, indicative of excellent reversibility. CV was performed at various scan rates in the 0.1–0.5 mV s−1 range to study the catalytic capability of these three sulfur hosts for LiPS transformation (Fig. 4a and S21†).36 Excellent electrochemical stability was observed, as evidenced by the unchanged shape of the redox peaks with increasing scan rate. The diffusion behaviors of Li+ were evaluated using the Randles–Sevick equation:
| Ip = (2.69 × 105)n1.5ADLi0.5CLiν0.5 |
 | ||
| Fig. 4 (a) CV curves at various scan rates of the FeCoNiP-2h-S electrode. The plots of peak current versus the square root of the scan rate (ν0.5) for (b) the transformation process from S8 to Li2S6/Li2S4 and (c) the transformation process from Li2S6/Li2S4 to Li2S2/Li2S. (d) The GCD profiles at 0.5C of the FeCoNiP-2h-S electrode. (e) Rate performance of the FeCoNiX-2h-S electrode at various current densities. (f) EIS spectra for the three PBA derivatives. (g) Cycle performance at 0.5C over 200 cycles. (h) Radar map of the initial and final capacities of all electrode materials at 0.5C. (i) Schematic diagram of the influence of FeCoNiO, FeCoNiS and FeCoNiP on the adsorption and catalytic ability of sulfur cathodes. | ||
The galvanostatic charge/discharge (GCD) curves under different cycles at 0.5C clearly manifested two discharge platforms, which corresponded to the S → Li2Sx → Li2S2/Li2S conversion process (Fig. 4d and S22†). The FeCoNiP-2h-S cathode delivered a higher initial specific discharge capacity (1356.2 mA h g−1) than FeCoNiO-2h-S (1317.4 mA h g−1) and FeCoNiS-2h-S (1347.5 mA h g−1) cathodes. For FeCoNiP-2h, the large discharge capacity in the sloping region at the end of discharge is mainly due to its acceleration of the transformation from Li2S2 to Li2S (Fig. 4d). Moreover, there is a deviation between the oxidation peak of CV curves and the voltage plateau of GCD curves, which may be due to different degrees of electrode polarization caused by different test conditions. The cycling measurement of the three TMCs as sulfur hosts at 0.5C further verified the superior battery performance of the FeCoNiP-2h-S electrode (Fig. 4g). A stable capacity of 525.8 mA h g−1 over 200 cycles was maintained for FeCoNiP-2h-S, far exceeding those of FeCoNiO-2h-S (278.2 mA h g−1) and FeCoNiS-2h-S (400.9 mA h g−1). Relative to previously reported results of metal compounds as sulfur hosts, the FeCoNiP-2h-S cathode displayed competitive electrochemical performance (Table S2†). Furthermore, it also achieved a superior coulombic efficiency of over 98% during cycling. Even under a higher current density of 1C, the FeCoNiP-2h-S cathode delivered a high specific capacity of 481.9 mA h g−1 after 200 cycles (Fig. S23†). The FeCoNi-PBA precursor as a sulfur carrier provided a low specific capacity of 351.1 mA h g−1 over 200 cycles, which further proved that sulfur can be well anchored in the FeCoNiP-2h-S electrode to heighten the cycling performance (Fig. S24†). In addition, the rate performance of FeCoNiX-2h-S was evaluated at 0.1–1.0C. FeCoNiP-2h-S exhibited an outstanding rate performance with specific capacities of 1272.6, 764.9, 632.0, and 504.1 mA h g−1 at 0.1, 0.2, 0.5, and 1.0C, respectively, suggestive of a high sulfur utilization rate (Fig. 4e and S25†). The specific capacity recovered to 743.8 mA h g−1 as the current density turned to 0.1C. The kinetic behavior of FeCoNiX-2h-S samples was assessed using electrochemical impedance spectroscopy (EIS). The semicircle in the high-frequency area and the inclined line in the low-frequency area represented the charge-transfer resistance (Rct) and the Warburg impedance (Wo), respectively.39 The Nyquist plots displayed in Fig. 4f showed that the FeCoNiP-2h-S electrode possessed the smallest Rct value, which was favorable for improving rate performance and reducing polarization. The Warburg tail slope of the FeCoNiP-2h-S electrode is the steepest, indicative of a rapid Li+ diffusion process.
Interestingly, we also studied how FeCoNiX calcination time affected Li–S battery performance (Fig. S26†). For FeCoNiS and FeCoNiP, the performance of FeCoNiX-2h was better than that of FeCoNiX-1h and FeCoNiX-3h (Fig. 4h). However, the performance of FeCoNiO-1h was better than that of FeCoNiO-2h and FeCoNiX-3h. The poor performance of FeCoNiX-3h was possibly ascribable to morphological collapse resulting from prolonged calcination. The destruction of the structure led to the lack of a relatively closed sulfur storage space, which resulted in the easy dissolution of sulfur into organic electrolyte and the decrease of the utilization rate. Overall, the performance followed the order: FeCoNiP > FeCoNiS > FeCoNiO. The excellent performance originated from the high adsorption ability and catalytic effect of FeCoNiP (Fig. 4i). In addition, FeCoNiO@FeCoNi-PBA outperformed both the FeCoNi-PBA precursor and FeCoNiO-2h (Fig. S27–S29†). However, FeCoNiS@FeCoNi-PBA and FeCoNiP@FeCoNi-PBA performed more poorly than FeCoNiS-2h and FeCoNiP-2h, respectively. This indicated that a lower degree of oxidation and higher degrees of sulfidation and phosphatization are beneficial for high performance.
To further assess the adsorption ability of metal oxides, sulfides and phosphides toward LiPS, visual LiPS adsorption tests were carried out for FeCoNiO-2h, FeCoNiS-2h and FeCoNiP-2h. The absorption peak at 417 nm belonged to the Li2S4 species (Fig. S30†).40 The brown Li2S4-containing solution became transparent because of chemical adsorption. The color of the Li2S4 solution containing FeCoNiP-2h significantly faded after 10 h, whereas the Li2S4 solution containing FeCoNiS-2h remained very turbid (Fig. S31†), which indicates that Li2S4 was highly absorbed by FeCoNiP-2h. The Li 1s XPS spectrum of FeCoNi-2h-P soaked in Li2S4 solution exhibited a characteristic peak at 55.1 eV that was attributable to the Li–S bond in Li2S4 (Fig. S32†). The upper Li2S4 solution was collected and examined by UV-vis spectroscopy. The FeCoNiP-2h supernatant exhibited the weakest absorption at 417 nm, while FeCoNiS-2h exhibited the strongest. These observations were consistent with the visible LiPS adsorption results. The strong chemical binding between FeCoNiP and LiPS was further verified by DFT calculations. Fig. 5g displays the optimized configurations of Li2S4 adsorbed on the main phases of FeCoNiO, FeCoNiS, and FeCoNiP, which showed that Fe3O4, Co3O4, NiO, FeS2, CoS2, NiS2, FeP, CoP, and Ni2P bind to Li2S4 molecules with calculated energies of −3.68, −3.91, −3.89, −2.03, −3.71, −3.40, −4.97, −4.15, and −4.87 eV, respectively (Fig. 5h). While Li2S4 maintained a relatively complete structure on the surfaces of FeCoNiO and FeCoNiS, it decomposed owing to stronger polar–polar interactions between Li2S4 and FeCoNiP. Overall, adsorption ability toward LiPS followed the order: FeCoNiP-2h > FeCoNiO-2h > FeCoNiS-2h. Furthermore, SEM revealed that the three TMCs were very stable in the Li2S4 solution (Fig. S33†). Surface morphologies before and after battery cycling were also studied using SEM (Fig. S34†), with all FeCoNiX-2h-S samples showing only slight changes after cycling, which was indicative of good electrochemical stability.
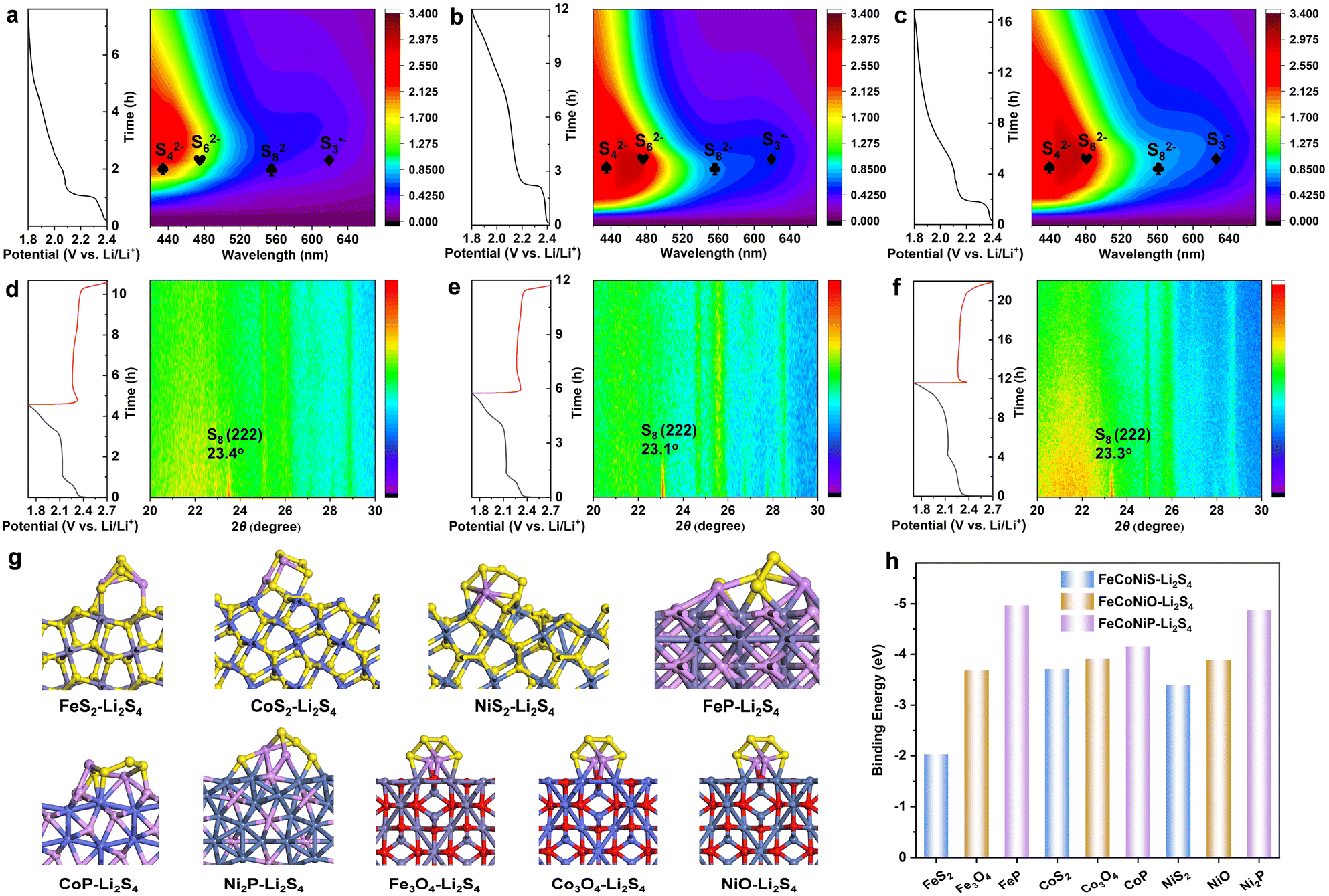 | ||
| Fig. 5 Contour maps of in situ UV-vis spectra and the corresponding discharge profile of (a) FeCoNiO-2h-S, (b) FeCoNiS-2h-S, and (c) FeCoNiP-2h-S electrodes. Contour map of in situ XRD patterns and the GCD profile of (d) FeCoNiO-2h-S, (e) FeCoNiS-2h-S, and (f) FeCoNiP-2h-S electrodes. (g) The adsorption configurations of the Li2S4 molecule adsorbed on the main phases of FeCoNiO, FeCoNiS, and FeCoNiP. (h) Comparison of the binding energies of Li2S4 to Fe3O4, Co3O4, NiO, FeS2, CoS2, NiS2, FeP, CoP, and Ni2P. | ||
We used in situ UV-vis spectroscopy with a visual cuvette during discharging at 0.05C to deeply probe the catalytic mechanism associated with LiPS conversion. Various types of LiPS can be confirmed qualitatively and long-chain or short-chain LiPS can be distinctly discriminated.41,42 The FeCoNiO-2h-S, FeCoNiS-2h-S, and FeCoNiP-2h-S electrodes were involved in multiple redox reactions in Li–S batteries and formed S82−, S62−, S42− and S3˙−, with S42− and S3˙− as steady and predominant reaction intermediates (Fig. 5a–c).43 In the contour map, the concentration of different polysulfide intermediates was displayed in different colors, with shades closer to purple and red representing lower and higher concentrations, respectively. In contrast to FeCoNiO-2h-S and FeCoNiS-2h-S electrodes, the UV-vis spectrum of the FeCoNiP-2h-S electrode showed the highest level of S82− and S3˙− free radicals in the Li–S electrolyte, which indicated that FeCoNiP-2h-S can efficiently enhance the reaction kinetics of LiPS conversion. Moreover, the FeCoNiS-2h-S electrode exhibited a higher level of S62− and S3˙− concentrations than FeCoNiO-2h-S, indicating a faster catalytic reaction from S62− to 2S3˙−. Therefore, the catalytic ability followed the order: FeCoNiP-2h > FeCoNiS-2h > FeCoNiO-2h. This was consistent with the observed performance order, indicating that the catalytic ability was more dominant than the adsorption ability. Overall, FeCoNiP offered enormous promise as a sulfur host for Li–S batteries, which originated from its high electrical conductivity, excellent LiPS adsorption and catalytic capabilities. The in situ XRD patterns of FeCoNiP-2h-S during the discharge process at 0.05C are displayed in Fig. 5f, in which the peak at 23.3° was assigned to the S8 molecule.44 With the increase of discharge depth, the peak intensity decreased. FeCoNiO-2h-S and FeCoNiS-2h-S exhibited similar in situ XRD patterns to FeCoNiP-2h-S (Fig. 5d and e).
Conclusion
In summary, FeCoNiO, FeCoNiS and FeCoNiP nanomaterials were synthesized systematically, and their effects on the adsorption and catalytic ability of sulfur cathodes were studied. Li2S4 adsorption tests and DFT calculations were used to study the adsorption ability, and in situ UV-vis spectroscopy and CV tests were used to study the catalytic ability. The reaction mechanism operating in the Li–S battery during the GCD process was investigated using in situ XRD. When utilized as a sulfur carrier, FeCoNiP was found to serve as a LiPS immobilizer and a catalyst to deliver superior electrochemical performance in terms of cycling stability and rate properties. In addition, according to the study of adsorption and catalytic strength, it is found that catalytic ability plays a leading role in determining battery performance. This work reveals the relationship between anionic species and performance, which provides a valuable reference for the selection of sulfur host materials. Additionally, the results of this study show that the construction of metal phosphides from MOFs is an instructive strategy for designing high-performance Li–S batteries with high conductivity, strong binding, and fast kinetics, which would bring about gigantic prospects for the development of energy storage systems.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Meng Du: experiment, investigation, data curation, and writing original draft. Jiakang Shi and Yuxiao Shi: formal analysis. Guangxun Zhang: methodology. Yan Yan: investigation. Pengbiao Geng: validation and review & editing. Ziqi Tian: software. Huan Pang: conceptualization and review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52371240) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX22_3460).Notes and references
- C. Zhou, Z. Li, X. Xu and L. Mai, Natl. Sci. Rev., 2021, 8, nwab055 CrossRef CAS PubMed.
- T. Chen, F. Wang, S. Cao, Y. Bai, S. Zheng, W. Li, S. Zhang, S. Hu and H. Pang, Adv. Mater., 2022, 34, 2201779 CrossRef CAS PubMed.
- N. Wang, X. Zhang, Z. Ju, X. Yu, Y. Wang, Y. Du, Z. Bai, S. Dou and G. Yu, Nat. Commun., 2021, 12, 4519 CrossRef CAS PubMed.
- M. Du, Q. Li, Y. Zhao, C.-S. Liu and H. Pang, Coord. Chem. Rev., 2020, 416, 213341 CrossRef CAS.
- T. Wang, J. He, Z. Zhu, X. Cheng, J. Zhu, B. Lu and Y. Wu, Adv. Mater., 2023, 35, 2303520 CrossRef CAS PubMed.
- G. Zhou, H. Chen and Y. Cui, Nat. Energy, 2022, 7, 312–319 CrossRef CAS.
- T. Chen, Z. Zhang, B. Cheng, R. Chen, Y. Hu, L. Ma, G. Zhu, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139, 12710–12715 CrossRef CAS PubMed.
- P. Geng, M. Du, C. Wu, T. Luo, Y. Zhang and H. Pang, Inorg. Chem. Front., 2022, 9, 2389–2394 RSC.
- H. Peng, G. Zhang, X. Chen, Z. Zhang, W. Xu, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- H. Xu, Q. Jiang, K. S. Hui, S. Wang, L. Liu, T. Chen, Y. Zheng, W. F. Ip, D. A. Dinh, C. Zha, Z. Lin and K. N. Hui, ACS Nano, 2024, 18, 8839–8852 CrossRef CAS PubMed.
- Y. Sun, J. Wang, T. Shang, Z. Li, K. Li, X. Wang, H. Luo, W. Lv, L. Jiang and Y. Wan, Angew. Chem., Int. Ed., 2023, 62, e202306791 CrossRef CAS PubMed.
- F. Liu, G. Sun, H. Bin Wu, G. Chen, D. Xu, R. Mo, L. Shen, X. Li, S. Ma, R. Tao, X. Li, X. Tan, B. Xu, G. Wang, B. S. Dunn, P. Sautet and Y. Lu, Nat. Commun., 2020, 11, 5215 CrossRef CAS PubMed.
- J. Wu, T. Ye, Y. Wang, P. Yang, Q. Wang, W. Kuang, X. Chen, G. Duan, L. Yu, Z. Jin, J. Qin and Y. Lei, ACS Nano, 2022, 16, 15734–15759 CrossRef CAS PubMed.
- Z. Ye, Y. Jiang, L. Li, F. Wu and R. Chen, Adv. Mater., 2020, 32, 2002168 CrossRef CAS PubMed.
- G. Chen, Y. Li, W. Zhong, F. Zheng, J. Hu, X. Ji, W. Liu, C. Yang, Z. Lin and M. Liu, Energy Storage Mater., 2020, 25, 547–554 CrossRef.
- C. Liu, Y. Bai, W. Li, F. Yang, G. Zhang and H. Pang, Angew. Chem., Int. Ed., 2022, 61, e202116282 CrossRef CAS PubMed.
- S. Zheng, Y. Sun, H. Xue, P. Braunstein, W. Huang and H. Pang, Natl. Sci. Rev., 2022, 9, nwab197 CrossRef CAS PubMed.
- Y. Chen, W. Zhang, D. Zhou, H. Tian, D. Su, C. Wang, D. Stockdale, F. Kang, B. Li and G. Wang, ACS Nano, 2019, 13, 4731–4741 CrossRef CAS PubMed.
- J. Xie, L. Ma, J. Li, X. Yin, Z. Wen, Y. Zhong, C. Li, Y. Liu, Z. Shen, W. Mai, G. Hong and W. Zhang, Adv. Mater., 2022, 34, 2205625 CrossRef CAS PubMed.
- G. Zhang, Y. Li, X. Xiao, Y. Shan, Y. Bai, H.-G. Xue, H. Pang, Z. Tian and Q. Xu, Nano Lett., 2021, 21, 3016–3025 CrossRef CAS PubMed.
- D. Su, M. Cortie, H. Fan and G. Wang, Adv. Mater., 2017, 29, 1700587 CrossRef PubMed.
- P. Geng, L. Wang, M. Du, Y. Bai, W. Li, Y. Liu, S. Chen, P. Braunstein, Q. Xu and H. Pang, Adv. Mater., 2022, 34, 2107836 CrossRef CAS PubMed.
- J. Ge, L. Fan, A. M. Rao, J. Zhou and B. Lu, Nat. Sustain., 2021, 5, 225–234 CrossRef.
- H. Xu, Q. Jiang, B. Zhang, C. Chen and Z. Lin, Adv. Mater., 2020, 32, 1906357 CrossRef CAS PubMed.
- F. Li, M. R. Kaiser, J. Ma, Z. Guo, H. Liu and J. Wang, Energy Storage Mater., 2018, 13, 312–322 CrossRef.
- H. Jiang, X. Liu, Y. Wu, Y. Shu, X. Gong, F. Ke and H. Deng, Angew. Chem., Int. Ed., 2018, 57, 3916–3921 CrossRef CAS PubMed.
- M. Du, P. Geng, C. Pei, X. Jiang, Y. Shan, W. Hu, L. Ni and H. Pang, Angew. Chem., Int. Ed., 2022, 61, e202209350 CrossRef CAS PubMed.
- Z. Liang, C. Qu, W. Zhou, R. Zhao, H. Zhang, B. Zhu, W. Guo, W. Meng, Y. Wu, W. Aftab, Q. Wang and R. Zou, Adv. Sci., 2019, 6, 1802005 CrossRef PubMed.
- Y. Deng, Y. Cao, Y. Xia, X. Xi, Y. Wang, W. Jiang, D. Yang, A. Dong and T. Li, Adv. Energy Mater., 2022, 12, 2202394 CrossRef CAS.
- Y. Zhong, L. Yin, P. He, W. Liu, Z. Wu and H. Wang, J. Am. Chem. Soc., 2018, 140, 1455–1459 CrossRef CAS PubMed.
- R. Sun, Y. Bai, M. Luo, M. Qu, Z. Wang, W. Sun and K. Sun, ACS Nano, 2021, 15, 739–750 CrossRef CAS PubMed.
- C. Guo, Y. Zheng, J. Ran, F. Xie, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2017, 56, 8539–8543 CrossRef CAS PubMed.
- Y. Guo, J. Tang, Z. Wang, Y.-M. Kang, Y. Bando and Y. Yamauchi, Nano Energy, 2018, 47, 494–502 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- J. Yu, J. Xiao, A. Li, Z. Yang, L. Zeng, Q. Zhang, Y. Zhu and L. Guo, Angew. Chem., Int. Ed., 2020, 59, 13071–13078 CrossRef CAS PubMed.
- W. Xia, Y. Chen, M. Han, X. Wu, H. Yang, K. Fu, M. Chen, X. Wang and H. Shu, Adv. Funct. Mater., 2024, 2400262 CrossRef.
- C. Zhang, J. J. Biendicho, T. Zhang, R. Du, J. Li, X. Yang, J. Arbiol, Y. Zhou, J. R. Morante and A. Cabot, Adv. Funct. Mater., 2019, 29, 1903842 CrossRef.
- D. Yang, C. Zhang, J. J. Biendicho, X. Han, Z. Liang, R. Du, M. Li, J. Li, J. Arbiol, J. Llorca, Y. Zhou, J. R. Morante and A. Cabot, ACS Nano, 2020, 14, 15492–15504 CrossRef CAS PubMed.
- Y. Wu, N. Wu, X. Jiang, S. Duan, T. Li, Q. Zhou, M. Chen, G. Diao, Z. Wu and L. Ni, Inorg. Chem., 2023, 62, 15440–15449 CrossRef CAS PubMed.
- Y. Liu, Z. Ma, G. Yang, Z. Wu, Y. Li, J. Gu, J. Gautam, X. Gong, A. N. Chishti, S. Duan, C. Chen, M. Chen, L. Ni and G. Diao, Adv. Funct. Mater., 2022, 32, 2109462 CrossRef CAS.
- Y. Tsao, M. Lee, E. C. Miller, G. Gao, J. Park, S. Chen, T. Katsumata, H. Tran, L.-W. Wang, M. F. Toney, Y. Cui and Z. Bao, Joule, 2019, 3, 872–884 CrossRef CAS.
- N. Xu, T. Qian, X. Liu, J. Liu, Y. Chen and C. Yan, Nano Lett., 2017, 17, 538–543 CrossRef CAS PubMed.
- L. Ni, J. Gu, X. Jiang, H. Xu, Z. Wu, Y. Wu, Y. Liu, J. Xie, Y. Wei and G. Diao, Angew. Chem., Int. Ed., 2023, 62, e202306528 CrossRef CAS PubMed.
- P. Chen, T. Wang, D. He, T. Shi, M. Chen, K. Fang, H. Lin, J. Wang, C. Wang and H. Pang, Angew. Chem., Int. Ed., 2023, 62, e202311693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary data. See DOI: https://doi.org/10.1039/d4sc01628a |
| This journal is © The Royal Society of Chemistry 2024 |