
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal nitride catalysts for selective conversion of oxygen-containing molecules
William N.
Porter
,
Kevin K.
Turaczy
,
Marcus
Yu
,
Hansen
Mou
and
Jingguang G.
Chen
 *
*
Department of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: jgchen@columbia.edu
First published on 22nd April 2024
Abstract
Earth abundant transition metal nitrides (TMNs) are a promising group of catalysts for a wide range of thermocatalytic, electrocatalytic and photocatalytic reactions, with potential to achieve high activity and selectivity while reducing reliance on the use of Pt-group metals. However, current fundamental understanding of the active sites of these materials and the mechanisms by which selective transformations occur is somewhat lacking. Recent investigations of these materials from our group and others have utilized probe molecules, model surfaces, and in situ techniques to elucidate the origin of their activity, strong metal–support interactions, and unique d-band electronic structures. This Perspective discusses three classes of reactions for which TMNs have been used as case studies to highlight how these properties, along with synergistic interactions with metal overlayers, can be exploited to design active, selective and stable TMN catalysts. First, studies of the reactions of C1 molecules will be discussed, specifically highlighting the ability of TMNs to activate CO2. Second, the upgrading of biomass and biomass-derived oxygenates over TMN catalysts will be reviewed. Third, the use of TMNs for H2 production via water electrolysis will be discussed. Finally, we will discuss the challenges and future directions in the study of TMN catalysts, in particular expanding on opportunities to enhance fundamental mechanistic understanding using model surfaces, the elucidation of active centers via in situ techniques, and the development of efficient synthesis methods and design principles.
 William N. Porter | William Porter is a PhD candidate at the Department of Chemical Engineering at Columbia University, with a DOE Graduate Fellowship. His research interests focus on using surface science and in situ characterization to develop fundamental understanding of bimetallic and transition metal nitride catalysts. He is currently studying ethylene hydroformylation and biomass valorization. |
 Kevin K. Turaczy | Kevin Turaczy is a PhD candidate at the Department of Chemical Engineering at Columbia University, with an NSF Graduate Research Fellowship. His research interests include using surface science techniques to characterize potential electrocatalysts for water electrolysis. |
 Marcus Yu | Marcus Yu is a PhD Candidate at the Department of Chemical Engineering at Columbia University, with an NSF Graduate Research Fellowship. His research is focused on studying the reaction pathways of various alcohols and alternative fuels, such as methanol and glycerol, on model surfaces using fundamental surface science techniques. By coupling surface science results with electrochemical experiments and DFT simulations, he aims to better understand trends between model surfaces and powder catalysts to improve future catalyst design. |
 Hansen Mou | Hansen Mou is a PhD graduate from the Department of Chemical Engineering at Columbia University, with an NSF Graduate Research Fellowship. His research interests include the development of earth-abundant electrocatalysts for the production and utilization of alternative fuels. He is currently a Hydrogen R&D Engineer at Linde Engineering. |
 Jingguang G. Chen | Jingguang Chen is the Thayer Lindsley Professor of Chemical Engineering at Columbia University, with a joint appointment at Brookhaven National Laboratory. His research interests include fundamental understanding of carbides, nitrides, and bimetallic catalysts for applications in thermocatalysis and electrocatalysis. His research group utilizes a combination of experimental studies, in situ characterization and density functional theory calculations. |
1. Introduction
Transition metal nitrides (TMNs) are synthesized by the integration of nitrogen atoms into the lattice of a parent transition metal. They typically display high melting temperatures, hardness, tensile strength, and electrical conductivity, making them promising for many applications in materials science, energy storage, and catalysis.1–9 In particular, the use of earth-abundant TMNs as catalysts represents a promising approach to exploit their properties in order to improve catalytic activity and selectivity, while also reducing reliance on platinum-group metals (PGM). Compared with PGMs, TMNs typically have lower cost10 and improved hardness and strength, often resembling that of ceramic materials.4 Comparisons of catalytic performance between TMNs and PGMs depend on the specific nitride itself, the exposed surface facet, the presence of defects, and the reaction chemistry.11 Some of the earliest demonstrations of the catalytic properties of TMNs were studies of ammonia synthesis and decomposition on Mo catalysts, where it was shown that the rate depended on the extent of nitridation of the surface layers.12,13 Subsequently, inspired by work indicating the ability of transition metal carbides (TMCs) to facilitate hydrogenolysis reactions, TMNs were investigated as promising catalysts for hydrodenitrogenation (HDN) reactions.14In general, TMNs and TMCs display similar structural and catalytic properties, which can be attributed to similarities in size and electronegativity between carbon and nitrogen atoms. When comparing TMCs to TMNs, Group 6 metal carbides tend to behave more like Group 5 nitrides than their nitride counterparts due to the extra electron of nitrogen.1 Since the early demonstrations of ammonia synthesis and HDN, significant progress has been made in designing TMN-based catalysts for a wide variety of reactions including hydrogenolysis of various carbon-heteroatom (C–N, C–S, and C–O) bonds, methane dry reforming, plastic upcycling, wastewater treatment, CO and CO2 upgrading, biomass conversion, and others. The superiority of TMNs versus TMCs is unique to the performance metric of interest, the reaction chemistry and conditions, and the parent metal. When comparing corresponding TMNs and TMCs for acetone condensation, the activity followed the trend of Mo2N > Mo2C, which was attributed to an increased base site density of Mo2N.15 On the other hand, MoCx has displayed improved activity toward biomass deoxygenation compared to MoNx.16 In addition, a study of TMN and TMC catalysts for Fischer–Tropsch synthesis found that for Mo and W parent metals, the activity of the carbides exceeded the nitride, but for V and Nb parent metals, the nitrides were more active than the carbides.17 Despite the broad range of catalytic reactions to which TMN and TMC materials have been applied, transition metal nitrides, carbides, and even phosphides and sulfides, are often grouped together in review articles. While it is often useful to consider them together, the diversity of both reaction chemistries as well as TMN structures and compositions calls for a focused perspective on utilizing these earth-abundant materials for sustainable fuel and chemical production.
This article will focus on the unique properties of TMNs by highlighting promising studies of TMN-based catalysts for thermocatalytic, electrocatalytic and photocatalytic reactions. Specifically, we will use three classes of reactions of oxygen-containing molecules—valorization of C1 molecules, selective conversion of biomass and biomass-derived oxygenates, and electrolysis of H2O—as representative case studies to offer insights into the reaction mechanisms. We will discuss recent progress on developing TMN-based catalysts for the selective transformation of C1 molecules including CO, CO2, CH3OH, and HCOOH. Selectively transforming biomass feedstocks to useful compounds is critical to transitioning to a sustainable chemical industry, so the conversion of biomass and biomass derivatives using TMNs will also be discussed, with a focus on identifying the active sites for selective bond scission reactions by using model surfaces, probe molecules, and in situ techniques. We will then summarize TMN-based catalysts for water electrolysis to produce H2, which is often required for the valorization of CO2 and biomass, the only two major sources of non-fossil-based carbon. We will conclude this Perspective by highlighting the challenges and future directions for research on using TMN-based catalysts for selective chemical transformations.
2. Transition metal nitride synthesis and characterization
This Perspective will outline investigations of TMNs on both model surfaces and powder catalysts. A range of different synthesis methods have been utilized to prepare these materials, dependent on the application and fundamental questions being studied. Previous reviews have summarized the synthesis and characterization of TMCs and TMNs in detail,18,19 but an overview of the methods that have been used for manufacturing TMN structures for catalytic reactions will be given here.In the powder form, TMNs have been typically synthesized through nitridation of a metal oxide with NH3 at elevated temperatures and pressures. The resulting nitride phase can be controlled by modifying reaction conditions including the heating rate, nitridation temperature, and space velocity.20,21 These parameters, especially space velocity, have a significant influence on the surface area of the resulting TMN.22,23 The temperature required to reach full nitridation varies depending on the identity of the transition metal and whether the precursor is a binary or ternary oxide.24 Due to the hazards and expense of the typical synthesis procedure using NH3, they have been limited to small, lab-scale applications, and methods that utilize a safer and less costly source of N have been investigated.25 Mixtures of N2 and H2 have been employed, but these methods have more strict space velocity and heating rate requirements compared with those that use NH3.26–28 Urea has been demonstrated as a source of either nitrogen or carbon to produce nearly pure TMNs or TMCs from solid metal chloride precursors.29,30 More novel methods relying on biomass-derived precursors have also been demonstrated. Chen et al. devised a method to react ground soybean powder with ammonium molybdate, as a Mo precursor, in water. Further calcination at 1073 K in Ar resulted in the synthesis of an active HER electrocatalyst containing both tetragonal γ-Mo2N and orthorhombic β-Mo2C as confirmed via XRD.31 Meng et al. similarly utilized soybean powder as a carbon and nitrogen source to generate a composite of W2C and WN where the carbide acted as the active catalyst and the nitride functioned as a catalyst stabilizer in acidic electrolyte.32 In order to develop industrially relevant TMN catalysts for the reactions highlighted in this Perspective, synthesis methods such as these should continue to be studied and refined.
Model surfaces of TMNs have also been used to perform fundamental mechanistic studies of reaction intermediates and pathways on specific active sites and crystal facets. These can be either single crystals or thin films. Single crystals have been prepared by using magnetron sputtering of pure Ti, V, or Nb in Ar/N2 to deposit epitaxial TiN, VN, or NbN on an MgO(111) surface.33 Studies of TMN single crystals synthesized via this method have typically focused on structural and mechanical properties rather than catalytic performance. On the other hand, investigations of TMN model surfaces for catalytic reactions have relied mainly on TMN thin films, which can be prepared ex situ by nitridation of a polycrystalline foil of the parent metal with N-containing compounds such as NH3 or N2/H2 mixtures, similarly to the traditional methods for synthesizing powder TMNs. Thin TMN films can also be synthesized via reactive sputtering of the parent metal surface in N2,34 or through atomic layer deposition to control film thickness and uniformity, which is used mainly for electronics applications.35 Recent work has utilized a combination of fundamental studies of TMN thin films in ultrahigh vacuum (UHV) with electrocatalytic evaluation of the same films or thermocatalytic testing of the corresponding powder catalysts to develop correlations between the configuration and binding strength of surface intermediates and performance under realistic conditions.36,37
The characterization of TMN catalysts has relied on both conventional techniques and advanced methods to measure the surface area, crystal structure, chemical state, surface composition and other properties of TMNs. The conventional techniques that are widely used include BET surface area measurements,38 as well as X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) that are used to characterize the crystal structure and oxidation states of the atoms, respectively. Symmetric XRD and glancing incidence XRD (GI-XRD) can be used in tandem to probe the bulk and surface crystal structure, respectively. GI-XRD, in combination with XPS, is particularly useful for characterizing the thickness of TMN thin films, as demonstrated in a study of an Mo2N model surface.39 XPS, as well as Auger electron spectroscopy (AES), can also be used to probe the stoichiometry and elemental composition of TMN powders and thin films.40,41 Ambient pressure XPS has been used to probe the oxidation state and surface composition of TMNs under different conditions.42 Scanning electron microscopy (SEM), transmission electron microscopy (TEM), and elemental mapping via energy dispersive X-ray spectroscopy (EDS)43 or electron energy loss spectroscopy (EELS)42 have been employed to provide detailed information about the surface morphology and elemental composition, as well as the particle size and dispersion of admetals. In addition to these more traditional tools, recent studies have utilized in situ techniques, especially X-ray absorption spectroscopy (XAS), to elucidate the chemical state and local structure of the metals under reaction conditions.42–44 The XAS techniques of X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) are particularly powerful for characterizing metal–support interactions between TMNs and admetals. By combining these material characterization methods with catalyst evaluation experiments and in situ measurements of reaction intermediates using techniques such as diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) or Raman spectroscopy, structure–performance relationships can be developed to guide the design of TMN catalysts with desired catalytic properties.45
3. Case studies of TMN-based catalysts
3.1 Reactions of C1 molecules
Selective transformations of C1 molecules have potential to enable the production of valuable chemicals from low-cost and waste feedstocks. This class of reactions also plays a critical role in mitigating climate change as many of the most prevalent greenhouse gases are C1 molecules. Due to their ability to facilitate strong metal–support interactions and to promote selective bond scissions, TMN catalysts have recently been utilized for catalytic conversions of C1 oxygenates. In addition, C1 molecules have been used as probes to elucidate metal–support interactions and the acid–base character of active sites of TMNs.In particular, the use of Mo2N as an active and stable support for several different reactions of CO2 is promising. Lin et al.43 have demonstrated the promising stability of an Mo2N-supported Ni catalyst for the RWGS reaction of CO2 to produce CO. They observed reverse thermal sintering, a process that involves a transition from larger 3D metal particles to sub nanometer or 2D particles as the temperature increases, due to interactions between the γ-Mo2N support and Ni. Using ab initio molecular dynamics (AIMD) (Fig. 1a) simulations, it was shown that a 3D Ni particle supported on Mo2N rapidly converted into a 2D raft-like structure, while this wetting phenomenon was not observed on a CeO2 support. XANES results (Fig. 1b) showed an increase in the peak height at the Ni K edge as the reduction temperature was increased from 753 K to 863 K, suggesting that there were beneficial electronic interactions taking place, specifically in the form of charge transfer from Ni to the Mo2N support. EXAFS analysis provided additional evidence of the structural transformation of Ni due to direct interactions with the Mo2N support, with the appearance of a unique Ni–Mo bond at 2.62 Å. Combined secondary electron and scanning tunneling electron microscopy measurements indicated the formation of surface Ni4N at 673 K, which spread into thin layers after heating to 793 K. Catalytic evaluation of the CO2 hydrogenation reaction (Fig. 1c) showed improved CO selectivity and yield over Ni/γ-Mo2N compared with Ni/CeO2 due to these under-coordinated surface Ni species formed via reverse thermal sintering. The inferior performance of the Ni/CeO2 catalyst could be attributed to a much larger particle size of Ni on Ni/CeO2 compared with Ni/Mo2N, as indicated from the EXAFS measurements.
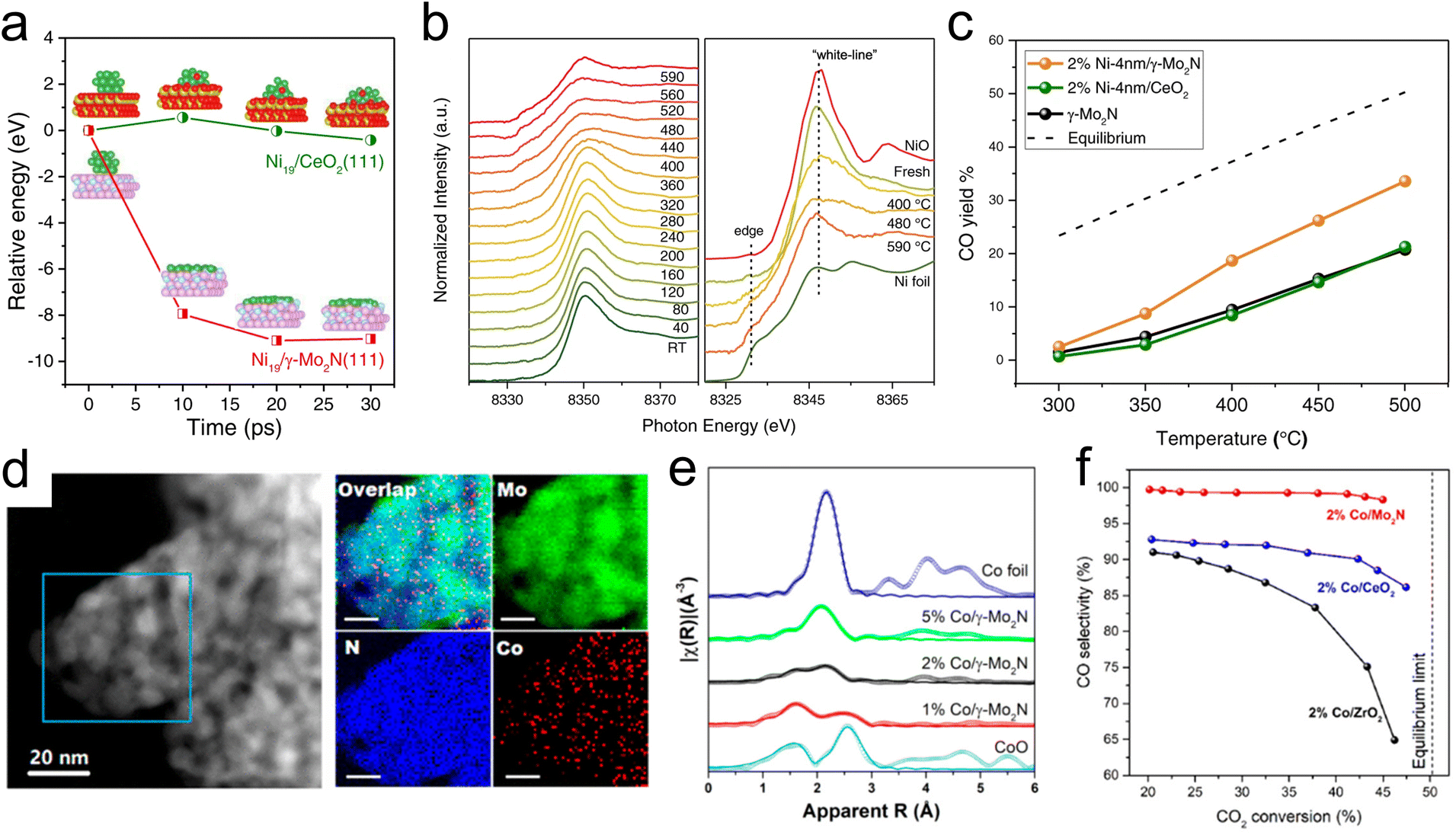 | ||
| Fig. 1 Studies of Ni-modified (a–c) and Co-modified (d–f) γ-Mo2N for thermocatalytic CO2 conversion. (a) Relative energy change of redispersion on Ni19/CeO2(111) (green) and Ni19/γ-Mo2N (red). (b) Ni K-edge X-ray absorption near edge spectra of 4% Ni-4 nm/γ-Mo2N catalyst, with right panel comparing Ni foil and NiO standards with the fresh catalyst and after reduction at 673 K, 753, and 863 K. (c) CO yield during the CO2 hydrogenation reaction at different temperatures over 2% Ni-4 nm/γ-Mo2N, 2% Ni-4 nm/CeO2, and γ-Mo2N catalysts. (d) TEM and STEM-EDS images of a Co/γ-Mo2N catalyst. (e) EXAFS of 1%, 2%, and 5% Co/γ-Mo2N catalysts with reference Co foil and CoO spectra. (f) Catalytic evaluation of 2% Co/ZrO2, Co/CeO2, and Co/γ-Mo2N catalysts. (a–c) Reprinted from ref. 43 with permission from Springer Nature, Copyright 2021. (d–f) Reprinted from ref. 42 with permission from the American Chemical Society, Copyright 2019. | ||
Further studies demonstrated unique metal–support interactions using a γ-Mo2N support for the RWGS reaction of CO2. Specifically, Co-modified Mo2N facilitated CO2 conversion, CO selectivity, and catalytic stability.42 Due to strong bonding between Co and γ-Mo2N, subnanometer Co particles were stabilized under reaction conditions. TEM and STEM-EELS (Fig. 1d) images taken after catalyst activation showed that Co was highly dispersed, indicating that the Mo2N support was able to anchor Co under high temperature conditions to prevent agglomeration. A comparison of the XANES results for a range of Co/γ-Mo2N catalysts showed decreasing white line intensity with increasing Co loading, indicating that low loadings led to improved charge transfer and reduced cluster size. EXAFS analysis of the reduced 1% Co/γ-Mo2N sample showed a component at approximately 2.0 Å that, due to the dissimilarity of the XANES profile with Co oxide, was attributed to the Co–N bond formation. Wavelet transformation analysis of the EXAFS profile further confirmed that this component was attributed to Co–N bonds (Fig. 1e). The coordination number was 2.5 for Co–N and 2.4 for Co–Co, suggesting a Co cluster size of <1 nm. The EXAFS and EELS results of a reference Co/CeO2 catalyst showed increased cluster size despite the CeO2 and γ-Mo2N supports having similar surface areas. These findings supported the existence of strong interactions between Co and γ-Mo2N that led to improved dispersion, thermal stability, and enhanced catalytic performance for CO2 hydrogenation. Compared with catalysts containing equivalent loadings of Co supported on CeO2 and ZrO2, the Co/γ-Mo2N catalyst showed the highest CO selectivity at similar conversions (Fig. 1f). Due to the presence of undercoordinated Co, DFT calculations predicted a lower lying d-band center of Co in Co/γ-Mo2N, which likely led to a decrease in CO binding energy, thus preventing C–O scission and increasing CO selectivity as was observed experimentally. In addition, Co/γ-Mo2N showed the best stability among the three catalysts. This study demonstrated that strong metal–support interactions between Co and γ-Mo2N, as well as improved Co dispersion, led to the Co/γ-Mo2N catalyst outperforming the more traditional Co/CeO2 and Co/ZrO2 in terms of CO selectivity (98.3% on Co/γ-Mo2N compared with 88.0% on Co/CeO2 and 68.2% on Co/ZrO2) and stability. The inferior performance of Co/CeO2 and Co/ZrO2 could be attributed to the extent of interactions between Co and the support, where the reducible oxide supports facilitated much weaker interactions between Co and CeO2 or ZrO2, while stronger metal–support interactions occurred on γ-Mo2N. This was suggested by the EXAFS and elemental mapping with EELS that showed larger particle sizes of Co on CeO2 and ZrO2.
Beyond the RWGS reaction, TMN supports have also been used to synthesize other C1 molecules from CO2. Recently, an Mo2N-supported PdMo catalyst showed activity for methanol synthesis at temperatures below 373 K, while a benchmark Cu/ZnO/Al2O3 catalyst did not.49 The PdMo/Mo2N catalyst showed higher activity and lower activation energy for CO2 hydrogenation compared with a Pd/Mo2N catalyst with a similar Pd loading. STEM and XRD analysis of the PdMo/Mo2N catalyst showed the presence of an intermetallic hcp-PdMo phase with alternating Pd and Mo layers, while the Pd/Mo2N catalyst facilitated the distribution of Pd nanoparticles that were not overlapping with regions of strong Mo and N signals. Methanol production from CO2 has also been shown using a TMN as the active material rather than a support. Wang et al. demonstrated superior TOF of a Co4N nanosheet for CO2 hydrogenation to methanol compared with both a Co nanosheet and a commercial Cu/ZnO/Al2O3 catalyst.50 Co4N has also been shown to be active for natural gas production from the hydrogenation of a mixture of CO and CO2.51 In addition, nitrides such as Fe2N52 and W2N3 (ref. 53) have been shown to possess activity for C–C coupling to produce C2+ molecules from CO2.
In addition to their utilization in thermocatalytic reactions, TMNs have also been used in photocatalytic and electrocatalytic reactions of CO2. Photo-assisted reduction of CO2 to CO has been demonstrated on an Mo2N catalyst, where it was shown that the active species was an Mo2NHx formed through photo-induced H2 heterolysis.54 This change in the H2 dissociation mode was suggested by in situ FTIR and electron paramagnetic resonance analysis, while kinetic isotope effect evaluation demonstrated a lower activation barrier for CO2 reduction due to this photo-assisted H2 heterolysis (Fig. 2a). Other investigations of photocatalytic and electrocatalytic CO2 reduction have elucidated a promotional effect of Co2N on the activity of a BiOBr catalyst,55 a photoelectrochemical conversion of CO2 to formate over 3D TiN nanoshells,56 and CO2 electroreduction over CuNNi3.57
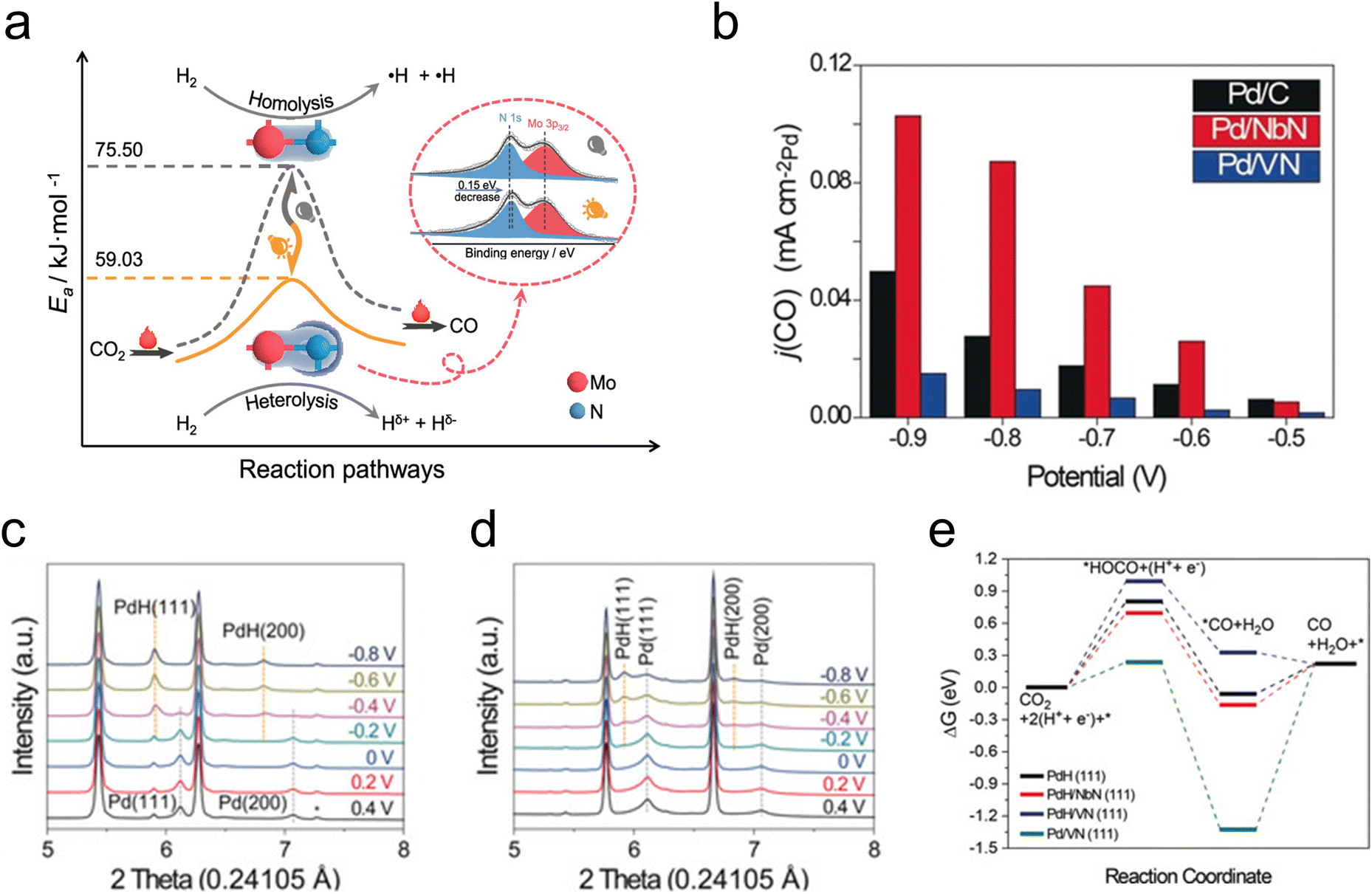 | ||
| Fig. 2 Photo- and electrocatalytic CO2 reduction. (a) Schematic showing the decrease in activation barrier due to photo-assisted H2 heterolysis on Mo2N. (b) CO partial current density for Pd/VN (blue), Pd/NbN (red), and commercial Pd/C (black). (c) In situ XRD profiles of Pd/NbN. (d) In situ XRD profiles of Pd/VN. (e) DFT-calculated free energy diagram of CO2 reduction. (a) Reprinted from ref. 54 with permission from the American Chemical Society, Copyright 2021. (b–e) Reprinted from ref. 58 with permission from John Wiley and Sons, Copyright 2020. | ||
The co-electrolysis of CO2 and H2O to produce syngas with controlled CO/H2 ratios has been investigated over TMN-supported Pd. The results showed that Pd/NbN facilitated a higher faradaic efficiency to syngas than Pd/VN and commercial Pd/C, owing to the more facile formation of the active PdH species under reaction conditions (Fig. 2b).58In situ XRD measurements demonstrated a clear peak shift at −0.2 V for Pd/NbN and Pd/C, corresponding to lattice expansion as H atoms diffused into the Pd lattice, indicative of a phase transformation of Pd to PdH (Fig. 2c). On the other hand, a mixture of Pd and PdH was observed for Pd/VN (Fig. 2d). DFT calculations (Fig. 2e) supported the XRD results, suggesting more favorable PdH formation on NbN than on VN, and reaction energy calculations demonstrated that activity for CO2 to CO depends on the adsorption energy of the *HOCO intermediate on PdH.
Several investigations have taken advantage of the Pt-like electronic properties of TMNs by using them as active supports to reduce Pt loadings for methanol electrooxidation. Qi et al. showed that supporting Pt on various TMNs (TiN, VN, NbN, Mo2N and TaN) increased the Pt-based mass activity towards the methanol oxidation reaction in alkaline conditions compared to commercial Pt/C.63 The catalysts were prepared using a solvothermal reduction method with sodium citrate and ethylene glycol as reducing agents that resulted in uniform 1.8–1.9 nm Pt nanoparticles at 3 wt% Pt loading. The authors evaluated the electrocatalytic activity using cyclic voltammetry in 1 M CH3OH and 1 M KOH at 50 mV s−1 and found promising mass activity for all of the Pt-TMN/C catalysts. The best performing catalyst was Pt–NbN/C, which exhibited a Pt-based mass activity that was an order of magnitude greater than that of Pt/C. The authors further examined the CO poisoning resistance of the Pt-TMN/C catalysts using CO stripping experiments in alkaline conditions and found that the onset potential was reduced compared to the commercial Pt/C catalyst. These results suggested that the presence of TMNs can improve the CO tolerance of Pt-based electrocatalysts for methanol oxidation.
Fundamental studies of the decomposition of methanol under UHV conditions have also been conducted. Recently, the reaction of methanol on Pt-modified Mo2N and TiN thin films was studied using temperature programmed desorption (TPD) to determine the gas-phase products and high-energy electron energy loss spectroscopy (HREELS) to identify the surface reaction intermediates64 (Fig. 3). Under UHV conditions, the decomposition of methanol can occur through either C–O bond cleavage to produce methane or selective C–H bond scission to produce CO, with the latter being preferred for DMFC applications. The clean Mo2N surface displayed high selectivity toward CO compared to methane, while TiN strongly favored C–O bond scission leading to methane production or complete decomposition. The addition of Pt onto the TMN surfaces increased the CO selectivity for both Mo2N and TiN. HREELS experiments provided insight into the surface intermediates from methanol decomposition, showing the formation of a methoxy intermediate on all surfaces at 200 K. One key observation was a blueshift of the C–O stretching mode on TiN as the surface was heated to higher temperatures. This shift was previously observed by Hwu et al. on C/W(111) and was attributed to a perpendicular orientation of the methoxy intermediate rather than an inclined position.65 This orientation was consistent with a reduced interaction between the TiN surface and the C–O and C–H bonds. As a result, most of the methoxy intermediate remained stable on TiN until 600 K, at which point C–O bond scission was observed. In comparison, nearly all the methoxy intermediates on the Pt/Mo2N, and Pt/TiN surfaces appeared to have desorbed or reacted by 500 K. This blueshift was not observed for Mo2N, which showed that the orientation of the methoxy intermediate remained to be inclined as the surface was heated. The difference in orientation and the resulting selectivity may be explained by a difference in the carbophilicity between Ti and Mo. While both are highly oxophilic, Mo has a higher carbophilicity than Ti such that the CH3 group on methanol interacted more strongly with Mo than with Ti.66 The C–H bonds were therefore more easily broken allowing for higher CO selectivity on Mo2N than on TiN.
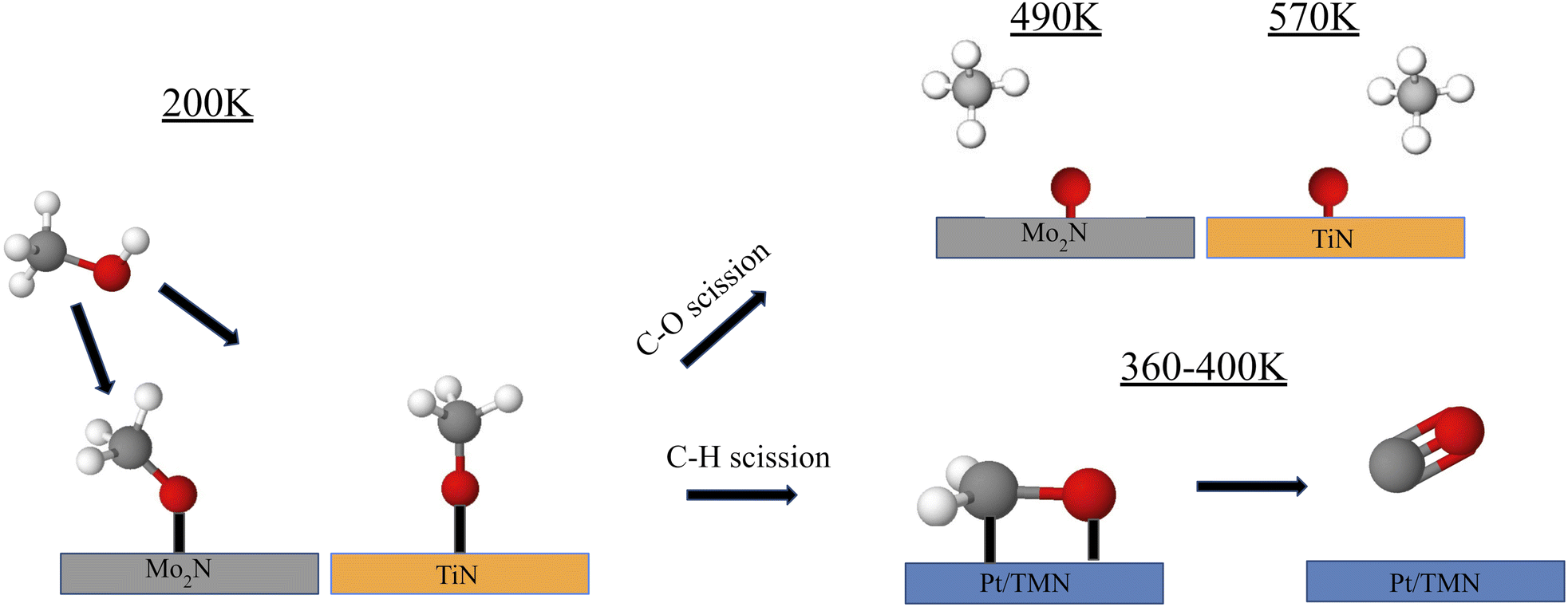 | ||
| Fig. 3 Schematic representation of reaction pathways for methanol on Mo2N and TiN. An inclined methoxy intermediate was formed on Mo2N while a perpendicular orientation was taken on TiN. C–H bond scission to form CO was favored on 1 monolayer (ML) Pt-covered surfaces and C–O bond scission was suppressed. | ||
Trends on TiN were similar to those on WC, which may be attributed to the formation of highly stable methoxy intermediates on both surfaces. DFT analysis of methanol decomposition on WC and Pt/WC surfaces showed that O–H bond scission to form the methoxy intermediate was highly favored on WC with a binding energy of −3.52 eV compared to −0.91 eV for the *CH2OH intermediate.67 Additionally, C–H scission of the methoxy intermediate had a large activation of barrier of 2.11 eV. In contrast, chemisorption via O–H bond scission was only slightly favored on Pt/WC over C–H bond scission. Further C–H scission of methoxy on Pt/WC had a lower activation barrier of 1.01 eV. The Pt layer over WC significantly reduced the surface oxophilicity, which prevented C–O scission while also enhancing C–H scission.
These results from well-characterized surfaces revealed that the presence of Pt on TMNs suppressed the C–O bond scission pathway that led to undesired methane formation, suggesting the potential of Pt/TMN catalysts for the selective electrooxidation of methanol to CO and CO2.
In addition to being a probe molecule for acid–base chemistry, formic acid can also be derived from biomass compounds to serve as a platform chemical for the synthesis of CO via dehydration, H2via dehydrogenation, or syngas via selective decomposition.68 Recent work has shown promising performance of Mo2N-based catalysts for selective formic acid dehydrogenation to produce H2 at lower temperatures than traditional solid-acid catalysts.69 In another study comparing γ-Mo2N synthesized at different temperatures, promising activity and selectivity for the dehydration of formic acid to produce CO was demonstrated. The authors showed that the activity change was consistent with differences in acidity and H content of the Mo2N catalysts. Furthermore, a Pd–Mo2N catalyst has shown enhanced performance compared to other Pd-based materials for formic acid electrooxidation, which can be applied in a direct formic acid fuel cell.70
3.2 Valorization of biomass and biomass-derived oxygenates
Aside from CO2, biomass is the other major non-fossil source of carbon. Increasing research focus has been placed on effectively utilizing biomass resources in order to decrease reliance on fossil fuels. In particular, the use of lignocellulosic biomass is appealing because it avoids the need to sacrifice farmland or crops to produce biobased fuels and chemicals.71,72 A major step of this process involves the removal of oxygen through hydrodeoxygenation (HDO).73,74 Further upgrading biomass derivatives requires selectively breaking bonds while preserving functional groups of interest.75 This concept of breaking down complex biomass molecules into useful platform chemicals represents a method of synthesizing compounds that is fundamentally different from the manufacture of petrochemicals, which generally proceeds through the addition of functional groups to platform compounds.76 TMNs have shown promising catalytic performance for the removal of heteroatoms, including O, N, and S, through hydrotreatment reactions.77,78 Therefore, TMNs have been applied recently to reactions of biomass molecules, although further elucidation of the active species and mechanisms is necessary. This section will describe recent progress in identifying promising TMN catalysts for upgrading biomass materials and biomass-derived oxygenates. Before describing the more commercially-relevant studies, recent investigations of reaction pathways and active sites that have relied on probe molecules and model surfaces will be discussed in the context of the current understanding of the electronic and structural properties of TMN-based materials.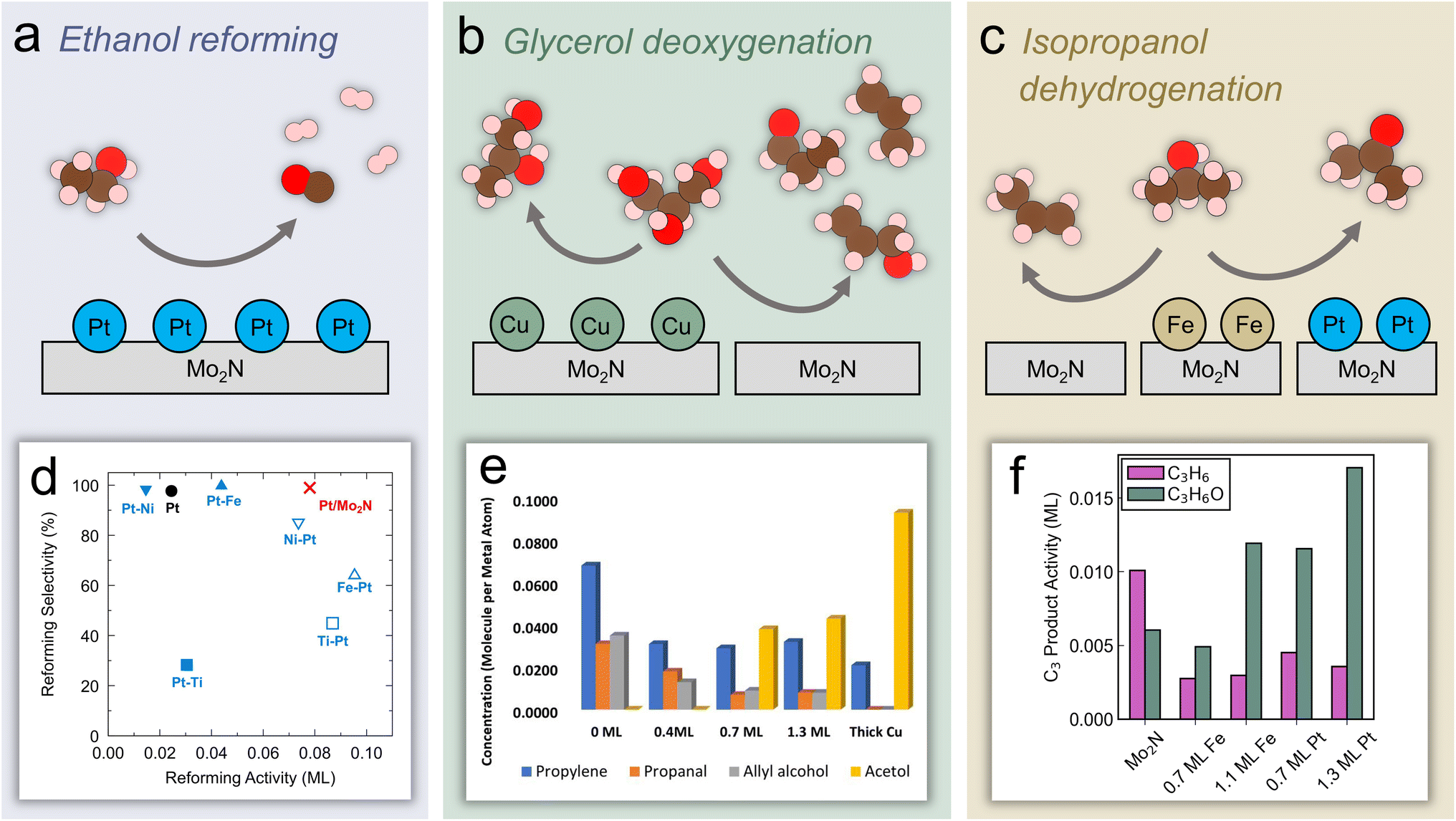 | ||
| Fig. 4 Experimental and theoretical studies using probe molecules on Mo2N-based model surfaces and catalysts. Reaction schemes over Mo2N: (a) ethanol reforming on Pt-modified Mo2N (b) glycerol HDO over Cu-modified Mo2N. (c) Isopropanol dehydration/dehydrogenation over Fe- and Pt-modified Mo2N. (d) Machine learning predicted ethanol reforming activity and selectivity over a range of catalysts. (e) Quantified TPD activity of glycerol HDO over Cu/Mo2N. (f) C3 product activity on Fe/Mo2N and Pt/Mo2N. (d) Reprinted from ref. 79 with permission from Elsevier, Copyright 2022. (e) Reprinted from ref. 39 with permission from the American Chemical Society. (f) Reprinted from ref. 36 with permission from the American Chemical Society. | ||
As the simplest molecule containing C–O, C–C, C–H, and O–H bonds, ethanol is a useful model compound to probe inherent bond scission selectivity of active catalytic sites. Furthermore, converting ethanol into hydrogen through the reforming reaction has potential as a route to produce fuel from renewable feedstocks (Fig. 4a). Denny et al. predicted high activity and selectivity of ethanol reforming on Pt/Mo2N using machine learning models that calculated these descriptors based on key reaction steps.79 It was shown that the reforming activity and selectivity increased as C–O bond scission (*CH3CH2O → *CH3CH2 + *O) became more thermodynamically favorable, but were inversely correlated with the feasibility of C–C cleavage (*CH3CHO → *CH3 + *CHO). In addition, fast kinetics and an increased thermodynamic driving force of other bond scission reactions (*CH2CH2O → *CH2 + *CH2O and *CH3CO → *CH3 + *CO, respectively) led to further increases in the reforming selectivity. From this analysis, monolayer Pt/Mo2N was predicted to facilitate three times higher reforming activity and similar selectivity compared with a pure Pt surface (Fig. 4d), suggesting the potential of the Mo2N support to allow a reduction of Pt loading with improved performance. In parallel, TPD studies of the Pt(111) and Pt-modified Mo2N model surfaces were conducted that confirmed the computational prediction, revealing a similar selectivity but significantly higher activity toward reforming on the monolayer Pt/Mo2N surface compared with Pt(111). To elucidate the origin of this effect, the density of states of bulk Pt and Pt in Pt/Mo2N were compared, showing additional occupied states at the valence band edge of Pt in Pt/Mo2N, leading to increased oxophilicity of this surface.79 This ability to bind more strongly to ethanol and ethoxy contributed to a lower-energy transition-state for C–O cleavage compared with C–C cleavage, while the opposite trend was observed on Pt. This method of combining computational predictions and fundamental understanding with experimental verification represents a powerful tool for developing promising catalysts with enhanced activity and selectivity.
Another recent study has used a similar approach to identify catalysts with high activity for a different reaction of biomass-derived alcohols.80 The dehydrogenation of alcohols is a method of producing valuable carbonyl molecules as well as H2 from renewable biomass resources. Methanol dehydrogenation to formaldehyde was used as an initial probe reaction to build a map of turnover frequency as a function of C and O binding energies on a series of transition metals. Combining this map with corrections based on experimental results led to an equivalent volcano plot for 2-octanol dehydrogenation over a range of transition metals, carbides, and nitrides (Fig. 5a), which showed two regions of high TOF corresponding to the alkoxy and hydroxyalkyl pathways. The trend in the predicted TOF was experimentally validated through flow reactor evaluations of a series of transition metals. Then, using this map, β-Mo2N(001) and γ-Mo2N(100) were predicted to be promising dehydrogenation catalysts. Microkinetic modeling further validated this by showing higher TOF on β-Mo2N(001) compared to Pt(111) in the presence of co-fed H2 (Fig. 5b). Experimental testing of this prediction showed that, although the TOF was higher overall on Pt compared to Mo2N, it decreased significantly on Pt at low H2 pressures due to coking, while the TOF on Mo2N was not as affected. The authors suggested that as Mo2N synthesis methods improve to preferentially expose the active (001) and (100) facets, the performance of Mo2N-based catalysts can be further enhanced.
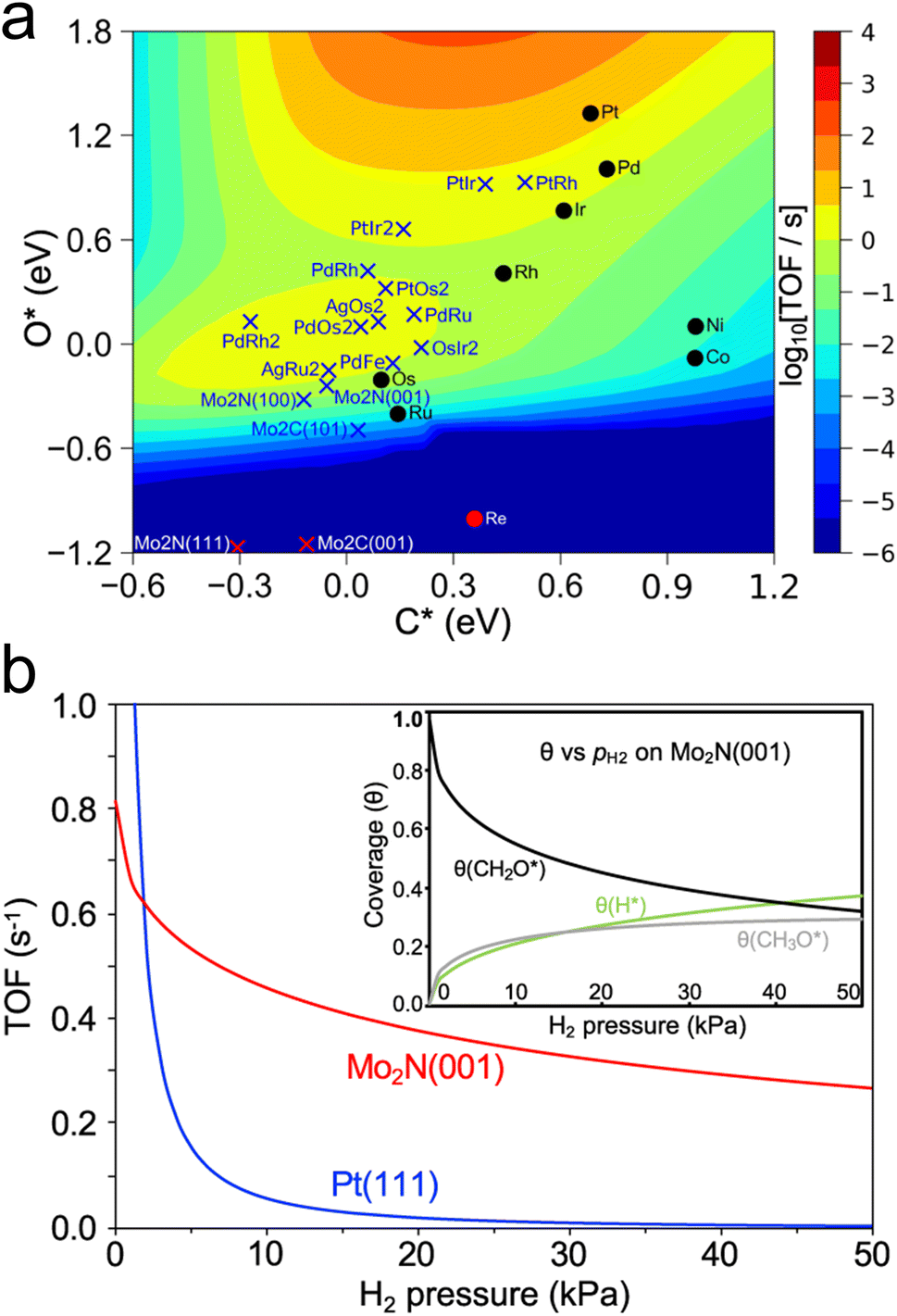 | ||
| Fig. 5 Screening of catalysts for alcohol dehydrogenation. (a) Turnover frequency of 2-octanol dehydrogenation to 2-octanone as a function of C and O adsorption energy. (b) Predicted turnover frequency of methanol dehydrogenation on Pt(111) and β-Mo2N(001) from microkinetic modelling of H2 co-feed at 453 K and pCH3OH = 14 kPa. Inset shows change in coverage of certain intermediates. Reprinted from ref. 80 with permission from Springer Nature, Copyright 2021. | ||
In order to further develop synthesis methods to enable high activity and selectivity, detailed understanding of how metal modification can influence the active sites of TMN surfaces is critical. Glycerol, an abundant byproduct of biodiesel production, has been used as a probe molecule to investigate trends in C–O bond scission on Cu/Mo2N model surfaces (Fig. 4b).39 Because approximately 10 kg of glycerol byproduct is formed per 1 kg of biodiesel, upgrading glycerol to valuable C3 oxygenates is of interest. Furthermore, glycerol can serve as a model compound to study HDO of other biomass-derived polyol molecules. Experimental results showed that the unmodified Mo2N surface was active for C–O scission, breaking two or three C–O bonds and leading to allyl alcohol, propanal, and propylene formation. However, by adding Cu to the surface, the activity shifted toward breaking a single C–O bond and production of acetol (Fig. 4e). The unique selectivity of the Cu/Mo2N surface toward a single C–O bond scission suggested its potential for the selective conversion of other biomass-derived oxygenates. DFT calculations of glycerol on a N-terminated Mo2N surface indicated that it interacted strongly with O atoms, even in the presence of neighboring surface N species, as evidenced by a high exothermicity of O–H and C–O cleavage. In fact, the surface N atoms were important species that activated the O–H bonds in glycerol and influenced the reaction mechanism and selectivity by blocking certain Mo sites to prevent a third C–O scission, leading to propanal and allyl alcohol formation in addition to propylene. On the Mo2N surface, DFT analysis of the reaction energies and activation barriers of key steps elucidated the mechanism of the glycerol bond scission pathways. Glycerol adsorbed via two oxygen atoms interacting with surface Mo sites with the corresponding H atoms hydrogen-bonding to surface N species. Following adsorption, scission of the secondary O–H bond occurred followed by scission of the terminal O–H bond. Then, the concerted C–O cleavage led to allyl alcohol formation and the C–H and O–H cleavage led to a CHOCHOCH2O* formation, which then produced acrolein through C–O cleavage. The allyl alcohol could further react on Mo2N to form acrolein, propanal, or propylene. Due to the potential for strongly bound oxygen atoms on Mo2N and other studies reporting the formation of oxynitride phases, the authors investigated the removal of surface O species with H2. They found that the formation of surface OH species through the reaction of adsorbed O with gaseous H2 was favored over surface H. This indicated that adsorbed O species on Mo sites could be removed by exposure to sufficient H2. Furthermore, a comparison of the energy of two adsorbed OH species reacting to form H2O suggested that oxygen removal on Mo2N was more facile than on Mo2C.
Porter et al. have recently used isopropanol as a probe molecule to study selective bond scission pathways on Fe- and Pt-modified Mo2N surfaces, and extended the results to an evaluation of corresponding powder catalysts (Fig. 4c).36 By focusing on the C3 products, this work showed a shift from propylene formation, or C–O scission, being favored on an unmodified Mo2N surface, to acetone formation, or C–H scission, being favored on Fe/Mo2N and Pt/Mo2N (Fig. 4f). In particular, the Pt/Mo2N surface showed the highest acetone selectivity and the lowest desorption temperature in TPD. Using a combination of vibrational studies and DFT calculations, it was shown that the change in selectivity was due to differences in reaction intermediates that formed on the surface. The unmodified Mo2N surface preferred to form an isopropoxide intermediate (*CH3CHOCH3) that led to C–O cleavage, while the Pt/Mo2N surface stabilized a different isomer (*CH3COHCH3) through α C–H bond scission of isopropanol, leading to preservation of the C–O bond and thus the production of acetone. This difference was ultimately attributed to differences in the projected density of states of Mo in Mo2N and Pt in Pt/Mo2N, where the former had a higher lying, more delocalized d band while the latter was centered at lower energy. This led to a higher oxygen affinity of the Mo2N surface. To extend these findings to more realistic conditions, corresponding Mo2N, Fe/Mo2N and Pt/Mo2N powder catalysts were synthesized via incipient wetness impregnation and evaluated for the reaction of isopropanol in a flow reactor.36 The steady-state selectivity toward acetone followed the order of Mo2N < Fe/Mo2N < Pt/Mo2N, and the product distribution agreed with the trend that was observed on the model surfaces. Finally, in situ XANES measurements showed that the oxidation state of Fe and Pt in these catalysts were relatively stable under reaction conditions. Overall, these results showed the feasibility of using model surface studies to aid in the design of powder catalysts. The simple isopropanol probe molecule allowed understanding of the active sites and pathways that lead to a preference for C–O or C–H bond scission. However, further work using larger biomass probe molecules such as phenol, guaiacol, and anisole should be undertaken to better understand how TMN model surfaces and catalysts could be used for valorization of oxygenates and bio-oil derived from lignocellulosic biomass.
One approach to upgrading biomass relies on thermal treatment such as gasification to produce syngas or pyrolysis to make bio-oil.10 Biofuels can then be produced by removing the abundant oxygen in bio-oil through hydrotreatment to improve the higher heating value, a metric based on the energy content of the fuel.82 TMN-based catalysts have been investigated for both the initial step of catalytic pyrolysis to produce bio-oil, as well as the second step that involves hydrodeoxygenation and decarboxylation of bio-oil and biomass compounds to produce value-added biofuels. Furthermore, TMN-based catalysts have also have shown promise for catalytic transfer hydrogenation of nitrobenzene,83 suggesting that these materials could be applied to the use of organic hydrogen carriers for hydrotreatment of biomass molecules.
Catalytic fast pyrolysis of biomass is a potential pathway to produce fuels from a renewable source of carbon, and has the benefit of being feasible at relatively moderate temperatures. Catalytic fast pyrolysis can be achieved non-catalytically, but utilizing catalysts for the reaction has been shown to improve the heating value of the products, which is generally low due to the significant fraction of oxygenated compounds. Catalysts used for this reaction can be either physically mixed with the raw biomass feedstock, or placed downstream from the pyrolysis reactor to upgrade the pyrolysis vapors. Recently, this has been demonstrated using TMN-based materials including Ti(SO4)2–Mo2N/HZSM-5,84,85 activated carbon-supported W2N and Mo2N,86 TiN, ZrN, and W2N,87 and MoN and VN.88 In general, the oil that is produced from catalytic fast pyrolysis cannot be used directly as a fuel, therefore the design of catalysts that can effectively cleave C–O bonds while promoting C–C coupling is necessary.89 Monometallic TMNs have been widely explored for hydrodeoxygenation and other hydrocracking reactions to directly upgrade biomass materials such as crude palm oil, as well as model biomass compounds like guaiacol. The catalysts investigated include bentonite-ZrN,90 bentonite NiN,91 unsupported Mo2N,92 carbon-supported Mo2N,93 MoNx on metal oxides (Al2O3, TiO2, or ZrO2),94 SBA-15-supported Mo2N–nitrogen-doped carbon catalysts,95 and CoNx supported on nitrogen-doped carbon.96 Recently, research efforts have been directed towards developing bimetallic TMN catalysts for the hydrodeoxygenation reactions to produce sustainable fuels. A recent study by Du et al.97 has shown promising performance of a Ni–Mo bimetallic nitride catalyst for the hydrodeoxygenation of palmitic acid (C15H31COOH) to produce C15 and C16 alkanes. The addition of Ni into the nitride can lead to a lower-lying Mo d-band, resulting in improved catalytic performance. In this work, a Ni3Mo3N alloy was synthesized using a novel method comprising pyrolysis of a nickel molybdate–ethylenediamine mixture, followed by a reduction treatment. It was shown that the Ni3Mo3N produced via this synthesis method facilitated higher conversion and alkane selectivity than the same alloy made using traditional NH3 treatment. The authors suggested that the use of the pyrolysis synthesis technique to form a nitrogenated nickel complex, which provided the N source, followed by a H2 reduction treatment, which led to metallic Ni and Mo formation, ultimately resulted in a more uniform distribution of N in the lattice compared with other synthesis techniques based on oxide precursors. Similar bimetallic nitride effects have been shown by using Co3Mo3N,98 Ni2Mo3N and Ni3Mo3N,99 and Ni–Mo carbonitride100 for the hydrodeoxygenation and selective bond scission of another fatty ester, methyl palmitate. In particular, the use of Ni–Mo carbonitride (NixMoCN) for hydrodeoxygenation and decarbonylation of methyl palmitate represents another promising application of bimetallic TMNs. In this case, including C in the composite led to unique catalytic activity. The synthesis relied on pyrolysis of an organic–inorganic precursor, yielding coexisting Ni2Mo3N and Mo2C phases in the resulting material (Fig. 6a). Compared with MoCN, the NixMoCN catalyst showed a significant increase in the conversion of methyl palmitate along with excellent hydrocarbon selectivity. It was shown that increasing Ni content shifted the reaction pathways to favor decarbonylation/decarboxylation over hydrodeoxygenation (Fig. 6b).
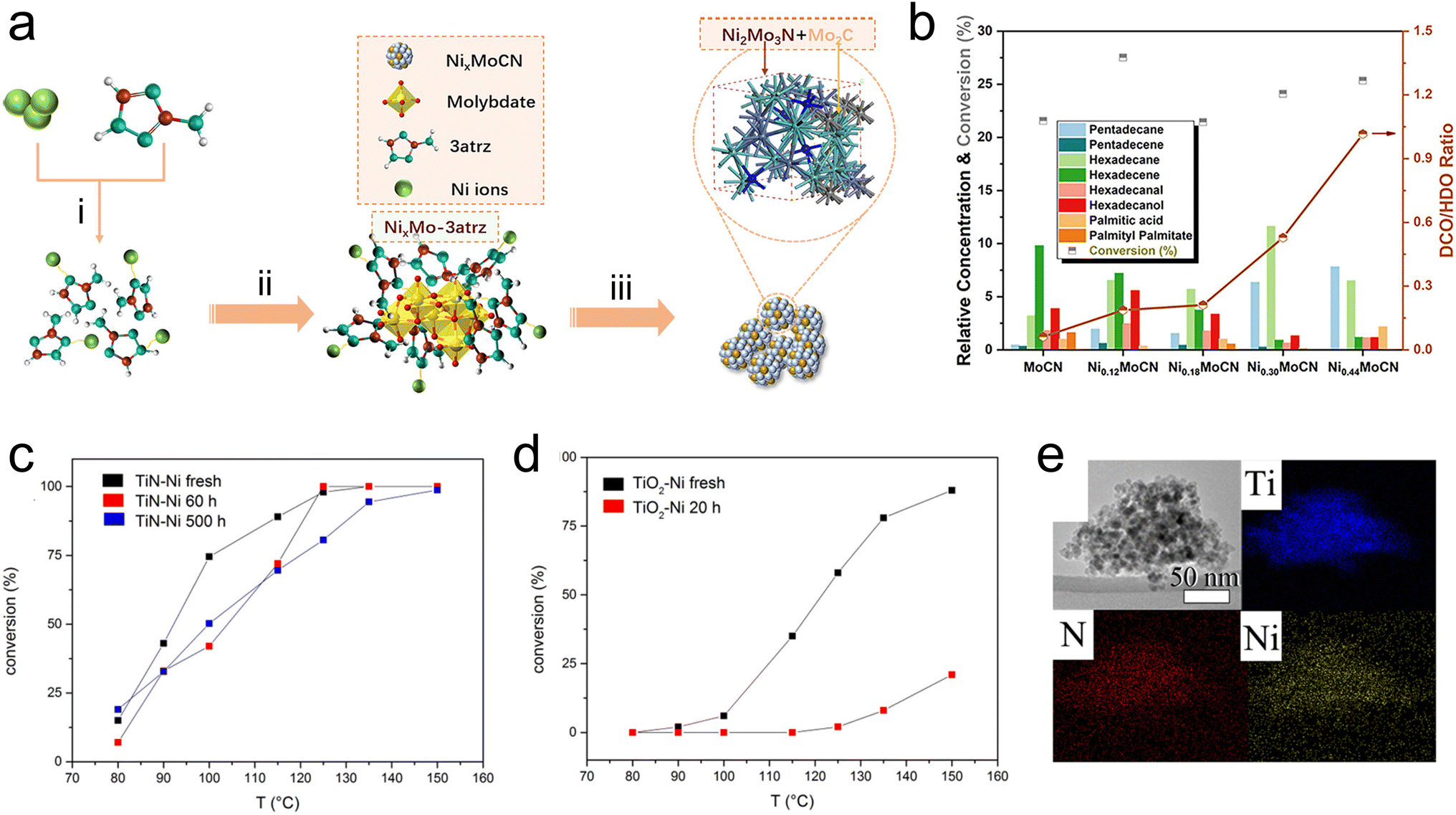 | ||
| Fig. 6 TMNs for biomass upgrading. (a) Scheme showing synthesis procedure of NixMoCN via (a) reaction of Ni nitrate and 3-amino-1,2,4-triazole (3atrz), (ii) addition of Mo to produce organic–inorganic precursor, and (iii) in situ pyrolysis. (b) Product distribution during the reaction of methyl palmitate. (c and d) Conversion of benzyl phenyl ether after continuous reaction of Kraft lignin for 20 h, 60 h, or 500 h. (e) XDS mapping of TiN–Ni catalyst. (a and b) Reprinted from ref. 100 with permission from the American Chemical Society, Copyright 2022. (c–e) Reprinted from ref. 106 with permission from the American Chemical Society, Copyright 2016. | ||
Beyond these more traditional methods of pyrolysis and hydrodeoxygenation to make syngas and fuels, some recent studies have explored lignin depolymerization to sustainably produce valuable chemical intermediates. Lignin is a biopolymer that exists in nature as part of a lignin–carbohydrate complex, which consists of lignin and hemicellulose that are covalently bonded via organic linkages.101 This network must be decomposed to obtain lignin, which itself is made up of a non-repeating pattern of p-hydroxyphenyl, guaiacyl, and syringyl monolignols. These lignin monomers can be obtained via depolymerization, where abundant oxygen content must be removed.102 Due to their activity for hydrodeoxygenation and selective C–O cleavage, TMN-based catalysts have been investigated for lignin depolymerization reactions. One such study proposed a two-step depolymerization method comprising a selective oxidation followed by hydrogenolysis over a TiN–Cu catalyst.103 In another, the authors screened TiN, NbN, Mo2N, and W2N for the depolymerization of soda lignin, showing that TiN and NbN produced promising monomer yield and facilitated similar reaction chemistry due to the presence of Lewis acid sites.104 In addition, Ni-modified TiN105 and Ni–Fe/TiN44 catalysts have also shown promise for depolymerization of α-O-4 lignin model compounds via selective C–O cleavage to produce aromatics. Another promising recent investigation of Ni-modified TiN catalysts for lignin depolymerization was focused on using a Ni-doped TiN (TiN–Ni) for the hydrogenolysis of Kraft lignin.106 Using XRD, the authors showed that the incorporation of Ni led to a doping effect whereby the cell size decreased due to the smaller diameter of Ni relative to Ti. EDX mapping demonstrated that the distribution of Ni was uniform throughout the TiN lattice (Fig. 6e). Benzyl phenyl ether was used as a probe molecule for the α-O-4 linkage of lignin, and improved conversion to phenol and toluene was observed over TiN–Ni compared to a corresponding oxide (TiO2–Ni). To demonstrate the practical feasibility of using the nitride catalyst, the conversion of benzyl phenyl ether was evaluated after 20 h, 60 h, or 500 h of the continuous reaction of Kraft lignin (Fig. 6c and d). The TiN–Ni catalyst clearly showed enhanced stability after 500 h, while the TiO2–Ni was significantly deactivated after only 20 h. The authors attributed the enhanced performance of TiN–Ni to improved dispersion of Ni into the TiN, as well as the different oxidation states of Ti (Ti3+ in TiN–Ni and Ti4+ in TiO2–Ni). In addition, superior stability of TiN–Ni over 500 h was observed when compared with other common lignin refining catalysts of Pd/C and Raney Ni, which underwent significant pressure drop and system blockage after only 12 h. This was attributed to the specific porosity of TiN–Ni, which prevented pore blockage due to coke deposition.106 While this study identified the promising potential of the TiN–Ni catalyst for lignin hydrogenolysis, further understanding of the unique electronic structure of this composite material and the origin of its enhanced stability is necessary, potentially through the use of in situ characterization.
3.3 Water electrolysis for hydrogen production
Because the upgrading of CO2 and many biomass feedstocks often rely on hydrogenation and hydrotreatment, it is imperative to utilize green H2 to minimize their carbon footprint. The production of H2via water electrolysis is a promising alternative to traditional, CO2-intensive methods such as steam-methane reforming.107,108 Electrocatalysts are employed to promote the hydrogen evolution reaction (HER) for water electrolysis. The choice of electrocatalyst depends on the electrolyte in which the reaction takes place, but Pt-based materials are the typical benchmark HER catalysts in both acidic and alkaline electrolytes. Due to its cost and scarcity, efforts have been made to either reduce the amount of Pt or supplant Pt entirely while maintaining high HER activity. Metal alloys, metal–organic frameworks, and various transition metal compounds have exhibited promising results to this end.109–113 In recent years, TMN-based catalysts have demonstrated low overpotentials and high HER activity rivaling that of Pt, especially under alkaline conditions.Under acidic conditions, HER is composed of two steps: the Volmer step, where a proton in aqueous solution adsorbs onto the catalyst, and then either the Heyrovsky step, where the adsorbed hydrogen, Hads, combines with a proton in solution to form H2, or the Tafel step, where Hads combines with another Hads and desorbs as H2 from the catalyst surface. In alkaline solution, water dissociates onto the surface to produce Hads. Therefore, HER catalysts must follow Sabatier's principle by exhibiting optimal binding strength to hydrogen. The HBE value has been demonstrated over many types of electrocatalysts to be an effective descriptor of HER activity.115–117 Ologunagba and Kattel determined HBE values for PGM/TMNs and found that both Pt/TiN and Pt/MnN exhibited HBE within 0.25 eV of a Pt(111) surface.114 When comparing the HBE values to the limiting potential, UL (the lowest applied potential where the reaction proceeds downhill on a free energy diagram), a volcano relationship was obtained with both Pt/MnN and Pt/TiN situated at the top (Fig. 7a). Interestingly, 1 ML of Pd on VN also resulted in a HBE within 0.25 eV of Pt, and a UL that was close to zero. These three PGM/TMN electrocatalysts minimized the necessary overpotential required to conduct HER while remaining stable.
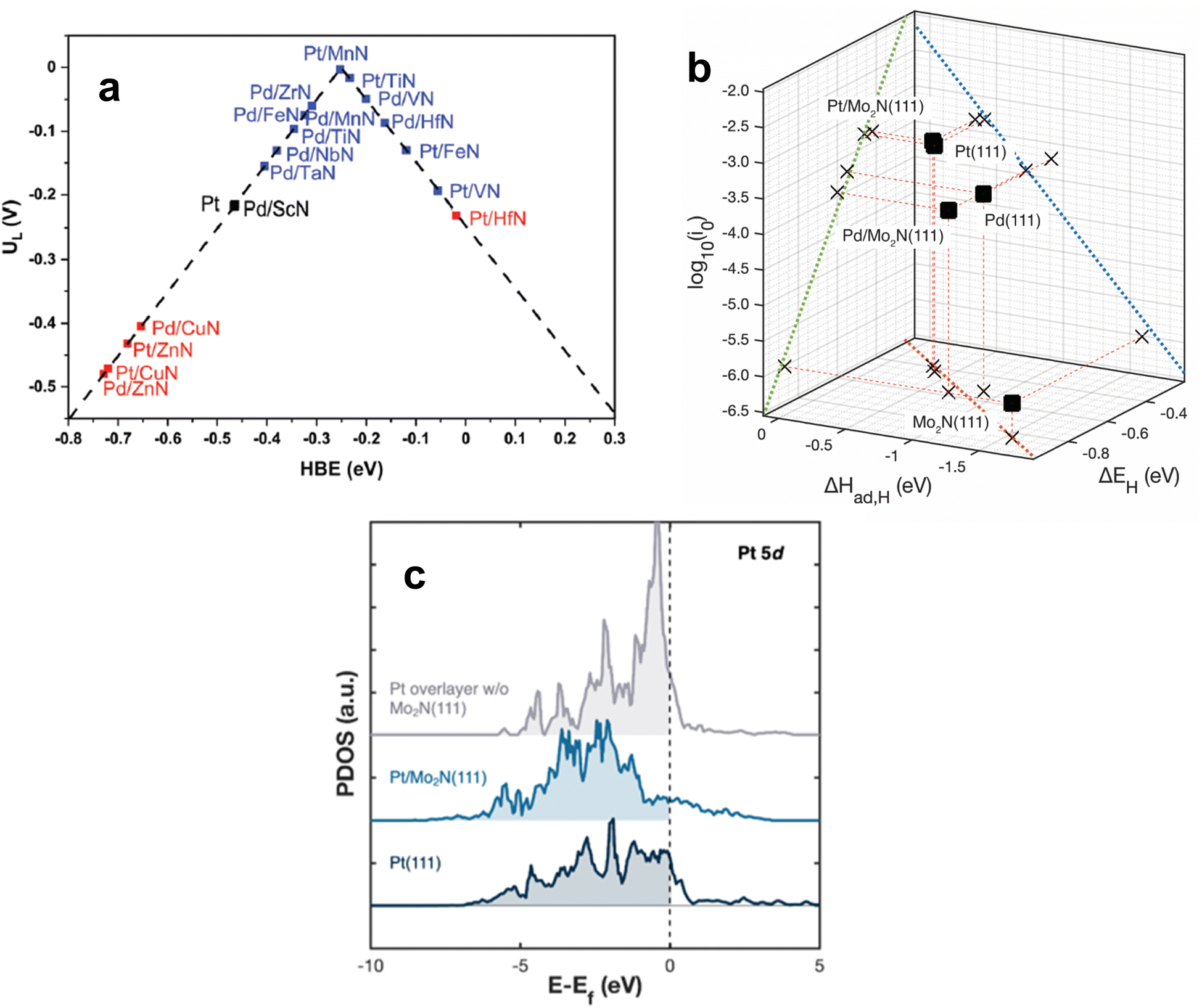 | ||
| Fig. 7 (a) Volcano relationship for Pt-modified TMNs. (b) Correlation between the HER activity (log10(i0)) and the binding strength of atomic hydrogen determined by TPD (ΔHad,H) and DFT (ΔEH). (c) Projected density of states (PDOS) for Pt and Pt/TMN surfaces. (a) Reprinted from ref. 114 with permission from the Royal Society of Chemistry, Copyright 2022. (b and c) Reprinted from ref. 37 with permission from the American Chemical Society, Copyright 2023. | ||
Catalyst screening typically entails comparison of DFT calculations on model surfaces with the HER activity of powder catalysts. Such comparison overlooks the different morphologies between well-characterized model surfaces and power catalysts that often contain multiple facets and defects. A recent investigation by Turaczy et al. directly correlated experimentally determined HBE values with HER activity over the same Mo2N thin films in order to reduce this discrepancy. It was demonstrated that by depositing 1 ML of Pt or Pd, the required overpotentials to achieve a current density of 10 mA cm−2 shifted to be similar to that of bulk Pt in acidic electrolyte.37 TPD measurements were used to experimentally determine HBE values for Pt- and Pd-modified Mo2N surfaces and correlate them with HER activity. After depositing 1 ML of Pt onto Mo2N via physical vapor deposition, desorption temperatures of H2 at varying heating rates were used to determine the enthalpy of atomic hydrogen adsorption or HBE. The values obtained for Pt and Pd were similar to those found in literature for Pt(111) and Pd(111) single crystals. The stronger binding energies of Pd and Pd-modified Mo2N resulted in slower HER kinetics and higher overpotentials while the binding energies of Pt and Pt-modified Mo2N corresponded to more optimal HER kinetics and overpotentials (Fig. 7b). These results confirmed the validity of using HBE values as a descriptor for TMN-supported HER catalysts. The high HER activity of the 1 ML Pt/Mo2N surface was attributed to an alteration of the density of states (DOS) of Pt. An unsupported Pt ML was predicted to have a high lying d-band center that hindered hydrogen adsorption, but introducing a Mo2N(111) support shifted this center to lower energy levels, resembling the DOS for Pt(111) (Fig. 7c).
Implementing ML PGM/TMN catalysts as core–shell nanoparticles (NPs) has been suggested as a potential method to alleviate the cost of Pt in HER.121 Traditional Pt/C can have an average cathode catalyst lifetime cost of $260 per m2, but reducing the amount of Pt to 1 ML can decrease this to an average lifetime cost of approximately $100 per m2. Pushing the limits of Pt integration to sub-monolayer coverages could further reduce this to an average of approximately $20 per m2 with an overall 12-fold decrease in costs.121 Reducing the PGM coverage to sub-monolayer requires further catalyst optimization, and the combination of DFT-calculated and experimentally-determined HBE screening can aid these efforts to identify promising TMN supports that stabilize low loadings of PGMs while maintaining high HER activity and stability.
In alkaline electrolyte, it is generally more difficult to achieve the overpotentials seen in acidic media. However, the feasibility of using TMNs to surpass the overpotentials of typical PGM electrocatalysts in alkaline media has been demonstrated. Turaczy et al. showed that for thin films in 0.1 M KOH, 1 ML Pt on Mo2N was able to shift the relatively high overpotential required to drive 10 mA cm−2 of current on clean Mo2N to that of Pt foil.37 Other studies have used Pt NPs on Ni3N that outperformed the benchmark Pt/C catalyst by electrodepositing 10 nm Pt NPs onto the surface of Ni3N nanosheets.122 The Pt/Ni3N catalyst exhibited higher HER activity than Pt/C at a current density of 50 mA cm−2. The enhancement became even more apparent when shifting to higher overpotentials, where Pt/Ni3N was able to achieve a current density of 200 mA cm−2 while Pt/C attained half of that value at the same overpotential. Though highly active, the Pt/Ni3N electrocatalyst exhibited a current retention of 82.5% after 24 h at a constant overpotential of 50 mV.
In addition to reducing loadings of PGMs using TMN supports, many studies have focused on replacing PGMs entirely with TMNs as HER electrocatalysts. Monometallic nitrides, such as MoxN, have been shown to be active for HER, and several studies have focused on morphologically altering the catalyst structure for improved activity.123,124 Xie et al. discovered that atomically-thin 1.3 nm nanosheets of MoN increased HER activity compared to bulk MoN and in the process, elucidated Mo as an active site for transforming protons to hydrogen.125 The MoN nanosheets exhibited 11.8 times higher normalized current density at 300 mV than bulk MoN. Furthermore, Zhu et al. engineered porous nitrogen-doped carbon with embedded MoN nano-octahedra, and they attributed the enhancement in HER activity in acidic electrolyte to the nitrogen doping of the carbon matrix, which could introduce more charge carriers, reduce charge transfer resistance, and prevent MoN nanoparticles from agglomerating.126
Nickel nitride is another promising monometallic nitride for HER, especially in alkaline electrolyte, where the anode electrocatalyst can be replaced with a non-PGM material. Shalom et al. showed that even without Pt NPs, Ni3N could achieve low overpotentials.127 These results were improved by only partially nitrifying the Ni foam (NF) support surface using different techniques. Shen et al. varied nitridation temperature for Ni(OH)2 on Ni foam in order to vary the nitrogen doping with an optimal temperature of 673 K for Ni/Ni–N0.28 synthesis on Ni foam.128 Accompanying DFT studies varied N content in a Nx–Ni(111) model and suggested that Ni/Ni–N0.28/NF had a ΔGH value close to zero. Song et al. took a different approach and electrodeposited Ni NPs on NF followed by thermal nitridation in ammonia at different temperatures.129 The optimal temperature of 573 K for 6 hours resulted in Ni3N/NF that outperformed an optimized loading of Pt/C powder. The partial nitridation led to numerous interfacial sites as shown by high resolution transmission electron microscopy (HRTEM) (Fig. 8a), which led to high HER activity (Fig. 8b). DFT results suggested that water preferred to bind at these interfacial sites (Fig. 8c), and the Ni3N/Ni had the lowest energy barrier for water dissociation compared to Ni or Ni3N alone. Though other monometallic TMN electrocatalysts have been demonstrated for HER using Fe,130 W,131,132 Ta,133 and Co,41,134 the most promising monometallic TMNs consist of either Mo or Ni.
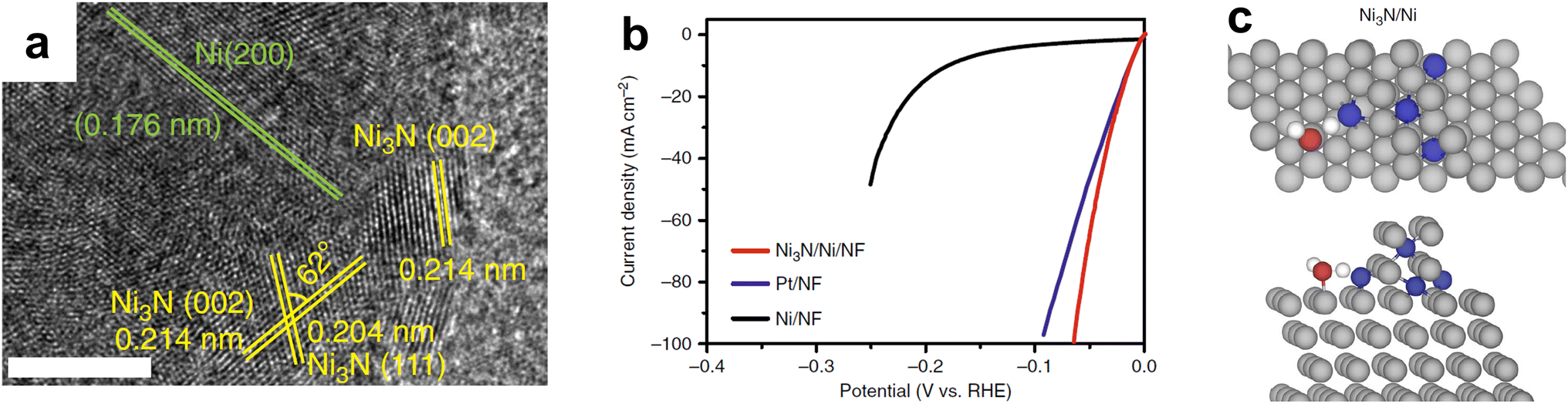 | ||
| Fig. 8 (a) HRTEM of interfacial Ni3N/Ni electrocatalyst with scale bar of 5 nm. (b) Linear sweep voltammetry (LSV) curves of Ni3N/Ni/NF, Pt/NF, and Ni/NF for HER in 1.0 M KOH with the current density normalized by their respective geometric electrode surface areas. (c) Transition state structures for water dissociation over Ni3N/Ni, showing both the top view (top of (c)) and side view (bottom of (c)). Color code: Ni – gray; N – blue; O – red; H – white. Reprinted from ref. 129 with permission from Springer Nature, Copyright 2018. | ||
Bimetallic TMNs have also been investigated as HER catalysts due to their tunability and multi-functionality. Typically, bimetallic TMNs for HER utilize either Mo or Ni as one of the metals. Jia et al. synthesized NiFe3N NPs via a microemulsion technique,135 which exhibited exceptional HER activity at higher current densities compared to 20% Pt/C, requiring only 416 mV to generate a current density of 200 mA cm−2 compared to 469 mV for Pt/C. The gap in overpotential widened as both electrocatalysts operated at higher current densities (>400 mA cm−2). Electrochemical impedance spectroscopy revealed that the NiFe3N NPs had a lower charge transfer resistance than Pt/C, resulting in faster electron transfer during HER. Yan et al. utilized an electrodeposition technique to deposit FeNi(OH)2, which was further nitrified using NH3 for HER.136 Yu et al. compared several bimetallic TMNs and identified the trend in HER activity following NiCoN > NiFeN > CoFeN.137 The ratio of Ni to Co in NiCoN was optimized by varying the ratio of the precursors used, demonstrating its ease of tunability. Among four different Ni/Co ratios, NiCo2N was shown to be the most active, displaying lower overpotentials and higher turnover frequencies than both CoN and Ni3N and indicating a synergistic effect due to the bimetallic interactions.
Though not as widely explored, Mo-based bimetallic TMNs have also displayed promising results for HER. Nickel nitrides are typically unstable under acidic conditions, while Mo is more tolerant of acidic media, but exhibits a low intrinsic HER activity compared to Pt/C. Cao et al. were able to tune the electronic states of Mo by using Co to introduce strain in the lattice as Co0.6Mo1.4N2, which significantly reduced the necessary overpotential for HER compared to MoN in acidic media.138 V has also been combined with Mo to synthesize a MoVN catalyst with promising performance for HER in both acidic and alkaline media, demonstrating a synergistic effect when compared to Mo2N and VN separately.139 Evaluating MoVN in alkaline media also showed improved HER activity, which was attributed to the MoVN catalyst being especially effective at reducing the energy barrier for the Volmer step.
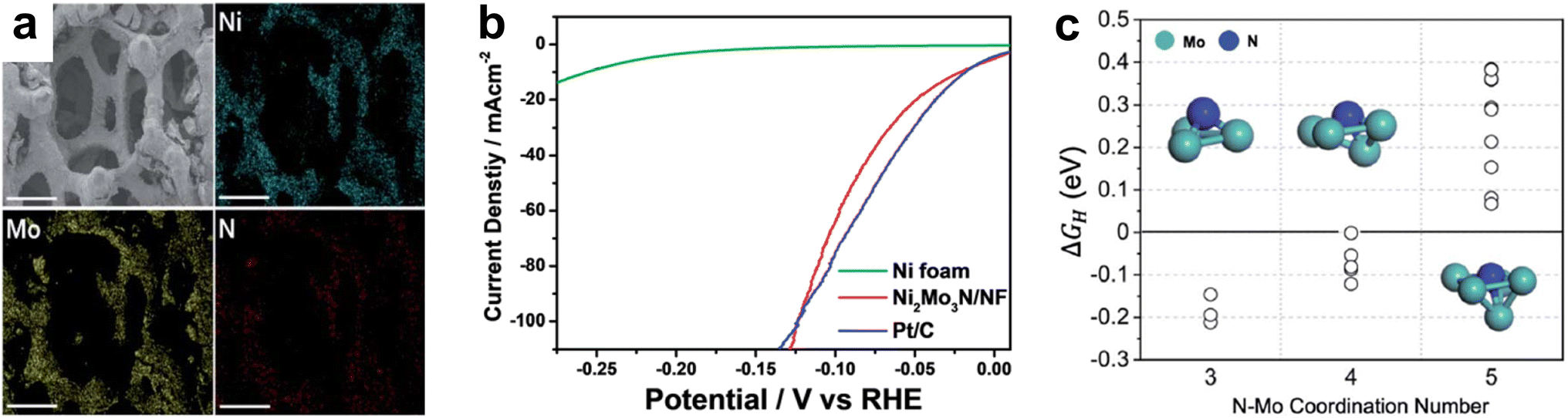
Since both Ni- and Mo-based catalysts have shown promising HER activity, recent studies have investigated Ni–Mo bimetallic TMNs for HER in both acidic and alkaline electrolytes. In acidic electrolyte, NiMo4N5 nanoparticles obtained current densities of 20 mA cm−2 with an overpotential of 43 mV, only 11 mV more than benchmark Pt/C, which was further reduced to 6 mV higher than Pt/C in alkaline electrolyte.140 Stability measurements showed a slight decrease in overpotential after 24 hours of continuous water electrolysis at a constant current density of 20 mA cm−2. Park et al. devised a simple method to grow nanostructured Ni2Mo3N in one pot that was both economical and environmentally conscious. Most methods utilize a hydrothermal reaction and/or a nitridation reaction with ammonia gas, however, this study annealed MoCl5 with Ni foam and urea under a N2 atmosphere to obtain 7 nm Ni–Mo bimetallic nitride particles supported on Ni foam (Fig. 9a).141 The electrocatalyst achieved similar overpotentials to commercial 20 wt% Pt/C at lower current densities (<100 mA cm−2), while at higher current densities, the Ni2Mo3N/NF catalyst outperformed Pt/C, suggesting its practicality on an industrial scale (Fig. 9b). Stability tests at both 50 and 100 mA cm−2 were conducted for 10 hours and showed minimal deviation in overpotential. XRD, SEM, EDS, and TEM did not show evidence of phase changes or agglomeration following these tests. DFT calculations suggested that the coordination number (CN) of nitrogen atoms played a major role in determining the HBE and therefore, the HER activity (Fig. 9c). Nitrogen was determined to be the active site for H adsorption and it was shown that a CN number for N of 3 or 5 resulted in too strong or too weak binding, respectively. A CN of 4 resulted in N having an average ΔGH = −0.07 eV, which was close to that for Pt(111) (−0.05 eV).141 Therefore, the CN of nitrogen atoms may be a possible parameter to adjust for optimizing other bimetallic TMN electrocatalysts for HER.
 | ||
| Fig. 9 (a) SEM-EDS of Ni2Mo3N/NF with scale bar of 300 μm. (b) LSV curves of Ni foam, Ni2Mo3N/NF, and Pt/C in 1.0 M KOH electrolyte. (c) The Gibbs free energy of adsorbed hydrogen (ΔGH) on different nitrogen adsorption sites with varying coordination number for N–Mo bonding. Reprinted from ref. 141 with permission from the Royal Society of Chemistry, Copyright 2021. | ||
4. Conclusions and future outlook
The recent studies described here have identified promising TMN catalysts for the selective conversion of the three classes of oxygen-containing molecules. They have also elucidated details about the active sites and mechanisms on these materials, showing that by incorporating interstitial N atoms into the crystalline lattice of transition metals, unique electronic and geometric properties arise that lead to enhanced catalytic performance. By focusing on the upgrading of C1 molecules, the selective conversion of biomass compounds, and the splitting of water to produce H2, we have highlighted how the use of TMN catalysts has been extended beyond the more traditional focuses related to hydrotreatment and heteroatom removal. Furthermore, by utilizing novel TMN compositions beyond typical monometallic nitrides, unique electronic and structural effects have been achieved that enable enhanced activity for these selective conversions. Transformations of C1 oxygenates demonstrated how strong metal–support interactions coupled with the unique properties of TMNs can be exploited to perform CO2 hydrogenation with improved yield and stability compared to more traditional metal oxide-based catalysts. Due to their stability and electrical conductivity, electrocatalytic and photocatalytic applications of TMNs have been investigated for CO2 electroreduction, as well as CH3OH oxidation for applications in DMFCs. Similar efforts have been undertaken for the valorization of biomass, where model surfaces and probe molecules were employed to demonstrate the inherent bond scission selectivity of metal-modified TMNs. Then, these results were extended to investigations of corresponding powder catalysts. The use of TMNs has also been demonstrated for catalytic fast pyrolysis of raw biomass feedstock, as well hydrodeoxygenation of bio-oil and other biomass derivatives. Much of this progress has been enabled by fundamental studies of TMN model surfaces, coupled with computational research and improvements in synthesis methods. Finally, for hydrogen production via water electrolysis, combined theoretical and experimental studies on TMN model surfaces were used to identify descriptors for HER activity, and electrocatalytic investigations of TMN films and powder catalysts demonstrated how these catalysts can reduce the loading of expensive PGMs for water electrolysis.Compared with TMCs that are often covered by surface carbon during carbide synthesis, TMN synthesis leaves an inherently clean surface due to the favorable desorption of N2. This allows direct investigation of the active sites on TMN model surfaces and extension of these results to corresponding supported powder catalysts, therefore enabling the fundamental investigations described in this Perspective. In order to further bridge the gap between these model surface studies and practical investigations of high surface area powder catalysts, and thus aid in the practical applications of TMNs for the selective transformations of oxygen-containing molecules described herein, the following key areas should be emphasized in future research.
First, the fundamental understanding of active sites that has been developed using well-characterized model surfaces and probe molecules should be extended to bimetallic TMNs and other more complex, multiphase TMNs. As specialized synthesis strategies continue to advance and allow the production of more complex TMN-based materials such as Co3Mo3N, Ni–TiN, and Ni–Mo carbonitrides with unique doping effects and the coexistence of different nitride/carbide phases, strategies in model surface preparation should advance in parallel. This will potentially allow the use of UHV techniques such as TPD and vibrational spectroscopy to be applied in understanding the reactivity at interfaces and the effects of different bimetallic ratios on catalytic performance. In addition, model surface studies of TMNs should seek to employ larger and more complex probe molecules to elucidate the bond scission pathways of compounds that may be more relevant in the application of these catalytic materials. In particular, the use of biomass model compounds such as guaiacol, phenol, and anisole should benefit the understanding of TMNs for biomass valorization.
Second, the use of in situ techniques to understand the active centers of TMN catalysts under reaction conditions should continue to be expanded. The investigations of TMNs for CO2 hydrogenation that were highlighted in this Perspective demonstrate important insights from in situ X-ray absorption spectroscopy to elucidate the origin of the enhanced catalytic performance of these materials. By tracking changes in the XANES and EXAFS regions, it was shown how temperature and reaction conditions could influence strong metal–support interactions and reverse sintering effects between admetals and TMN supports. While these results were highly beneficial for the utilization of metal-modified TMNs for CO2 activation, these principles should be used to develop these catalysts for other classes of reactions including those described here (biomass upgrading and water electrolysis) as well as new applications of TMNs such as methane pyrolysis, plastic upcycling, and wastewater treatment. In addition, other synchrotron techniques beyond X-ray absorption, such as in situ XRD and pair distribution function (PDF) analysis, high energy resolution fluorescence detected (HERFD) XAS, and resonant inelastic X-ray scattering (RIXS) should be employed to investigate the structures and electronic interactions of TMN-based catalysts. Furthermore, in situ FTIR analysis can be coupled with these synchrotron-based characterization techniques to understand both the reaction intermediates and pathways along with the chemical state of the active centers over TMN catalysts.
Third, design principles for TMN synthesis should be developed to enable larger-scale manufacturing in a sustainable way. Early studies of Mo2N included systematic investigations of the influence of temperature, space velocity, and heating rate during synthesis with NH3.23 Similar approaches should be used to develop design principles for the use of N2/H2 mixtures, and other N sources such as urea or guanidine. The urea glass method, as well as the biomass-derived mixed carbide/nitride synthesis that have been highlighted in this Perspective, are promising routes to produce TMNs from lower-cost and renewable nitrogen sources. Based on the development of synthesis methods using alternative nitrogen sources, improving the scalability of TMN manufacturing should also be required for the commercial application of these materials. For example, the use of N2/H2 mixtures with a fluidized bed reactor has shown potential over a fixed bed setup.142 Further studies should seek to fill gaps in knowledge of manufacturing techniques using similar reaction engineering approaches. Beyond this, it has been suggested that a sol–gel method is one of the most promising synthesis techniques for large scale applications due to its flexibility and simplicity.143
Fourth, in tandem with the focus on systematic studies to develop design principles and structure–performance relationships for TMNs manufactured using these methods, in-depth investigations of stability and regeneration methods should also be undertaken to enable prolonged use of TMNs under commercially relevant reaction conditions. Previous work on NO reduction has shown improved stability and resistance to oxidation of a bimetallic nitride (Co3Mo3N) compared with monometallic Mo2N that was attributed to differences in crystal structure.144 Another study has shown a correlation between the Mo loading in a mesoporous zeolite (ZSM-5) supported Mo2N catalyst and its stability.145 Furthermore, a comparison of several nitrides for NO reduction found that the stability followed the trend of Co4N > Fe3N > Mo2N > VN.11,146 The principles and methods used in these studies of NO reduction can be applied in the thermocatalytic reactions of C1 molecules and biomass compounds that were highlighted in this Perspective. In particular, improved understanding of the support effect and the relationships between crystallographic structure and stability should be developed.
Fifth, the use of TMNs for CO valorization should be expanded. The utilization of CO can allow the formation of certain commodity chemicals more cleanly than current processes. Specifically, by reacting CO with H2 (syngas), hydrocarbons, alcohols, and olefins can be produced. Catalysts for these reactions must be able to activate CO and H2, cleave C–O bonds, and facilitate C–C coupling and hydrogenation.17 While Fe-, Co-, Ni-, and Ru-based materials are the most common catalysts for Fischer–Tropsch synthesis, studies over the past decade have identified several TMN catalysts with promising activity and selectivity for CO utilization reactions.17,147–152 In particular, studies showing production of C2+ alcohols from syngas on Fe2N, Fe3N, and Fe4N catalysts147 and C2+ olefin synthesis from syngas on γ-Mo2N149 represent promising demonstrations of the C–C coupling activity of TMN catalysts. Additionally, CO has been utilized in investigations to probe the Pt-like properties of the surface sites of Mo2N.152,153 These studies have identified the promising activity of TMNs for the upgrading of CO, but significant efforts are needed to improve the yield and selectivity of desired products.
Finally, while PGMs remain the benchmark HER catalysts via water electrolysis, there are strong efforts to reduce or replace these otherwise expensive and scarce materials. To this end, HBE calculations are useful methods to screen potential catalysts. Monolayer coverage of Pt can significantly reduce Pt loadings while still maintaining high HER activity in acidic media. Ultimately, TMNs of earth-abundant metals such as Ni, Fe, or Mo have shown promise to replace PGMs for HER in alkaline electrolyte. Ni-based bimetallic TMN electrocatalysts especially show promise to become durable and highly active materials that can operate at high current densities that are relevant for industrial applications. The challenges for these TMNs as alternative HER catalysts are improving stability at these high current densities as well as developing inexpensive and simple methods for industrial-scale synthesis.
Data availability
The experimental or computational data associated with this article is available from the cited references.Author contributions
William N. Porter: writing – original draft preparation. Kevin K. Turaczy: writing – original draft preparation. Marcus Yu: writing – original draft preparation. Hansen Mou: writing – rview & editing. Jingguang G. Chen: writing – review & editing, supervision.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the United States Department of Energy, Office of Basic Energy Sciences, Catalysis Science Program (Grant No. DE-FG02-13ER16381). William N. Porter acknowledges partial support by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division (DE-SC00234430), and by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program, which is administered by the Oak Ridge Institute for Science and Education for the DOE under contract number DE-SC0014664. Kevin K. Turaczy, Marcus Yu, and Hansen Mou acknowledge support by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 2036197.References
- L. E. Toth, Transition Metal Carbides and Nitrides, Academic Press, 1971, vol. 7 Search PubMed.
- G. C. Bond, G. Webb, S. T. Oyama and G. L. Haller, in Catalysis, ed. G. C. Bond and G. Webb, The Royal Society of Chemistry, 1982, vol. 5 Search PubMed.
- S. T. Oyama, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- The Chemistry of Transition Metal Carbides and Nitrides, ed. S. T. Oyama, Springer Netherlands, Dordrecht, 1996 Search PubMed.
- L. I. Johansson, Surf. Sci. Rep., 1995, 21, 177–250 CrossRef CAS.
- J. G. Chen, Chem. Rev., 1996, 96, 1477–1498 CrossRef CAS PubMed.
- J. G. Chen, Surf. Sci. Rep., 1997, 30, 1–152 CrossRef CAS.
- Y. Wang, Y. Nian, A. N. Biswas, W. Li, Y. Han and J. G. Chen, Adv. Energy Mater., 2021, 11, 2002967 CrossRef CAS.
- D. Tian, S. R. Denny, K. Z. Li, H. Wang, S. Kattel and J. G. Chen, Chem. Soc. Rev., 2021, 50, 12338–12376 RSC.
- K. J. Smith, Curr. Opin. Green Sustainable Chem., 2020, 22, 47–53 CrossRef.
- J. S. J. Hargreaves, Coord. Chem. Rev., 2013, 257, 2015–2031 CrossRef CAS.
- S. Tsuchiya and A. Ozaki, Bull. Chem. Soc. Jpn., 1969, 42, 344–348 CrossRef CAS.
- L. Volpe and M. Boudart, J. Phys. Chem., 1986, 90, 4874–4877 CrossRef CAS.
- L. T. Thompson, C. W. Colling, D. Choi, B. G. Demczyk and J.-G. Choi, in Studies in Surface Science and Catalysis, ed. L. Guczi, F. Solymosi and P. Tétényi, Elsevier, 1993, vol. 75, pp. 941–954 Search PubMed.
- S. K. Bej and L. T. Thompson, Appl. Catal., A, 2004, 264, 141–150 CrossRef CAS.
- J. Horáček, U. Akhmetzyanova, L. Skuhrovcová, Z. Tišler and H. de Paz Carmona, Appl. Catal., B, 2020, 263, 118328 CrossRef.
- J. A. Schaidle and L. T. Thompson, J. Catal., 2015, 329, 325–334 CrossRef CAS.
- Z. Lin, S. R. Denny and J. G. Chen, J. Catal., 2021, 404, 929–942 CrossRef CAS.
- Z. N. Jaf, H. A. Miran, Z.-T. Jiang and M. Altarawneh, Rev. Chem. Eng., 2023, 39, 329–361 CrossRef CAS.
- A. Lilić, L. Cardenas, A. Mesbah, E. Bonjour, P. Jame, C. Michel, S. Loridant and N. Perret, J. Alloys Compd., 2022, 924, 166576 CrossRef.
- J. Choi, R. Curl and L. Thompson, J. Catal., 1994, 146, 218–227 CrossRef CAS.
- R. Kapoor and S. T. Oyama, J. Solid State Chem., 1992, 99, 303–312 CrossRef CAS.
- J. G. Choi, J. R. Brenner, C. W. Colling, B. G. Demczyk, J. L. Dunning and L. T. Thompson, Catal. Today, 1992, 15, 201–222 CrossRef CAS.
- J. B. Claridge, A. P. E. York, A. J. Brungs and M. L. H. Green, Chem. Mater., 2000, 12, 132–142 CrossRef CAS.
- Q. Gao, C. Giordano and M. Antonietti, Small, 2011, 7, 3334–3340 CrossRef CAS PubMed.
- X. Gao, H. E. Bush, J. E Miller, A. Bayon, I. Ermanoski, A. Ambrosini and E. B. Stechel, Chem. Mater., 2023, 35, 5864–5875 CrossRef CAS.
- D. Mckay, J. S. J. Hargreaves, J. L. Rico, J. L. Rivera and X.-L. Sun, J. Solid State Chem., 2008, 181, 325–333 CrossRef CAS.
- J. O. Conway and T. J. Prior, J. Alloys Compd., 2019, 774, 69–74 CrossRef CAS.
- C. Giordano, C. Erpen, W. Yao and M. Antonietti, Nano Lett., 2008, 8, 4659–4663 CrossRef CAS PubMed.
- C. Giordano, C. Erpen, W. Yao, B. Milke and M. Antonietti, Chem. Mater., 2009, 21, 5136–5144 CrossRef CAS.
- W. F. Chen, S. Iyer, S. Iyer, K. Sasaki, C. H. Wang, Y. M. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818–1826 RSC.
- F. Meng, E. Hu, L. Zhang, K. Sasaki, J. T. Muckerman and E. Fujita, J. Mater. Chem. A, 2015, 3, 18572–18577 RSC.
- P. B. Mirkarimi, M. Shinn and S. A. Barnett, J. Vac. Sci. Technol., A, 1992, 10, 75–81 CrossRef CAS.
- I. Jauberteau, A. Bessaudou, R. Mayet, J. Cornette, J. L. Jauberteau, P. Carles and T. Merle-Méjean, Coatings, 2015, 5, 656–687 CrossRef CAS.
- M. Juppo, M. Ritala and M. Leskelä, J. Electrochem. Soc., 2000, 147, 3377–3381 CrossRef CAS.
- W. N. Porter, H. A. Mera, W. Liao, Z. Lin, P. Liu, J. R. Kitchin and J. G. Chen, ACS Catal., 2024, 14, 1653–1662 CrossRef CAS.
- K. K. Turaczy, W. Liao, H. Mou, N. N. Nichols, P. Liu and J. G. Chen, ACS Catal., 2023, 13, 14268–14276 CrossRef CAS.
- C. H. Jaggers, J. N. Michaels and A. M. Stacy, Chem. Mater., 1990, 2, 150–157 CrossRef CAS.
- Z. Lin, S. C. Ammal, S. R. Denny, S. A. Rykov, K.-E. You, A. Heyden and J. G. Chen, JACS Au, 2022, 2, 367–379 CrossRef CAS PubMed.
- S. R. Denny, B. M. Tackett, D. Tian, K. Sasaki and J. G. Chen, Int. J. Hydrogen Energy, 2020, 45, 22883–22892 CrossRef CAS.
- Z. Y. Chen, Y. Song, J. Y. Cai, X. S. Zheng, D. D. Han, Y. S. Wu, Y. P. Zang, S. W. Niu, Y. Liu, J. F. Zhu, X. J. Liu and G. M. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS.
- S. Yao, L. Lin, W. Liao, N. Rui, N. Li, Z. Liu, J. Cen, F. Zhang, X. Li, L. Song, L. Betancourt De Leon, D. Su, S. D. Senanayake, P. Liu, D. Ma, J. G. Chen and J. A. Rodriguez, ACS Catal., 2019, 9, 9087–9097 CrossRef CAS.
- L. L. Lin, J. J. Liu, X. Liu, Z. R. Gao, N. Rui, S. Y. Yao, F. Zhang, M. L. Wang, C. Liu, L. L. Han, F. Yang, S. Zhang, X. D. Wen, S. D. Senanayake, Y. C. Wu, X. N. Li, J. A. Rodriguez and D. Ma, Nat. Commun., 2021, 12, 6978 CrossRef CAS PubMed.
- T. S. Nguyen, M. T. Le and V. H. Nguyen, Biomass Bioenergy, 2023, 174, 106821 CrossRef CAS.
- Z. S. Zhang, Q. Fu, K. Xu, W. W. Wang, X. P. Fu, X. S. Zheng, K. Wu, C. Ma, R. Si, C. J. Jia, L. D. Sun and C. H. Yan, J. Am. Chem. Soc., 2020, 142, 13362–13371 CrossRef CAS.
- V. Dasireddy, D. Vengust, B. Likozar, J. Kovac and A. Mrzel, Renewable Energy, 2021, 176, 251–261 CrossRef CAS.
- H.-X. Liu, J.-Y. Li, X. Qin, C. Ma, W.-W. Wang, K. Xu, H. Yan, D. Xiao, C.-J. Jia, Q. Fu and D. Ma, Nat. Commun., 2022, 13, 5800 CrossRef CAS PubMed.
- K. Feng, J. M. Tian, J. J. Zhang, Z. W. Li, Y. X. Chen, K. H. Luo, B. Yang and B. H. Yan, ACS Catal., 2022, 12, 4696–4706 CrossRef CAS.
- H. Sugiyama, M. Miyazaki, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2023, 145, 9410–9416 CrossRef CAS PubMed.
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 2, 869–876 CrossRef CAS.
- R. Razzaq, C. S. Li, M. Usman, K. Suzuki and S. J. Zhang, Chem. Eng. J., 2015, 262, 1090–1098 CrossRef CAS.
- B. H. Zhao, M. Y. Sun, F. P. Chen, Y. M. Shi, Y. F. Yu, X. G. Li and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4496–4500 CrossRef CAS PubMed.
- B. Li, B. Ma, S. Y. Wang, M. M. Yu, Z. Q. Zhang, M. J. Xiao, H. Zhang, J. F. Wu, Y. Peng, Q. Wang and H. L. Zhang, Nanoscale, 2022, 14, 9736–9742 RSC.
- M. Y. Sun, B. H. Zhao, R. Yang, F. P. Chen, Y. F. Yu and B. Zhang, ACS Energy Lett., 2021, 6, 2024–2029 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, P. Song, M. Duan, J. Xiong, R. Long, M. Xu, L. Kang, S. Guo, S. Chen, H. Chen, Z. Chi, Y.-X. Weng, H. Li, L. Song, M. Wu, Q. Yan, S. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CrossRef CAS.
- S. K. Kuk, Y. Ham, K. Gopinath, P. Boonmongkolras, Y. Lee, Y. W. Lee, S. Kondaveeti, C. Ahn, B. Shin, J.-K. Lee, S. Jeon and C. B. Park, Adv. Energy Mater., 2019, 9, 1900029 CrossRef.
- Q. Liang, Y. Zhao, J. D. Chen, J. J. Dai, X. Y. Ding, Z. Tong, S. J. Xie, J. Zhang, Z. H. Zhou, J. T. Li, J. F. Li and Y. Zhou, Chem. Mater., 2022, 34, 5607–5620 CrossRef CAS.
- Y. Liu, D. Tian, A. N. Biswas, Z. Xie, S. Hwang, J. H. Lee, H. Meng and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 11345–11348 CrossRef CAS PubMed.
- F. Schorn, J. L. Breuer, R. C. Samsun, T. Schnorbus, B. Heuser, R. Peters and D. Stolten, Adv. Appl. Energy, 2021, 3, 100050 CrossRef CAS.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Nat. Commun., 2021, 12, 5235 CrossRef CAS.
- A. S. Moura, J. L. C. Fajín, M. Mandado and M. N. D. S. Cordeiro, Catalysts, 2017, 7, 47 CrossRef.
- M. A. Abdelkareem, T. Wilberforce, K. Elsaid, E. T. Sayed, E. A. M. Abdelghani and A. G. Olabi, Int. J. Hydrogen Energy, 2021, 46, 23529–23547 CrossRef CAS.
- X. Qi, N. Ye, R. Zhang, Z. Jiang and T. Fang, Fuel, 2023, 350, 128773 CrossRef CAS.
- M. Yu, H. Mou, J. J. Jeong and J. G. Chen, in preparation.
- H. H. Hwu, B. D. Polizzotti and J. G. Chen, J. Phys. Chem. B, 2001, 105, 10045–10053 CrossRef CAS.
- G. O. Kayode and M. M. Montemore, J. Mater. Chem. A, 2021, 9, 22325–22333 RSC.
- A. L. Stottlemyer, P. Liu and J. G. Chen, J. Chem. Phys., 2010, 133, 104702 CrossRef PubMed.
- Z. Yu, X. An, I. Kurnia, A. Yoshida, Y. Yang, X. Hao, A. Abudula, Y. Fang and G. Guan, ACS Catal., 2020, 10, 5353–5361 CrossRef CAS.
- Z. Yu, Y. Yang, S. Yang, J. Zheng, X. Hao, G. Wei, H. Bai, A. Abudula and G. Guan, Appl. Catal., B, 2022, 313, 121445 CrossRef CAS.
- H. Yan, Y. Jiao, A. Wu, C. Tian, L. Wang, X. Zhang and H. Fu, J. Mater. Chem. A, 2018, 6, 7623–7630 RSC.
- I. U. Haq, K. Qaisar, A. Nawaz, F. Akram, H. Mukhtar, X. Zohu, Y. Xu, M. W. Mumtaz, U. Rashid, W. A. W. A. K. Ghani and T. S. Y. Choong, Catalysts, 2021, 11, 1–26 Search PubMed.
- D. Martin Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- W. Jin, L. Pastor-Pérez, D. K. Shen, A. Sepúlveda-Escribano, S. Gu and T. Ramirez Reina, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- S. Gazi, Appl. Catal., B, 2019, 257, 117936 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- D. J. Sajkowski and S. T. Oyama, Appl. Catal., A, 1996, 134, 339–349 CrossRef CAS.
- S. Ramanathan and S. T. Oyama, J. Phys. Chem., 1995, 99, 16365–16372 CrossRef CAS.
- S. R. Denny, Z. Lin, W. N. Porter, N. Artrith and J. G. Chen, Appl. Catal., B, 2022, 312, 121380 CrossRef CAS.
- T. Wang, J. Sha, M. Sabbe, P. Sautet, M. Pera-Titus and C. Michel, Nat. Commun., 2021, 12, 5100 CrossRef CAS PubMed.
- T. W. Walker, A. H. Motagamwala, J. A. Dumesic and G. W. Huber, J. Catal., 2019, 369, 518–525 CrossRef CAS.
- C. Sheng and J. L. T. Azevedo, Biomass Bioenergy, 2005, 28, 499–507 CrossRef CAS.
- S. Luo, Y. Long, K. Liang, X. Sun, J. Qin, G. Yang and J. Ma, Mol. Catal., 2022, 531, 112684 CrossRef CAS.
- Q. Lu, Z. X. Wang, H. Q. Guo, K. Li, Z. X. Zhang, M. S. Cui and Y. P. Yang, Fuel, 2019, 243, 88–96 CrossRef CAS.
- Q. Liu, J. Wang, J. Zhou, Z. Yu and K. Wang, J. Anal. Appl. Pyrolysis, 2021, 153, 104964 CrossRef CAS.
- Q. Lu, M. X. Zhou, W. T. Li, X. Wang, M. S. Cui and Y. P. Yang, Catal. Today, 2018, 302, 169–179 CrossRef CAS.
- T. Iqbal, Q. Lu, Z.-X. Zhang, Z. Ali, K. Li, S.-W. Ma, A. Abbas and C.-Q. Dong, J. Biobased Mater. Bioenergy, 2019, 13, 870–905 CrossRef CAS.
- X. Chen, H. Yang, Y. Chen, W. Chen, T. Lei, W. Zhang and H. Chen, J. Anal. Appl. Pyrolysis, 2017, 127, 292–298 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. F. Iii, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- H. Hasanudin, W. R. Asri, I. S. Zulaikha, C. Ayu, A. Rachmat, F. Riyanti, F. Hadiah, R. Zainul and R. Maryana, RSC Adv., 2022, 12, 21916–21925 RSC.
- H. Hasanudin, W. R. Asri, U. Permatahati, W. Purwaningrum, F. Hadiah, R. Maryana, M. Al Muttaqii and M. Hendri, AIMS Energy, 2023, 11, 197–212 CAS.
- B. M. Wyvratt, J. R. Gaudet, D. B. Pardue, A. Marton, S. Rudic, E. A. Mader, T. R. Cundari, J. M. Mayer and L. T. Thompson, ACS Catal., 2016, 6, 5797–5806 CrossRef CAS.
- I. T. Ghampson, C. Sepulveda, R. Garcia, L. R. Radovic, J. L. G. Fierro, W. J. DeSisto and N. Escalona, Appl. Catal., A, 2012, 439, 111–124 CrossRef.
- H. De Paz Carmona, J. Horáček, Z. Tišler and U. Akhmetzyanova, Fuel, 2019, 254, 115582 CrossRef CAS.
- F. Wang, C. Wen, M. Lu, P. Zhang, J. Zhu, M. Li, Y. Shan and C. Song, Biomass Bioenergy, 2023, 168, 106680 CrossRef CAS.
- X. Liu, L. Xu, G. Xu, W. Jia, Y. Ma and Y. Zhang, ACS Catal., 2016, 6, 7611–7620 CrossRef CAS.
- X. Z. Du, X. M. Lei, L. Y. Zhou, Y. Peng, Y. Zeng, H. R. Yang, D. Li, C. W. Hu and H. Garcia, ACS Catal., 2022, 12, 4333–4343 CrossRef CAS.
- J. Q. Wang, X. Chen, X. Z. Chen, C. X. Zhao, Y. Ling and C. H. Liang, Sustainable Energy Fuels, 2022, 6, 3025–3034 RSC.
- C. Zhao, J. Wang, X. Chen and C. Liang, Eur. J. Inorg. Chem., 2023, 26, e202300073 CrossRef CAS.
- X. Z. Chen, X. Chen, J. Q. Wang and C. H. Liang, ACS Appl. Nano Mater., 2022, 5, 14987–14998 CrossRef CAS.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS PubMed.
- N. Ji, P. Ri, X. Y. Diao, Y. Rong and C. Kim, Catal. Sci. Technol., 2023, 13, 2618–2637 RSC.
- M. Rashidi, M. Konarova, W. Aslam and J. N. Beltramini, ChemistrySelect, 2018, 3, 3379–3385 CrossRef CAS.
- L. Chen, T. I. Koranyi and E. J. M. Hensen, Chem. Commun., 2016, 52, 9375–9378 RSC.
- L.-L. Bie, F.-J. Liu, Z.-M. Zong, G.-H. Liu, J.-P. Guo, Z.-X. Li, Z.-H. Ma, W.-W. Yan and X.-Y. Wei, Fuel Process. Technol., 2020, 209, 106523 CrossRef CAS.
- V. Molinari, G. Clavel, M. Graglia, M. Antonietti and D. Esposito, ACS Catal., 2016, 6, 1663–1670 CrossRef CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. Chen, G. T. K. K. Gunasooriya, I. McCrum, R. Mom, N. López and M. T. M. Koper, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS.
- Y. Yu, S. J. Lee, J. Theerthagiri, Y. Lee and M. Y. Choi, Appl. Catal., B, 2022, 316, 121603 CrossRef CAS.
- S. Naik Shreyanka, J. Theerthagiri, S. J. Lee, Y. Yu and M. Y. Choi, Chem. Eng. J., 2022, 446, 137045 CrossRef CAS.
- Y. Yu, S. J. Lee, J. Theerthagiri, S. Fonseca, L. M. C. Pinto, G. Maia and M. Y. Choi, Chem. Eng. J., 2022, 435, 134790 CrossRef CAS.
- Y. Oh, J. Theerthagiri, M. L. Aruna Kumari, A. Min, C. J. Moon and M. Y. Choi, J. Energy Chem., 2024, 91, 145–154 CrossRef CAS.
- Y. Oh, J. Theerthagiri, A. Min, C. J. Moon, Y. Yu and M. Y. Choi, Carbon Energy, 2024, e448 CrossRef.
- D. Ologunagba and S. Kattel, Phys. Chem. Chem. Phys., 2022, 24, 12149–12157 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- Q. Zhang, Z. Jiang, B. M. Tackett, S. R. Denny, B. Tian, X. Chen, B. Wang and J. G. Chen, ACS Catal., 2019, 9, 2415–2422 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- H. Mou, J. J. Jeong, B. Lamichhane, S. Kattel, Z. Zhuang, J. H. Lee, Q. Chang and J. G. Chen, Chem Catal., 2024, 4, 100867 CrossRef CAS.
- J. Peng, J. J. Giner-Sanz, L. Giordano, W. P. Mounfield, G. M. Leverick, Y. Yu, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2023, 7, 150–167 CrossRef CAS.
- C. T. Campbell, Annu. Rev. Phys. Chem., 1990, 41, 775–837 CrossRef CAS.
- S. T. Hunt, M. Milina, Z. Wang and Y. Román-Leshkov, Energy Environ. Sci., 2016, 9, 3290–3301 RSC.
- Y. Wang, L. Chen, X. Yu, Y. Wang and G. Zheng, Adv. Energy Mater., 2017, 7, 1601390 CrossRef.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- H. Y. Jin, X. Liu, Y. Jiao, A. Vasileff, Y. Zheng and S. Z. Qiao, Nano Energy, 2018, 53, 690–697 CrossRef CAS.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- Y. Zhu, G. Chen, X. Xu, G. Yang, M. Liu and Z. Shao, ACS Catal., 2017, 7, 3540–3547 CrossRef CAS.
- M. Shalom, D. Ressnig, X. Yang, G. Clavel, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 8171–8177 RSC.
- J. Shen, X. Zheng, L. Peng, G. I. N. Waterhouse, L. Tan, J. Yang, L. Li and Z. Wei, ACS Appl. Nano Mater., 2020, 3, 11298–11306 CrossRef CAS.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef PubMed.
- R. Qiang, H. Wang, K. Xu, Q. Yuan, Y. Yu, L. Li, J. Wang, L. Zheng, P. C. Sherrell, J. Chen and X. Bi, Adv. Mater. Interfaces, 2021, 8, 2100070 CrossRef CAS.
- J. Shi, Z. Pu, Q. Liu, A. M. Asiri, J. Hu and X. Sun, Electrochim. Acta, 2015, 154, 345–351 CrossRef CAS.
- H. Jin, H. Zhang, J. Chen, S. Mao, Z. Jiang and Y. Wang, J. Mater. Chem. A, 2018, 6, 10967–10975 RSC.
- S. Wirth, F. Harnisch, M. Weinmann and U. Schröder, Appl. Catal., B, 2012, 126, 225–230 CrossRef CAS.
- Z. Chen, Y. Ha, Y. Liu, H. Wang, H. Yang, H. Xu, Y. Li and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 7134–7144 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- F. Yan, Y. Wang, K. Li, C. Zhu, P. Gao, C. Li, X. Zhang and Y. Chen, Chem.–Eur. J., 2017, 23, 10187–10194 CrossRef CAS PubMed.
- L. Yu, S. Song, B. McElhenny, F. Ding, D. Luo, Y. Yu, S. Chen and Z. Ren, J. Mater. Chem. A, 2019, 7, 19728–19732 RSC.
- B. F. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- B. Wei, G. Tang, H. Liang, Z. Qi, D. Zhang, W. Hu, H. Shen and Z. Wang, Electrochem. Commun., 2018, 93, 166–170 CrossRef CAS.
- T. Wang, X. Wang, Y. Liu, J. Zheng and X. Li, Nano Energy, 2016, 22, 111–119 CrossRef CAS.
- S. H. Park, T. H. Jo, M. H. Lee, K. Kawashima, C. B. Mullins, H. K. Lim and D. H. Youn, J. Mater. Chem. A, 2021, 9, 4945–4951 RSC.
- L. Kong, J. Wu, B. Lü, Q. Li and T. Xiong, Ind. Eng. Chem. Res., 2008, 47, 1779–1783 CrossRef CAS.
- A. K. Tareen, G. S. Priyanga, S. Behara, T. Thomas and M. Yang, Prog. Solid State Chem., 2019, 53, 1–26 CrossRef CAS.
- C. Shi, A. M. Zhu, X. F. Yang and C. T. Au, Catal. Lett., 2004, 97, 9–16 CrossRef CAS.
- C. Shi, A. M. Zhu, X. F. Yang and C. T. Au, Appl. Catal., A, 2005, 293, 83–90 CrossRef CAS.
- Z. Yao, A. Zhang, Y. Li, Y. Zhang, X. Cheng and C. Shi, J. Alloys Compd., 2008, 464, 488–496 CrossRef CAS.
- L. G. Li, M. Qing, X. W. Liu, H. Wang, S. Y. Liu, Y. Zhang, H. L. Wan, X. D. Wen, Y. Yang and Y. W. Li, ChemCatChem, 2020, 12, 1939–1943 CrossRef CAS.
- C. Wang, J. Zhang and J. G. Chen, ChemistrySelect, 2020, 5, 3953–3958 CrossRef CAS.
- S. F. Zaman, A. A. Alzahrani, S. Podila and Y. Al Hamed, Asia-Pac. J. Chem. Eng., 2020, 15, e2516 CrossRef CAS.
- S. F. Zaman, N. Pasupulety, A. A. Al-Zahrani, M. A. Daous, S. S. Al-Shahrani, H. Driss, L. A. Petrov and K. J. Smith, Appl. Catal., A, 2017, 532, 133–145 CrossRef CAS.
- S. F. Zaman, N. Pasupulety, A. A. Al-Zahrani, M. A. Daous, H. Driss, S. S. Al-Shahrani and L. Petrov, Can. J. Chem. Eng., 2018, 96, 1770–1779 CrossRef CAS.
- J. Zhang, W. C. Wu, S. Yan and D. Zhang, China Pet. Process. Petrochem. Technol., 2010, 12, 43–45 CAS.
- S. Yang, C. Li, J. Xu and Q. Xin, Chem. Commun., 1997, 573, 1247–1248 RSC.
| This journal is © The Royal Society of Chemistry 2024 |