
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelical polyisocyanide-based macroporous organic catalysts for asymmetric Michael addition with high efficiency and stereoselectivity†
Xun-Hui
Xu
ad,
Run-Tan
Gao
b,
Shi-Yi
Li
b,
Li
Zhou
 *a,
Na
Liu
c and
Zong-Quan
Wu
*b
*a,
Na
Liu
c and
Zong-Quan
Wu
*b
aDepartment of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, and Anhui Province Key Laboratory of Value-Added Catalytic Conversion and Reaction Engineering, Hefei University of Technology, Hefei 230009, Anhui Province, China. E-mail: lizhou@hfut.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, Jilin 130012, China
cThe School of Pharmaceutical Sciences, Jilin University, 1266 Fujin Road, Changchun, Jilin 130021, P. R. China
dBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 1st July 2024
Abstract
Porous materials have attracted interest due to their high specific surface area and rich functionality. Immobilizing organocatalysts onto porous polymers not only boosts enantioselectivity but also improves the reaction rates. In this work, a series of porous polymers C-poly-3ms with rigid polyisocyanide-carrying secondary amine pendants as building blocks were successfully prepared. And the pore size and optical activity of C-poly-3ms can be controlled by the length of the polyisocyanide blocks due to their rigid and helical backbone. C-poly-3150 demonstrated a preferred left-handed helix with a θ364 value of −8.21 × 103. The pore size and SBET of C-poly-3150 were 17.52 nm and 7.98 m2 g−1, respectively. The porous C-poly-3150 catalyzes the asymmetric Michael addition reaction efficiently and generates the target products in satisfactory yield and excellent enantioselectivity. For 6ab, an enantiomeric excess (ee) and a diastereomeric ratio (dr) up to 99% and 99/1 could be achieved, respectively. The recovered catalyst can be recycled at least 6 times in the asymmetric Michael addition reaction while maintaining activity and stereoselectivity.
Introduction
In the past few decades, porous materials have attracted considerable attention from academia and industry due to their wide range of potential applications in storage, separation, catalysis, and other fields.1–5 A variety of new functional porous materials have been developed, such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and porous organic polymers (POPs).6–10 Among them, POPs have been widely studied because of their facile synthesis and diverse synthesis methods.11–14 Generally, the pore sizes of the reported porous polymers are not uniform and cover a wider range due to the limitations of the synthetic methods.15 Therefore, it is meaningful to develop a strategy for preparing porous polymers with controllable pore size through living polymerization.16,17 Among many kinds of polymers, polyisocyanides have aroused the continuous interest of researchers because of their facile available materials, convenient polymerization methods and controllable structures.18–21 The polyisocyanide consists of a carbon backbone with carbon–carbon single bonds, and each carbon atom in the backbone carries substituent groups. Due to the steric effect of the pendants on each atom of the main chain, polyisocyanide adopts a rod-like structure and twists into a helical shape.22,23 As one of the rigid organic polymers, the persistence length of isotactic helical polyisocyanide can be reached to ca. 200 nm.24,25 Therefore, polyisocyanide is a desirable building block with sufficient rigidity to support the frameworks for the construction of porous materials.Asymmetric organocatalysis has developed rapidly since the early 2000s, and as this method has matured, it was adopted in industry to produce chiral drugs, natural products, and bioactive molecules.26–31 The early development of asymmetric organocatalysis used only small organic molecules as chiral catalysts, such as alkaloid derivatives, amino acid derivatives and phosphoric acid.32–34 The subsequently developed catalysts with polymer-supported groups showed strong competitiveness.35,36 Polymer-supported catalysts solve the problems of uneconomical and difficult product purification since this type of catalyst can be easily recovered from the reaction mixture and reused.37,38 In the past few decades, a variety of polymer backbones such as polystyrene, polyacetylene and polymethacrylate. have been developed to support catalytic groups.39–41 Artificial helical polymers, one of the special species of polymer materials, have broad applications in many fields, such as chiral separation, chiral recognition, asymmetric catalysis, liquid crystallization and circularly polarized light generation due to their helical backbone.42–46 Among them, polyisocyanide exhibits unique advantages because of its rigid helical structure. The static helical orientation of the polyisocyanide backbone can provide an additional asymmetric environment, which exhibits synergistic effects, amplifying effects such as stereoselectivity of asymmetric reactions.47–49
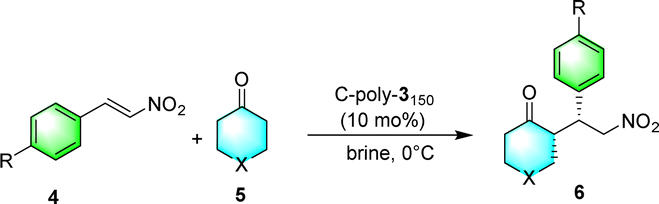
Then, polyisocyanide-based porous polymers should have significant advantages in asymmetric organocatalysis. In this work, we not only establish a strategy for constructing a porous polymer framework using optically active rigid polyisocyanides as structural units but also explore the application of this framework in asymmetric Michael addition reactions. First, a four-arm polymer 4-poly-1ms was obtained by polymerizing isocyanide monomer 1 using the four-arm catalyst 4-Pd(II) as the initiator (Scheme 1). Then, through crosslinking the chain ends of 4-poly-1ms, the porous polymers C-poly-1ms were generated (Scheme 1). The excellent iodine adsorption capacity and variable pore size of C-poly-1ms have also been verified.17 In this project, the protective tert-butoxycarbonyl group (BOC) was removed from the porous polymers to obtain novel porous polymers, C-poly-3ms (Scheme 1). Previously, we proved that linear polymer poly-3ms could serve as an organocatalyst for asymmetric Michael addition reactions with satisfactory stereoselectivity and a lower reaction rate.50 We speculate that the porous polymer catalyst may be beneficial for not only improving the stereoselectivity but also accelerating the reaction rate. Because of the preferred left-handed helix of the polyisocyanide main chain, the resulting C-poly-3ms also showed high optical activity. C-poly-3ms show excellent performance in catalyzing asymmetric Michael addition reactions. Under optimized reaction conditions, the asymmetric Michael addition reaction catalyzed by C-poly-3150 gave the desired product in high yields (>80%) with a diastereomeric ratio (dr) of 99/1 and an enantiomeric excess (ee) of 99% for the main product. Importantly, the dr and ee values of the catalytic product catalyzed by C-poly-3150 were significantly higher than those achieved under catalysis of the linear polymer poly-3150. Taking advantage of its heterogeneous catalysis, C-poly-3150 can be facilely recovered from the reaction mixture and could catalyze the asymmetric Michael addition for at least 6 cycles while maintaining selectivity and activity.
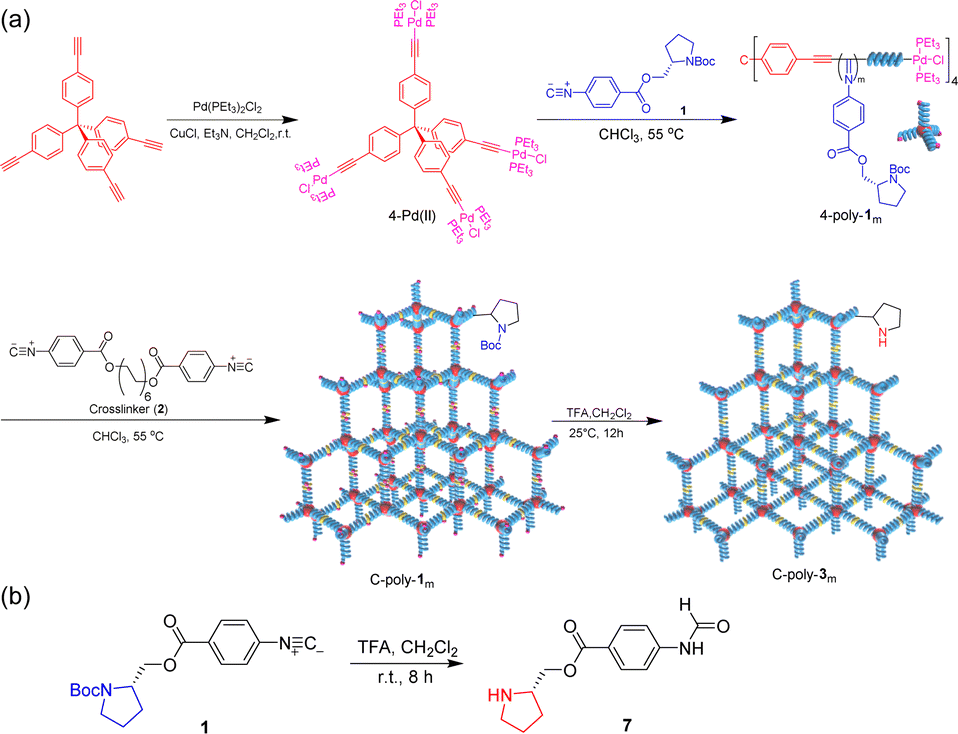 | ||
| Scheme 1 (a) Fabrication of porous organic frameworks using polyisocyanides as frame blocks. (b) Synthesis route of compound 7. | ||
Results and discussion
Macroporous organic catalysts C-poly-3ms were synthesized and prepared according to the previous work reported by our group.17 First, the polymer 4-poly-120 was generated through the polymerization of monomer 1 using the four-arm initiator 4-Pd(II). Size exclusion chromatography (SEC) analysis of 4-poly-120 showed a single modal and narrow elution peak. The Mn and Mw/Mn of 4-poly-120 determined by SEC were 13.8 kDa and 1.19, respectively (Fig. 1a). Further, a series of polymerizations were performed at different ratios of 1 to 4-Pd(II). The generated 4-poly-1ms showed single-peak SEC curves, and the Mw/Mn values were less than 1.24, proving that the polymerization process followed a living/controlled polymerization mechanism. Furthermore, there was a linear relationship between Mn and the feed ratio of 1 to 4-Pd(II) (Fig. 1b). Therefore, a series of 4-poly-1ms with different Mn values and narrow Mw/Mn could be obtained by adjusting the ratio of 1 to the 4-Pd(II) catalyst (Table S1 in the ESI†). Then, the 1H nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FT-IR) analyses were performed to confirm the structures of the synthesized polyisocyanides. The expected resonances of the phenyl rings and the BOC pendants at 8.15–6.67 and 1.45 ppm could be clearly distinguished in the 1H NMR spectrum of 4-poly-120 (Fig. S1, ESI†). In the FT-IR spectrum of 4-poly-120, a characteristic vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds was observed at about 1605 cm−1 (Fig. S2, ESI†). The 31P NMR spectrum could be used to corroborate the successful polymerization. A new characteristic signal shift in the 31P NMR spectrum of 4-poly-120 appeared at 13.5 ppm, accompanied by the complete disappearance of the 31P NMR signal of 4-Pd(II) at 17.9 ppm (Fig. 1c). All of these results showed that the four Pd(II) units on the 4-Pd(II) catalyst had the same activity and participated in the process of the polymerization of monomer 1. Furthermore, the optical activity of 4-poly-1ms was investigated by circular dichroism (CD) and UV-vis spectroscopy. The isolated 4-poly-1ms showed intense negative CD in the absorption region of the polyisocyanide backbone around 364 nm, suggesting the formation of a preferred left-handed helix, and the maximum intensity was recorded for the polymer with a degree of polymerization (DP) value close to 150 (Fig. S3, ESI†).
N bonds was observed at about 1605 cm−1 (Fig. S2, ESI†). The 31P NMR spectrum could be used to corroborate the successful polymerization. A new characteristic signal shift in the 31P NMR spectrum of 4-poly-120 appeared at 13.5 ppm, accompanied by the complete disappearance of the 31P NMR signal of 4-Pd(II) at 17.9 ppm (Fig. 1c). All of these results showed that the four Pd(II) units on the 4-Pd(II) catalyst had the same activity and participated in the process of the polymerization of monomer 1. Furthermore, the optical activity of 4-poly-1ms was investigated by circular dichroism (CD) and UV-vis spectroscopy. The isolated 4-poly-1ms showed intense negative CD in the absorption region of the polyisocyanide backbone around 364 nm, suggesting the formation of a preferred left-handed helix, and the maximum intensity was recorded for the polymer with a degree of polymerization (DP) value close to 150 (Fig. S3, ESI†).
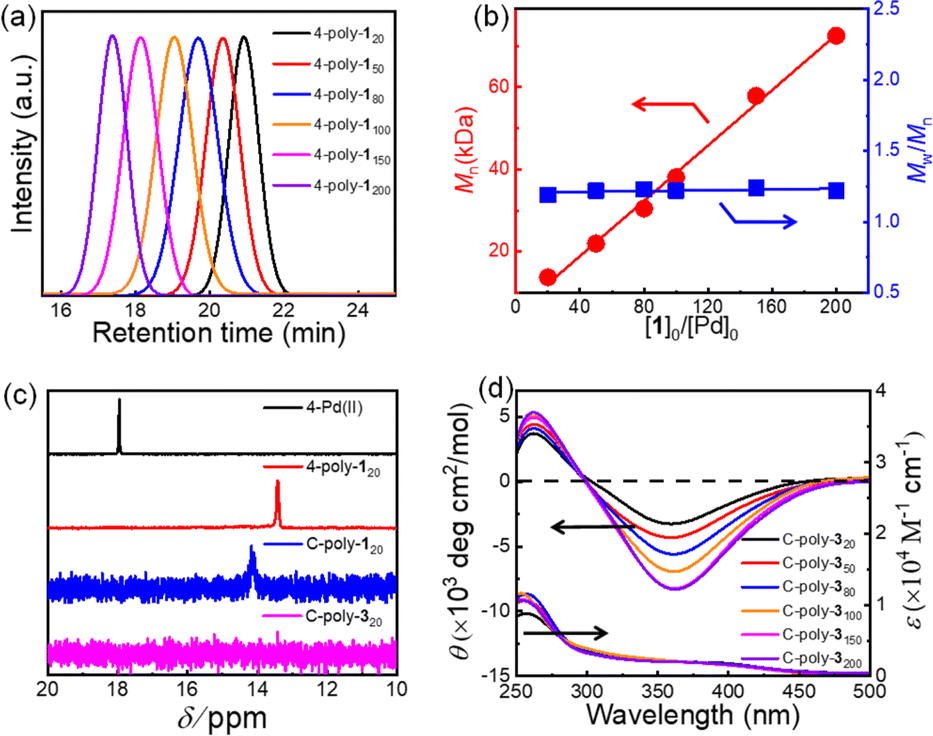 | ||
| Fig. 1 (a) SEC curves for 4-poly-1ms obtained by initiating with 4-Pd(II) with various initial feed ratios of monomer 1 to the initiator. (b) Plots of Mn and Mw/Mn values of 4-poly-1ms as a function of the initial 1-to-Pd(II) ratio. The values of Mn and Mw/Mn were recorded by SEC with polystyrene standards using tetrahydrofuran (THF) as eluent at 40 °C. (c) 31P NMR spectra of 4-Pd(II), 4-poly-120, C-poly-120 and C-poly-320 recorded in CDCl3 at 25 °C. (d) The CD and UV-vis spectra of film state C-poly-3ms at 25 °C. | ||
Subsequently, the active Pd(II) complexes on the chain ends of 4-poly-1ms were crosslinked through the insertion of the bifunctional isocyanide monomer 2 at 55 °C in CHCl3, as depicted in Scheme 1 ([2]0/[Pd]0 = 1/2). Thus, the porous polymers C-poly-1ms were obtained in a gel state and could be precipitated in methanol, after which they were collected in high yields by centrifugation (run 1, 3, 5, 7, and 9, Table 1). The possibility of intramolecular crosslinking had been proved to be difficult to take place due to the limited length of crosslinker 2 according to density functional theory (DFT) calculations in our previous work.17 The 1H NMR and FT-IR spectra of the crosslinked polymer C-poly-120 were similar to those of the 4-poly-120 precursor (Fig. S2 and S4, ESI†), indicating that the chemical structures of the polyisocyanide segments were maintained. In the 31P NMR spectrum of the porous polymer C-poly-120, the signal at 13.5 ppm completely disappeared, and a new resonance signal appeared at 14.1 ppm (Fig. 1c). The CD and UV-vis spectra of C-poly-1ms film showed that C-poly-1ms had similar properties to the precursor 4-poly-1ms. The ellipticity of the C-poly-1ms backbone at 364 nm (θ364) increased with the increase in DP, and the θ364 value remained constant when DP was higher than 150 (Fig. S5, ESI†).
| Run | Polymer | Yieldb (%) | S BET (m2 g−1) | V pore (cm3 g−1) | Pore-sizec (nm) | θ 364 (× 103) |
|---|---|---|---|---|---|---|
| a The porous polymers were fabricated according to Scheme 1. b Isolated yields. c These data were estimated from N2 adsorption isotherms. d The CD intensity of film state C-poly-1ms and C-poly-3ms at 364 nm. | ||||||
| 1 | C-poly-120 | 90 | 30.37 | 8.3 × 10−2 | 4.13 | –6.10 |
| 2 | C-poly-320 | 85 | 41.20 | 1.2 × 10−1 | 5.62 | –3.24 |
| 3 | C-poly-150 | 89 | 15.26 | 8.0 × 10−2 | 8.38 | –9.32 |
| 4 | C-poly-350 | 82 | 27.97 | 1.1 × 10−1 | 10.68 | –4.34 |
| 5 | C-poly-180 | 86 | 9.58 | 7.5 × 10−2 | 10.49 | –11.60 |
| 6 | C-poly-380 | 81 | 19.17 | 9.9 × 10−2 | 12.74 | –5.63 |
| 7 | C-poly-1100 | 85 | 8.93 | 7.2 × 10−2 | 14.12 | –13.04 |
| 8 | C-poly-3100 | 82 | 10.26 | 8.9 × 10−2 | 15.42 | –6.93 |
| 9 | C-poly-1150 | 87 | 7.03 | 6.2 × 10−2 | 16.01 | –14.33 |
| 10 | C-poly-3150 | 83 | 7.98 | 6.8 × 10−2 | 17.52 | –8.21 |
Next, the protecting BOC groups and Pd(II)-terminals of C-poly-1ms were removed using a mixture of trifluoroacetic acid (TFA) and dichloromethane at room temperature. After removing the BOC groups and neutralizing with ammonia solution, C-poly-3ms with poor solubility in most common organic solvents except CH3OH were obtained by precipitation in n-hexane (Scheme 1). However, the lower yield of the C-poly-3ms may be attributed to the incomplete neutralization of the ammonium trifluoroacetate on the pendants in the interior of the polymer network structure. These resulted in a small amount of the polymer ammonium dissolving in water, leading to a slightly reduced yield. The disappearance of the tert-butyl resonance at 1.45 ppm in the 1H NMR spectrum of C-poly-320 also confirmed the removal of the BOC groups (Fig. S6, ESI†). Furthermore, it was demonstrated that the resonance signal of CH2 on the crosslinker 2 appeared at 1.31 ppm, which was another piece of evidence for the successful crosslinking of 4-poly-120 (Fig. S6, ESI†). Because of the newly formed N–H bond on the pendant, a new resonance signal appeared at 6.85 ppm. Furthermore, a new broad vibration band at 3417 cm−1 attributed to the N–H bond was observed in the FT-IR spectrum of C-poly-320 (Fig. S2, ESI†). The Pd residue in C-poly-120 and C-poly-320 was evaluated by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). The Pd residue of C-poly-120 and C-poly-320 was 0.8 wt% and 0.04 wt%, respectively. As expected, no obvious signals were observed in the 31P NMR spectrum of the porous polymer C-poly-320 (Fig. 1c). Therefore, during the TFA treatment, the Pd(II) terminal groups have been simultaneously removed. Due to the preferred left-handed helix of the main chain, the isolated porous polymer C-poly-3150 showed a strong negative CD in the region of 450–300 nm. The ellipticity of C-poly-3ms also increased with the increase in DP until it reached 150 (Fig. 1d and S7, ESI†).
The uncrosslinked 4-poly-1ms had been proved not to contain pores according to the results of XRD and BET experimental results in our previous work.17 Then, to investigate the porosity of the porous polymers C-poly-1ms and C-poly-3ms, nitrogen adsorption–desorption isotherms were recorded (Fig. 2a and S8, ESI†). C-poly-320 adsorbed nitrogen in the medium-pressure region, and the desorption curve lagged behind the adsorption curve, indicating the presence of numerous mesopores in C-poly-320. The adsorption curves of C-poly-350, C-poly-380, C-poly-3100, and C-poly-3150 increased significantly in the high-pressure region (P/P0 = 0.8–1.0), indicating mainly macroporous structures in these porous polymers. The Brunauer–Emmett–Teller (BET) specific surface area (SBET) and pore volume (Vpore) of C-poly-320 were 41.20 m2 g−1 and 0.12 mL g−1, respectively, calculated by a nonlocal density functional theory (DFT) approach (run 2, Table 1 and Fig. S9a, ESI†). The SBET values decreased to 27.97 m2 g−1 for C-poly-350, 19.17 m2 g−1 for C-poly-380, 10.26 m2 g−1 for C-poly-3100, and 7.98 m2 g−1 for C-poly-3150, as the length of polyisocyanide blocks increased (run 4, 6, 8, and 10, Table 1 and Fig. S9a, ESI†). Furthermore, a relationship between the pore size and the length of the polyisocyanide arms was observed. For instance, the pore size of C-poly-320 was 5.62 nm and increased to 10.68, 12.74, 15.42, and 17.52 nm for C-poly-350, C-poly-380, C-poly-3100, and C-poly-3150 with narrow distributions, respectively (run 2, 4, 6, 8, and 10, Table 1, Fig. 2b and S9b, ESI†). However, the pore size of C-poly-3150 deviated slightly from the linear relationship, which may be attributed to the chain interpenetration between the longer polymer chains, which has an influence on the defect of the frameworks of the porous polymer.51,52 Compared to the porous polymer C-poly-1ms, the pore diameter, SBET, and Vpore of C-poly-3ms increased correspondingly (Table 1). This result may be attributed to the removal of the large BOC groups. The powder X-ray diffraction (PXRD) pattern of the porous polymers C-poly-3ms exhibited an extensive diffraction at 2θ of 19° and a sharp diffraction in the 2θ range of 3°–6° (Fig. S10, ESI†). The 2θ value of C-poly-3ms decreased apparently with increasing main chain length of the polyisocyanides (Fig. S11, ESI†). These results not only supported that ordered porous polymers formed through the connection of rigid polyisocyanide chain ends but also explained that the pore size was determined by the length of polyisocyanide blocks.
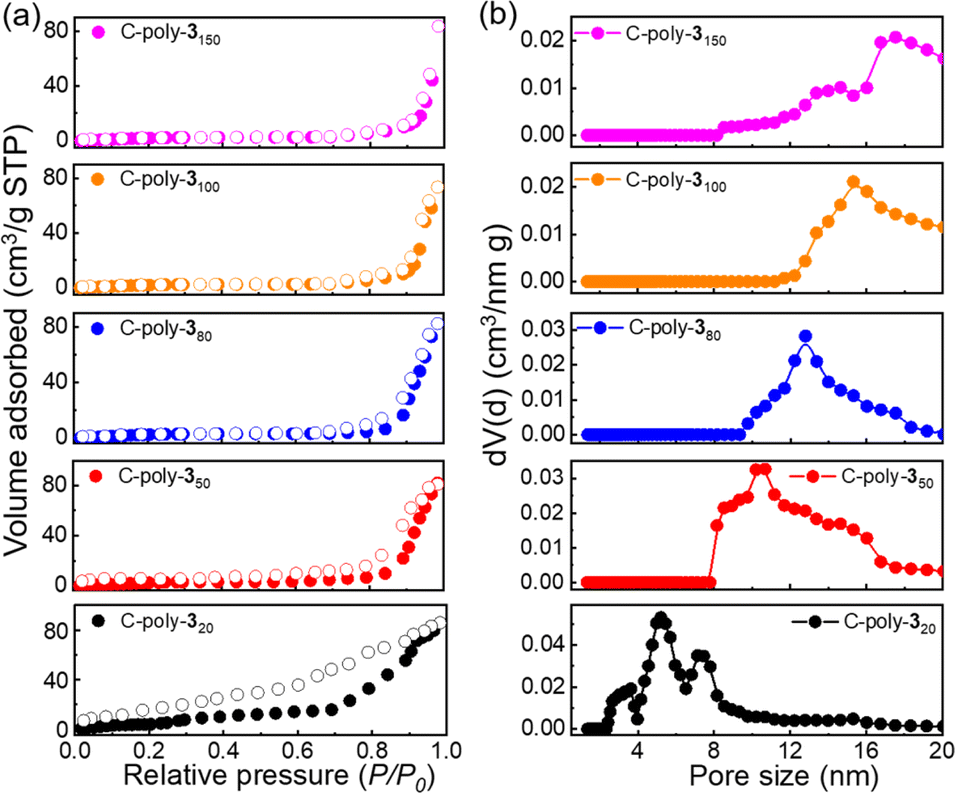 | ||
| Fig. 2 Nitrogen sorption isotherms (a), and pore size distributions (b) of C-poly-3ms were recorded at 77 K. Notably, the filled symbols denote gas adsorption while the empty symbols denote gas desorption in (a). | ||
Scanning electron microscopy (SEM) was used to observe the loose and porous morphology of porous polymers. As depicted in Fig. 3, C-poly-3ms had different morphologies with different lengths of polyisocyanide arms. In Fig. 3a, a rough surface was observed for C-poly-320 with relatively short polyisocyanide arms. With the increase in the length of the polyisocyanide arms, the surfaces of C-poly-350 and C-poly-3100 gradually transformed into flat surfaces (Fig. 3b and c). According to the SEM image, C-poly-3150 with longer polyisocyanide arms had a sponge-like morphology (Fig. 3d). These observations suggested that the morphologies of these porous polymers were controlled by the length of the polyisocyanide arms.
 | ||
| Fig. 3 SEM images of the porous polymers of C-poly-320 (a), C-poly-350 (b), C-poly-3100 (c), and C-poly-3150 (d). | ||
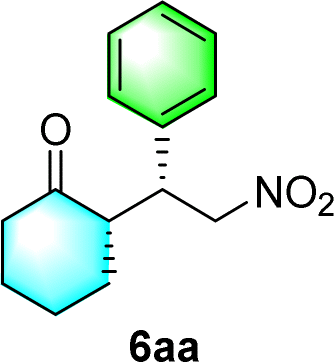
Chiral secondary amines are excellent catalysts that are widely used in asymmetric Michael addition reactions for constructing the skeleton of natural products and drugs.53–55 With the optical porous polymers C-poly-3ms bearing abundant secondary amine pendants in hand, we attempted to screen their catalytic abilities in asymmetric organocatalysis. Inspired by the previous work on the asymmetric catalytic ability of linear polymer catalyst poly-3150 in the asymmetric Michael addition,50 we decided to investigate the capacity of C-poly-3150 in the asymmetric Michael addition between cyclohexanone (5a) and trans-nitrostyrene (4a). The reaction was conducted in dichloromethane at 25 °C with a catalyst loading of 30%. The reaction produced the expected compound 6aa in 86% yield. The dr and ee values of the target product were 91/9 and 93%, respectively, as determined by high performance liquid chromatography (HPLC) (run 8, Table 2). In addition, the same Michael addition reaction was conducted under identical conditions with the linear polymer catalysts poly-3150 and 4-poly-3150. The dr and ee values of the product catalyzed by poly-3150 were 82/18 and 75%, which were much lower than those obtained with the porous polymer catalyst C-poly-3150 (run 2 and 8, Table 2). However the dr and ee values catalyzed by 4-poly-3150 were almost the same as those of 4-poly-3150 (run 3 and 8, Table 2). Furthermore, an analogue compound 7 with a similar structure to the repeating unit of C-poly-3150 was then generated (Scheme 1b and Fig. S12, ESI†) and employed in the same reaction with identical 30% loading. As anticipated, the expected product could be isolated in 75% yield. The dr value of the product was 79/21, and the ee of the main product was 43% (run 1, Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Run | Catalyst | Solvent | Temp. (°C) | X (mol%) | syn/antib | eeb (%) | Yieldc (%) |
| a Unless otherwise denoted, all reactions were carried out with 4a (0.20 mmol), 5a (0.80 mmol) in a specific solvent (1.0 mL). b The dr and ee values were determined by HPLC analysis using a chiral stationary phase. c Isolated yields. | |||||||
| 1 | 7 | CH2Cl2 | r.t. | 30 | 78/22 | 43 | 75 |
| 2 | Poly-3150 | CH2Cl2 | r.t. | 30 | 82/18 | 75 | 82 |
| 3 | 4-Poly-3150 | CH2Cl2 | r.t. | 30 | 90/10 | 92 | 87 |
| 4 | C-poly-320 | CH2Cl2 | r.t. | 30 | 65/35 | 53 | 85 |
| 5 | C-poly-350 | CH2Cl2 | r.t. | 30 | 72/28 | 60 | 87 |
| 6 | C-poly-380 | CH2Cl2 | r.t. | 30 | 80/20 | 68 | 86 |
| 7 | C-poly-3100 | CH2Cl2 | r.t. | 30 | 85/15 | 75 | 85 |
| 8 | C-poly-3150 | CH2Cl2 | r.t. | 30 | 91/9 | 93 | 86 |
| 9 | C-poly-3200 | CH2Cl2 | r.t. | 30 | 91/9 | 94 | 85 |
| 10 | C-poly-3150 | THF | r.t. | 30 | 89/11 | 91 | 88 |
| 11 | C-poly-3150 | CH3OH | r.t. | 30 | 93/7 | 95 | 82 |
| 12 | C-poly-3150 | Toluene | r.t. | 30 | 85/15 | 92 | 80 |
| 13 | C-poly-3150 | Water | r.t. | 30 | 95/5 | 94 | 89 |
| 14 | C-poly-3150 | Brine | r.t. | 30 | 90/10 | 95 | 89 |
| 15 | C-poly-3150 | Brine | 0 | 30 | 93/7 | 97 | 88 |
| 16 | C-poly-3150 | Brine | −10 | 30 | 92/8 | 99 | 85 |
| 17 | C-poly-3150 | Brine | −10 | 20 | 92/8 | 99 | 84 |
| 18 | C-poly-3150 | Brine | −10 | 10 | 92/8 | 99 | 84 |
Following the reaction with C-poly-3150, a series of porous polymers C-poly-3ms with different pore sizes were used in the asymmetric Michael addition reaction between cyclohexanone and trans-nitrostyrene under the same experimental conditions. The results are summarized in Table 2. After carefully analying the ee values in Table 2, it was found that the ee values of the isolated products were quite different and seemed to correlate with the pore size of C-poly-3ms. For example, the ee values of the major Michael addition product 6aa catalyzed by C-poly-320, C-poly-350, C-poly-380, and C-poly-3100 were 53%, 60%, 68%, and 75%, respectively (run 4–7, Table 2 and Fig. 4a). As shown in Fig. 4a, the dr and ee values of 6aa increased gradually with the increase in DP of the porous polymer catalysts until DP reached 150 (run 4–8, Table 2 and Fig. 4a). Despite the further increase in the DP of the catalyst, no apparent increase in these values was noted. The ee and dr values of 6aa obtained under C-poly-3200 catalysis were similar to those synthesized with C-poly-3150 (run 8–9, Table 2 and Fig. 4a). The correlation between stereoselectivity and pore sizes may be attributed to the Mn-dependent helicity of the polyisocyanide blocks of the porous polymers.49,50 Furthermore, the different DP of porous polymers C-poly-3ms had little effect on the yield (Fig. S13, ESI†).
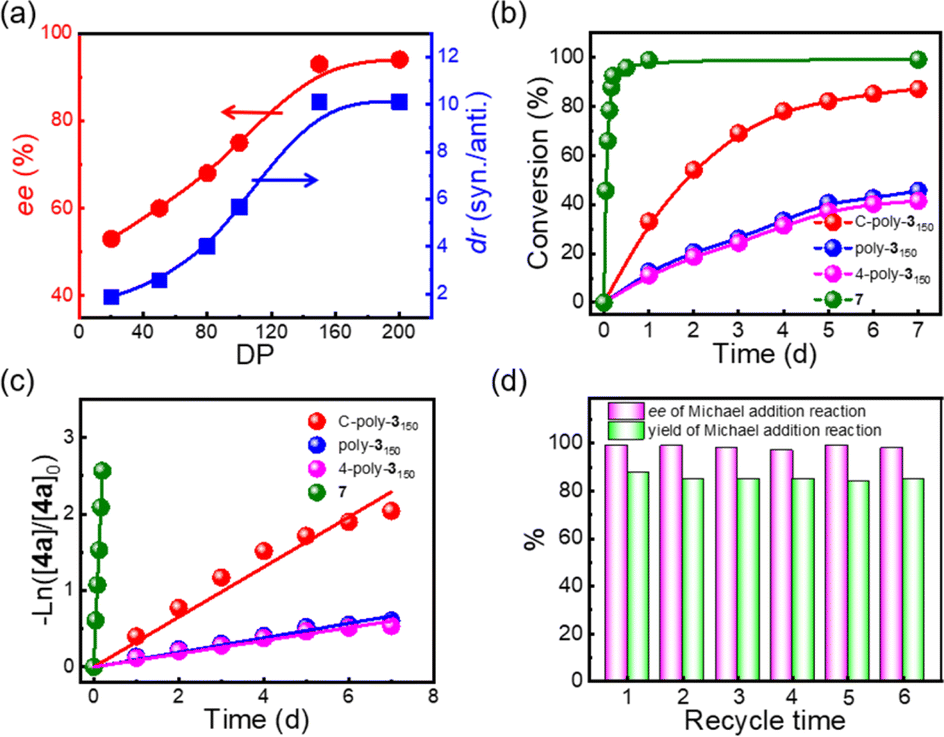 | ||
| Fig. 4 (a) Plots of ee and dr values of the product 6aavs. DP of the C-poly-3ms. The plots of time-dependent conversions of 4a (b) and −ln([4a]/[4a]0) (c) vs. reaction time using C-poly-3150, poly-3150 4-poly-3150, and 7 as catalysts, respectively. (d) Yield and ee values of the reaction of 4a (0.20 mmol) and 5a (0.80 mmol) in brine (1.0 mL) at −10 °C catalyzed by recycled C-poly-3150 (with 10% loading). | ||
The asymmetric Michael addition reaction catalyzed by the porous polymers C-poly-3ms was investigated in more detail. First, the solvents including tetrahydrofuran (THF), toluene, methyl alcohol (CH3OH), water, and brine were screened. Using THF and toluene as solvents resulted in lower dr and ee values of the product (run 10 and 12, Table 2). Strong polar solvents such as CH3OH, water, and brine may be beneficial for enhancing the stereoselectivity of the reaction (run 11, 13–14, Table 2), and polar solvents may facilitate the proton transfer and imide hydrolysis of the intermediate according to the reported mechanism catalyzed by the pyrrolidine-based catalyst.56,57 The dr values of the product reached 93/7 and 90/10, respectively, using CH3OH and brine as solvents, while they had the same ee value of 95%. However, a yield of 89% was achieved in brine, which was much higher than the product yield in CH3OH (run 11 and 14, Table 2). Hence, brine was chosen as the best reaction medium for the asymmetric Michael addition reaction. Interestingly, the ee and dr values of the desired product improved to 99% and 92/8 without apparent loss of catalyst efficiency when lowering the reaction temperature from room temperature to −10 °C (run 16, Table 2). Importantly, reducing the loading of C-poly-3150 from 30 mol% to 10 mol% did not have any negative effect on the yield or the ee and dr values of the product (run 16 and 18, Table 2). Therefore, the optimal conditions comprised a C-poly-3150 loading of 10 mol%, brine as the solvent, and a reaction temperature of −10 °C.
As commonly known, porous polymers have a higher specific surface area and inherent porosity than linear polymers. In order to confirm that the porous polymer catalysts maintained a higher specific surface, the time-domain NMR was used to investigate the wetted surface area of C-poly-3ms in water. It could be observed that the spin–spin relaxation time (T2) increased with the increased DP of C-poly-3ms. The wetted surfaces could be calculated to 129.89 m2 g−1, 102.72 m2 g−1 −1 and 58.21 m2 g−1 of C-poly-320, C-poly-380 and C-poly-3150, respectively (Fig. S14, ESI†).58,59 In our previous studies, the linear polymer poly-3150 has also been explored to catalyze asymmetric Michael addition reactions with satisfactory ee values and longer reaction time.50 Compared to the linear polymer poly-3150, the porous polymer C-poly-3150 may have higher catalytic efficiency. Therefore, the difference in kinetics of the test reaction catalyzed by C-poly-3150 and poly-3150 was assessed by HPLC (Fig. S15, ESI†). According to Fig. 4b, the trans-nitrostyrene conversion catalyzed by C-poly-3150 was about twice that observed under poly-3150 catalysis within 7 d. However the conversion of substrate 4a was over 90% within 5 h when catalyzed by compound 7 (Fig. 4b and S15d, ESI†). Furthermore, the reaction rate constants of the Michael addition reaction catalyzed by compound 7, poly-3150, 4-poly-3150 and C-poly-3150 were 5.21 × 10−1 h−1, 3.66 × 10−3 h−1, 3.27 × 10−3 h−1 and 1.36 × 10−2 h−1, respectively (Fig. 4c). These results quantitatively confirmed that the reaction rate of the asymmetric Michael addition could be accelerated when using porous polymers instead of linear polymers as the catalyst. That's probable because the mesoporous and macroporous structures facilitated the diffusion of organic substrates during the reaction process.60,61 In addition, C-poly-3150 could be easily recovered by precipitation and centrifugation after the completion of the reaction due to its poor solubility in brine. Then, the recovered C-poly-3150 could be recycled and successfully used in further reactions. The yield and ee values of the isolated product 6aa were plotted over the recycle times, as displayed in Fig. 4d. C-poly-3150 was recycled and used at least six times without significant loss in activity and stereoselectivity (Fig. 4d and Table S2, ESI†).
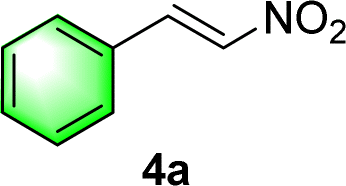
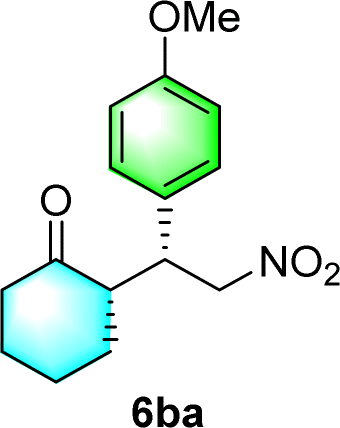
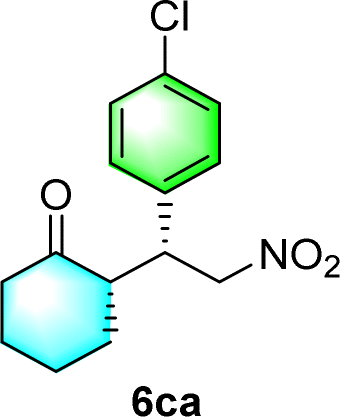
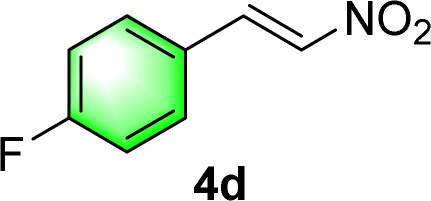
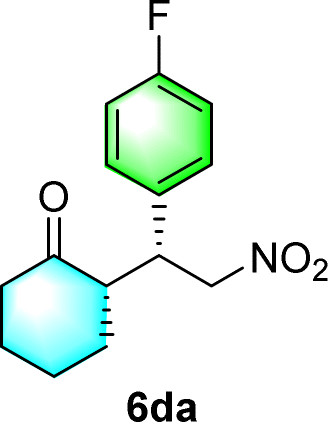
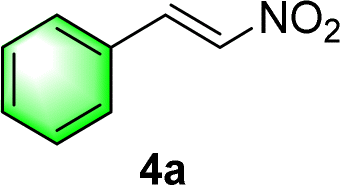
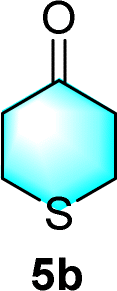
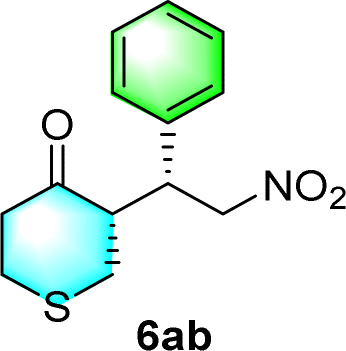
After the establishment of the optimal conditions for the asymmetric Michael addition reaction catalyzed by C-poly-3150, the scope of substrates with different substituents was screened. As summarized in Table 3, the optimized reaction conditions were applied to different substituted trans-nitrostyrenes and cycloketones. When using substrate 4b with an electron-donating OMe group, the products 6ba were isolated with 99% ee despite a slight decrease in the dr value (90/10) (run 2, Table 3). The reaction of substrates 4c and 4d with halogen substituents such as Cl and F proceeded smoothly with ee values of 99% and dr values exceeding 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 (run 3 and 4, Table 3). Notably, the product 6ab of the reaction between 4a and tetrahydro-4H-thiopyran-4-one (5b) was generated with both higher dr and ee values of 99/1 and 99%, respectively (run 5, Table 3). It could be demonstrated that all the stereoselectivity of the main products listed in Table 3 could be up to ca. 99%. Among these products, the stereoselectivity of 6aa, 6ba and 6ca catalyzed by C-poly-3150 was improved apparently compared with those catalyzed by linear polymer catalysts in previous work.50
:6 (run 3 and 4, Table 3). Notably, the product 6ab of the reaction between 4a and tetrahydro-4H-thiopyran-4-one (5b) was generated with both higher dr and ee values of 99/1 and 99%, respectively (run 5, Table 3). It could be demonstrated that all the stereoselectivity of the main products listed in Table 3 could be up to ca. 99%. Among these products, the stereoselectivity of 6aa, 6ba and 6ca catalyzed by C-poly-3150 was improved apparently compared with those catalyzed by linear polymer catalysts in previous work.50
|
|
||||||
|---|---|---|---|---|---|---|
| Run | 4 | 5 | 6 | Yieldb (%) | syn/antic | eec (%) |
| a Reaction conditions: 4 (0.20 mmol), 5 (0.80 mmol), and C-poly-3150 (10% loading). The reaction was carried out in brine (1.0 mL) at 0 °C. b Isolated yields. c The dr and ee values were determined by HPLC analysis using a chiral stationary phase. | ||||||
| 1 |
|

|
|
84 | 92/8 | 99 |
| 2 |
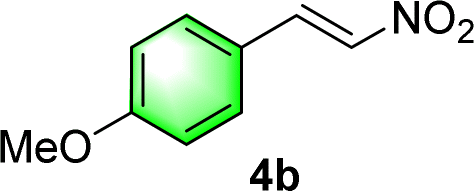
|
|
|
83 | 90/10 | 99 |
| 3 |
|
|
|
86 | 97/3 | 99 |
| 4 |
|
|
|
86 | 94/6 | 99 |
| 5 |
|
|
|
80 | 99/1 | 99 |
Conclusions
In summary, a series of novel porous polymer catalysts C-poly-3ms with secondary amine pendants were designed and synthesized using a four-arm Pd(II) catalyst. The pore size of the porous polymers can be controlled by adjusting the DP of the polyisocyanide blocks. Among the C-poly-3ms compounds, C-poly-3150 had a higher optical activity with a θ364 value of −8.21 × 103, indicating that its main chain adopted a preferred left-handed helix. The SBET and pore size of the C-poly-3150 were 7.98 m2 g−1 and 17.52 nm, respectively. Compared to the linear polymer poly-3150, the porous polymer C-poly-3150 exhibited excellent activity and selectivity in organocatalyzed asymmetric Michael addition reactions. Under the optimized reaction conditions, various Michael addition reaction products were synthesized in high yields and with satisfactory stereoselectivity. The ee and dr values of 6ab could reach 99% and 99/1, respectively. The recovered catalyst can be recycled at least six times in the asymmetric Michael addition reaction while maintaining activity and stereoselectivity. In the future, we will focus on the studies for constructing POP polymers with uniform aperture distribution through a reversible cross-linking method. We believe that this study not only develops a novel porous polymer organocatalyst for asymmetric Michael addition reactions, but also provides a direction for the future design of novel topological polymer-supported chiral organocatalysts.Data availability
All experimental procedures, characterisation data, SEC spectra, CD and UV-vis spectra, NMR spectra, and HPLC spectra can be found in the ESI.†Author contributions
X.-H. Xu performed the synthesis of polymers, characterized the structures of the polymers and investigated the Michael reaction. R.-T. Gao and S.-Y. Li took part in the reaction development. L. Zhou and Z.-Q. Wu prepared the manuscript, guided and supervised the project. N. Liu reviewed and edited the manuscript. All of the authors discussed the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China for financial support (NSFC, No. 92256201, 52273204, 52273006, 22071041, 21971052, 51903072 and 21871073). L. Zhou also thanks the Anhui Provincial Natural Science Foundation (Grant No. 2008085MB51) and the Fundamental Research Funds for the Central Universities of China (Grant No. PA2021GDSK0064). X.-H. Xu acknowledges the financial support from the China Postdoctoral Science Foundation (No. 2023M730048).Notes and references
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- M. Dincă and J. R. Long, Chem. Rev., 2020, 120, 8037–8038 CrossRef PubMed.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Nature, 2019, 565, 213–217 CrossRef CAS PubMed.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267–8302 CrossRef CAS PubMed.
- C. Gropp, T. Ma, N. Hanikel and O. M. Yaghi, Science, 2020, 370, eabd6406 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- D.-H. Yang, Y. Tao, X. Ding and B.-H. Han, Chem. Soc. Rev., 2022, 51, 761–791 RSC.
- X. Yang, Z. Ullah, J. F. Stoddart and C. T. Yavuz, Chem. Rev., 2023, 123, 4602–4634 CrossRef CAS PubMed.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- Y. Zhao, Y. He and T. M. Swager, ACS Macro Lett., 2018, 7, 300–304 CrossRef CAS PubMed.
- B. Aguila, Q. Sun, H. C. Cassady, C. Shan, Z. Liang, A. M. Al-Enizic, A. Nafadyc, J. T. Wright, R. W. Meulenberg and S. A Ma, Angew. Chem., Int. Ed., 2020, 59, 19618–19622 CrossRef CAS PubMed.
- S. Bhattacharjee, A. Tripathi, R. Chatterjee, R. Thapa, T. E. Mueller and A. Bhaumik, ACS Catal., 2024, 14, 718–727 CrossRef CAS.
- Y. Xia, T. Di, Z. Meng, T. Zhu, Y. Lei, S. Chen, T. Li and L. Li, Macromolecules, 2021, 54, 4682–4692 CrossRef CAS.
- E. Altay, D. Nykypanchuk and J. Rzayev, ACS Nano, 2017, 11, 8207–8214 CrossRef CAS PubMed.
- X.-H. Xu, Y.-X. Li, L. Zhou, N. Liu and Z.-Q. Wu, Chem. Sci., 2022, 13, 1111–1118 RSC.
- Y.-X. Xue, Y.-Y. Zhu, L.-M. Gao, X.-Y. He, N. Liu, W.-Y. Zhang, J. Yin, Y. Ding, H. Zhou and Z.-Q. Wu, J. Am. Chem. Soc., 2014, 136, 4706–4713 CrossRef CAS PubMed.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Noltea and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- N. Liu, L. Zhou and Z.-Q. Wu, Acc. Chem. Res., 2021, 54, 3953–3967 CrossRef CAS PubMed.
- L. Zhou, K. He, S.-M. Kang, X.-Y. Zhou, H. Zou, N. Liu and Z.-Q. Wu, Angew. Chem., Int. Ed., 2023, 62, e202310105 CrossRef CAS PubMed.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS PubMed.
- H. Yuan, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Angew. Chem., Int. Ed., 2020, 59, 2720–2724 CrossRef CAS PubMed.
- T. Miyabe, H. Iida, M. Banno, T. Yamaguchi and E. Yashima, Macromolecules, 2011, 44, 8687–8692 CrossRef CAS.
- M. Banno, Z.-Q. Wu, K. Nagai, S. Sakurai, K. Okoshi and E. Yashima, Macromolecules, 2010, 43, 6553–6561 CrossRef CAS.
- G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC.
- T. Gatzenmeier, M. van Gemmeren, Y. Xie, D. Höfler, M. Leutzsch and B. List, Science, 2016, 351, 949–952 CrossRef CAS PubMed.
- T. Govender, P. I. Arvidsson, G. E. M. Maguire, H. G. Kruger and T. Naicker, Chem. Rev., 2016, 116, 9375–9437 CrossRef CAS PubMed.
- X. Zhang, M. Liu, H. Ge and Z. Zhang, ACS Catal., 2023, 13, 8273–8280 CrossRef CAS.
- M. Wang, Z. Zhang and W. Zhang, Acc. Chem. Res., 2022, 55, 2708–2727 CrossRef CAS PubMed.
- B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC.
- S. Li, S. Wang, J. Li, Y. Qi, C. Wang, L. Zong and C.-H. Tan, ACS Catal., 2021, 11, 15141–15148 CrossRef CAS.
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed.
- D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme and H. Zhu, Chem. Sci., 2020, 11, 4381–4390 RSC.
- N. Zhang, Z. Sun and C. Wu, ACS Catal., 2022, 12, 4777–4783 CrossRef CAS.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2020, 142, 18317–18323 CrossRef CAS PubMed.
- S. Cañellas, C. Ayats, A. H. Henseler and M. A. Pericàs, ACS Catal., 2017, 7, 1383–1391 CrossRef.
- S.-M. Kang, S.-S. Han, Y.-Y. Zhu and Z.-Q. Wu, ACS Catal., 2021, 11, 13838–13847 CrossRef CAS.
- T. Ikai, M. Ando, M. Ito, R. Ishidate, N. Suzuki, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2021, 143, 12725–12735 CrossRef CAS PubMed.
- N. Madhavan, C. W. Jones and M. Weck, Acc. Chem. Res., 2008, 41, 1153–1165 CrossRef CAS PubMed.
- H. Zhang and J. Deng, Chem. Phys., 2016, 217, 880–888 CAS.
- J. Shen and Y. Okamoto, Chem. Rev., 2016, 116, 1094–1138 CrossRef CAS PubMed.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- S. Wang, S. Xie, H. Zeng, H. Du, J. Zhang and X. Wan, Angew. Chem., Int. Ed., 2022, 61, e202202268 CrossRef CAS PubMed.
- H. Dai, R. Hong, Y. Ma, X. Cheng and W. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202314848 Search PubMed.
- X. Wang, B. Zhao and J. Deng, Adv. Mater., 2023, 35, 2304405 CrossRef CAS PubMed.
- L. Xu, L. Zhou, Y.-X. Li, R.-T. Gao, Z. Chen, N. Liu and Z.-Q. Wu, Nat. Commun., 2023, 14, 7287 CrossRef CAS PubMed.
- Z.-Q. Wu, X. Song, Y.-X. Li, L. Zhou, Y.-Y. Zhu, Z. Chen and N. Liu, Nat. Commun., 2023, 14, 566 CrossRef CAS PubMed.
- L. Zhou, B.-F. Chu, X.-Y. Xu, L. Xu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2017, 6, 824–829 CrossRef CAS.
- L. Shen, L. Xu, X.-H. Hou, N. Liu and Z.-Q. Wu, Macromolecules, 2018, 51, 9547–9554 CrossRef CAS.
- G. Barin, G. W. Peterson, V. Crocellà, J. Xu, K. A. Colwell, A. Nandy, J. A. Reimer, S. Bordiga and J. R. Long, Chem. Sci., 2017, 8, 4399–4409 RSC.
- H. Cao, S. Wang, Y. Wang, H. Lyu, R. Krishna, Z. Lu, J. Duan and W. Jin, CrystEngComm, 2017, 19, 6927–6931 RSC.
- S. N. Reddy, V. R. Reddy, S. Dinda, J. B. Nanubolu and R. Chandra, Org. Lett., 2018, 20, 2572–2575 CrossRef CAS PubMed.
- A. Kunzendorf, G. Xu, J. J. H. van der Velde, H. J. Rozeboom, A.-M. W. H. Thunnissen and G. J. Poelarends, ACS Catal., 2021, 11, 13236–13243 CrossRef CAS PubMed.
- X. Gu, T. Guo, Y. Dai, A. Franchino, J. Fei, C. Zou, D. J. Dixon and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 10249–10253 CrossRef CAS PubMed.
- D.-Z. Xu, S. Shi and Y. Wang, Eur. J. Org Chem., 2009, 28, 4848–4853 CrossRef.
- Q. Zhu, H. Shen, Z. Yang and H. Ji, Chin. J. Catal., 2016, 37, 1227–1234 CrossRef CAS.
- T. Cosgrove, S. Stebbing, M. Ackroyd, D. Fairhurst, K. Sanderson and S. W. Prescott, Powder Technol., 2023, 414, 118065 CrossRef CAS.
- D. Fairhurst, T. Cosgroveb and S. W. Prescottc, Magn. Reson. Chem., 2016, 54, 521–526 CrossRef CAS PubMed.
- Y. Qin, X. Han, Y. Li, A. Han, W. Liu, H. Xu and J. Liu, ACS Catal., 2020, 10, 5973–5978 CrossRef CAS.
- X. Li, L. Fan, B. Xu, Y. Shang, M. Li, L. Zhang, S. Liu, Z. Kang, Z. Liu, X. Lu and D. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 53892–53903 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01316f |
| This journal is © The Royal Society of Chemistry 2024 |