
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acid-catalyzed diastereoselective formal ene reaction of thioindolinones/thiolactams with bicyclobutanes†
Avishek
Guin
 *,
Shiksha
Deswal
,
Mahesh Singh
Harariya
and
Akkattu T.
Biju
*
*,
Shiksha
Deswal
,
Mahesh Singh
Harariya
and
Akkattu T.
Biju
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: avishekguin@iisc.ac.in; atbiju@iisc.ac.in; Web: https://atbiju.in/
First published on 2nd July 2024
Abstract
Bicyclo[1.1.0]butanes (BCBs), featuring two fused cyclopropane rings, have found widespread application in organic synthesis. Their versatile reactivity towards radicals, nucleophiles, cations, and carbenes makes them suitable for various reactions, including ring-opening and annulation strategies. Despite this versatility, their potential as enophiles in an ene reaction remains underexplored. Considering this and given the challenges of achieving diastereoselectivity in ring-opening reactions of BCBs, herein, we present a unique method utilizing BCBs as enophiles in a mild and diastereoselective Sc(OTf)3-catalyzed formal ene reaction with thioindolinones/thiolactams, delivering 1,3-disubstituted cyclobutane derivatives in high yields and excellent regio- and diastereoselectivity. Notably, structurally different thiolactam derivatives underwent diastereoselective addition to BCBs, affording the corresponding cyclobutanes. The synthesized thioindole-substituted cyclobutanes could serve as a versatile tool for subsequent functional group manipulations.
Introduction
Through the conceptual evolution of strain release, chemists have harnessed the innate reactivity stemming from thermodynamic push and destabilization. These “spring-loaded” molecules offer an excellent framework for accessing complex molecular entities under mild conditions, eliminating the need for additional energy sources.1 Among these, bicyclo[1.1.0]butanes (BCBs) have emerged as one of the most structurally compact cyclic compounds known, owing to their small size and high strain energy (66.3 kcal mol−1). Notably, BCBs are a unique type of donor–acceptor (D–A) cyclopropane with higher strain energy compared to typical D–A cyclopropanes. Consequently, they often exhibit similar reactivity to D–A cyclopropanes in many cases.2 BCBs offer a versatile platform for constructing a range of intricate bicyclic frameworks, predominantly via annulation reactions.3 BCBs can be engaged in dearomatization reactions as well with aromatic counterparts such as thiophenes or phenols, resulting in the formation of decorated bicyclo derivatives.4 Complex bicyclic scaffolds can also be achieved through functionalization of BCBs without cleaving the central C–C bond.5 In recent years, significant progress has been made in the development of heterolytic and homolytic ring-opening processes involving BCBs, facilitating the foundation of structurally intricate cyclobutane derivatives.Notably, cyclobutanes serve as crucial intermediates in chemical synthesis and found in a diverse array of natural products and other biologically relevant compounds.6 Specifically, 1,3-substituted cyclobutanes hold particular importance, being frequently assessed in drug discovery due to their advantageous electronic, steric, and conformational characteristics.7 Nevertheless, because of the challenges associated with synthesizing these small four-membered rings, the available methods for producing cyclobutanes featuring a diverse array of synthetically pertinent and pharmaceutically relevant functional groups are restricted.8 Hence, chemists have recently turned their attention to highly strained bicyclic systems like BCBs, aiming to discover innovative protocols to prepare these significant motifs. In 2016, Baran and co-workers reported strain-release-driven amination of BCBs for the synthesis of cyclobutyl amines, which primarily revolutionized the field.9 Subsequently, BCBs were used routinely for synthesizing functionalized cyclobutane derivatives. However, majority of these reactions proceed via radical pathway, resulting in very poor diastereoselectivity.10 Notably, Aggarwal and co-workers explored the nucleophilic reactivity of BCBs through the in situ formation of highly strained BCB boronates. These strained BCB boronates can react with various nucleophiles for the synthesis of trisubstituted cyclobutane derivatives with very high diastereoselectivity.11 Recently, Glorius and co-workers developed photoredox-mediated thio-alkenylation of BCBs, resulting in the synthesis of functionalized cyclobutanes in high diastereoselectivity.12 We have also devised an operationally simple Lewis acid-catalyzed protocol for the diastereoselective carbofunctionalization of bicyclobutanes using naphthols (Scheme 1, eqn (1)).13 Although attaining high diastereoselectivity in BCB ring-opening presents a challenge, the strategic selection of appropriate reaction partners/conditions can promote high diastereoselectivity in the process.
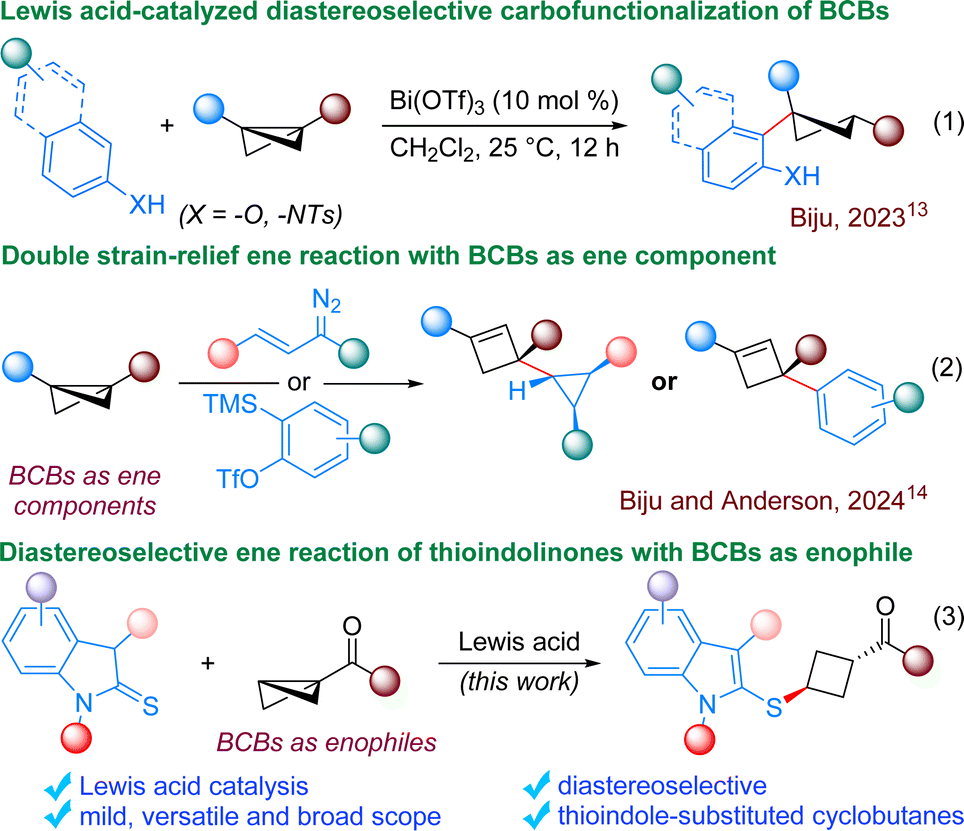 | ||
| Scheme 1 BCBs in ring-opening and ene reactions. | ||
Very recently, the Anderson group and our group demonstrated a dual strain-relief-driven ene-like reaction involving BCBs, where the in situ generated cyclopropenes or benzynes function as the enophiles, while BCBs serve as the ene component (eqn (2)).14 This strategy provides access to cyclopropanes or arene substituted highly decorated cyclobutenes. As enophiles are inherently electrophilic species and given the fact that BCBs are also electrophilic species, theoretically BCBs should engage as the enophile in the ene reaction. We speculated that a strategically selected reaction partner with a tendency to act as the ene component could compel BCBs to function as the enophile, contrary to the previously outlined approach.14 Literature precedents indicate that 2-thioindolinones have the potential to serve as an ene component, wherein aromaticity can serve as an additional driving force for the formation of the indole-substituted products.15 Hence, 2-thioindolinones were selected as the preferred reaction partner for the intended ene reaction. Our previous experience on the ability of Lewis acids to control the diastereoselectivity inspired us to formulate the process catalyzed by a Lewis acid.13 We envisioned that the coordination of BCBs by the Lewis acid could enhance the feasibility of thioindolinone addition, while the transfer of a proton from the same side of the thioindolinones could yield the desired product with high diastereoselectivity (eqn (3)). Considering the importance of 1,3-substituted cyclobutanes in mind, it was planned to use monosubstituted BCBs for our investigation.7 It is worth noting that the synthesis of 1-thio-3-keto disubstituted cyclobutanes from BCBs has been previously utilized in chemical biology tools, though the products were obtained with very poor diastereoselectivity.16
Results and discussion
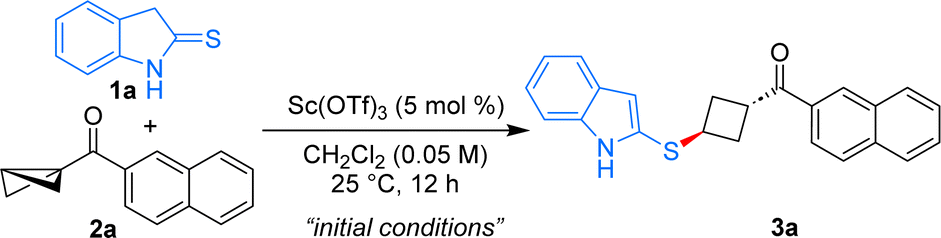
With the conceived idea in mind, the present studies were initiated by the treatment of 2-thioindolinones 1a with the keto-naphthyl substituted BCB 2a in the presence of Sc(OTf)3 (5 mol%) in CH2Cl2 at 25 °C. Using these conditions, the anticipated 1,3-disubstituted cyclobutane 3a was formed in 63% yield as a single diastereomer (Table 1, entry 1). Under the present reaction conditions, Bi(OTf)3 was found to be ineffective to improve the yield of the product further (entry 2). Notably, employing Cu(OTf)2 or TMSOTf as Lewis acid catalysts resulted in the formation of the expected product with lower yield and poor diastereomeric ratio (entries 3 and 4). Augmenting the catalyst loading did not enhance the yield of product 3a (entry 5). Changing the stoichiometry of 1a and 2a resulted in a decreased yield of product formation (entries 6 and 7). Conducting the reaction at 0 °C led to an improvement in the yield of 3a to 71% (entry 8). Additionally, carrying out the reaction at 0 °C to rt furnished product 3a in a similar yield (entry 9). Varying the solvents was not beneficial in improving the yield of the product (entries 10, 11). Hence, entry 9 was selected as the optimal condition for this diastereoselective formal ene reaction.17|
|
|||
|---|---|---|---|
| Entry | Variation of the initial conditionsa | Yield of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
drc |
| a Initial conditions: 1a (0.10 mmol), 2a (0.12 mmol), Sc(OTf)3 (5 mol%), CH2Cl2 (2.0 mL), 25 °C for 12 h. b The 1H NMR yield of the crude products was determined using CH2Br2 as the internal standard and the isolated yield was given in parenthesis. c dr value was determined from 1H NMR of the crude reaction mixture. d The reaction was performed at 0 °C to rt. | |||
| 1 | None | 63 | >20:1 |
| 2 | Bi(OTf)3 instead of Sc(OTf)3 | 40 | >20:1 |
| 3 | Cu(OTf)2 instead of Sc(OTf)3 | 60 | 2:1 |
| 4 | TMSOTf instead of Sc(OTf)3 | 47 | 1:1 |
| 5 | 10 mol% Sc(OTf)3 instead of 5 mol% | 62 | >20:1 |
| 6 | 1.4 equiv of 2a instead of 1.2 equiv | 57 | >20:1 |
| 7 | 1.2 equiv of 1a | 51 | >20:1 |
| 8 | 0 °C instead of 25 °C | 71 | >20:1 |
| 9 | 0 °C to rt instead of 25 °C | 71 (70) | >20:1 |
| 10d | DCE instead of CH2Cl2 | 60 | >20:1 |
| 11d | THF instead of CH2Cl2 | 69 | 1:1 |
With the identified reaction conditions in hand, we examined the scope and drawbacks of this diastereoselective formal ene reaction involving thioindolinones and BCBs (Scheme 2). First, the variation in BCBs was evaluated. Different keto containing bicyclo[1.1.0]butanes, featuring substitutions at various positions on the benzene ring, smoothly underwent the ring-opening reactions, resulting in the diastereoselective formation of the desired thioindole-substituted cyclobutanes in good yields (3a–3d). The scalability of the developed ene reaction was demonstrated by isolating 3a in a 72% yield when the reaction was conducted on a 2.0 mmol scale. Moreover, keto BCBs with disubstituted arene or heteroaryl ring were well tolerated under the present conditions, yielding the anticipated cyclobutane derivatives in good yields (3e-3f). BCB with butyl substitution also performed effectively, delivering the desired product 3g in 92% yield as a single diastereomer. This diastereoselective ene reaction could be extended beyond keto BCBs as enophiles; ester substituted BCBs also furnished the desired ene product. For instance, BCBs derived from benzyl, cinnamyl, and cyclohexyl alcohols delivered the anticipated product in good to excellent yields (3h–3j). BCBs derived from natural products such as (−)-menthol and geraniol also furnished the desired ene products (3k-3l). In the case of menthol, the product was obtained in 12:1 diastereomeric ratio. Notably, BCB substituted with phenyl and ester did react under the present reaction conditions, and the desired product 3m was formed in 88% yield and 2:1 dr.18
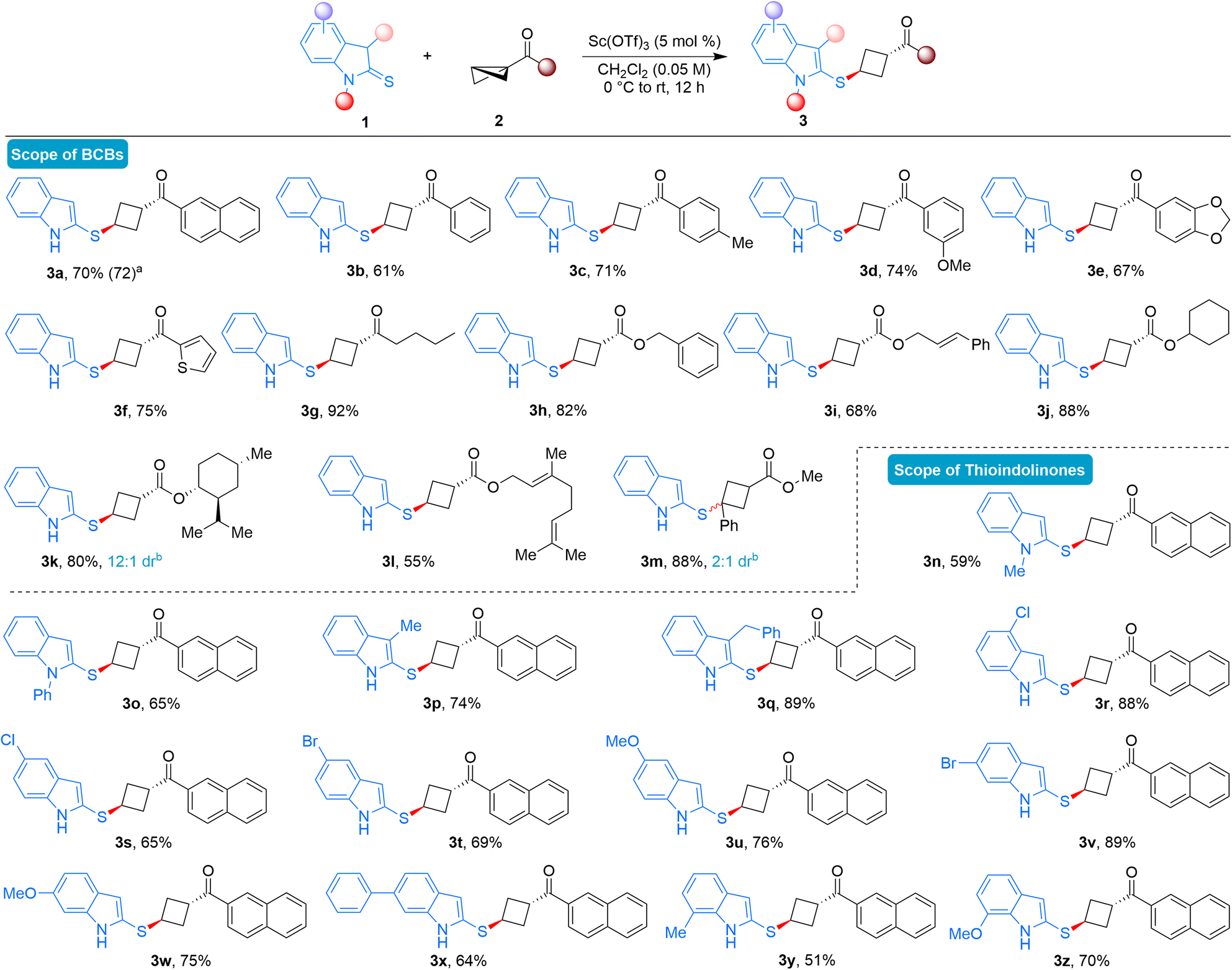 | ||
| Scheme 2 Substrate scope of the reaction. Reaction conditions: 1 (0.20 mmol), 2 (0.24 mmol), Sc(OTf)3 (5 mol%), CH2Cl2 (4.0 mL), 0 °C to rt for 12 h. Provided are yields of isolated products. aYield of the experiment conducted on a 2.0 mmol scale. bUnless otherwise noted, dr > 20:1 and the dr value was determined from 1H NMR of the crude reaction mixture. | ||
The reaction scope was subsequently assessed by employing various substituted thioindolinones. N-protected thioindolinones served as tolerable substrates under the present reaction conditions and the products were formed in moderate yields (3n-3o). Therefore, successful outcomes in this ene reaction are not always dependent on the presence of free N–H substituted thioindolinones. Thioindolinones substituted at the 3-position yielded the desired products in good yields (3p-3q). Various 4- and 5-substituted thioindolinones could also be used as substrates to obtain the corresponding 1,3-disubstituted cyclobutanes under the optimized reaction conditions (3r–3u). Several thioindolinones substituted at the 6-position proved to be effective substrates in this diastereoselective ene reaction, furnishing the expected products in high yields (3v–3x). Finally, 7-substitution also yielded the anticipated products in good yields (3y-3z), demonstrating that the developed BCB ring-opening reactions are effective with thioindolinones bearing substitutions at every position.
When pyrrolidine-2-thione was employed as the ene component under the present reaction conditions, the product 5a bearing imine was formed in 68% yield in single diastereomer (Scheme 3). The distinct reactivity of indoline-2-thione compared to pyrrolidine-2-thione can be explained by the additional driving force of aromatization present in the product of indoline-2-thione, which is lacking in the latter case. This reaction was also scalable, as evident by a 1 mmol scale reaction that produced the desired product with a 66% yield. The generality of this reaction was tested with different thiones and BCBs. Thiones with different ring size delivered the anticipated products in good yields (5b–5e). Aliphatic ketone substituted BCB also furnished the desired product 5f in 73% yield, thereby broadening the scope of the reaction.
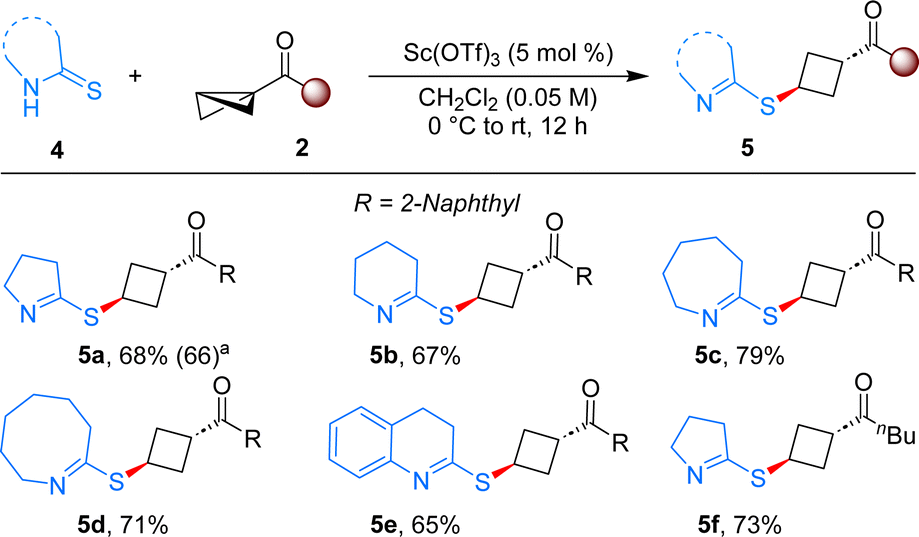 | ||
| Scheme 3 Reaction with structurally different thioamides. Reaction conditions: 4 (0.20 mmol), 2 (0.24 mmol), Sc(OTf)3 (5 mol%), CH2Cl2 (4.0 mL), 0 °C to rt for 12 h. Provided are yields of isolated products. aYield of the experiment conducted on a 1.0 mmol scale. The dr value determined from 1H NMR of the crude reaction mixture was >20:1 in all cases. | ||
A few mechanistic experiments were conducted to gain insight into the mechanism of the described ene reaction. When the product 3a was treated in the presence of LDA followed by quenching with D2O, it underwent epimerization, resulting in the formation of a 1:1 diastereomeric mixture of 3a and 3a′ (Scheme 4, eqn (4)). This study reveals that the observed diastereoselectivity in the described ene reaction is not governed by thermodynamic control. If thermodynamic parameters were decisive in determining diastereoselectivity, then minimal change would be expected in diastereoselectivity after the deprotonation and re-protonation steps.13 Furthermore, performing the reaction in the presence of D2O also delivered a 1:1 diastereomeric mixture of 3a and 3a′ (eqn (5)). This experiment suggests that the introduction of an external proton source could modify the diastereoselectivity of the product. Given the fact that the presence of such a proton source can influence diastereoselectivity, it is likely that intramolecular proton transfer occurs under the optimized reaction conditions, possibly originating from the same side of the thioindolinones.19
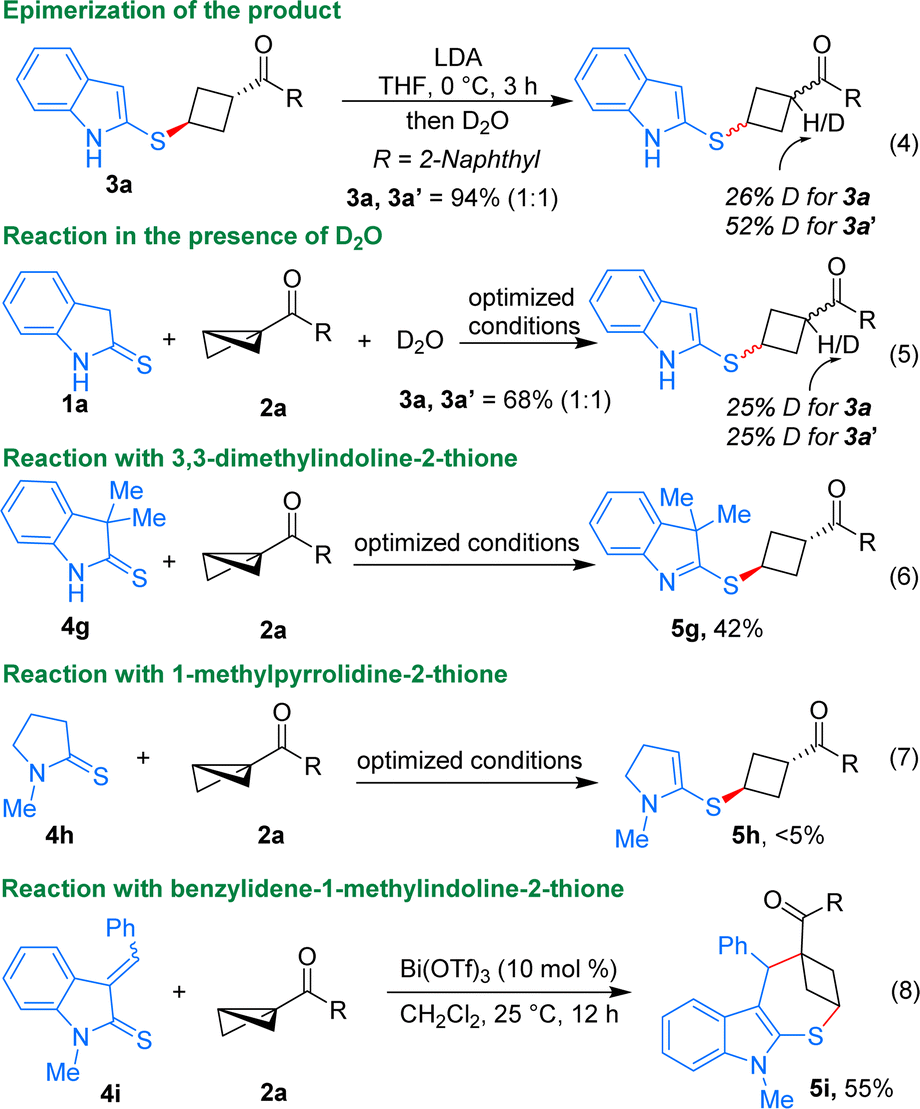 | ||
| Scheme 4 Mechanistic studies. | ||
The reaction involving 3,3-dimethylindoline-2-thione 4g resulted in the formation of product 5g in 42% yield, suggesting that even without the favourable aromatization process, thiones can still undergo the reaction (eqn (6)). Notably, 1-methylpyrrolidine-2-thione did not participate in the reaction to form any products due to the absence of free N–H and favourable aromatization in the process (eqn (7)). Interestingly, the reaction performed using benzylidene-1-methylindoline-2-thione 4i (which stays in a dimeric form via self-Diels–Alder reaction) resulted in the formation of the (4 + 3) annulated product 5i in 55% yield. While the addition of thioamide to BCB could potentially yield the ring-opened product in this case, it is the driving force of aromatization that propels the reaction towards the formation of the annulated product.20
Based on the mechanistic studies and DFT calculations a catalytic cycle for the formal diastereoselective ene reaction of thioindolinones is presented in Scheme 5. The reaction begins with the coordination of BCB 2a with Sc(OTf)3 to form the intermediate A′. Now, the 2-thioindolinone 1a attacks the intermediate A′ at C3 position to generate the intermediate B. From this intermediate several pathways can be followed, but the major pathway goes via the TS(B-C).17 Depending upon the position of triflate, two different diastereomers can be formed. Triflate t1 abstracts the proton from thioindolinone viaTS(B-C) to from intermediate C, while proton abstraction by triflate t2viaTS(B-C′) leads to intermediate C′.17 In the final step, the proton is transferred from triflate to the C1 position of the BCB to give the product. This proton transfer is barrierless and very facile. Since the diastereomeric product formation depends upon the proton abstraction by triflate, the formation of CviaTS(B-C) becomes the diastereo determining step of the reaction. After subsequent steps, proton abstraction by triflate t1 leads to the major product while abstraction by triflate t2 gives the minor product. The TS(B-C) is more stable than TS(B-C′) by 1.6 kcal mol−1. Furthermore, the energy difference between diastereomeric products 3a and 3a′ was found to be only 0.2 kcal mol−1, which suggests that the diastereoselectivity is not governed by thermodynamic stability of the products. The mechanistic study also validates this result where 1:1 mixture of products 3a and 3a′ is found when the reaction is carried out in the presence of LDA and D2O. From the free energy values and mechanistic studies, it is likely that the reaction is kinetically controlled.
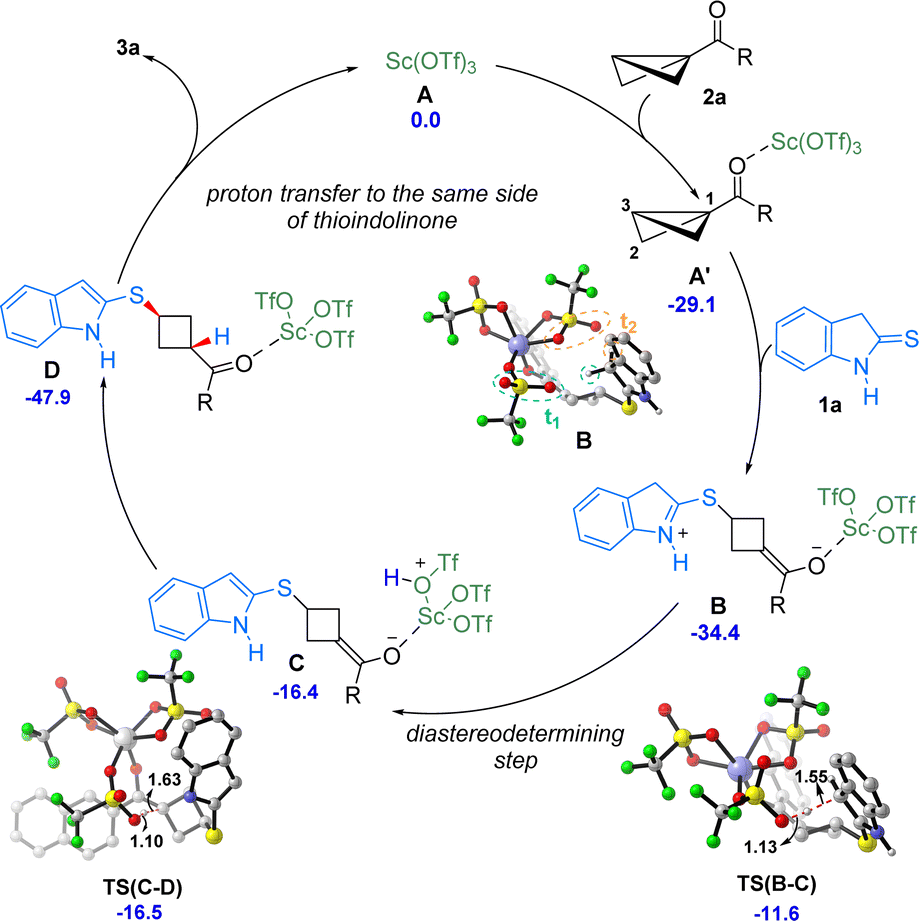 | ||
| Scheme 5 Proposed catalytic cycle. Relative Gibbs free energies (ΔG) are given in kcal mol−1. Distances are given in Å. | ||
The 1,3-substituted cyclobutane derivatives generated from the diastereoselective ene process provide multiple synthetic linchpins for further post synthetic modifications (Scheme 6). Treatment of 3a with N-bromosuccinimide resulted in the formation of 3,3-dibromo substituted product 6 in 81% yield via dearomatization of the indole moiety. The sulfur centre in product 3a was selectively converted to the sulfoxide 7 in the presence of 0.5 equivalent of oxone. Moreover, through the aid of m-CPBA, selective transformation of sulfone 8 was achieved in 97% yield. For sulfone 8, the product structure was confirmed using single-crystal X-ray analysis.21,22 Reduction of the ketone moiety using NaBH4 delivered the secondary alcohol derivative 9 in 98% yield with excellent dr. When 3a was treated in the presence of vinyl magnesium bromide, the anticipated tertiary alcohol 9 was formed in 63% yield and excellent dr. In the presence of thiourea catalyst, 3a underwent addition to para-quinone methide 11via the third position of indole, leading to the formation of highly decorated indole scaffold 12 in 97% yield and excellent dr.23 Notably, efforts to diversify the product under base treatment were unsuccessful as it only compromised the diastereoselectivity of 3a. Interestingly, product 5a could be further functionalized: m-CPBA oxidation yielded compound 13 by oxidizing both the sulfur and nitrogen centers. The addition of vinyl magnesium bromide to 5a produced the tertiary alcohol 14 as a single diastereomer in 86% yield, thereby further enhancing the synthetic utility of this method.
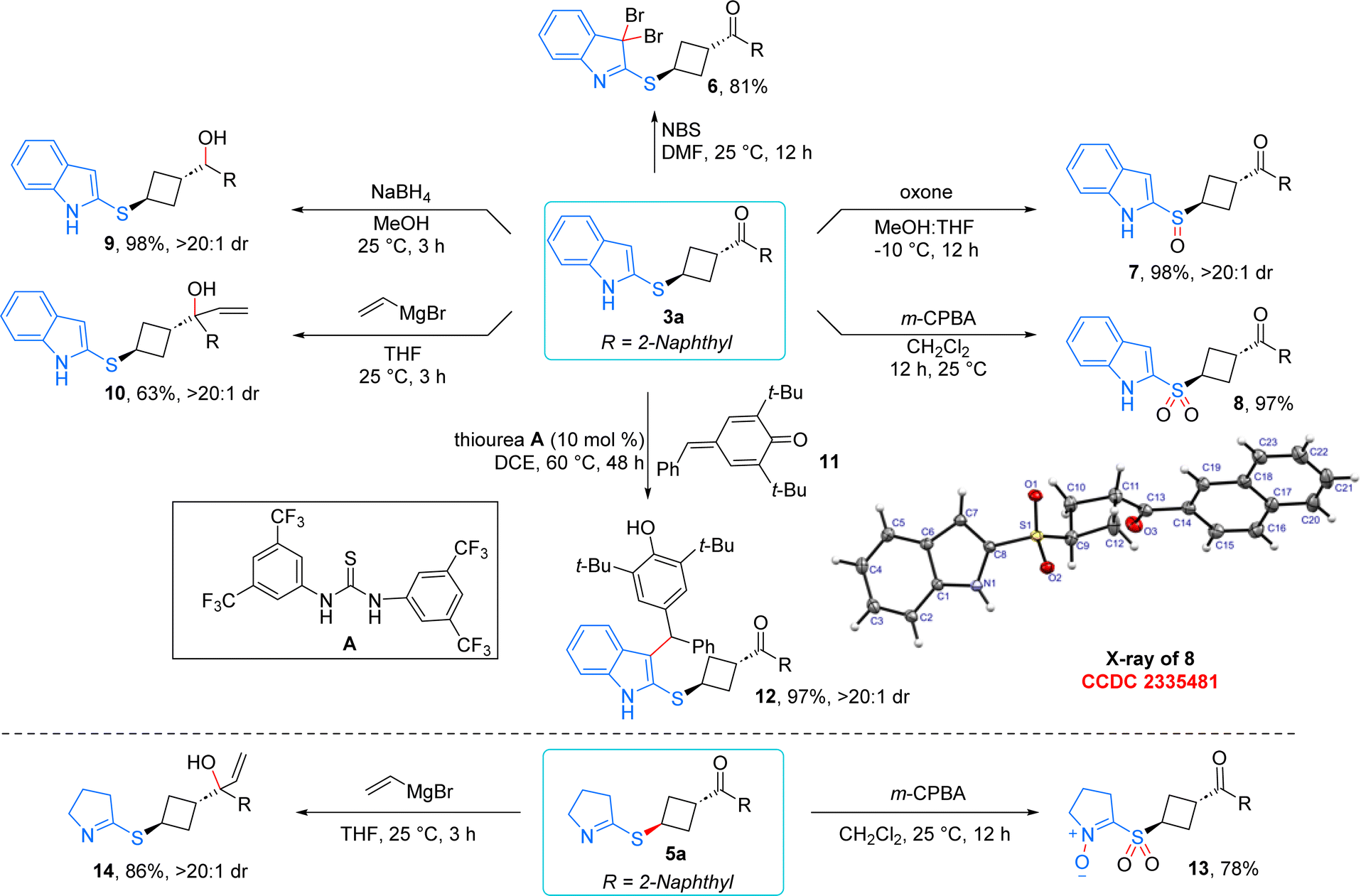 | ||
| Scheme 6 Synthetic utility of the product. | ||
Conclusions
In conclusion, Lewis acid-catalyzed diastereoselective formal ene reaction of thioindolinones/thiolactams employing BCBs as enophiles for the synthesis of 1,3-trans disubstituted cyclobutanes has been realized.24 The diastereoselectivity arises during the proton transfer step, presumably involving proton transfer from the same side of the thioindolinones. The present reaction is operationally simple, advances smoothly under mild conditions, and can be fine-tuned for every portion of both substrates. This reaction is not restricted to thioindolinones as the ene component; other thiones also reacted efficiently to form the functionalized cyclobutanes with excellent diastereoselectivity. Mechanistic experiments and DFT studies were performed to get insight into the possible course of the reaction. Functional group interconversions were carried out to reveal synthetic handles for additional synthetic transformations.Data availability
Details on experimental procedures, mechanistic experiments, characterization data of all the 1,3-disubstituted cyclobutanes and X-ray data of 8. The details are available in the ESI† of the manuscript.Author contributions
A. G. designed the project, and performed the optimization studies, substrate scope analysis and mechanistic studies. S. D. helped in the substrate scope analysis and mechanistic studies. M. S. H. performed the DFT studies. A. G. wrote the first draft of the manuscript, which was edited by A. T. B. All authors have given approval to the final version of the manuscript. A. T. B. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Science and Engineering Research Board (SERB), Government of India (File Number: CRG/2021/001803) is greatly acknowledged. A.G. and S.D. thank MHRD (for PMRF) and M. S. H. thank CSIR (for SRF) for the research fellowship. We thank Prof. Garima Jindal for the help in DFT studies, Sanjeevni Harikumar for preparing some of the substrates, Mr Kishorkumar Sindogi (IISc) for collecting the X-ray data of 8.Notes and references
- (a) K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312 CrossRef; (b) J. Turkowska, J. Durkaab and D. Gryko, Chem. Commun., 2020, 56, 5718 RSC; (c) J. A. Milligan and P. Wipf, Nat. Chem., 2016, 8, 296 CrossRef CAS PubMed; (d) A. Luque, J. Paternoga and T. Opatz, Chem.–Eur. J., 2021, 57, 4500 CrossRef PubMed; (e) A. Fawcett, Pure Appl. Chem., 2020, 92, 751 CrossRef CAS; (f) M. A. A. Walczak, T. Krainz and P. Wipf, Acc. Chem. Res., 2015, 48, 1149 CrossRef CAS PubMed; (g) C. B. Kelly, J. A. Milligan, L. Tilley and T. M. Sodano, Chem. Sci., 2022, 13, 11721 RSC.
- (a) P. Singh, R. K. Varshnaya, R. Dey and P. Banerjee, Adv. Synth. Catal., 2020, 362, 1447 CrossRef CAS; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (c) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385 CrossRef CAS PubMed; (d) A. U. Augustin and D. B. Werz, Acc. Chem. Res., 2021, 54, 1528 CrossRef CAS PubMed; (e) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227 CrossRef CAS PubMed; (f) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC; (g) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (h) A. Kreft, A. Lücht, J. Grunenberg, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2019, 58, 1955 CrossRef CAS PubMed.
- (a) R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477 CrossRef CAS PubMed; (b) Y.-J. Liang, R. Kleinmans, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2022, 144, 20207 CrossRef CAS PubMed; (c) R. Guo, Y. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988 CrossRef CAS PubMed; (d) Y. Zheng, W. Huang, R. K. Dhungana, A. Granados, S. Keess, M. Makvandi and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 23685 CrossRef CAS PubMed; (e) S. Agasti, F. Beltran, E. Pye, N. Kaltsoyannis, G. E. M. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 535 CrossRef CAS PubMed; (f) D. Ni, S. Hu, X. Tan, Y. Yu, Z. Li and L. Deng, Angew. Chem., Int. Ed., 2023, 62, e202308606 CrossRef CAS PubMed; (g) N. Radhoff, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2023, e202304771 CAS; (h) Y. Liang, F. Paulus, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2023, 62, e20230504 Search PubMed; (i) J. Zhang, J.-Y. Su, H. Zheng, H. Li and W.-P. Deng, Angew. Chem., Int. Ed., 2024, e202318476 CAS; (j) K. Dhake, K. J. Woelk, J. Becia, A. Un, S. E. Jenny and D. C. Leitch, Angew. Chem., Int. Ed., 2022, 61, e202204719 CrossRef CAS PubMed.
- (a) H. Wang, H. Shao, A. Das, S. Dutta, H. T. Chan, C. Daniliuc, K. N. Houk and F. Glorius, Science, 2023, 381, 75 CrossRef CAS PubMed; (b) S. Dutta, D. Lee, K. Ozols, C. G. Daniliuc, R. Shintani and F. Glorius, J. Am. Chem. Soc., 2024, 146, 2789 CrossRef CAS PubMed.
- (a) R. E. McNamee, A. L. Thompson and E. A. Anderson, J. Am. Chem. Soc., 2021, 143, 21246 CrossRef CAS PubMed; (b) R. E. McNamee, M. M. Haugland, J. Nugent, R. Chan, K. E. Christensen and E. A. Anderson, Chem. Sci., 2021, 12, 7480 RSC.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC; (c) M. Wrobleski, G. Reichard, S. Paliwal, S. Shah, H. Tsui, R. Duffy, J. Lachowicz, C. Morgan, G. Varty and N. Shih, Bioorg. Med. Chem. Lett., 2006, 16, 3859 CrossRef CAS PubMed; (d) K. C. Nicolaou, D. Vourloumis, S. Totokotsopoulos, A. Papakyriakou, H. Karsunky, H. Fernando, J. Gavrilyuk, D. Webb and A. F. Stepan, ChemMedChem, 2016, 11, 31 CrossRef CAS PubMed.
- (a) M. L. Conner and M. K. Brown, J. Org. Chem., 2016, 81, 8050 CrossRef CAS PubMed; (b) K. Majima and M. Yamano, J. Org. Chem., 2021, 86, 11464 CrossRef CAS PubMed.
- (a) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485 CrossRef CAS PubMed; (b) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (c) S. Poplata, A. Troster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed.
- (a) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241 CrossRef CAS PubMed; (b) J. M. Lopchuk, K. Fjelbye, Y. Kawamata, L. R. Malins, C.-M. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209 CrossRef CAS PubMed.
- (a) C. J. Pratt, R. A. Aycock, M. D. King and N. T. Jui, Synlett, 2020, 31, 51 CrossRef CAS PubMed; (b) G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J. Cintrat, Angew. Chem., Int. Ed., 2020, 59, 2618 CrossRef CAS PubMed; (c) M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355 CrossRef CAS PubMed; (d) X. Yu, M. Lübbesmeyer and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 675 CrossRef CAS PubMed; (e) K. Das, A. Pedada, T. Singha and D. P. Hari, Chem. Sci., 2024, 15, 3182 RSC.
- (a) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766 CrossRef CAS PubMed; (b) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117 CrossRef CAS PubMed; (c) M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511 CrossRef CAS PubMed.
- H. Wang, J. E. Erchinger, M. Lenz, S. Dutta, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 23771 CrossRef CAS PubMed.
- A. Guin, S. Bhattacharjee, M. S. Harariya and A. T. Biju, Chem. Sci., 2023, 14, 6585 RSC.
- A. Dasgupta, S. Bhattacharjee, Z. Tong, A. Guin, R. E. McNamee, K. E. Christensen, A. T. Biju and E. A. Anderson, J. Am. Chem. Soc., 2024, 146, 1196 CrossRef CAS PubMed.
- (a) P. Tang, Y.-Y. Wei, H.-J. Ma, Y. Yang and Y. Jiang, J. Org. Chem., 2022, 87, 10890 CrossRef CAS PubMed; (b) X. Chen, Z.-H. Qi, S.-Y. Zhang, L.-P. Kong, Y. Wang and X.-W. Wang, Org. Lett., 2015, 17, 42 CrossRef CAS PubMed.
- (a) B. D. Schwartz, M. Y. Zhang, R. H. Attard, M. G. Gardiner and L. R. Malins, Chem.–Eur. J., 2020, 26, 2808 CrossRef CAS PubMed; (b) K. Tokunaga, M. Sato, K. Kuwata, C. Miura, H. Fuchida, N. Matsunaga, S. Koyanagi, S. Ohdo, N. Shindo and A. Ojida, J. Am. Chem. Soc., 2020, 142, 18522 CrossRef CAS PubMed; (c) A. Kaur, W. Lin, V. Dovhalyuk, L. Driutti, M. L. D. Martino, M. Vujasinovic, J.-M. Löhr, M. E. Sellin and D. Globisch, Chem. Sci., 2023, 14, 5291 RSC.
- See the ESI† for details..
- Disappointingly, amide BCBs did not work under the present reaction conditions..
- Notably, under optimized conditions, the reaction doesn't yield 1H-indole-2-thiol in the absence of BCB 2a, thereby eliminating the possibility of 1H-indole-2-thiols forming first and then adding to BCBs..
- For a related reaction of BCBs with thioindolinones that appeared while the manuscript was under review, see: J.-J. Wang, L. Tang, Y. Xiao, W.-B. Wu, G. Wang and J.-J. Feng, Angew. Chem., Int. Ed., 2024, 63, e202405222 Search PubMed.
- CCDC 2335481† (8) contains the supplementary crystallographic data for this paper..
- The relative stereochemistry of all products 3 and 5 was determined based on the stereochemistry observed in the crystal structure of derivative 8..
- The relative configuration of the newly generated stereocentre in 7, 9, 10, 12 and 14 was not determined..
- A. Guin, S. Deswal and A. T. Biju, ChemRxiv, preprint, 2024, DOI:10.26434/chemrxiv-2024-gf80l.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental procedures, characterization data of all compounds. CCDC 2335481. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02194k |
| This journal is © The Royal Society of Chemistry 2024 |