
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiachronic evolvement from tetra-icosahedral to quasi-hexagonal close-packed bimetal clusters†
Shisi
Tang‡
a,
Endong
Wang‡
b,
Yanzhen
Wu‡
c,
Tongxin
Song
a,
Meng
Zhou
 *c,
Xiao
Cai
a,
Yi
Gao
*b,
Weiping
Ding
a and
Yan
Zhu
*a
*c,
Xiao
Cai
a,
Yi
Gao
*b,
Weiping
Ding
a and
Yan
Zhu
*a
aSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhuyan@nju.edu.cn
bPhonon Science Research Center for Carbon Dioxide, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China. E-mail: gaoyi@sari.ac.cn
cHefei National Research Center for Physical Sciences at the Microscale, Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, China. E-mail: mzhou88@ustc.edu.cn
First published on 8th May 2024
Abstract
Here we report a diachronic evolvement from tetra-icosahedral Au30Ag12(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)24 to quasi-hcp (hexagonal close-packed) Au47Ag19(CCR)32via a one-step reduction, in which the size/structure conversion of the two clusters is not a typical Oswald growth process, but involves interface shrinking followed by core rearrangement and surface polymerization. Au30Ag12(CCR)24 has an aesthetic Au18Ag8 kernel that is composed of four interpenetrating Au10Ag3 icosahedra, while Au47Ag19(CCR)32 has a twisted Au19 core capped by a Au12Ag19 shell that are stacked in a layer-by-layer manner with a quasi-hcp pattern. The discovery of the two clusters not only provides further evidence for icosahedral clusters with longer excited-state lifetime compared to hcp-like clusters, but also discloses a double increase in catalytic reactivity for electrocatalytic oxidation of ethanol over quasi-hcp clusters in comparison with icosahedral clusters. This work provides the rationale for reversing the bottom-up growth process to remake bimetal clusters.
CR)24 to quasi-hcp (hexagonal close-packed) Au47Ag19(CCR)32via a one-step reduction, in which the size/structure conversion of the two clusters is not a typical Oswald growth process, but involves interface shrinking followed by core rearrangement and surface polymerization. Au30Ag12(CCR)24 has an aesthetic Au18Ag8 kernel that is composed of four interpenetrating Au10Ag3 icosahedra, while Au47Ag19(CCR)32 has a twisted Au19 core capped by a Au12Ag19 shell that are stacked in a layer-by-layer manner with a quasi-hcp pattern. The discovery of the two clusters not only provides further evidence for icosahedral clusters with longer excited-state lifetime compared to hcp-like clusters, but also discloses a double increase in catalytic reactivity for electrocatalytic oxidation of ethanol over quasi-hcp clusters in comparison with icosahedral clusters. This work provides the rationale for reversing the bottom-up growth process to remake bimetal clusters.
Introduction
Atomically precise metal clusters with crystallographically solved structures are ideally composed of an exact number of metal atoms.1–4 The knowledge gained from the study of these clusters has provided valuable information about the evolution from the metallic state to the molecular exciton state,5,6 the origin of the chiral structure,7 the fundamental understanding of heterogeneous catalysis8 and so on, which are currently not available from typical nanoparticles. Recently, significant advances have been made in the precise synthesis of metal clusters, thereby building the library of metal clusters with rich size and structure diversity. The icosahedron is perhaps the most popular structure in nanoclusters. Among reported clusters, many clusters are formed from single or multiple-twinned icosahedra, such as Au25(SR)18, Au38(SR)24, Au133(SR)52, Au144(SR)60, Au25(PPh3)10(SR)5Cl2, Au37(PPh3)10(SR)10Cl2, Au55(PPh3)12Cl6, Au60Se2(PPh3)10(SeR)15, Au12Ag60(SR)31Br9(Dppp)6, etc.9–22 Interestingly, hcp or hcp-like structures are rarely observed in the library of clusters.23–26 Currently, the understanding of the formation mechanism of clusters is still lacking, as it is difficult to directly observe dynamic processes of such small metal ions and ligands in the solution phase. As a result, manipulating the assembly of atoms into the target structure via the bottom-up process remains a challenge.One alternative way to solve the issue is the interconversion strategy from the parent clusters to target structures such as hcp-like clusters, in which the atomically precise starting point with clusters can permit tracing the growth process via experiments coupled with theory. In fact, the size/structure transformation of metal clusters into another new ones is well precedented.27–34 For example, Xie and colleagues reported a three-stage size growth mechanism from [Au25(SR)18]− to [Au44(SR)26]2− monitored by time-course electrospray ionization mass spectrometry (ESI-MS): kinetically controlled accumulation of Au25 reaction with AuSR; Au25-mediated size growth with bottom-up Lamer and volcano-shaped growth patterns; thermodynamically driven size-focusing into Au44.35 Jin and colleagues proposed a shuttling-out mechanism for the formation of [Au24(PPh3)10(SR)5Cl2]+ from its parent [Au25(PPh3)10(SR)5Cl2]2+ based on density functional theory (DFT) simulations and ESI-MS: adsorption of a PPh3 onto a gold atom located at the waist position to initiate the reaction; Au–S bond breaking and the gold atom at the center dislocating toward the surface; the Au(PPh3)2 moiety formed on the surface eventually detaching from the Au25 to result in hollow Au24.36 To enrich the metal clusters with tailored structure and functionality, a fundamental understanding of the critical growth event from non-hcp to hcp-like clusters is thus most needed.
In this study, we initially present the synthesis of an icosahedron-based Au30Ag12(CCR)24 cluster (where HCCR is 1-ethynyl-2,4-dimethylbenzene) through the co-reduction of the AuCCR and AgCCR by tert-butylamine borane (C4H11NBH3). Au30Ag12(CCR)24 contains a Au18Ag8 core that is fused from four Au10Ag3 icosahedral units. Notably, the Au30Ag12(CCR)24 cluster is then reduced by C4H11NBH3 and further transformed into a new Au47Ag19(CCR)32 cluster with quasi-hcp structure, which is composed of a layered-like Au19 core encapsulated by a Au12Ag19 shell. The transformation from tetra-icosahedral Au30Ag12(CCR)24 to quasi-hcp Au47Ag19(CCR)32 proceeds via a stepwise mechanism, with the surface staples shrinking sequentially with the core growing. More notably, tetra-icosahedral Au30Ag12(CCR)24 and quasi-hcp Au47Ag19(CCR)32 show diversity in physicochemical properties pursued by our studies.
Results and discussion
The total structure of the Au30Ag12(CCR)24 (hereafter Au30Ag12) cluster was solved by single-crystal X-ray diffraction (Table S1†). As shown in Fig. 1, Au30Ag12 has a S4 symmetry and can be viewed as a tetra-icosahedral structure. The Au18Ag8 kernel in Au30Ag12 can be seen as an assembly of four icosahedral Au10Ag3 building blocks (Fig. 1a and S1†). Interestingly, the central atoms of the four icosahedra that are respectively marked with 1, 2, 3, 4 compose a Au4 tetrahedron. The four icosahedra are assembled through interpenetration sharing a M7 pentagonal bipyramid (Fig. S1†), and they are slightly distorted, which may be ascribed to the asymmetric introduction of Ag atoms into the icosahedra. The Au18Ag8 kernel is further stabilized by four trimeric RCC–Au–C2(R)–Ag–C2(R)–Au–RCC (motifs A and B in Fig. 1b) and four monomeric RCC–Au–CCR (motif C in Fig. 1b). In motif A, all four alkyne ligands interact with metal atoms via σ–π bonds and the Ag atom in the middle interacts with two alkyne ligands via π bonds, while in motif B, one alkyne ligand adopts a bridge mode with only a σ interaction μ2-η1(Au), η2(Ag). The chemical formula of Au30Ag12(CCR)24 is further confirmed by ESI-MS (Fig. S2†).
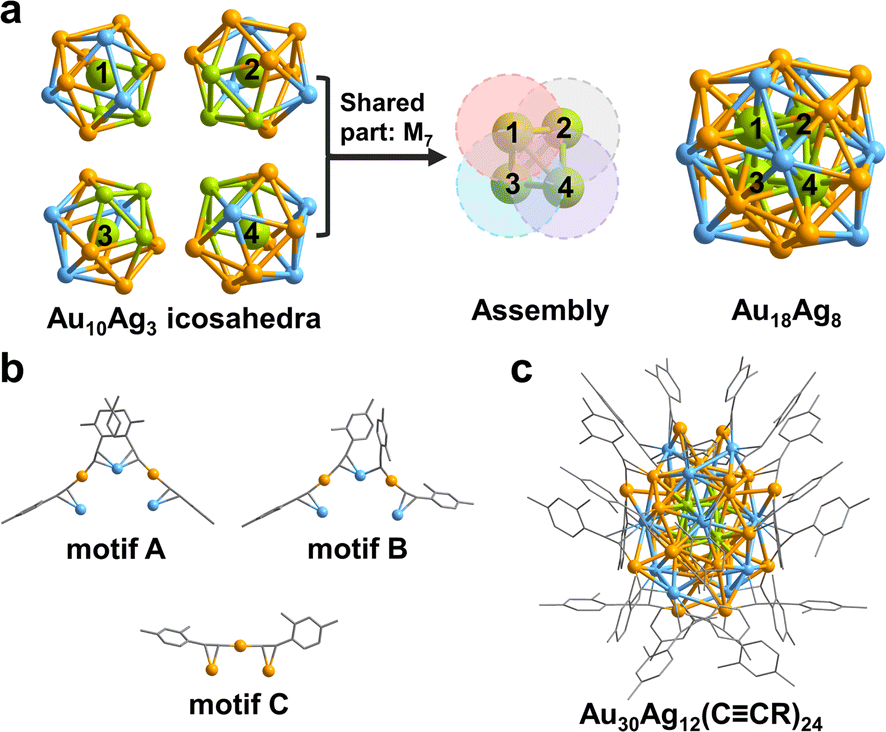 | ||
| Fig. 1 (a) Au18Ag8 kernel formed by four interpenetrating Au10Ag3 icosahedra. (b) Three different motifs. (c) Total structure of Au30Ag12(CCR)24. Color codes: orange/green, Au; blue, Ag; gray, C. The H atoms are omitted for clarity. | ||
Next, the Au30Ag12(CCR)24 cluster can be further converted to another new cluster using C4H11NBH3. Adding thimbleful reductant into the dichloromethane solution of Au30Ag12 clusters, the green solution turned purple tardily. We monitored the transformation process using time-resolved UV-vis adsorption spectroscopy. From Fig. 2a and S3,† after the addition of the reducing agent, the three characteristic peaks at 444, 594 and 687 nm of Au30Ag12 became weak, accompanied by the redshift of the peak at 444 nm, indicating that the cluster structure gradually dissociated. After 6 h, a peak appeared around 550 nm, gradually intensified and redshifted. At 20 h, the peak offset to around 450 nm began to intensify and continued to redshift. After 28 h, deduced from the optical absorption from the reaction solution, no significant change was observed in the range of 300–900 nm, which suggested that a new cluster was formed in the solution and finally exhibited two distinct peaks at 460 and 554 nm (Fig. 2a and S4†).
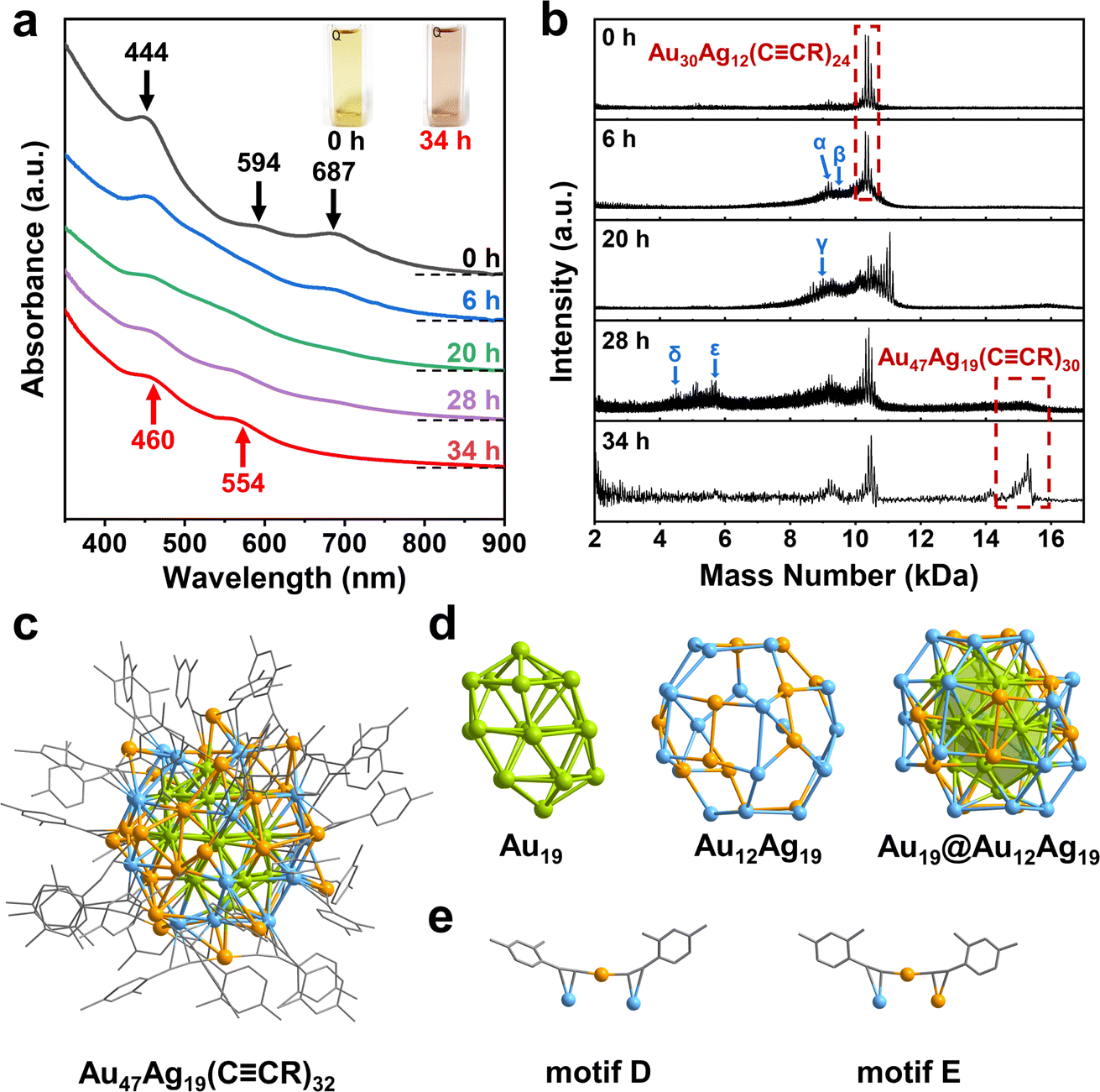 | ||
| Fig. 2 (a) Time-resolved UV-vis spectra and (b) MALDI-TOF-MS spectra obtained from the transformation from Au30Ag12(CCR)24 to Au47Ag19(CCR)32. Formulae of intermediates captured in MALDI-TOF-MS: α, Au28Ag10(CCR)20; β, Au29Ag11(CCR)20; γ, Au26Ag12(CCR)20; δ, Au18Ag8; ε, Au22Ag8(CCR)4. (c) Total structure of Au47Ag19(CCR)32. (d) Au19 core, Au12Ag19 shell and Au19@Au12Ag19. (e) Two different motifs. Color codes: orange/green, Au; blue, Ag; gray, C. The H atoms are omitted for clarity. | ||
We separated and purified the transformation product to obtain the novel cluster, whose chemical formula was analyzed by matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF-MS). The two dominant broad peaks at 15.184 and 10.377 kDa were assigned to Au47Ag19(CCR)30 (expected m/z = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 184) and Au39Ag19(CCR)5 (expected m/z = 10378), respectively (Fig. S5†). Therefore, we speculated that the new cluster might contain 47 Au atoms and 19 Ag atoms (short for Au47Ag19). Moreover, MALDI-TOF-MS was performed to identify stable intermediate species during the cluster transformation. For pure Au30Ag12, MALDI-TOF-MS gave a main peak at 10.302 kDa, which matched the formula weight of Au30Ag12(CCR)24. And two slight peaks at 9.177 and 6.230 kDa were assigned to Au28Ag10(CCR)20 and Au22Ag8(CCR)8, respectively (Fig. 2b and S6†). At 6 h after the addition of the reducing agent, the main peaks still existed, but a series of new peaks appeared around 9.3 kDa, corresponding to Au28Ag10(CCR)20, Au29Ag11(CCR)20, Au28Ag12(CCR)21 and so on, indicating that the surface structural units of Au30Ag12 began to dissociate (Fig. 2b and S7†). At 20 h, the characteristic peak of Au30Ag12 disappeared, and peaks with smaller molecular weight appeared around 9.0 kDa and larger substances were generated: the peak at 11.059 kDa is assigned to Au32Ag13(CCR)26, and the peak around 15.2 kDa is assigned to Au47Ag19(CCR)30 (Fig. 2b and S8†). At 28 h, peaks around 9.0 kDa decreased and were replaced by a series of peaks around 5.5 kDa (Fig. 2b, S9–S12†). At 34 h, the characteristic peaks of the cluster and a bunch of peaks below 2.5 kDa were left, indicating that the cluster and Au-alkyne complexes were finally present in the solution.
184) and Au39Ag19(CCR)5 (expected m/z = 10378), respectively (Fig. S5†). Therefore, we speculated that the new cluster might contain 47 Au atoms and 19 Ag atoms (short for Au47Ag19). Moreover, MALDI-TOF-MS was performed to identify stable intermediate species during the cluster transformation. For pure Au30Ag12, MALDI-TOF-MS gave a main peak at 10.302 kDa, which matched the formula weight of Au30Ag12(CCR)24. And two slight peaks at 9.177 and 6.230 kDa were assigned to Au28Ag10(CCR)20 and Au22Ag8(CCR)8, respectively (Fig. 2b and S6†). At 6 h after the addition of the reducing agent, the main peaks still existed, but a series of new peaks appeared around 9.3 kDa, corresponding to Au28Ag10(CCR)20, Au29Ag11(CCR)20, Au28Ag12(CCR)21 and so on, indicating that the surface structural units of Au30Ag12 began to dissociate (Fig. 2b and S7†). At 20 h, the characteristic peak of Au30Ag12 disappeared, and peaks with smaller molecular weight appeared around 9.0 kDa and larger substances were generated: the peak at 11.059 kDa is assigned to Au32Ag13(CCR)26, and the peak around 15.2 kDa is assigned to Au47Ag19(CCR)30 (Fig. 2b and S8†). At 28 h, peaks around 9.0 kDa decreased and were replaced by a series of peaks around 5.5 kDa (Fig. 2b, S9–S12†). At 34 h, the characteristic peaks of the cluster and a bunch of peaks below 2.5 kDa were left, indicating that the cluster and Au-alkyne complexes were finally present in the solution.
Furthermore, the total structure of Au47Ag19(CCR)32 was determined by single-crystal X-ray diffraction (Table S2†). As shown in Fig. 2c, the cluster is protected by 32 alkyne ligands and hence its chemical formula can be unambiguously identified as Au47Ag19(CCR)32. Different from the parent Au30Ag12 cluster, Au47Ag19 can be viewed as a core–shell structure arranged along a C2 axis. The kernel of Au47Ag19 is a twisted Au19 that is dissected in a layer-by-layer manner (Fig. 2d), in which the middle layer is a regular hexagon Au7, the two layers adjacent to the Au7 hexagon are pentagons, and two vertex atoms at both ends. The Au19 kernel is surrounded by the Au12Ag19 shell. Au47Ag19 includes only monomeric RCC–Au–CC(R) staples that adopt two coordination modes: μ2-η1(Au), η2(Au) and μ2-η1(Au), η2(Ag) (Fig. 2e). Generally, nanoclusters as large as Au47Ag19 contain basic polyhedral structural units which can be tetrahedron, icosahedron or based on fcc, hcp or bcc (body-centered cubic) arrangements. And the basic units grow into larger structures through fusion, interpenetration, shell-by-shell and layer-by-layer modes. Strictly speaking, the basic units mentioned above are not found in Au47Ag19. The Au19 nucleus is somewhat similar to the hcp structure, even though there is a torsion Angle between the two Au5 layers. Therefore, the Au19 core can be viewed as a quasi-hcp structure whose layers are stacking in the ABABA pattern with distortion (Fig. S13†). This indicates that the tetra-icosahedral Au30Ag12 grows towards hcp during the transformation process. The two clusters are quite robust (Fig. S14†) and the in-lattice connection modes of the two clusters are shown in Fig. S15 and S16† in detail.
To elucidate the growth from Au30Ag12 to Au47Ag19, we conducted density functional theory (DFT) calculations to model the structural transformation based on the MALDI-TOF-MS experiment results. Fig. 2b illustrates that, except for the bunch of signals relating to the Au30Ag12(CCR)24 cluster persisting throughout the entire transformation process, the intermediate size decreased gradually from 0–28 h, evident in the shift of the peaks. The transition from 0–28 h might signal the size decreasing period. During this period, several intermediates, including the Au28Ag10(CCR)20 and Au29Ag11(CCR)20 formed at 6 h, Au26Ag12(CCR)20 formed at 20 h, and Au22Ag8(CCR)4 and Au18Ag8 formed at 28 h, were notably observed with apparent intensity signals, indicating their key role in the size-decreasing process. As shown in Fig. 3a, starting from the full Au30Ag12(CCR)24 cluster (isomer 1), four HCCAuCC(H)Ag fragments were removed one by one with the energy preference sequence, producing the isomers from 2–5, as depicted in Fig. 3a. Among them, isomer 2 (Au29Ag11(CCR)22) can transform into Au29Ag11(CCR)20via abscission of two CCR fragments and isomer 3 was consistent with the intermediate Au28Ag10(CCR)20 characterized at 6 h. After that, the HCCAuCCH motifs were further removed stepwise, finally leaving the metal Au18Ag8 kernel. Among the isomers of 6–13, Au24Ag8(CCR)12 (isomer 7), Au22Ag8(CCR)8 (isomer 9), and Au20Ag8(CCR)4 (isomer 11) can transform into Au24Ag8(CCR)0, Au22Ag8(CCR)4, and Au20Ag8(CCR)1 by CCR abscission. These isomers confirmed the rationality of the proposed pathway.
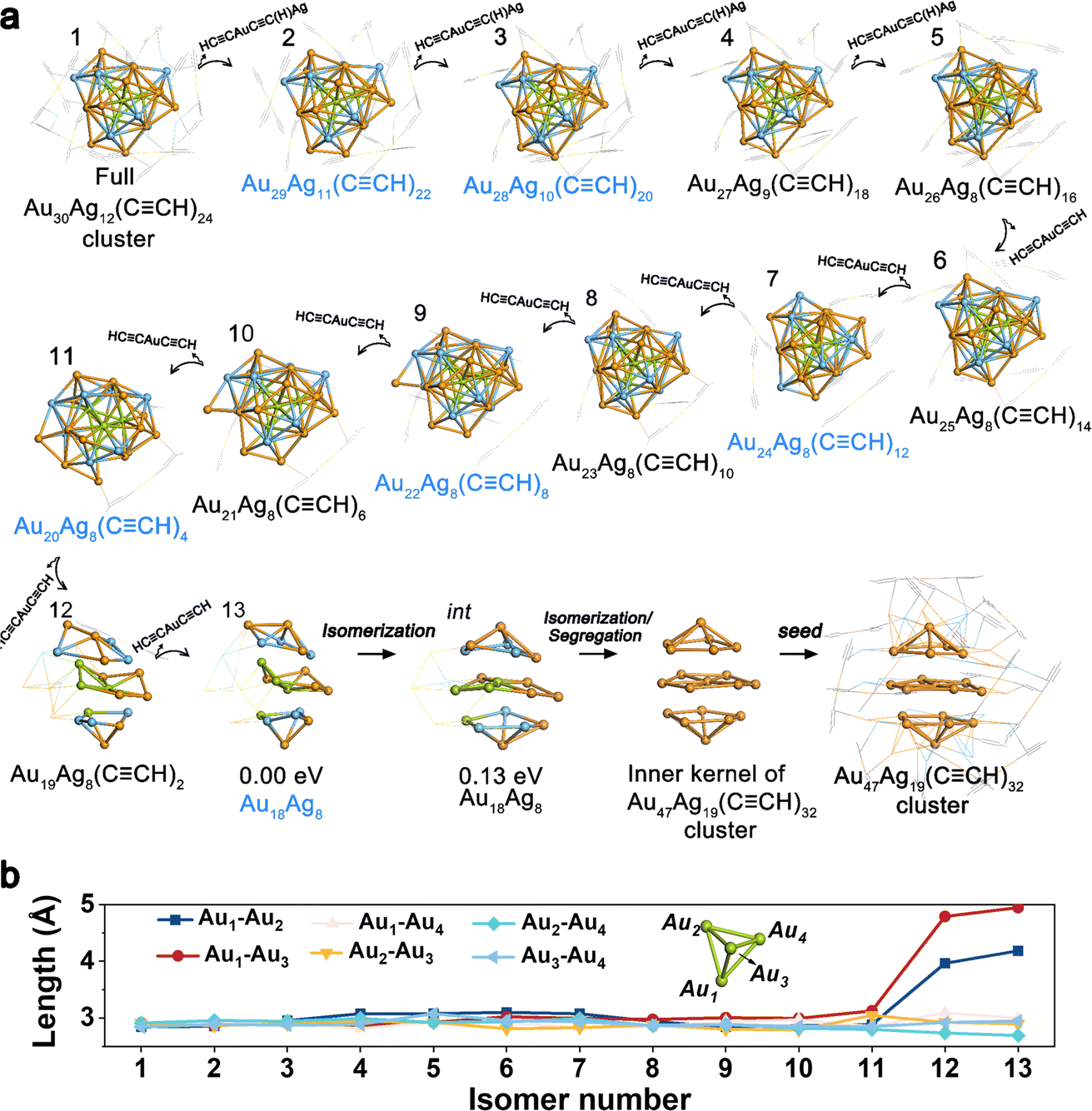 | ||
| Fig. 3 (a) The proposed transformation pathway from Au30Ag12(CCR)24 to the seed of the Au47Ag19(CCR)32 cluster. (b) Au–Au length of the Au4 fragment of the Au30Ag12(CCR)24 cluster. Color codes: orange/green, Au; blue, Ag; gray, C. The H atoms are omitted for clarity. | ||
From 28 h to 34 h, the peaks relating to the intermediates of Au18Ag8, Au22Ag8(CCR)4 disappeared, and the peak signaling the production of the Au47Ag19 cluster emerged, implying that the generation of the Au47Ag19 cluster at 34 h originated from the intermediates of Au18Ag8 and Au22Ag8(CCR)4 at 28 h. As mentioned above, the kernel of Au30Ag12 can be regarded as the fusion of four M13 fragments. The four Au atoms from each of the four M13 fragments constituted a central Au4 pyramid core. Fig. 3b shows the distances of the six Au–Au bonds of the Au4 core. For isomers 1–11, the Au–Au length did not change significantly upon the removal of the outer motifs. However, the transformation from isomer 11 to isomer 12 showed significant structural distortions. The Au1–Au2 and Au1–Au3 lengths of isomer 12 in Fig. 3b increased to 3.96 and 4.79 Å, indicating the dissociation of the central Au4 core of the cluster and the consequent destruction of the tetra-icosahedral pattern. The transformation from isomer 12 to isomer 13, i.e., naked Au18Ag8 isomer, involved the continued lengthening of the Au1–Au2 (4.18 Å) and Au1–Au3 lengths (4.95 Å). The naked Au18Ag8 intermediate (isomer 13) can easily isomerize to an isomer intermediate with similar stability, which possessed the same packing mode as the inner kernel of the Au47Ag19 cluster. Next, the Au13Ag6 part of the isomer intermediate can undergo segregation37,38 or Au–Ag exchange with the species in the solvation environment, generating the Au19 species or bigger intermediates containing the Au19 species. This can be seen as a seed for the birth of the Au47Ag19 cluster.
The Kohn–Sham (KS) orbital energy levels and atomic orbital components in each orbital of the two clusters are shown in Fig. S17.† The main absorption peak of Au30Ag12 lies at 1.70 eV (Fig. S18†), which corresponds to the orbital transitions including HOMO−3 → LUMO+2, HOMO−1 → LUMO+3, HOMO−2 → LUMO+4 (HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital). For Au47Ag19, the main absorption peaks are located at 1.41 and 2.31 eV, which include the orbital transitions of HOMO−6 → LUMO+2, HOMO−2 → LUMO+5 for 1.41 eV, and HOMO−48 → HOMO, HOMO−35 → LUMO+3 for 2.31 eV (Fig. S17a†), respectively. It can also be seen that the metal atoms contribute considerably to the population of the frontier orbital. Furthermore, for the Au30Ag12 cluster, the energies of the HOMO and LUMO orbitals are −5.38 and −4.41 eV, which lead to the HOMO–LUMO gap of 0.97 eV; for the Au47Ag19 cluster, the energy of the LUMO orbital slightly increases by 0.09 eV and the energy of the HOMO orbital elevates a little bit more by 0.20 eV, which eventually lowers the gap to 0.86 eV (Fig. S17b†). The transitions of molecular orbital topologies from the HOMO to the LUMO of the two clusters show different trends for their populations. For the Au30Ag12 cluster, the population of the orbital seems to narrow on the metal core from the HOMO orbital to the LUMO orbital. However, for the Au47Ag19 cluster, the LUMO orbital slightly extends to the outer ligands. From the UV-vis-NIR absorption spectra in the photon energy scale, the optical energy gaps of the Au30Ag12 and Au47Ag19 are determined as 0.89 and 0.82 eV, respectively (Fig. S19†). For the Square wave voltammetry (SWV) curves, there is an oxidation peak at 0.70 V (O1) and two reduction peaks at −0.05 V (R1) and −0.75 V (R2) for Au30Ag12, and there is an oxidation peak at 0.77 V (O1) and two reduction peaks at 0.07 V (R1) and −0.79 V (R2) for Au47Ag19. Therefore, the electrochemical energy gaps are calculated to be 0.75 eV for Au30Ag12 and 0.70 eV for Au47Ag19 (Fig. S19†).
The Au30(SR)18 with a hcp pattern was previously reported to give rise to a very short excited-state lifetime due to the close packing structure.39 With the two clusters in hand, a question was whether quasi-hcp Au47Ag19 was used to further justify the shorter TA lifetime of hcp-like clusters than non-hcp clusters. We investigated the excited-state dynamics of the two clusters by performing time-resolved transient absorption spectroscopy (TAS). Fig. 4a and b show the femtosecond (fs) TA data map of Au30Ag12 and Au47Ag19 with excitation at 380 nm (3.26 eV). Between 0.1 and 1 ps, one can observe ultrafast relaxation of excited state absorption (ESA) in both the NCs, which was rapid relaxation from higher to lower excited states. Between 1 ps and 2 ns, the TA signals (both ESA and GSB) of Au30Ag12 decayed to half of its maximum while the TA signals of Au47Ag19 decayed to zero. Fig. 4c shows the fs-TA kinetic traces and fittings of the two clusters probed at 630 nm, and the fitting parameters are shown in Table S3.† Since the TA signal of Au30Ag12 did not decay to zero at 2 ns, we performed nanosecond TA under the same experimental conditions (Fig. S20†). In contrast to Au30Ag12 that showed single exponential decay after 5 ps, Au47Ag19 exhibited two-exponential nanosecond decay in both GSB and ESA kinetics. The average excited state lifetime was fitted to be 2.5 ns for Au30Ag12 and 0.86 ns for Au47Ag19 (Fig. 4c and d), which was in full agreement with reported results. The shorter exciton lifetime in Au47Ag19 should also be ascribed to the stronger overlap of the HOMO and LUMO because of the close packed mode of the metal core.40,41
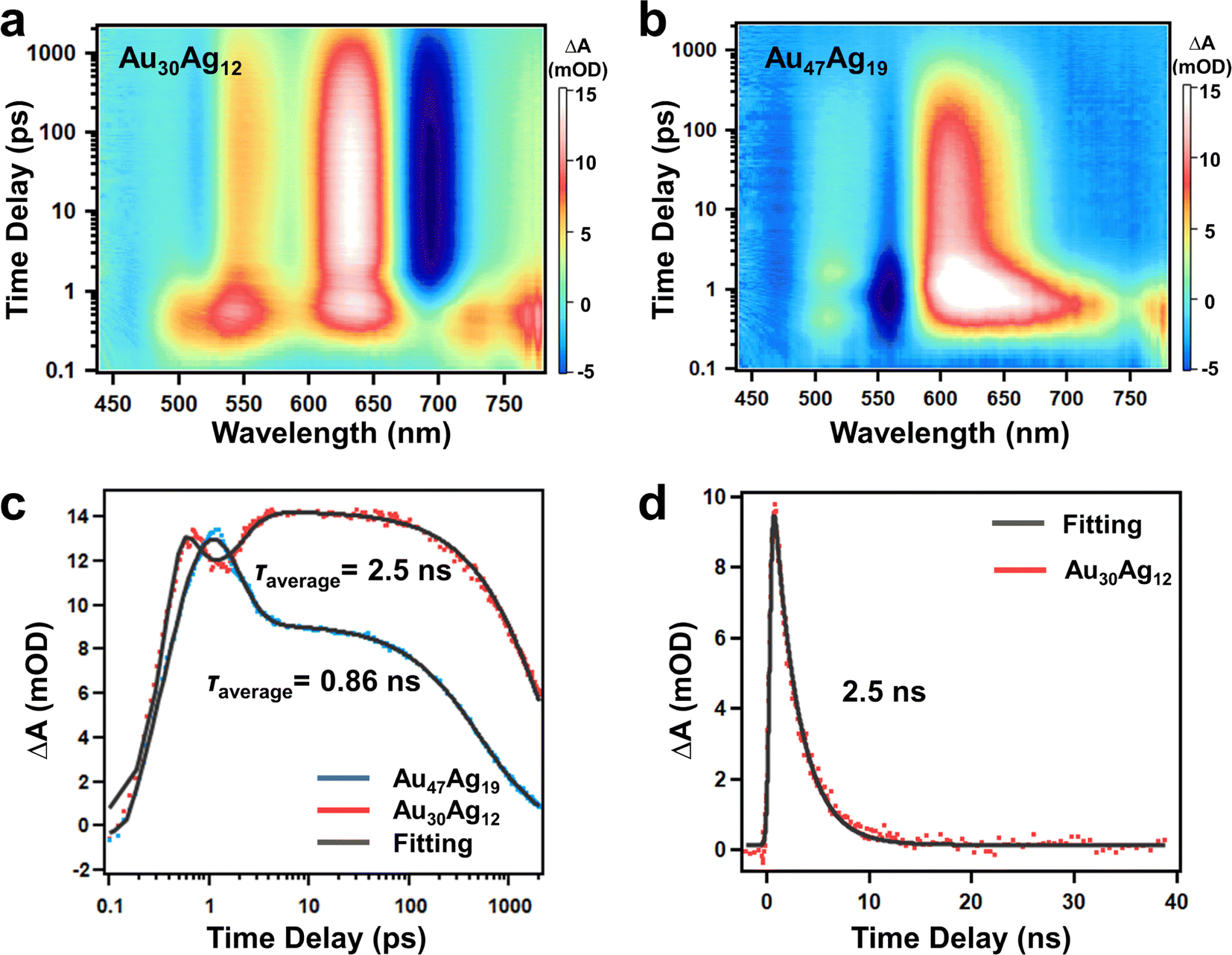 | ||
| Fig. 4 Fs-TA data map of (a) Au30Ag12 and (b) Au47Ag19 with excitation at 380 nm. (c) Comparison of TA kinetic traces and fittings probed at 630 nm of two NCs. (d) Ns-TA kinetics traces of Au30Ag12 probed at 630 nm and corresponding fit. | ||
We next sought to explore the catalytic properties of Au30Ag12 and Au47Ag19. As presented in Fig. 5a, using electrocatalytic oxidation of ethanol (EOR) toward CH3COOH as a probe reaction (Fig. S21†), the Au47Ag19 catalyst exhibited over 2-fold increase in the EOR current in 1 M KOH + 1 M CH3CH2OH solution, compared to the Au30Ag12 catalyst. The mass activities of Au30Ag12 and Au47Ag19 were 111.2 and 242.5 mA/mgAuAg at a cell voltage of 0.08 V vs. Ag/AgCl, respectively. As for the onset potential, Au47Ag19 also showed a significant negative shift (Fig. 5a). The electrochemically active surface areas (ECSAs) of Au30Ag12 and Au47Ag19 were measured to be 8.57 and 17.39 cm2/mgAuAg, respectively, based on the cyclic voltammogram curves (CVs) in Fig. 5b. Thus, the specific activities of EOR at 0.08 V vs. Ag/AgCl by normalizing ECSAs were 13.94 mA cm−2 for Au47Ag19 and 12.97 mA cm−2 for Au30Ag12 (Fig. S22†). Electrochemical impedance spectroscopy (EIS) was carried out at 0.08 V vs. Ag/AgCl. In Fig. 5c, Au47Ag19 possessed a much smaller semicircle diameter than Au30Ag12, suggesting that the former had better conductivity and smaller electron transfer resistance, which was consistent with better EOR performance of the Au47Ag19 catalyst. Fig. 5d shows that the Tafel slope of Au47Ag19 was obviously lower than that of Au30Ag12, indicating that the current density of Au47Ag19 increased faster, thereby showing better efficiency in EOR.
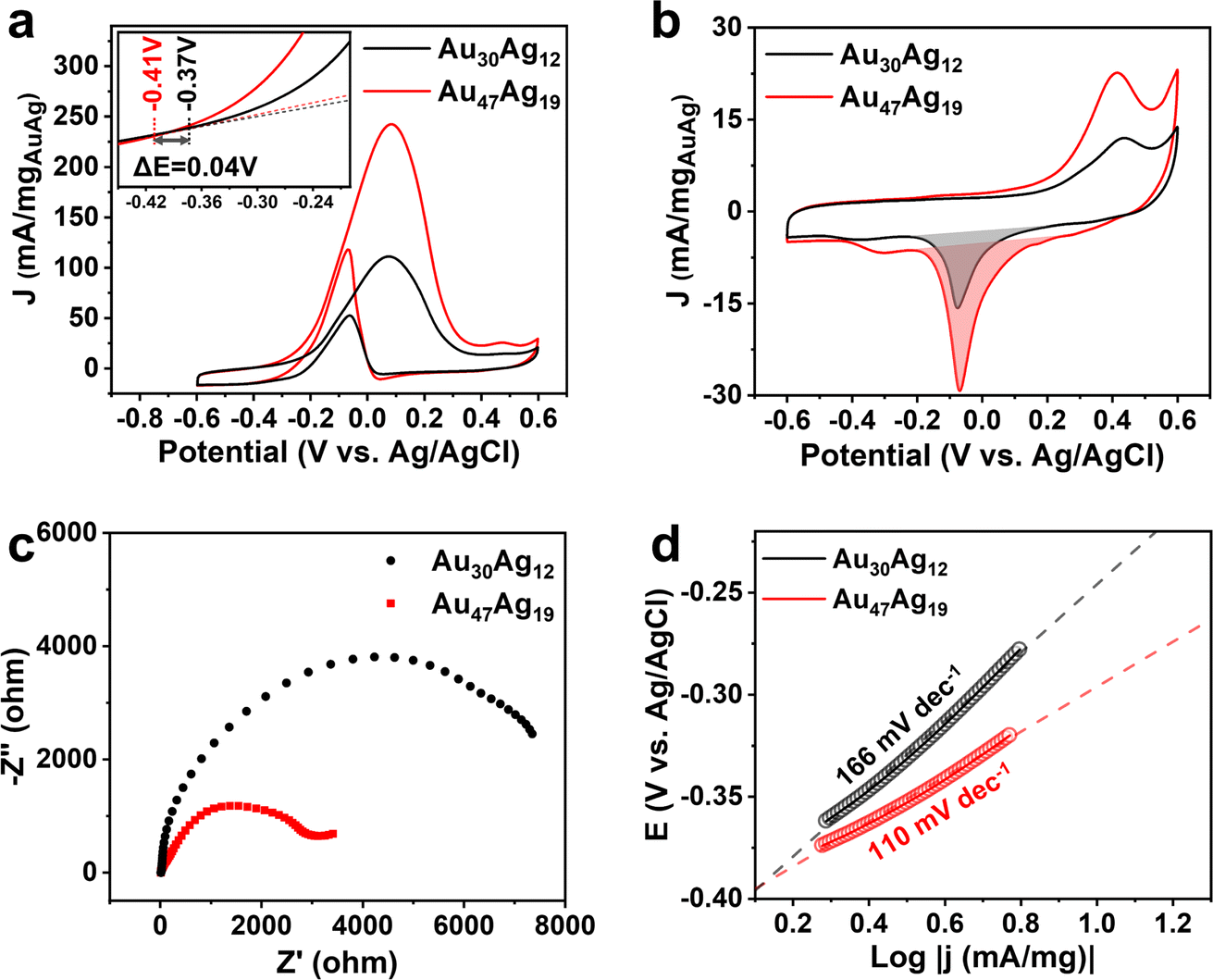 | ||
| Fig. 5 (a) CVs of the Au30Ag12 and Au47Ag19 catalysts in 1 M KOH and 1 M CH3CH2OH solution. (b) CVs of the two catalysts in 1 M KOH solution and the shaded areas are used to calculate the ECSAs. (c) Electrochemical impedance spectra and (d) Tafel plots for EOR obtained on the two catalysts. | ||
DFT calculations were performed to further shed light on the mechanism of the electrocatalytic oxidation of ethanol on the two catalysts. Previous studies have suggested that the oxidation reaction from CH3CH2OH to CH3COOH can proceed via the adsorption of the α-C atom or O atom of CH3CH2OH.42,43 The adsorption of the O atom was adopted in this work because the adsorption of the α-C atom, which was similar to quasi-horizontal placement of the CH3CH2OH, needs to overcome a larger steric effect with respect to the adsorption of the O atom, i.e., vertical placement of CH3CH2OH, due to the presence of the surface ligands. Due to the symmetry of Au30Ag12, five possible reactive sites can be found as in Fig. S23a.† The O–H scission was considered, which had the largest Gibbs free energy change (ΔG; Fig. S24†). Considering the computational cost, the electronic energy change of this step was used for screening the reactive sites (Fig. S23a†). Upon the adsorption of the CH3CH2OH on the surface of the Au30Ag12 cluster, the reaction initiated via the removal of the H atom from the –OH group of the adsorbed CH3CH2OH molecule, which had 0.79 eV increment of ΔG (Fig. S24†). Then, the H atom from the α-C atom was removed with the decrease of ΔG to 0.36 eV, which led to the formation of the CH3HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O@cluster. Then, the H2O molecule was added producing the CH3HC(OH)2@cluster, which further lowered ΔG to −0.01 eV. Next, H atom removal from one of the two –OH groups produced the CH3HCO(OH)@cluster. Similar to the aforementioned step, the H of the α-C atom was removed finally leading to the CH3COOH@cluster with the release of ΔG of 1.06 eV. After the desorption of CH3COOH from the Au30Ag12 cluster, the CH3CH2OH was oxidized to CH3COOH. Considering the symmetry of the Au47Ag19 cluster, 17 possible reactive sites existed. As shown in Fig. S23b,† through calculating the electronic energy change of the removal of the H atom from the O–H group of the adsorptive CH3CH2OH, the reactive site with lower energy alternation was applied. Following the identical reaction steps of the Au30Ag12 cluster, the whole reaction path leading to CH3COOH on Au47Ag19 can be obtained as depicted via the red line of Fig. S24.† The largest ΔG increment of the CH3CH2OH oxidation reaction was 0.52 eV, which was lower than that on Au30Ag12. This meant that the catalytic performance on Au47Ag19 was better, which agreed with the experimental results.
O@cluster. Then, the H2O molecule was added producing the CH3HC(OH)2@cluster, which further lowered ΔG to −0.01 eV. Next, H atom removal from one of the two –OH groups produced the CH3HCO(OH)@cluster. Similar to the aforementioned step, the H of the α-C atom was removed finally leading to the CH3COOH@cluster with the release of ΔG of 1.06 eV. After the desorption of CH3COOH from the Au30Ag12 cluster, the CH3CH2OH was oxidized to CH3COOH. Considering the symmetry of the Au47Ag19 cluster, 17 possible reactive sites existed. As shown in Fig. S23b,† through calculating the electronic energy change of the removal of the H atom from the O–H group of the adsorptive CH3CH2OH, the reactive site with lower energy alternation was applied. Following the identical reaction steps of the Au30Ag12 cluster, the whole reaction path leading to CH3COOH on Au47Ag19 can be obtained as depicted via the red line of Fig. S24.† The largest ΔG increment of the CH3CH2OH oxidation reaction was 0.52 eV, which was lower than that on Au30Ag12. This meant that the catalytic performance on Au47Ag19 was better, which agreed with the experimental results.
Conclusions
In summary, we report the synthesis mechanism and crystal structures of two intermetallic clusters protected by alkyne ligands: Au30Ag12(CCR)24 exhibits a fantastic Au18Ag8 kernel that is fused by four interpenetrating Au10Ag3 icosahedra, while Au47Ag19(CCR)32 exhibits a layered Au19 core capped by a Au12Ag19 shell. Our investigation demonstrates that one stable icosahedron-based cluster transforming into another hcp-like cluster by simple reduction is feasible, which can enrich the library of metal clusters. We also demonstrate that the atom-packing matching and cooperative effect of intermetallic compounds within the two different clusters play a pivotal role in tailoring the molecular exciton state and catalytic properties. We foresee that this work provides new design rules for atomically precise intermetallic nanomaterials.
Data availability
The data are provided in the ESI.†Author contributions
Y. Z. conceived the work. S. T. prepared the clusters and solved their single-crystals. E. W. and Y. G. performed the calculations. Y. W. and M. Z. performed the TA measurement. T. S., X. C. and W. D. gave helpful suggestions. All authors interpreted the data and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (22125202, 21932004, 22302091, 12174408, and 12204213), Natural Science Foundation of Jiangsu Province (BK20220033), Programs for high-level entrepreneurial and innovative talents introduction of Jiangsu Province, and China Postdoctoral Science Foundation (2022M721551).References
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Z. Lei, X. Wan, S. Yuan, Z. Guan and Q. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- R. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS PubMed.
- T. Laaksonen, V. Ruiz, P. Liljeroth and B. Quinn, Chem. Soc. Rev., 2008, 37, 1836–1846 RSC.
- Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 32, 1905488 CrossRef CAS.
- X. Cai, G. Li, W. Hu and Y. Zhu, ACS Catal., 2022, 12, 10638–10653 CrossRef CAS.
- M. Zhu, C. Aikens, F. Hollander, G. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- M. Heaven, A. Dass, P. White, K. Holt and R. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- H. Qian, W. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef PubMed.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. Rosi, R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. Calis and W. Vandervelden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. Rosi, R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS PubMed.
- Y. Tan, Y. Lv, L. Xu, Q. Li, J. Chai, S. Yang, H. Yu and M. Zhu, J. Am. Chem. Soc., 2023, 145, 4238–4245 CrossRef CAS PubMed.
- M. Zhou, K. Li, P. Wang, H. Zhou, S. Jin, Y. Pei and M. Zhu, Nanoscale, 2023, 15, 2633–2641 RSC.
- J. Hu, M. Zhou, K. Li, A. Yao, Y. Wang, Q. Zhu, Y. Zhou, L. Huang, Y. Pei, Y. Du, S. Jin and M. Zhu, Small, 2023, 19, 2301357 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS PubMed.
- T. Higaki, C. Liu, C. Zeng, R. Jin, Y. Chen, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2016, 55, 6694–6697 CrossRef CAS PubMed.
- Y. Li, Y. Song, X. Zhang, T. Liu, T. Xu, H. Wang, D. Jiang and R. Jin, J. Am. Chem. Soc., 2022, 144, 12381–12389 CrossRef CAS PubMed.
- B. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS.
- P. Pan, C. Zhou, H. Li, C. Zhu, C. Chen, X. Kang and M. Zhu, Nanoscale, 2021, 13, 17162–17167 RSC.
- Z. Guan, F. Hu, J. Li, Z. Wen, Y. Lin and Q. Wang, J. Am. Chem. Soc., 2020, 142, 2995–3001 CrossRef CAS PubMed.
- W. Fan, Y. Yang, Q. You, J. Li, H. Deng, N. Yan and Z. Wu, J. Phys. Chem. C, 2023, 127, 816–823 CrossRef CAS.
- Q. Li, S. Yang, T. Chen, S. Jin, J. Chai, H. Zhang and M. Zhu, Nanoscale, 2020, 12, 23694–23699 RSC.
- J. Wang, Z. Wang, S. Li, S. Zang and T. Mak, Angew. Chem., Int. Ed., 2021, 60, 5959–5964 CrossRef CAS PubMed.
- S. Chen, L. Xiong, S. Wang, Z. Ma, S. Jin, H. Sheng, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2016, 138, 10754–10757 CrossRef CAS PubMed.
- Q. Li, T. Luo, M. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- S. Zhang, Y. Li, L. Feng, Q. Xue, Z. Gao, C. Tung and D. Sun, Nano Res., 2021, 14, 3343–3351 CrossRef CAS.
- Q. Yao, X. Yuan, V. Fung, D. Leong, D. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef PubMed.
- S. Wang, H. Abroshan, C. Liu, T. Luo, M. Zhu, H. Kim, N. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef PubMed.
- G. Guisbiers, R. Mendoza-Cruz, L. Bazán-Díaz, J. Velázquez-Salazar, R. Mendoza-Perez, J. Robledo-Torres, J. Rodriguez-Lopez, J. Montejano-Carrizales, R. Whetten and M. José-Yacamán, ACS Nano, 2016, 10, 188–198 CrossRef CAS PubMed.
- L. Deng, W. Hu, H. Deng, S. Xiao and J. Tang, J. Phys. Chem. C, 2011, 115, 11355–11363 CrossRef CAS.
- M. Zhou, T. Higaki, G. Hu, M. Sfeir, Y. Chen, D. Jiang and R. Jin, Science, 2019, 364, 279–282 CrossRef CAS PubMed.
- W. Tian, W. Si, S. Havenridge, C. Zhan, Z. Wang, C. Aikens, C. Tung and D. Sun, Sci. Bull., 2024, 69, 40–48 CrossRef CAS PubMed.
- W. Si, C. Zhang, M. Zhou, W. Tian, Z. Wang, Q. Hu, K. Song, L. Feng, X. Huang, Z. Gao, C. Tung and D. Sun, Sci. Adv., 2023, 9, 3587 CrossRef PubMed.
- D. Hibbitts and M. Neurock, J. Catal., 2013, 299, 261–271 CrossRef CAS.
- E. Monyoncho, S. Steinmann, C. Michel, E. Baranova, T. Woo and P. Sautet, ACS Catal., 2016, 6, 4894–4906 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2220590–2220518. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00942h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |