
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light TiO2-catalyzed synthesis of dihydrobenzofurans by oxidative [3 + 2] annulation of phenols with alkenyl phenols†
Jingze
Wu
 ,
Yaning
Liu
and
Marisa C.
Kozlowski
*
,
Yaning
Liu
and
Marisa C.
Kozlowski
*
Department of Chemistry, Roy and Diana Vagelos Laboratories, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: marisa@sas.upenn.edu
First published on 28th March 2024
Abstract
An oxidative strategy for the preparation of dihydrobenzofurans via heterogeneous photocatalysis is reported. This method leverages the surface interaction between the alkenyl phenol and the TiO2 solid surface, which enables direct activation by visible light without the need for pre-functionalization or surface modification. The resulting alkenyl phenoxyl radical is proposed to be selectively captured by a neutral phenol nucleophile, rendering β-5′ coupling with excellent chemo- and regio-selectivity. The reaction proceeds under benign conditions, using an inexpensive, nontoxic, and recyclable photocatalyst under visible light irradiation with air as the terminal oxidant at room temperature.
Introduction
Phenols and their derivatives function as essential building blocks for various classes of complex molecules, playing crucial roles in biological systems.1 Lignans, for example, are phenol-derived natural products abundant in nature that exhibit unique structural patterns and versatile biological functions, sparking considerable attention toward their controlled synthesis. Within this class, neo-lignans derived from alkenyl phenols comprise a range of natural products (Fig. 1).2 Such structures arise from oxidation of alkenyl phenols by peroxidases or laccases to the radical intermediates that are stabilized through resonance (Scheme 1).3 Subsequently, radical couplings among these resonance forms have the potential to generate a diverse array of distinct structures via selective dimerization. In recent years, radical-based reactions have gained prominence as powerful tools for bond formation. And in this context, the reaction between alkenyl phenoxyl radicals and neutral alkenyl phenol nucleophiles offers intriguing opportunities for the construction of lignan homodimers. In 2019, the Kozlowski group reported a vanadium catalyzed homo-coupling of alkenyl phenols to selectively generate β–β and β-O homodimers.4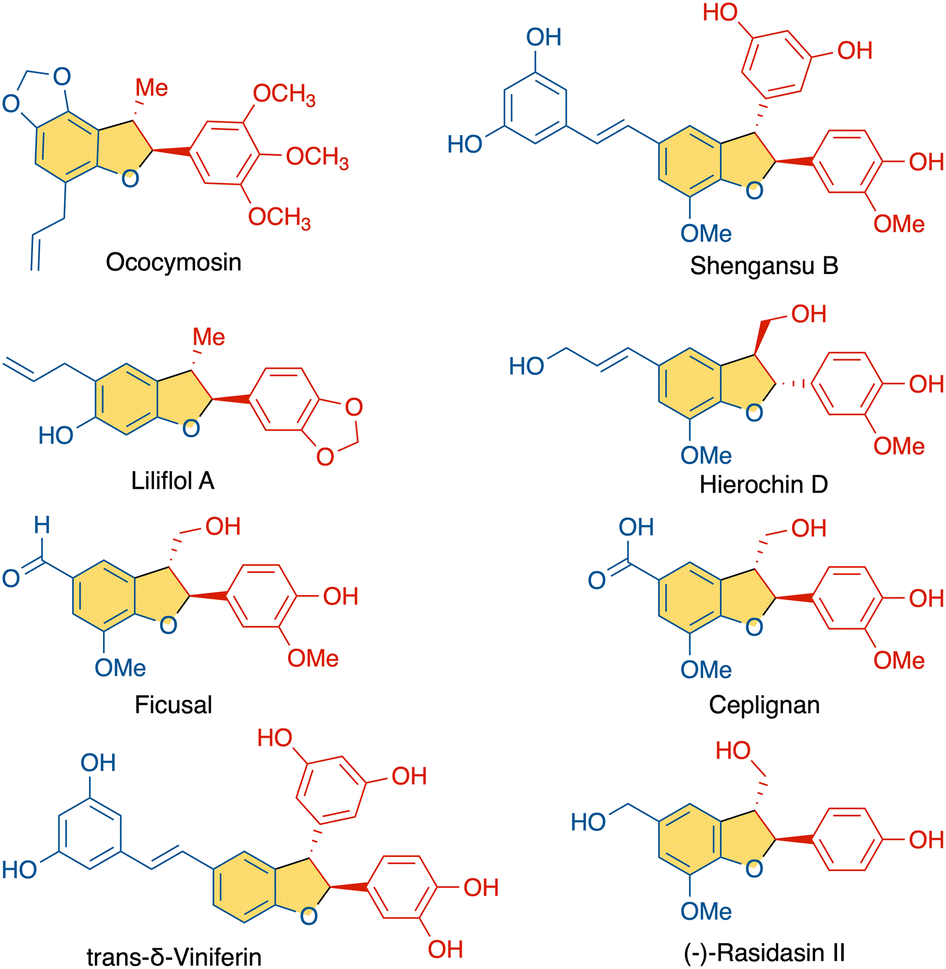 | ||
| Fig. 1 Naturally occurring β-5′ neolignans (selected examples). | ||
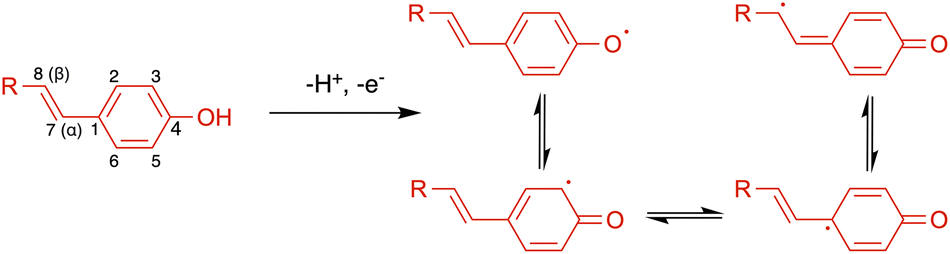 | ||
| Scheme 1 Resonance forms of phenoxyl radical from by one-electron oxidation. | ||
Within the realm of lignan scaffolds, the heterodimers possessing a dihydrobenzofuran core through β-5′ coupling stand out as subjects of significant interest due to a diverse spectrum of bioactivities, encompassing antioxidant, anti-inflammatory, antiparasitic, neuroprotective, and anticancer activities, among others.5 As a highly reactive species, the alkenyl phenoxyl radical possesses the potential to engage in selective C–C bond formation with neutral phenol partners. Achieving such selectivity, however, remains challenging due to the inherent complexity of radical reactions and the competing pathways of homo-coupling versus cross-coupling.6
Previous attempts for selective generation of β-5′ lignan heterodimers have been reported by means of enzymatic processes or bio-mimicking strategies.7 Recently, cross-couplings of alkenyl anisoles and phenols for selective β-5′ coupling have been reported by several groups (Scheme 2a–c), however, these reported methods require use of O-protected alkenyl anisoles.8–11 Furthermore, the accessibility of bespoke covalent organic framework (COF) catalysts poses a challenge for general use (Scheme 2c).
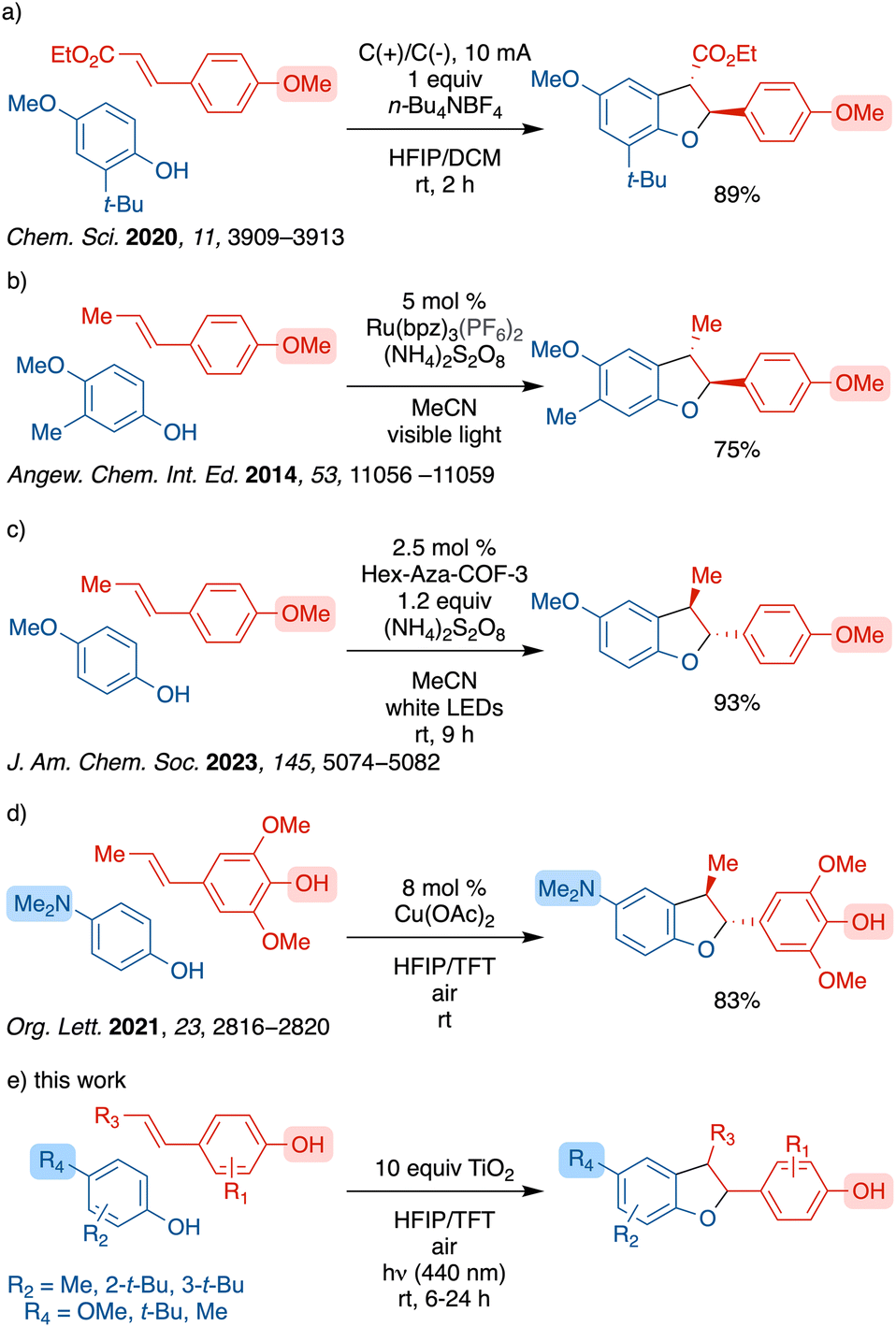 | ||
| Scheme 2 Previous strategies for lignan dihydrobenzofuran synthesis, (a) electrochemical method, (b) photocatalytic method using a ruthenium photocatalyst, (c) photocatalytic method using a COF catalyst, (d) copper acetate method, (e) this work. | ||
Selective cross-coupling of alkenyl phenols with other alkenyl phenols or with phenols is challenging due to several reactive sites as shown in Scheme 1 and the potential for homo-coupling of either partner. As such there are limited reports of such coupling. One approach to this challenge has been the use of one far more oxidizable partner such as a para-aminophenol as reported by the Liu group using catalytic copper acetate (Scheme 2d).12 However, there has been no general selective cross-couplings of alkenyl phenols in less activated systems.
The application of titanium dioxide (TiO2) as a photocatalyst is much less common in organic synthesis relative to metalorganic and organic photocatalysts. Most TiO2 catalyzed reactions typically utilize UV light, employ an adsorbate as the sensitizer to improve visible light absorption, or apply surface doping to lower the band gap.13 In contrast, there are limited examples where the substrate enables visible light absorption by forming an LMCT complex. Heteroatom-bearing substrates, such as amines,14 sulfides,15 diazoniums16 and benzyl alcohols,17 have been shown to form such LMCT complexes with unmodified TiO2 to achieve oxidation. However, TiO2–LMCT complexes with phenolic substrates remain underexplored.18 In 2023, we reported an oxidative synthesis of diphenols through a heterogeneous titanium dioxide (TiO2) photocatalyst where the phenol served a dual role as both the substrate and the photosensitizer.19 Further, we discovered that the dihydrobenzofuran core of the iso-eugenol homodimer, licarin A, can be selectively formed using TiO2 under blue light irradiation with excellent yield and stereoselectivity.19 We hypothesized that an interaction of the hydroxy group of the alkenyl phenol with TiO2 would enable selective generation of the lignan dihydrobenzofuran cores through cross-coupling between alkenyl phenols and phenols with a broader functional group tolerance. Herein, we report the first TiO2 photocatalyzed selective oxidative β-5′ cross-coupling of alkenyl phenols and phenols for the generation of lignan dihydrobenzofuran cores.
Results and discussion
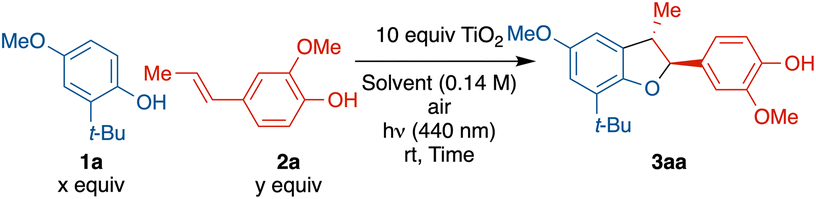
Exploration of the cross-coupling reaction between phenols and alkenyl phenols began with the readily oxidized substrate and easily available iso-eugenol (2a) with 2-tert-butyl-4-methoxyphenol (1a) (see in ESI† for full optimization table). An excess of TiO2 powder (10.0 equiv.) was used considering the limited surface area as reactions will only occur on the TiO2 solid surface (0.1 equiv. of TiO2 gave <10% conversion after 12 h). The exceptionally low cost and facile recyclability of TiO2 by centrifugation offsets this high loading. Hexafluoroisopropanol (HFIP) was used as it generally facilitates phenolic couplings.20–26 Initial reaction began with an excess of 2a (3.0 equiv.), however, only 2% of cross coupled 3aa was isolated, and 79% of 2a underwent homocoupling to generate licarin A, and 83% of the unreacted 1a was recovered (entry 1). Reducing the equivalence of 2a from 3.0 to 1.5 improved the yield of heterodimer 3aa significantly, from 2% to 32%, and 63% of the unreacted 1a was recovered (entry 2). Doubling the reaction time from 6 to 12 h only slightly increased the yield, from 32% to 38% (entry 3). Further reduction from 1.5 to 1.0 equivalents of 2a improved the yield from 32% to 42% (entry 4). A significant improvement in yield of 3aa (59%) was observed with an excess 1a (2.0 equiv.) (entry 5). Use of an HFIP/MeCN mixture offered no advantage (entry 6) but use of HFIP with trifluorotoluene (TFT) did improve the yield (entry 7) with the balance of limiting reagent 2a forming homodimer. Finally, use of an even greater excess of 1a (5.0 equiv.) was not effective (entry 8). Longer reaction times with optimal conditions (entry 7) resulted in overoxidation products (see Fig. S25 and S26†). Examination of a homogenous photocatalyst (MesAcr+BF4−), in analogy to the work of Yoon and coworkers,9 resulted in 14% yield with poor chemo-selectivity and poor mass balance (see ESI† for details).Control experiments without light (Table 2, entry 1), air/oxygen (entry 2), or TiO2 (entry 3) all failed to provide any product, showing all three components are necessary.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | 1a (x equiv.) | 2a (y equiv.) | Time (h) | 2a homodimerb (%) | Yield 3aab (%) |
| a Reaction conditions: 1a (0.56 mmol), 4.0 mL solvent, 6–12 h. b Isolated yield. | ||||||
| 1 | HFIP | 1.0 | 3.0 | 6 | 81 | 2 |
| 2 | HFIP | 1.0 | 1.5 | 6 | 50 | 32 |
| 3 | HFIP | 1.0 | 1.5 | 12 | 53 | 38 |
| 4 | HFIP | 1.0 | 1.0 | 6 | 46 | 42 |
| 5 | HFIP | 2.0 | 1.0 | 6 | 24 | 59 |
| 6 | HFIP/MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
2.0 | 1.0 | 6 | 19 | 51 |
| 7 |
HFIP/TFT (1:1) |
2.0 | 1.0 | 6 | 21 | 66 |
| 8 | HFIP/TFT (1:1) |
5.0 | 1.0 | 6 | 14 | 37 |
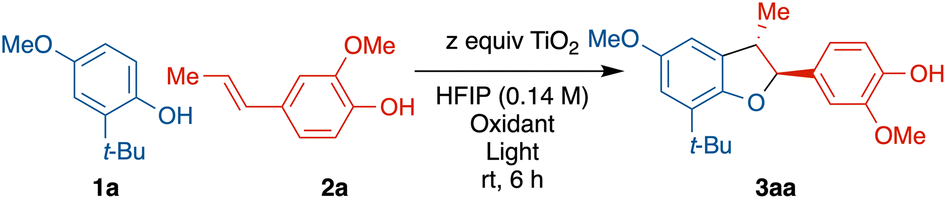
With the optimized conditions established, we then proceeded to evaluate the scope of this protocol (Scheme 3). Gram-scale synthesis using the same light source provided 3aa in a lower yield (39%), likely due to limited light penetration of the larger vessel. For the phenol component 1, use of a smaller ortho-group (Me vs. t-Bu) led to 3ca with a similar yield (57%) relative to 3aa (66%). In comparison to prior work (Scheme 2d) which required very electron-rich phenols, this method was successful with electron-neutral phenols, albeit with lower yields (comparing 3db 24% with 3ab 71% and 3eb 32% with 3cb 60%). meta-Substitution of the phenol in 3bb was also tolerated but resulted in lower yield (43%) compared to the ortho-congener 3ab (71%). N-Acetyl indole derived phenols did not react whereas a carbazole derived phenol (1f) underwent rapid oxidation to multiple products, giving diminished yield of 3fb in 27% with a poor diastereomeric ratio. In addition, more complex phenols such as piperazine derived phenol, aryl substituted phenol and dibenzofuran derived phenol all showed reactivity toward cross coupling with an alkenyl phenol, giving 3eg (65%), 3ig (88%) and 3hg (38%).
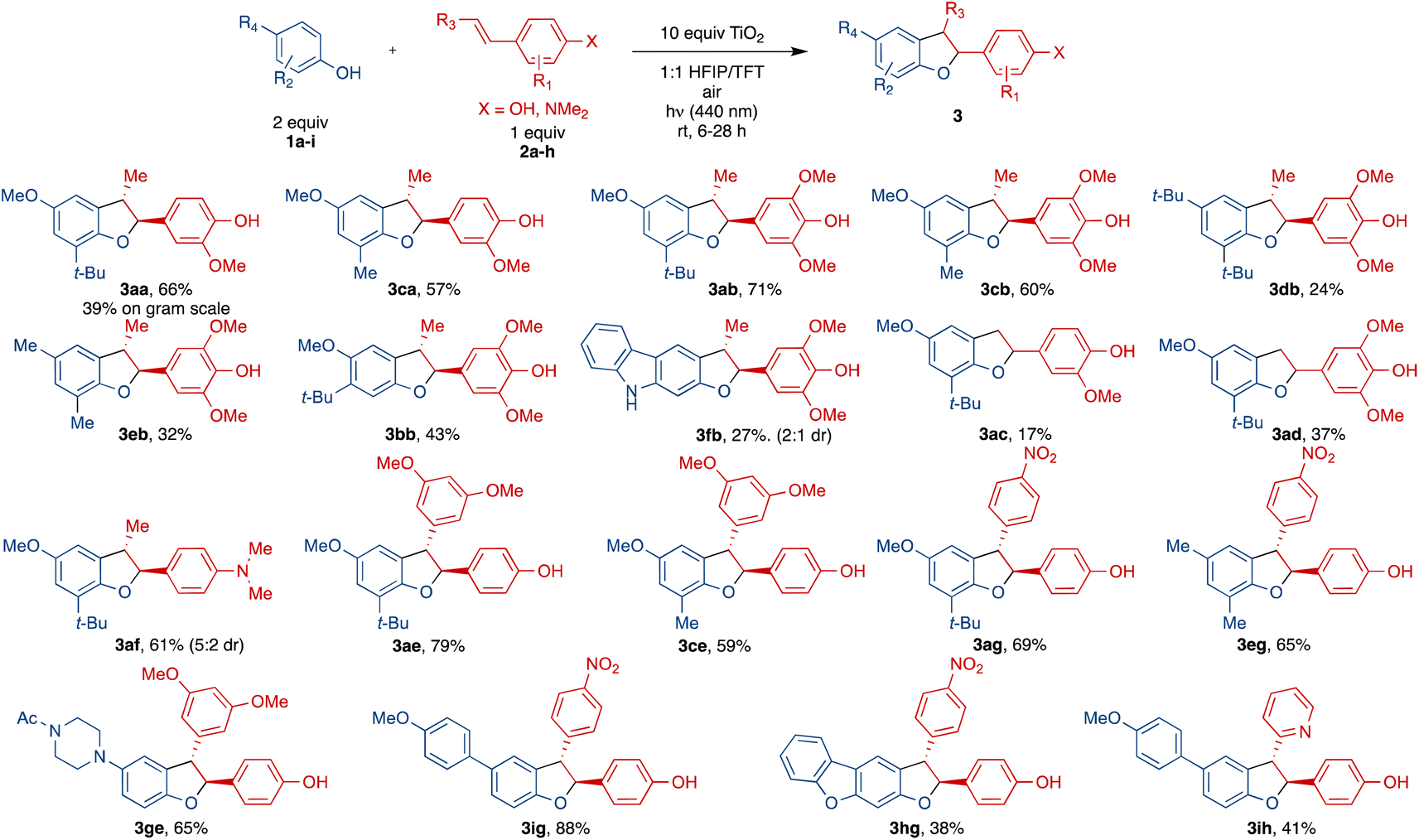 | ||
| Scheme 3 Substrate scope. See ESI† for reaction conditions. | ||
For the aryl ring of alkenyl phenol component 2, additional methoxy substitution in 3ab (71%) and 3cb (60%) led to slightly improved yields relative to 3aa (66%) and 3ca (57%). Replacing the phenol of this portion entirely with a dimethylamino arene results in good yield of 3af (61%) relative to the methoxyphenol 3aa (66%) or the dimethoxyphenol 3ab (71%). For alkenyl phenol component 2, substitution on the alkenyl terminus exerted a profound influence on the reaction outcomes. Specifically, a lack of substitution as in 3ad and 3ac resulted in significantly diminished yields (37% and 17%). On the other hand, alkenes with a dimethoxyphenyl or a nitrophenyl substituent led to pronounced improvements, most likely due to greater stabilization of the intermediate radial, forming 3ae and 3ig in 79% and 88% yield, respectively. Notably, this method is complementary to that using Cu(OAc)2 (ref. 12) from Scheme 2d which gave no reactivity with the substrates for 3ig (see ESI† for details). Changing the electronic characteristics of the alkenyl phenyl substituent at the β-position from electron rich to electron poor such as 3ig, 3ag, 3eg and 3ih resulted in 88%, 69%, 65% and 41% yield. Other alkenyl phenols with alkyl substitution on the aromatic ring were not stable (decomposed upon storage at −20 °C) and gave complex mixtures with no cross-coupling observed (see ESI† for details).
Visible light activation of TiO2 can be achieved by means of surface interaction with adsorbates that do not absorb visible light themselves via ligand to metal charge transfer (LMCT) from the highest occupied molecular orbital (HOMO) of the adsorbates to the conduction band (CB) of TiO2.27 Next, the oxidized adsorbate may either undergo further reactions with an electron donor or regenerate the neutral adsorbate via back electron transfer (BET) (Fig. 2).28 In this reaction, photocatalytic reactivity was observed under 440 nm light irradiation, while neither reaction component exhibits visible light absorption.29 To explain this phenomenon, we hypothesized that the surface interaction between the TiO2 semiconductor and phenols plays a crucial role in phenol activation and the subsequent reactivity.
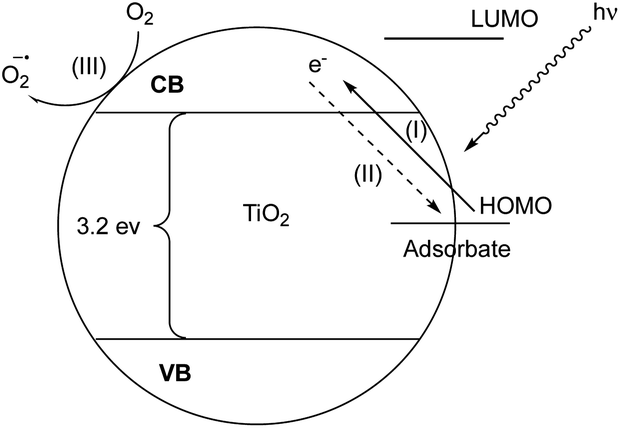 | ||
| Fig. 2 TiO2 visible-light LMCT sensitization: (I) visible-light induced LMCT transfer, (II) charge recombination, (III) electron transfer to a terminal oxidant, valence band (VB), conduction band (CB). | ||
Adding alkenyl phenol 2a to white TiO2 powder causes an immediate colour change indicating a red shift in the absorption and emission spectra (Fig. 3a). Fig. 3b shows the Raman spectra of 2a and TiO2–2a. The sharp and intense peak at 1265.7 cm−1 of 2a was assigned to the alkenyl phenol C–O and/or C–O–H vibrations in accord with prior literature measurements.30 Binding of 2a to the solid surface leads to a significant drop in intensity of this signal, indicating substantive structural changes around the hydroxy group which results in changes in polarity. The new intense peak at 1276.98 cm−1 in TiO2–2a was attributed to a Ti–O–C linkage.31 To further characterize the structural changes upon surface attachment, FT-IR spectra comparing 4-methylphenol and the TiO2-bound version were obtained (Fig. 3c). 4-Methylphenol was chosen instead of 2a as it gave a clearer signal for the O–H vibrations. Frequency analyses were performed [UB3LYP-D3/6-31G(d,p) see ESI† for details] for visualization and vibrational assignments. 4-Methylphenol shows an intense peak around 1222 cm−1, which was assigned to O–H bending vibrations. Upon surface binding, the broad peak around 1244 cm−1 was attributed to vibrations that resulted from a Ti–O–C linkage. The sharp peak around 1520 cm−1 for a methyl rotation did not undergo significant shifting upon surface binding. Similarly, the two adjacent peaks around 1600 cm−1 and 1630 cm−1 that were assigned to the symmetric and asymmetric stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds also remained unchanged. Combined, these findings indicate significant structural changes around the phenol hydroxy group, resulting from the formation of a Ti–O–C linkage, consistent with previous reports.31
C bonds also remained unchanged. Combined, these findings indicate significant structural changes around the phenol hydroxy group, resulting from the formation of a Ti–O–C linkage, consistent with previous reports.31
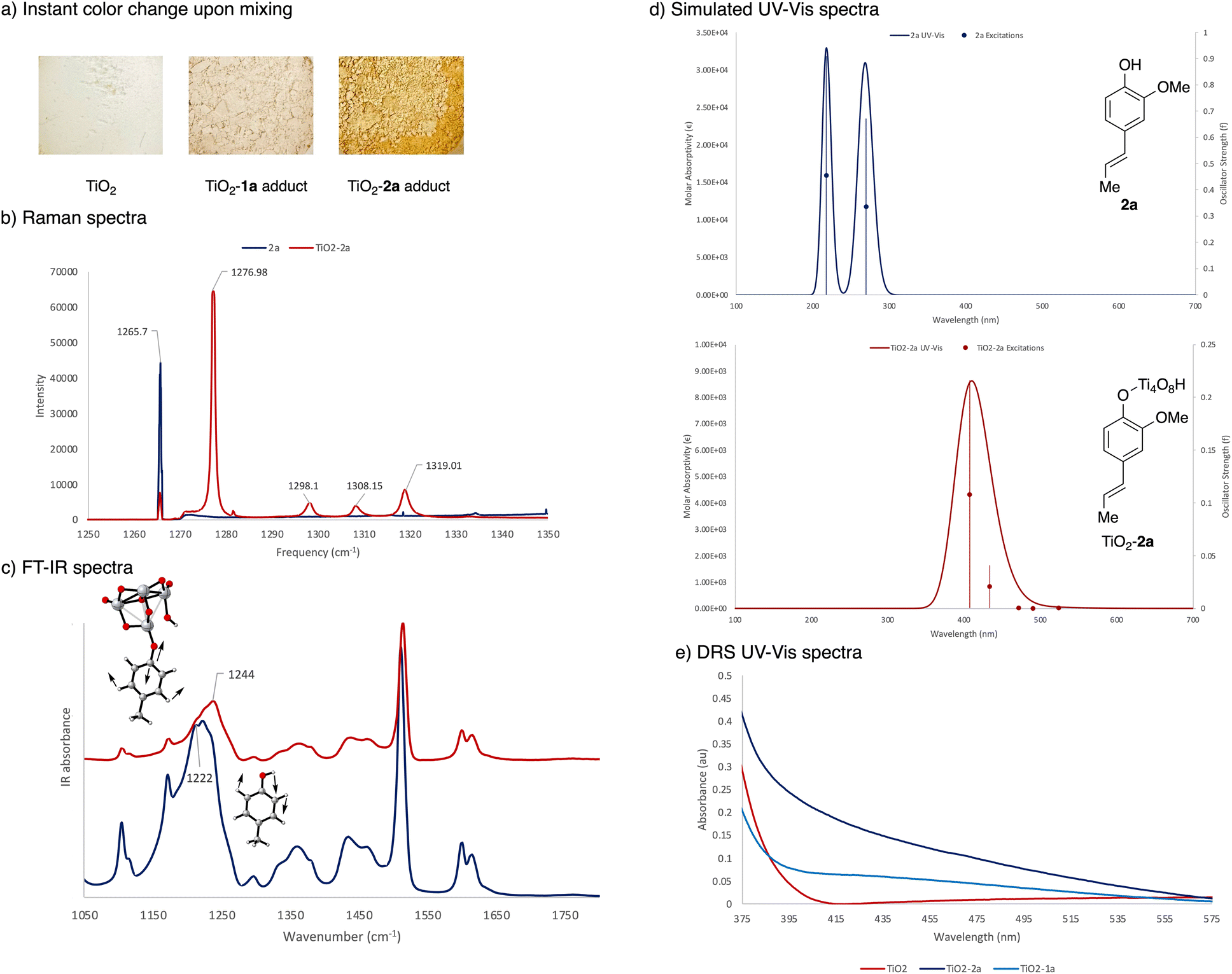 | ||
| Fig. 3 (a) Color change upon addition of 2a to TiO2, (b) Raman spectra comparing 2a and TiO2-bound 2a, (c) FT-IR spectra comparing 4-methylphenol and the TiO2-bound version, vibrational modes assigned using UB3LYP-D3/6-31G(d,p), (d) simulated UV-Vis spectra of 2a and TiO2-bound 2a generated using TD-UB3LYP-D3/6-31G(d,p)-SDD(Ti), (e) Kubelka–Munk transformations of diffuse reflection data of 2a and TiO2-bound 2a. | ||
Electronic excitation spectra and spatial electron distribution profiles were generated using TD-DFT [TD-UB3LYP-D3/6-31G(d,p)-SDD(Ti), see ESI† for details] with an anatase Ti tetramer cluster model complex.32 The TiO2 cluster was frozen during geometry optimization to maintain the anatase structure while all other atoms were allowed to relax. The simulated electronic excitation spectra in Fig. 3d shows two major excitations in the UVB and UVC region for unbound 2a. There is a significant red shift of this signal upon formation of an adduct with TiO2, consistent with both the colour change observed in the reaction and with the experimental diffuse reflectance data (Fig. 3e). Notably, this new absorption band from 350–525 nm coincides with the wavelength of light (440 nm Kessil lamp) used in the photocatalysis. In comparing the surface association of 1avs.2a, TiO2–2a showed a stronger absorption signal around 440 nm, consistent with the more intense color change observed, indicating that the TiO2–2a adduct is being activated preferentially under reaction conditions.
Fig. 4a shows the calculated energy levels of 2a and 1a, alongside with the valence and conduction band of TiO2.33 Both the photocatalyst and the substrates were found to exhibit wide band gap (3.2 eV) and HOMO–LUMO gaps (4.6 eV and 5.3 eV) outside of the visible range, excluding the possibility for direct excitation of either component. Aligned with our hypothesis, the energy gap between 2a HOMO and TiO2 conduction band is significantly lowered (1.5 eV) upon surface attachment, within the wavelength of light (440 nm Kessil lamp) used in the reaction.
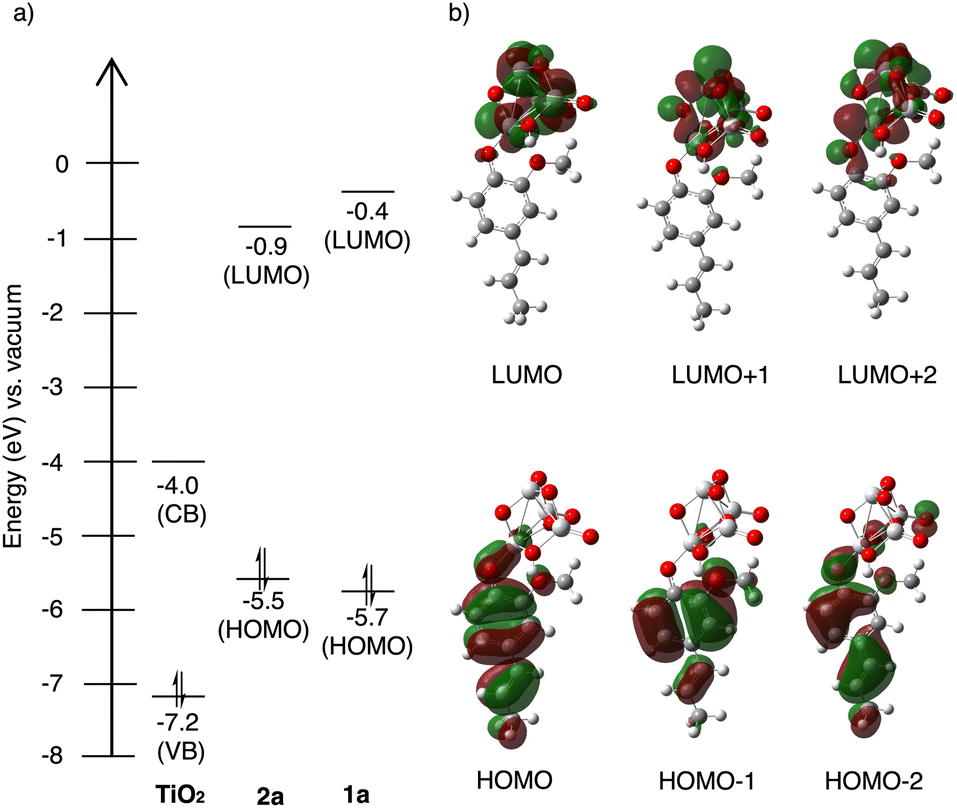 | ||
| Fig. 4 (a) energy level diagram of 2a and 1a calculated using RB3LYP/6-311+(d,p), alongside with the valence and conduction band of TiO2, (b) spatial electron distributions of HOMO, HOMO−1, HOMO−2, LUMO, LUMO+1, LUMO+2 of the TiO2–2a adduct calculated using TD-UB3LYP-D3/6-31G(d,p)-SDD(Ti). | ||
The spatial distribution of the highest, second, and third highest occupied molecular orbitals (HOMO, HOMO−1, HOMO−2), as well as the lowest, second, and third lowest unoccupied molecular orbitals (LUMO, LUMO+1, LUMO+2) in Fig. 4b, reveals the direction of the charge transfer. The electron density in HOMO, HOMO−1, and HOMO−2 is distributed throughout the alkenyl phenol π-conjugation. The LUMO, LUMO+1, and LUMO+2, which correspond to the conduction band of TiO2, on the other hand, have electron density predominantly concentrate on the TiO2 cluster moiety. Thus, it becomes clear that the photoinduced LMCT occurs across the interface joined by the alkenyl phenol HOMO and TiO2 conduction band.35 This surface attachment effectively bypasses the wide band gap of the semiconductor and allows for direct photoactivation by visible light, achieving similar effects as doping without the need for surface modification.
To gain further insights into the reaction mechanism and the observed selectivity, kinetics experiments and DFT calculations were undertaken (Fig. 5). In order to pinpoint which component is being activated predominately, decay of starting materials was monitored over the course of the reaction (Fig. 5a). After three hours, over half of 2a was consumed, whereas the concentration of 1a and 1b stayed mostly unchanged. After seven hours, full consumption of 2a was overserved, while more than 50% of 1a and 1b remained unreacted, consistent with the absence of 1a homodimer observed in the model reaction. As the low conversion efficiency in phenol systems was attributed to the fast back electron transfer (BET) in previous reports,34,35 the rapid charge recombination was believed to be responsible for the lack of reactivity in the phenol component.
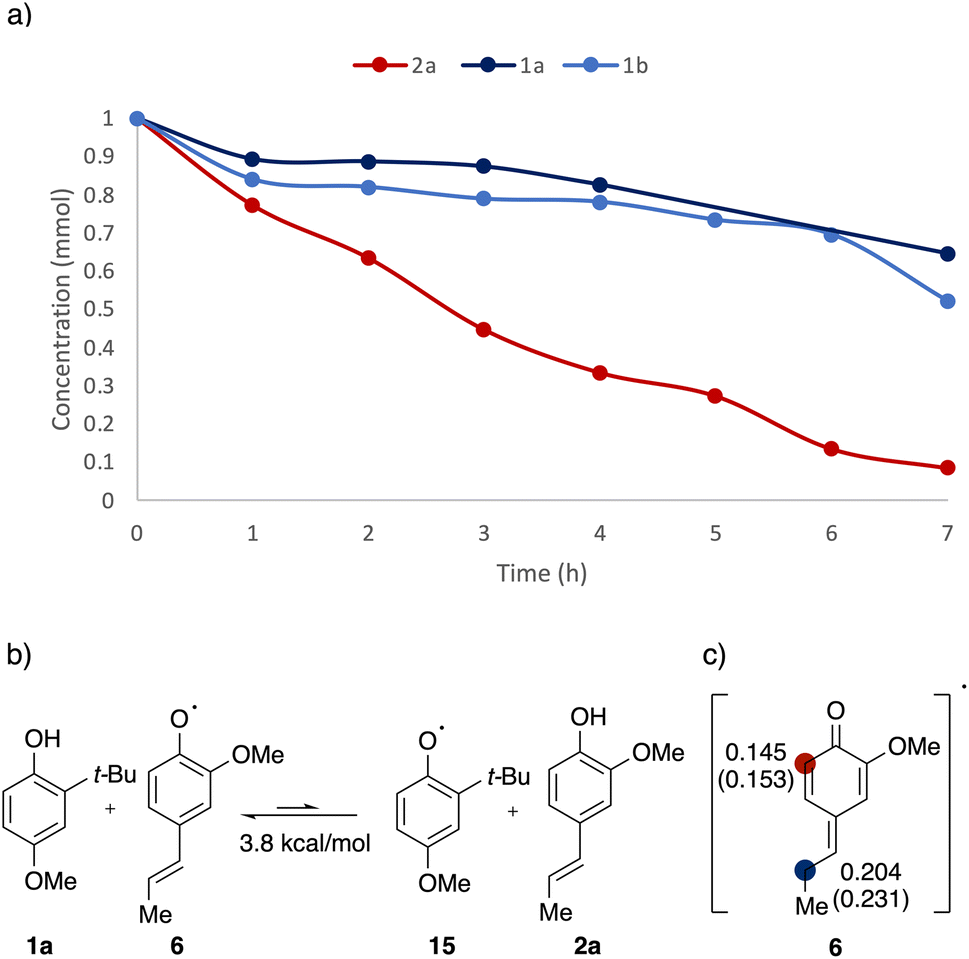 | ||
| Fig. 5 (a) Kinetics of 2avs.1avs.1b. Yield for 2a determined via UPLC using 4-ethylanisole as the internal standard; yield for 1a and 1b determined via UPLC using 1,3,5-trimethoxybenzene as the internal standard, (b) thermodynamic stability of 6vs.15. Free energies were computed using UM06/6-311++G(d,p)-CPCM(HFIP)//UB3LYP-D3/6-31G(d,p). (c) Spin density distribution of ortho- vs. β-sites calculated using population analysis. Numbers outside parentheses are derived from NBO spin, numbers inside are from Mulliken spin. | ||
The pronounced kinetic difference observed indicates a selectivity, that the alkenyl phenol (2a) was activated and underwent subsequent transformation with neutral phenol partners. Relative radical stability was used to rationalise the selectivity (Fig. 5b). The alkenyl phenoxyl radical 6 was found to be thermodynamically more stable by 3.8 kcal mol−1 than the comparative phenoxyl radical 15, most likely due to the extended conjugation enabled by the alkenyl chain. Combined, the kinetic evidence and DFT calculations confirmed that the TiO2–alkenyl phenol adduct was being photoactivated in the reaction, generating the reactive radical species.
Further investigation on spin density distribution was performed on the alkenyl phenoxyl radical 6 (Fig. 5c). Both NBO and Mulliken spin population indicated that the radical character is more concentrated on the β-carbon compared to the ortho-site, resulting in a regioselective coupling at the β-site.
To confirm the predicted regioselectivity, a radical trapping experiment was conducted (Fig. 6). Notably, the reaction was markedly inhibited by the addition of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). Upon surface binding and photoactivation, the resulting alkenyl phenoxyl radical delocalized to the β position was captured by TEMPO. The isolated adduct arising from nucleophilic addition of the solvent to the quinone methide intermediate was characterized, in support of a regioselective radical process. Further, the presence of peroxide formation was confirmed by a positive KI test (see ESI† for details).
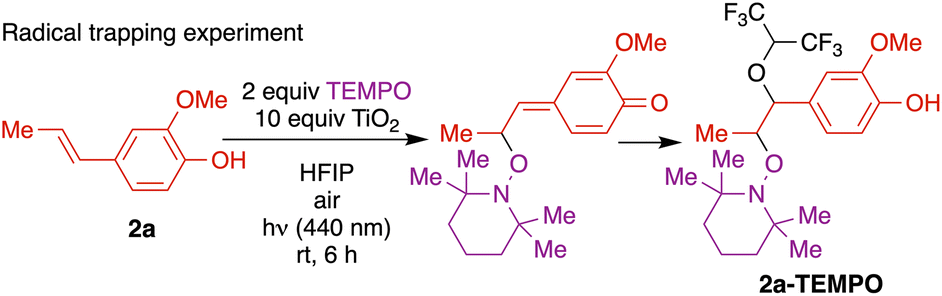 | ||
| Fig. 6 Mechanistic investigations: radical trapping experiment with TEMPO. | ||
To rationalize the absence of a β–β adduct from a potential radical–radical mechanism, the bond dissociation enthalpy (BDE) of the resulting di-para-quinone methide was calculated using DFT [UB3LYP-D3/6-31G(d,p), see ESI† for details]. The exceptionally weak β–β C–C bond (9.6 kcal mol−1) indicates the highly unstable nature of the β–β adduct, which could easily undergo rapid homolysis back to the corresponding phenoxyl radical 6.36
On the basis of the above mechanistic studies, a plausible catalytic cycle for this transformation was proposed in Scheme 4. First, the alkenyl phenol binds to the TiO2 solid surface through the hydroxy group, forming a Ti–O–C linkage.37 The resulting complex can then be directly activated under visible light via ligand-to-metal charge transfer (LMCT), where an electron is promoted from the alkenyl phenol HOMO to the TiO2 conduction band. The terminal oxidant, dioxygen, subsequently collects the extra electron from the TiO2 conduction band, producing superoxide. The regeneration of the photocatalyst is achieved via deprotonation of the titanium-hydroxo adduct by superoxide. The resulting alkenyl phenoxyl radical can then be captured by a neutral phenol nucleophile, forming the pivotal C–C bond. Subsequent hydrogen abstraction by the peroxyl radical rearomatizes the phenol scaffold. And finally, intramolecular nucleophilic attack to the quinone methide constructs the benzofuran ring, which, upon tautomerization affords the final cross-coupling product.
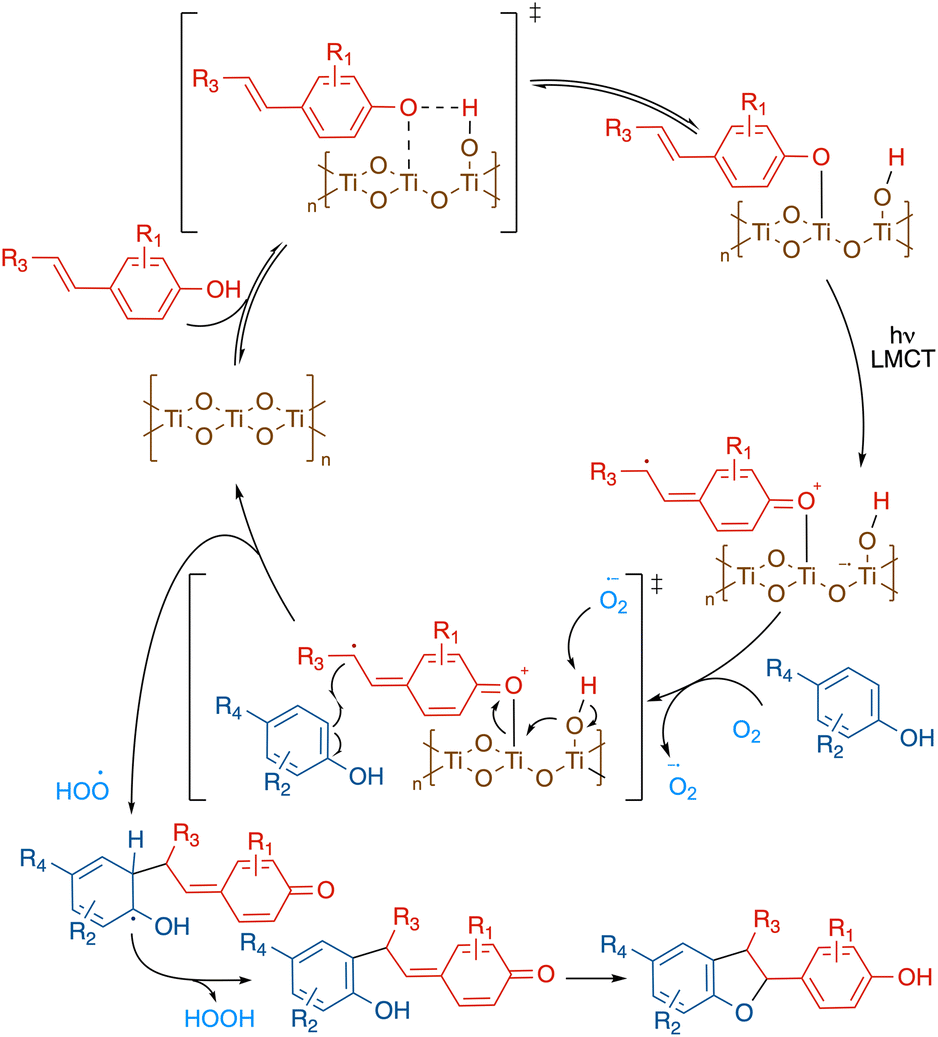 | ||
| Scheme 4 Proposed catalytic cycle. | ||
We deployed density functional theory (DFT) calculations [UM06/6-311++G(d,p)-SDD(Ti)-CPCM(HFIP)//UB3LYP-D3/6-31G(d,p)-SDD(Ti), see ESI† for details] to pinpoint key mechanistic steps and to unravel factors governing chemoselectivity. Specifically, the reaction between iso-eugenol (2a) and 2-tert-butyl-4-methoxyphenol (1a) were modeled to represent the general reaction scheme. Conformational searches were performed manually on each structure to locate the lowest energy profiles and only the lowest energy pathways are shown. First, we explored the selectivity in catalyst binding (Fig. 7a). The surface interaction occurs via a slightly-uphill transition state TS-E1 (4.2 kcal mol−1), where the bridging oxygen in TiO2 deprotonates substrate 2a while simultaneously forming a Ti–O bond in intermediate 5. Aided by the coordination of the adjacent methoxy group to the oxophilic Ti, the process was rendered overall downhill in energy (−17.3 kcal mol−1). Comparatively, binding of substrate 1a to TiO2viaTS-E2 was found to be overwhelmingly disfavored, both kinetically (9.9 kcal mol−1vs. 2.7 kcal mol−1) and thermodynamically (−12.2 kcal mol−1vs. −17.3 kcal mol−1), most likely due to the steric hindrance around the t-Bu group (see ESI† for details). Photoinduced LMCT of 5 generates the common intermediate alkenyl phenoxyl radical 6, which is uphill energetically (26.1 kcal mol−1) consistent with the lack of reactivity observed in the absence of light (Table 2, entry 1).
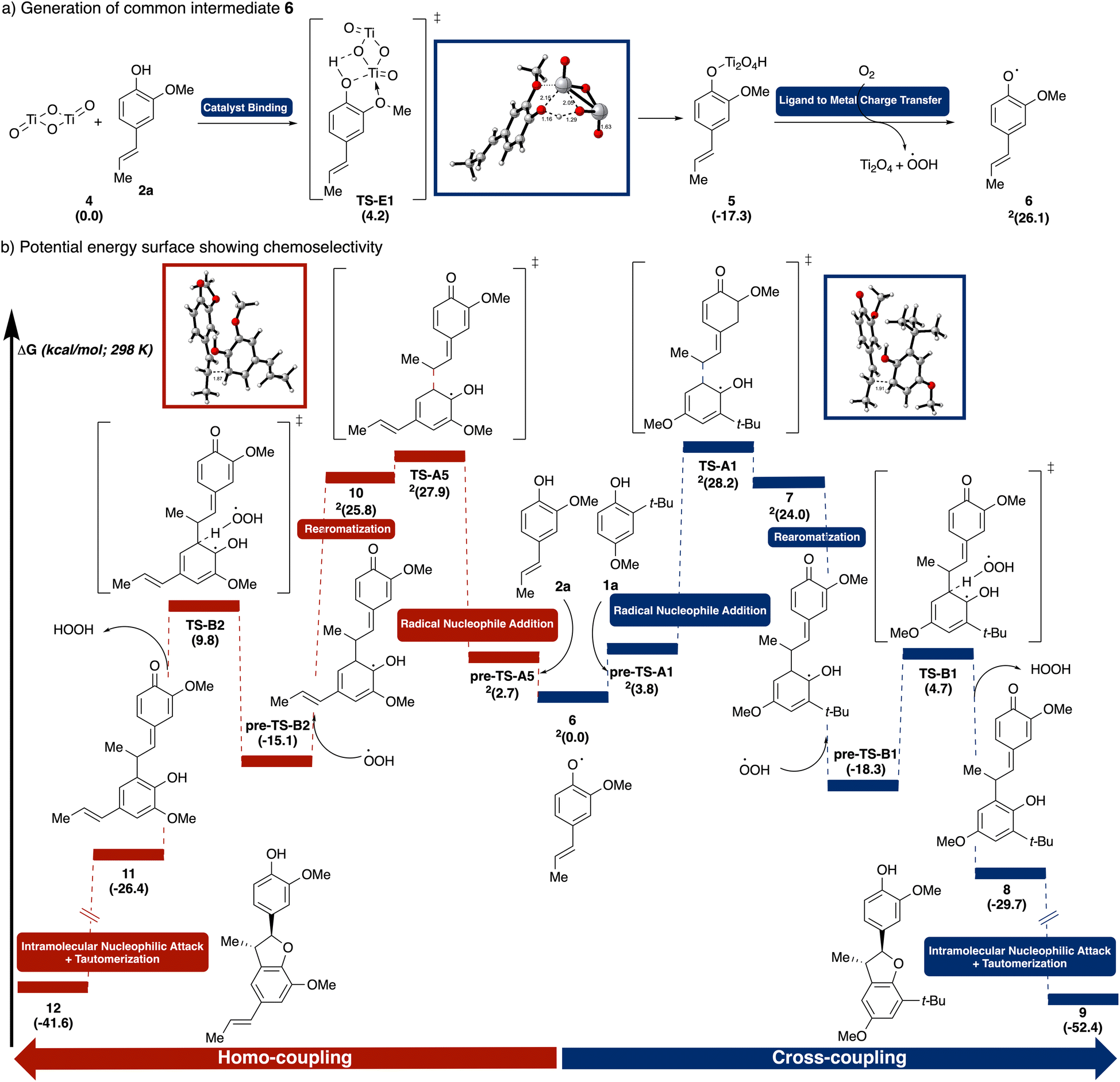 | ||
| Fig. 7 (a) Formation of common intermediate 6, (b) formation of cross-coupling and homo-coupling products from 6. Free energies were computed using UM06/6-311++G(d,p)-SDD(Ti)-CPCM(HFIP)//UB3LYP-D3/6-31G(d,p)-SDD(Ti). | ||
Next, from the common intermediate 6, we investigated the selectivity in coupling (Fig. 7b). Both homo-coupling and cross-coupling pathways were computed as both were observed experimentally. Nucleophilic addition from the ortho-position of 1a to the resonance-stabilized allyl radical 6 proceeds via transition state TS-A1 forming the β-5′ C–C bond. This transition state is stabilized by a CH–π interactions between the ortho–tert–butyl group of 1a and the aromatic ring of 6.
In comparing the cross-coupling and homo-coupling pathways, both the transition states, TS-A1 (28.2 kcal mol−1) and TS-A5 (27.9 kcal mol−1), and the resulting intermediates, 7 (24.0 kcal mol−1) and 10 (25.8 kcal mol−1), were found to be close in energy. This finding is consistent with the product mixture observed in the 1:1 1a:2a reaction (Table 1, entry 4). Rearomatization via peroxyl radical mediated hydrogen atom abstraction of 7viaTS-B1 forms the more stable 8 (−29.7 kcal mol−1). Subsequent intramolecular attack by the phenol to the quinone methide followed by tautomerization in product 9 drives the energy further downhill (−52.4 kcal mol−1).
To further validate the computational model on product distribution prediction, the key coupling step for another phenol/alkenyl phenol pair was computed. Specifically, the coupling between pterostilbene (2e) and 2-tert-butyl-4-methoxyphenol (1a) was chosen as the reaction afforded only cross-coupling product (see ESI† for details). The absence of homo-coupling can be rationalized via both the higher activation barrier (37.8 kcal mol−1) and the higher product free energy (36.6 kcal mol−1) for the homo-coupling.
As radical–nucleophile addition was identified to be the product determining step, we hypothesized that the cross-coupling chemoselectivity would be improved by employing an excess of 1a. For 1a, the homo-coupling pathway is far slower due to both less favorable initial coordination to the TiO2 and faster BET (see above). Indeed, with a 2:1 ratio of 1a:2a, cross-coupling yield improved significantly (Table 1, entry 5 vs. 4).
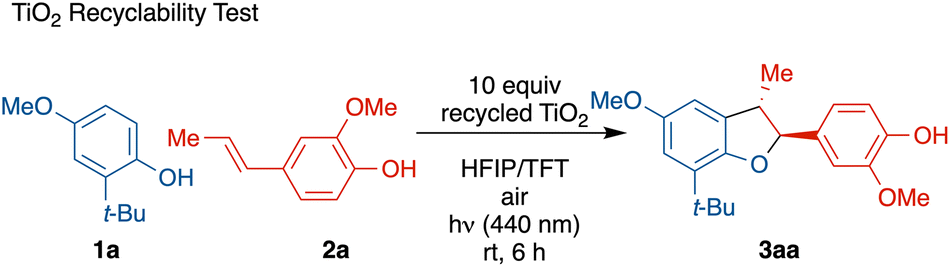
Finally, to assess the reusability of the heterogeneous photocatalyst, a TiO2 recyclability test was conducted through centrifugal separation (Table 3). After five rounds of recycling, no catalyst degradation or loss of reactivity was observed. Upon washing, the recycled TiO2 showed comparable activity, FT-IR spectrum, and morphology to fresh TiO2 samples, demonstrating the recyclability and reusability of the photocatalyst (see ESI† for details).
Conclusions
In summary, we developed a novel and single-step method for the synthesis of dihydrobenzofurans with an inexpensive, nontoxic, and recyclable TiO2 photocatalyst via oxidative cross-coupling of phenols with alkenyl phenols. This method leverages the surface interaction between alkenyl phenols and the TiO2 solid surface which enables direct activation under visible light, obviating the need for pre-functionalization or surface modification. Mechanistic studies including Raman and DRS UV-Vis spectroscopy, kinetics and radical trapping experiments, as well as DFT calculations, support the generation of an alkenyl phenoxyl radical which is captured by a phenol nucleophile. Notably, the reaction proceeds under benign conditions, with air as the terminal oxidant at room temperature, and is applicable to a broad range of substrates including phenols, anilines, and heterocycles. Moreover, the method is complementary to those in Scheme 2 and functions well with unprotected alkenyl phenols. The resulting 2,3-dihydrobenzofurans are the building blocks of bioactive natural products, and thus are of value in synthetic and pharmaceutical chemistry.Data availability
The data underlying this study are available in the published article and the ESI.†Author contributions
JW conceived the topic. JW and YL carried out experiments. YL performed computational studies. All authors reviewed and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the NSF (CHE2102626) and the NIH (R35 GM131902) for financial support of this research and to ACCESS (TG-CHE120052) for computational support. Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, CHE-1827457, 3R01GM118510-03S1, 3R01GM087605-06S1) as well as the Vagelos Institute for Energy Science and Technology and the Singh Center for Nanotechnology. Nicole Bohn (UPenn) is acknowledged for obtaining scanning electron microscope images. Dr Charles W. Ross III (UPenn) is acknowledged for obtaining accurate mass data. Dr Joshua Williams (Drexel University) is acknowledged for obtaining DRS UV-Vis spectra.Notes and references
- (a) A. Karioti, F. Carta and C. T. Supuran, Molecules, 2016, 21, 1649 CrossRef CAS PubMed; (b) Z. Huang and J.-P. Lumb, ACS Catal., 2019, 9, 521 CrossRef CAS; (c) W.-T. Wu, L.-M. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC.
- (a) L. B. Davin and N. G. Lewis, Phytochem. Rev., 2003, 2, 257 CrossRef CAS; (b) J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251 RSC; (c) R. B. Teponno, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2016, 33, 1044 RSC; (d) E. Fuss, Phytochem. Rev., 2003, 2, 307 CrossRef CAS.
- (a) E. Mnich, N. Bjarnholt, A. Eudes, J. Harholt, C. Holland, B. Jørgensen, F. H. Larsen, M. Liu, R. Manat, A. S. Meyer, J. D. Mikkelsen, M. S. Motawia, J. Muschiol, B. L. Møller, S. R. Møller, A. Perzon, B. L. Petersen, J. L. Ravn and P. Ulvskov, Nat. Prod. Rep., 2020, 37, 919 RSC; (b) L. B. Davin, H.-B. Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362 CrossRef CAS PubMed; (c) N. G. Lewis and L. B. Davin, ACS Symp. Ser., 1994, 562, 202 CrossRef CAS.
- W. C. Neuhaus, A. L. Jemison and M. C. Kozlowski, ACS Catal., 2019, 9, 11067–11073 CrossRef CAS PubMed.
- (a) Q.-B. Liu, X.-X. Huang, M. Bai, X.-B. Chang, X.-J. Yan, T. Zhu, W. Zhao, Y. Peng and S.-J. Song, J. Agric. Food Chem., 2014, 62, 7796 CrossRef CAS PubMed; (b) Y.-Y. Lu, Y.-B. Xue, J.-J. Liu, G.-M. Yao, D.-Y. Li, B. Sun, J.-W. Zhang, Y.-F. Liu, C.-X. Qi, M. Xiang, Z.-W. Luo, G. Du and Y.-H. Zhang, J. Nat. Prod., 2015, 78, 2205 CrossRef CAS PubMed; (c) L. H. Rakotondraibe, P. R. Graupner, Q.-B. Xiong, M. Olson, J. D. Wiley, P. Krai, P. J. Brodie, M. W. Callmander, E. Rakotobe, F. Ratovoson, V. E. Rasamison, M. B. Cassera, D. R. Hahn, D. G. I. Kingston and S. J. Fotso, Nat. Prod., 2015, 78, 431 CrossRef CAS PubMed; (d) L. Zhou, G.-D. Yao, L.-W. Lu, X.-Y. Song, B. Lin, X.-B. Wang, X.-X. Huang and S.-J. Song, J. Agric. Food Chem., 2018, 66, 11390 CrossRef CAS PubMed; (e) D. T. Tshitenge, D. Feineis, S. Awale and G. J. Bringmann, Nat. Prod., 2017, 80, 1604 CrossRef CAS PubMed; (f) T. Morikawa, I. Hachiman, K. Matsuo, E. Nishida, K. Ninomiya, T. Hayakawa, O. Yoshie, O. Muraoka and T. J. Nakayama, Nat. Prod., 2016, 79, 2005 CrossRef CAS PubMed; (g) L. Xiong, C.-G. Zhu, Y.-R. Li, Y. Tian, S. Lin, S.-P. Yuan, J.-F. Hu, Q. Hou, N.-H. Chen, Y.-C. Yang and J.-G. Shi, J. Nat. Prod., 2011, 74, 1188 CrossRef CAS PubMed; (h) L. Rao, Y.-X. You, Y. Su, Y. Fan, Y. Liu, Q. He, Y. Chen, J. Meng, L. Hu, Y.-Z. Li, Y.-K. Xu, B. Lin and C.-R. Zhang, J. Agric. Food Chem., 2020, 68, 8825 CrossRef CAS PubMed.
- (a) Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782 CrossRef CAS PubMed; (b) H. Shalit, A. Libman and D. J. Pappo, J. Am. Chem. Soc., 2017, 139, 13404 CrossRef CAS PubMed; (c) Y. Nieves-Quinones, T. J. Paniak, Y. E. Lee, S. M. Kim, S. Tcyrulnikov and M. C. Kozlowski, J. Am. Chem. Soc., 2019, 141, 10016 CrossRef CAS PubMed; (d) J. Wu and M. C. Kozlowski, ACS Catal., 2022, 12, 6532–6549 CrossRef CAS PubMed.
- F. Saliu, E. L. Tolppa, L. Zoia and M. Orlandi, Tetrahedron Lett., 2011, 52, 3856–3860 CrossRef CAS.
- Q. Zhao, J.-K. Jin, J. Wang, F.-L. Zhang and Y.-F. Wang, Chem. Sci., 2020, 11, 3909–3913 RSC.
- T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056–11059 CrossRef CAS PubMed.
- P. T. Parvatkar, S. Kandambeth, A. C. Shaikh, I. Nadinov, J. Yin, V. S. Kale, G. Healing, A. H. Emwas, O. Shekhah, H. N. Alshareef, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2023, 145, 5074–5082 CrossRef CAS PubMed.
- L. Guo, G. Chen, H. Li, C.-H. Tung and Y. Wang, Green Chem., 2023, 25, 7102 RSC.
- K. Dong, C.-Y. Zhao, X.-J. Wang, L.-Z. Wu and Q. Liu, Org. Lett., 2021, 23, 2816–2820 CrossRef CAS PubMed.
- (a) X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC; (b) D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC; (c) H. Cheng and W. Xu, Org. Biomol. Chem., 2019, 17, 9977–9989 RSC; (d) P. Riente and T. Noel, Catal. Sci. Technol., 2019, 9, 5186–5232 RSC; (e) S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456–475 CrossRef CAS.
- (a) X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem.–Eur. J., 2011, 18, 2624–2631 CrossRef PubMed; (b) X. Lang, W. R. Leow, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 1075–1082 RSC; (c) M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem.–Eur. J., 2012, 18, 3478–3481 CrossRef CAS PubMed; (d) C. Vila and M. Rueping, Green Chem., 2013, 15, 2056–2059 RSC.
- (a) F. Parrino, A. Ramakrishnan and H. Kisch, Angew. Chem., Int. Ed., 2012, 47, 7107–7109 CrossRef PubMed; (b) V. T. Bhat, P. A. Duspara, S. Seo, N. S. B. A. Bakar and M. F. Greaney, Chem. Commun., 2015, 51, 4383–4385 RSC.
- J. Zoller, D. C. Fabry and M. Rueping, ACS Catal., 2015, 5, 3900–3904 CrossRef CAS.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS.
- J. Zhang, H. Chen, M. Liu, T. Lu, B. Gao, X. Yang, L. Zhou, H. Li and Y. Su, J. Photochem. Photobiol., A, 2022, 431, 114005 CrossRef CAS.
- J. Wu and M. C. Kozlowski, Org. Lett., 2023, 25, 907–911 CrossRef CAS PubMed.
- M. Ben-Lulu, E. Gaster, A. Libman and D. Pappo, Angew. Chem., Int. Ed., 2020, 59, 4835–4839 CrossRef CAS PubMed.
- K. Niederer, P. Gilmartin and M. C. Kozlowski, ACS Catal., 2020, 10, 14615–14623 CrossRef CAS PubMed.
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS PubMed.
- T. Quell, N. Beiser, K. M. Dyballa, R. Franke and S. R. Waldvogel, Eur. J. Org Chem., 2016, 4307–4310 CrossRef CAS.
- K. Morimoto, K. Sakamoto, T. Ohshika, T. Dohi and Y. Kita, Angew. Chem., Int. Ed., 2016, 55, 3652–3656 CrossRef CAS PubMed.
- N. Y. More and M. Jeganmohan, Org. Lett., 2015, 17, 3042–3045 CrossRef CAS PubMed.
- M. Ziolek, I. Tacchini, M. T. Martınez, X. Yang, L. Sund and A. Douhal, Phys. Chem. Chem. Phys., 2011, 13, 4032–4044 RSC.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC.
- S. Kaniyankandy, S. Rawalekar, A. Sen, B. Ganguly and H. N. Ghosh, J. Phys. Chem. C, 2012, 116, 98–103 CrossRef CAS.
- (a) V. C. Vespe and D. F. Boltz, Anal. Chem., 1952, 24, 664–666 CrossRef CAS; (b) N. Hosaka, T. Sekiya, M. Fujisawa, C. Satoko and S. J. Kurita, J. Electron Spectrosc. Relat. Phenom., 1996, 78, 75–78 CrossRef CAS.
- B. Z. Chowdhry, J. P. Ryall, T. J. Dines and A. P. Mendham, J. Phys. Chem. A, 2015, 119, 11280–11292 CrossRef CAS PubMed.
- J. Fujisawa, S. Matsumura and M. Hanaya, Chem. Phys. Lett., 2016, 657, 172–176 CrossRef CAS.
- Z.-W. Qu and G.-J. Kroes, J. Phys. Chem. B, 2006, 110, 8998–9007 CrossRef CAS PubMed.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC.
- M. H. Keylor, B. S. Matsuura, M. Griesser, J.-P. R. Chauvin, R. A. Harding, M. S. Kirillova, X. Zhu, O. J. Fischer, D. A. Pratt and C. R. J. Stephenson, Science, 2016, 354, 1260–1265 CrossRef CAS PubMed.
- J. Fujisawa, S. Matsumura and M. Hanaya, Chem. Phys. Lett., 2022, 803, 139833 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectral copies (PDF), FAIR data, including the primary NMR FID files for compounds (ZIP) and computational coordinates (XYZ). See DOI: https://doi.org/10.1039/d4sc00723a |
| This journal is © The Royal Society of Chemistry 2024 |