
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronically and geometrically complementary perylenediimides for kinetically controlled supramolecular copolymers†
Alfonso J.
Schwalb
,
Fátima
García
and
Luis
Sánchez
 *
*
Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: lusamar@ucm.es
First published on 30th April 2024
Abstract
The synthesis of 3,4,9,10-benzo[d,e]isoquinolino[1,8-g,h]quinoline-tetracarboxylic diimide (BQQDI) 1 endowed with peripheral trialkoxybenzamide fragments is reported and its self-assembling features investigated. The peripheral benzamide moieties generate metastable monomeric species that afford a kinetically controlled supramolecular polymerization. The electron-withdrawing character of 1 in comparison with previously reported PDIs 2, together with the similar geometry, makes this dye an optimal candidate to perform seeded supramolecular copolymerization yielding four different supramolecular block copolymers. Whilst heteropolymers poly-1-co-2a, poly-2a-co-1 and poly-1-co-2b present an H-type arrangement of the monomeric units, heteropolymer poly-2b-co-1, prepared by seeding the chiral, metastable monomers of 2b with achiral seeds of 1, produces chiral, J-type aggregates. Interestingly, the monosignated CD signal of pristine poly-2b changes to a bisignated CD signal most probably due to the formation of columnar domains around the seeds of 1 which implies the blocky nature of the supramolecular copolymers formed.
Introduction
Natural coassembly, a process by which different elements come together spontaneously to form a complex structure, is ubiquitous in life sciences. Cell membranes, in which lipids, proteins and some other molecules form a functional bilayer structure, are remarkable examples of biological coassembly.1 Regarding chemical coassembled systems, block copolymers are exciting examples, which are constituted by covalently linked immiscible homopolymer blocks that give rise to well defined nanostructures, holding applicability in different areas like drug delivery, coatings, adhesives, and as templates for the synthesis of nanomaterials. In these block copolymers, as in the case of conventional polymers, the junction between the monomeric units are covalent bonds.2 In the last decades, a number of supramolecular polymers, i.e., macromolecules in which the monomeric units are held together by non-covalent interactions,3 has arisen as functional materials finding out applicability as biomaterials,4 elastomers,5 photocatalysts,6 adhesives,7 self-healing materials,8 or as active layer in optoelectronic devices.9 The development of supramolecular polymers relies, among some other factors, on the reported mass-balance model that allows deriving a complete set of thermodynamic parameters associated to the supramolecular polymerization process.10Despite a large number of supramolecular polymers are thermodynamically controlled and governed by an isodesmic or a cooperative (or nucleation–elongation) mechamism,3 the last years have witnessed the growth of kinetically controlled supramolecular polymerizations in which the self-assembly is retarded. In these kinetically controlled examples, competitive or consecutive processes yielding different supramolecular aggregated species can take place.3b,11 Furthermore, in good analogy with living crystallization driven self-assembly reported for covalent block copolymers,12 kinetically controlled processes have been utilized to perform living (LSP) and seeded (SSP) supramolecular polymerizations that decreases the polydispersity in the supramolecular polymers.13
Both thermodynamically and kinetically controlled supramolecular polymerizations have been harnessed to prepare supramolecular block copolymers (SBCPs) in which two or more different monomeric species non-covalently bonded yield new adaptive and functional materials.14 A key issue in the further development of block supramolecular polymers is controlling the microstructure (self-sorted, block, blocky, periodic, statistical or alternated) of the final coassembled species. In thermodynamically controlled supramolecular copolymers, it is difficult to control such microstructure but the recently reported mass-balance model reported by ten Eikelder and coworkers15 has allowed to unravel the microstructure of thermodynamically controlled heteropolymers once they are formed.16 On the other hand, kinetically controlled supramolecular copolymerizations have been reported to yield alternating supramolecular copolymers. The sequential addition of metastable monomers, able to undergo nucleation–elongation growth, is crucial to produce seeded supramolecular copolymerizations affording such alternating coassemblies. An elegant example of kinetically controlled SBCPs is that reported by Sugiyasu and co-workers based on Zn and Cu porphyrins in which while the Zn-core homopolymer is unstable, the heteropolymer constituted by fragments of both the Zn and the Cu-core porphyrins presents an enhanced stability.17 Another example of kinetically controlled SBCPs is the stereoselective coassembly of two napthalendiimides with the same point chirality at the binaphtyl-based linker and with electronically complementary character.18 Of special interest due to their intriguing characteristics and applications,19 is the kinetically controlled coassembly of electronically complementary perylenediimides (PDIs) described by F. Würthner and coworkers to form (AnBB)m alternating species.20 Kinetically controlled coassembly allows for the deliberate design and control of the assembly process, influencing the structures and properties of the resulting supramolecular polymers and, hence, new insights into the requirements to achieve SBCPs with controlled microstructure are necessary.
Herein, we envision that 3,4,9,10-benzo[d,e]isoquinolino[1,8-g,h]quinolinetetracarboxylic diimide (BQQDI)21 could be an excellent complementary scaffold to perform a seeded supramolecular copolymerization with previously reported PDIs 2 due to its enhanced electron-withdrawing character. Thus, we have synthesized the BQQDI 1 endowed with peripheral trialkoxybenzamide fragments (Fig. 1a). The π-surface of the BQQDI 1 together with the benzamide units allows its efficient supramolecular polymerization, governed by a cooperative mechanism, to afford H-type aggregates. At the same time, this peripheral benzamide groups provoke the formation of metastable monomeric species, M*, that retards the self-assembly of 1 to form the homopolymer poly-1. The autocatalytic time-dependent growth of 1 homopolymers can be accelerated by adding seeds. As stated before, the electron-withdrawing features of 1 together with its planar geometry makes this BQQDI an excellent scaffold to prepare kinetically controlled SBCPs with previously reported PDIs 2a and 2b, due to their geometric and electronic complementarity (Fig. 1a).13c,22 Whilst achiral 2a has been reported to form kinetically controlled H-type aggregates (poly-2a),13c chiral 2b yields J-type aggregated species (poly-2b) upon self-assembly (Fig. 1b).22a,22b Harnessing the dissimilar self-assembling features of the PDIs 2 and by using a SSP strategy, we have been able to prepare poly-1-co-2a, poly-1-co-2b, poly-2a-co-1 and poly-2b-co-1 supramolecular block copolymers of tailored H- or J-type character, thus shedding new insights to achieve electroactive heteropolymers (Fig. 1c).
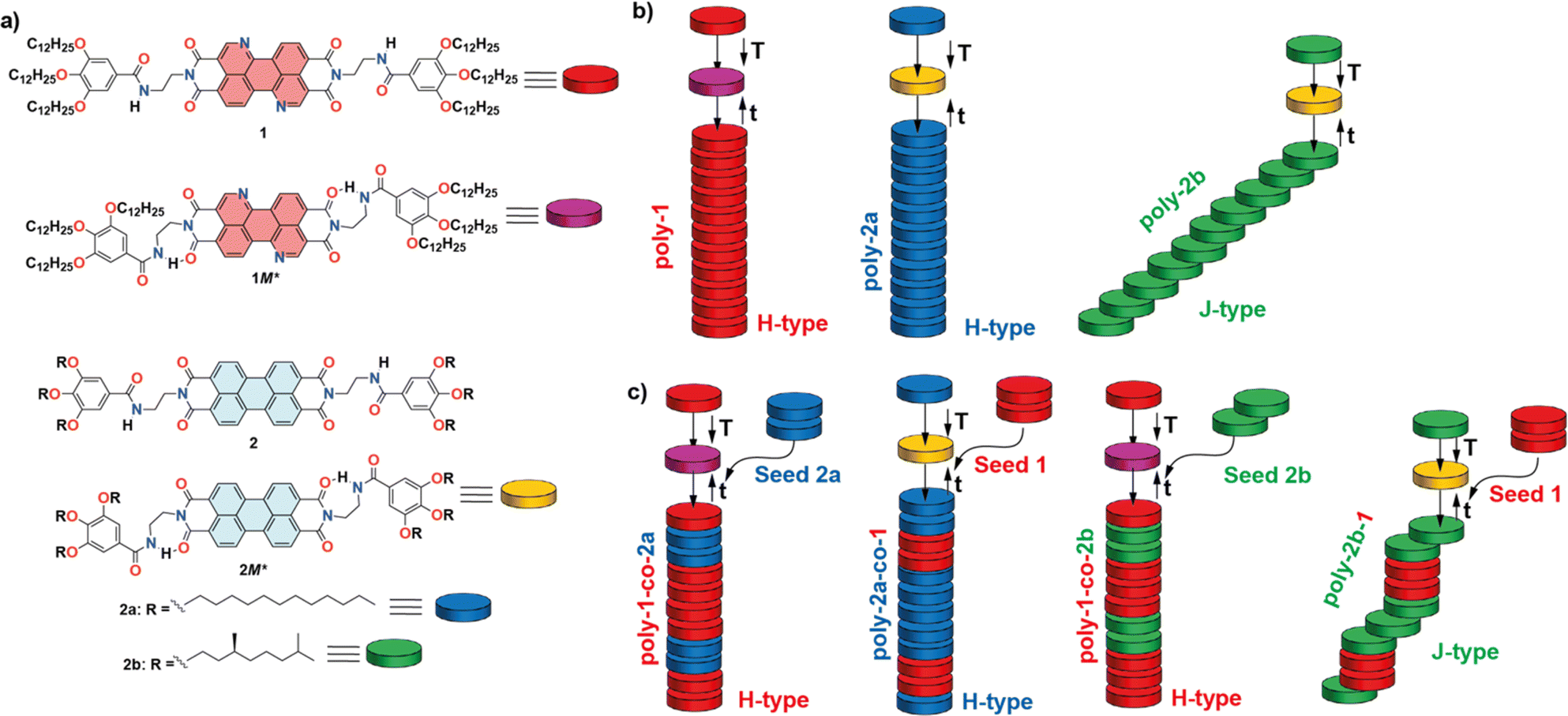 | ||
| Fig. 1 (a) Chemical structure of BQQDI 1 and the electronically and geometrically complementary PDIs 2 depicting the chemical structure of the intramolecularly H-bonded pseudocycles (M*); (b) schematic representation of the formation of kinetically controlled homopolymers poly-1, poly-2a and poly-2b through the metastable monomeric species M*; (c) schematic illustration of the seeded supramolecular polymerization yielding heteropolymers poly-1-co-2a, poly-2a-co-1, poly-1-2b and poly-2b-co-1. | ||
Results and discussion
Synthesis and homopolymerization of BQQDI 1
The synthesis of BQQDI 1 has been accomplished by following a convergent strategy in which dianhydride 9 was prepared by following the multistep protocol reported by Okamoto et al. in 2020.21a The synthesis starts with two aromatic nucleophilic substitutions in 1,5-dinitroanthraquinone followed by condensation in acidic conditions that yields diester 4 with nitrogen doped perylene scaffold present in target BQQDI 1. Treatment of 4 with N-phenyl-bis(trifluoromethanesulfonimide), followed by reduction with formic acid, bromination with NBS, Pd(0) coupling and treatment with p-TsOH at high temperatures yields dianhydride 9 (see Scheme S1 in ESI†).21a On the other hand, the peripheral benzamide moiety was prepared by following previously reported protocols.22 Finally, a two-fold condensation reaction under microwave irradiation between dianhydride 9 and benzamide 10 in the presence of propionic acid and o-dichlorobenzene as solvent yields BQQDI 1 in 21% yield (Scheme S1†). The chemical structure of the BQQDI 1 has been elucidated by the standard techniques (see ESI†).The self-assembling features of 1 in solution have been investigated by different spectroscopic techniques. To our surprise, 1 is sparingly soluble in most common organic solvents at relatively high concentrations. In fact, BQQDI 1 in CDCl3 at total concentration (cT) above 10 mM precipitates which impedes performing concentration dependent 1H NMR experiments that could contribute to clarify the non-covalent forces involved in the supramolecular polymerization. However, heating up a solution of 1 in CDCl3 at cT = 4 mM shows that, whilst the aromatic protons shield moderately upon decreasing the temperature, the opposite effect is observed for the amide protons. This is indicative of the π-stacking of the aromatic backbones and the formation of intermolecular H-bonding interactions between the amide groups, respectively (Fig. S1†). FTIR in solution has been utilized to demonstrate the formation of intermolecular H-bonds between the amide functional groups in non-polar solvents like methylcyclohexane (MCH). The stretching NH and amide I bands of 1 in MCH appears at 3260 and 1636 cm−1, respectively (Fig. 2a). These values are ascribable to the formation of intermolecular H-bonded amides.23 In CHCl3, a good solvent that favors the complete solvation of the monomeric species, two stretching NH bands centered at 3454 and 3414 cm−1 are observed, ascribable to free NH and intramolecular NH H-bonds, due to the presence of completely free monomeric species and seven-membered H-bonded pseudocycles formed by the intramolecular interaction between the NH of the amide groups and one of the carbonyls of the imide moiety (Fig. 1a and 2a).23 The formation of these pseudocycles has been corroborated by registering 1H NMR spectra at cT = 1 mM and at different temperatures. In these experiments, unlike in the previous studies carried out a 4 mM, none of the resonances corresponding to the aromatic protons experience any shift upon modifying the temperature. However, the triplet corresponding to the NH proton shifts upfield upon increasing the temperature, diagnostic of the rupture of the intramolecular H-bonding interactions (Fig. 2b). The formation of such metastable monomeric species has been reported to provoke kinetically controlled supramolecular polymerizations.23 In fact, variable temperature (VT) UV-Vis studies confirm that these dormant species retard the formation of the homopolymers poly-1.
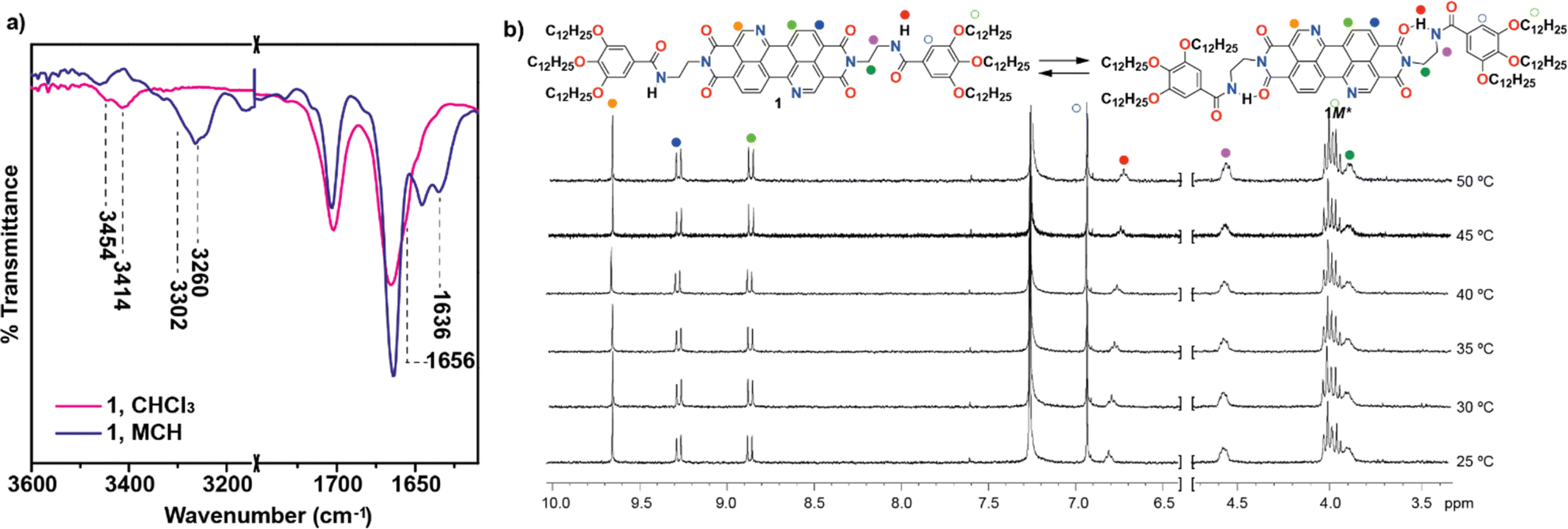 | ||
| Fig. 2 (a) Partial FTIR of 1 in different solvents (cT = 1 mM) showing the stretching NH and amide I bands; (b) partial 1H NMR spectra of 1 recorded at different temperatures showing the aromatic and some of the aliphatic protons (cT = 1 mM; CDCl3; 300 MHz). The upper part of panel (b) shows the equilibrium between the free and the metastable M* monomeric species. | ||
In an attempt to derive the thermodynamic parameters associated to the supramolecular polymerization of 1, we first utilized diluted solutions of 1 in MCH. In pristine MCH, the UV-Vis spectrum of BQQDI 1 at 20 °C shows maxima centered at λ = 469, 503 and 536 nm. These absorption bands are less intense than those observed in CHCl3 with maxima centered at λ = 454, 485 and 520 nm, ascribable to the characteristic A0–1/A0–0 transitions of pyrene-based scaffolds (Fig. S2†).19 These changes observed in the UV-Vis spectra implies the H-type nature of the supramolecular polymers formed by 1 in which the monomeric units are arranged in a face-to-face fashion. The UV-Vis spectrum of 1 in MCH upon heating up the solution at 90 °C does not match with that registered in CHCl3 and ascribable to the monomeric species. Therefore, only a partial disassembly of the H-type aggregates formed at low temperatures is achieved by heating up this MCH solution (Fig. S2b†). To decrease the stability of the supramolecular polymers formed by 1, we have utilized toluene (Tol) as solvent. In this case, the UV-Vis solution in Tol at 20 °C displays an identical absorption pattern to that registered in MCH at 20 °C diagnostic of the formation of the H-type aggregates (Fig. 3a). Heating up the Tol solution of 1 to 90 °C provokes a complete disassembly of these aggregated species, as demonstrate the UV-Vis absorption pattern coincident with that registered in CHCl3 (Fig. 3a and S2b†). Plotting the variation of the absorbance at λ = 520 nm against temperature upon cooling at 1 °C min−1 yields a non-sigmoidal curve that could be diagnostic of a cooperative mechanism. However, a global fitting of these curves by applying the one-component equilibrium model yields inaccurate results, most probably due to the operation of a kinetic process provoked by the formation of the metastable monomeric species 1M* (Fig. 1a, b and S3†). In fact, the stability of the supramolecular polymers formed by 1 in Tol strongly depends on the cooling rate utilized to form the H-type aggregates from the monomeric species (Fig. 3b). Thus, the temperature of elongation, Te, temperature at which the nucleation regime changes to the elongation one, decreases upon increasing the cooling rate (Fig. 3b). This effect is cancelled when the disassembly process is taken into account, i.e. heating a solution of the supramolecular polymers of 1 to provoke the depolymerization of poly-1 to form the monomeric species does not show a noticeable difference in the 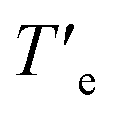 values (Fig. 3b). Furthermore, a clear hysteresis between the cooling and heating curves is observed diagnostic of a kinetically controlled supramolecular polymerization.13b,24 Noteworthy, during the heating and cooling processes, the absorption patterns of the involved species are identical which implies that in the kinetically controlled supramolecular polymerization of 1, the formation of more than one aggregated species is not detected (Fig. S4†).
values (Fig. 3b). Furthermore, a clear hysteresis between the cooling and heating curves is observed diagnostic of a kinetically controlled supramolecular polymerization.13b,24 Noteworthy, during the heating and cooling processes, the absorption patterns of the involved species are identical which implies that in the kinetically controlled supramolecular polymerization of 1, the formation of more than one aggregated species is not detected (Fig. S4†).
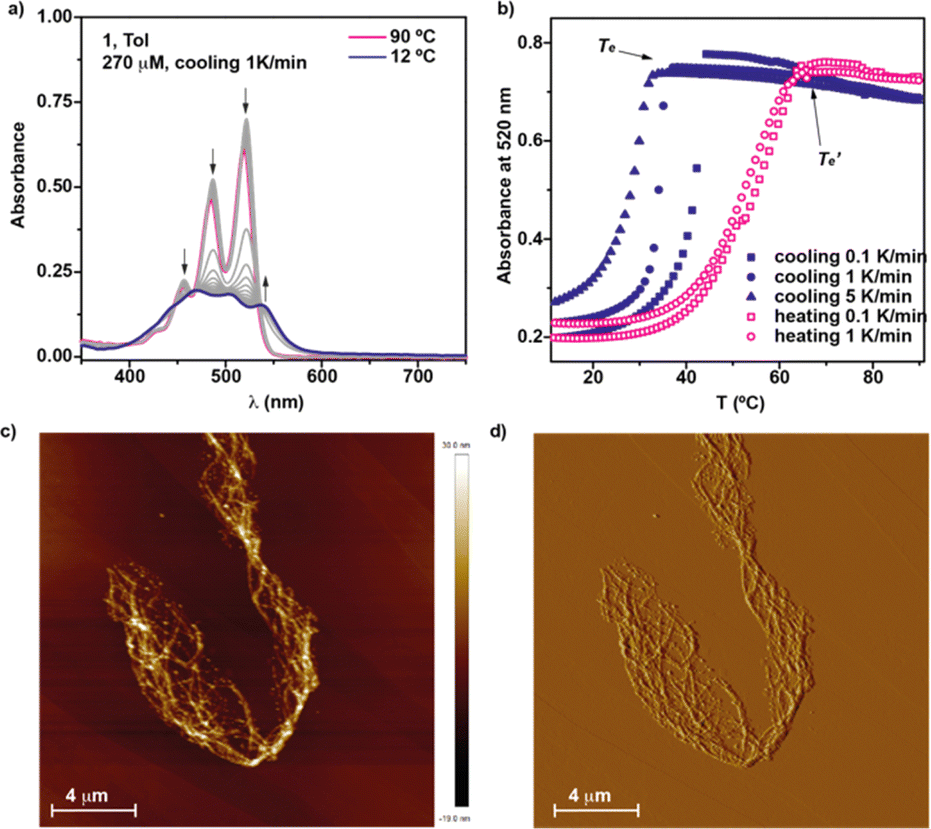 | ||
| Fig. 3 (a) UV-Vis spectra of 1 in Tol at different temperatures (cT = 10 M); (b) plot of the variation of the absorbance at 520 nm versus temperature at different cooling or heating rates; arrows in panel (a) indicate the spectral changes upon decreasing the temperature; height (c) and phase (d) AFM images of the fibrillar aggregates formed by 1 in MCH (z scale = 30 nm; cT = 10 M; HOPG as surface). | ||
The stability of the supramolecular polymers formed by 1 has been quantified by performing a solvent denaturation (SD) experiment in which CHCl3 and MCH are used as good and bad solvents to favor the disassembly or the aggregation, respectively. In these experiments, only monomeric and aggregated species are involved and, hence, it is possible to extract the thermodynamic parameters associated to the supramolecular polymerization of 1 by applying the SD model reported by E. W. Meijer and coworkers.25 As expected, the addition of increasing amounts of CHCl3 to the MCH solution provokes the gradual disassembly of the aggregates species to yield the molecularly dissolved monomers (Fig. S5a†). Plotting the variation of the degree of aggregation (α) versus the molar fraction of the good solvent results in a quasi–sigmoidal curve that can be fitted to the SD model. This fit shed values for the Gibbs free energy of −41.9 kJ mol−1, an m parameter, that expresses the influence of the good solvent in the disassembly process, of 27.5, and a degree of cooperativity, σ, of 0.07 (Fig. S5b†). The strong trend of BQQDI 1 to form supramolecular polymers has been visualized by atomic force microscopy (AFM) imaging. Spin-coating a diluted solution of 1 (cT = 10 μM) onto highly oriented pyrolytic graphite (HOPG) shows a dense network of intertwined fibrillar structures (Fig. 3c and d).
The presence of nitrogen atoms in BQQDI 1 produces a remarkable decreasing of the LUMO level of this core in comparison to PDIs.21 On the other hand, the molecular geometry of BQQDI 1 is similar to those of PDIs 2.22 The electronic and geometric features of BQQDIs make them excellent candidates to carry out copolymerization studies with PDIs 2. Noteworthy, the aggregation mode of both PDIs 2a and 2b has been reported to be dissimilar. Whilst achiral 2a self-assembles giving rise to H-type aggregates (Fig. 1b);22 chiral 2b affords dark-green, J-type supramolecular aggregates (Fig. 1b).22 Furthermore, the supramolecular polymerization of 2a has been reported to be kinetically controlled and can be accelerated by SSP.20b However, this is not the case of chiral 2b that, as far as we are aware, it is not reported whether or not the J-type aggregates of 2b are kinetically controlled and can be accelerated by SSP. Therefore, and prior to investigate the formation of the supramolecular co-polymers, we have demonstrated that chiral 2b also follows a kinetically controlled supramolecular polymerization. Unfortunately, this PDI is scarcely soluble in Tol and we have used a mixture of MCH and 1,2-dichloroethane (DCE) in a 7/3 ratio to perform the corresponding studies for the self-assembly of 2b. In this mixture, it is possible to achieve the absorption patterns previously reported for both the aggregated (20 °C) and the monomeric species (80 °C) of 2b as well as a circular dichroism (CD) spectrum identical to that previously reported (Fig. S6†).22 The cooling and heating curves of a 10 μM solution of 2b in MCH/DCE 7/3 mixture shows a hysteresis with values of Te and 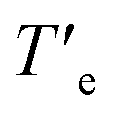 of 38 and 64 °C, respectively, that implies the trapping of the metastable M* species between these temperatures (Fig. S7a†). These metastable M* species evolve to the thermodynamically J-type aggregates at 39 °C upon 90 min without any lag phase (Fig. 4a). To accelerate the conversion of the M* species of 2b into the supramolecular polymers we have added 3% of seeds of the J-type aggregates, prepared by sonicating a 10 μM solution of 2b in MCH/DCE 7/3 for 5 min. The addition of the seeds allows completing the conversion of the M* monomers into the J-type aggregates in 20 min (Fig. 4a).
of 38 and 64 °C, respectively, that implies the trapping of the metastable M* species between these temperatures (Fig. S7a†). These metastable M* species evolve to the thermodynamically J-type aggregates at 39 °C upon 90 min without any lag phase (Fig. 4a). To accelerate the conversion of the M* species of 2b into the supramolecular polymers we have added 3% of seeds of the J-type aggregates, prepared by sonicating a 10 μM solution of 2b in MCH/DCE 7/3 for 5 min. The addition of the seeds allows completing the conversion of the M* monomers into the J-type aggregates in 20 min (Fig. 4a).
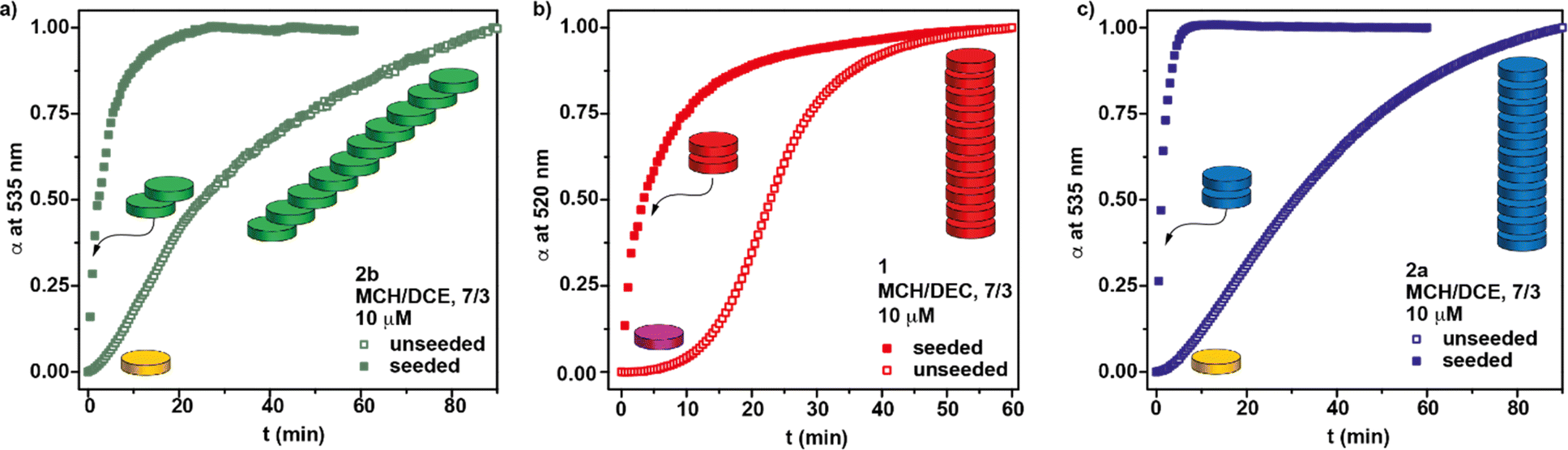 | ||
| Fig. 4 Kinetic profiles of aggregate formation without and with seeds of poly-2b at 39 °C (a), poly-1 at 30 °C (b); and poly-2a at 48 °C (c). The yellow and purple disk represents the 2M* and 1M* species; the green, red and blue disks represent the seeds and the homopolymers formed by 2b, 1, and 2a, respectively. | ||
Similar studies have been carried out for BQQDI 1 and PDI 2a in the same MCH/DEC 7/3 mixture. Thus, a clear hysteresis is observed for the cooling and heating curves of both dyes in this solvent mixture, thus confirming the formation of the corresponding 1M* and 2aM* species, respectively (Fig. S7b and c†). Interestingly, the kinetic evolution of the 1M* monomers into the corresponding H-type aggregates presents a clear lag phase ascribable to an autocatalytic process in which the full conversion into the thermodynamically controlled poly-1 is completed upon 60 min at 30 °C (Fig. 4b). This kinetic change can be accelerated by adding seeds of poly-1, the completion achieved upon 30 min (Fig. 4b). The kinetic profile of 2aM* species is very similar to those observed for 2b and the metastable monomeric species are completely transformed into the H-type poly-2a species upon 90 min at 48 °C (Fig. 4c). This conversion can be accelerated by adding a 3% of seeds of these H-type aggregates and the full conversion of the 2aM* species is completed in less than 10 min (Fig. 4c).
Seeded supramolecular polymerization and co-assembly of BQQDI 1 with PDIs 2
Upon investigating the seeded homopolymerization of all the three studied dyes 1 and 2 in the MCH/DCE 7/3 solution, we have performed a heteropolymerization study by combining the three scaffolds. Thus, we have investigated the formation of four different supramolecular block copolymers: poly-1-co-2a, poly-2a-co-1, poly-1-2b and poly-2b-co-1. The schematic experimental protocol followed to prepare the kinetically controlled co-polymers is depicted in Fig. 1c.First, we have studied the two-component seeded co-polymerization of the BQQDI 1 and the PDI 2a, that yields H-type aggregates by the corresponding homopolymerization. To achieve co-polymer poly-1-co-2a, a 10 μM solution of 1 in MCH/DCE 7/3 is heated up to 80 °C to produce the complete disassembly of poly-1 and rapidly cooled it down to 30 °C to generate kinetically trapped 1M* species. These metastable monomers present the same absorption pattern to that observed for 1 at 80 °C, thus confirming the monomeric nature of these species (Fig. S8a†). After the addition of the seeds of PDI 2a in a 3% ratio, the depletion of the absorption band at λ = 520 nm, ascribable to the monomeric species, was monitored. The addition of the seeds provokes the rapid transformation of the 1M* species into the supramolecular copolymer poly-1-co-2a that is completed upon 15 min (Fig. 5a).
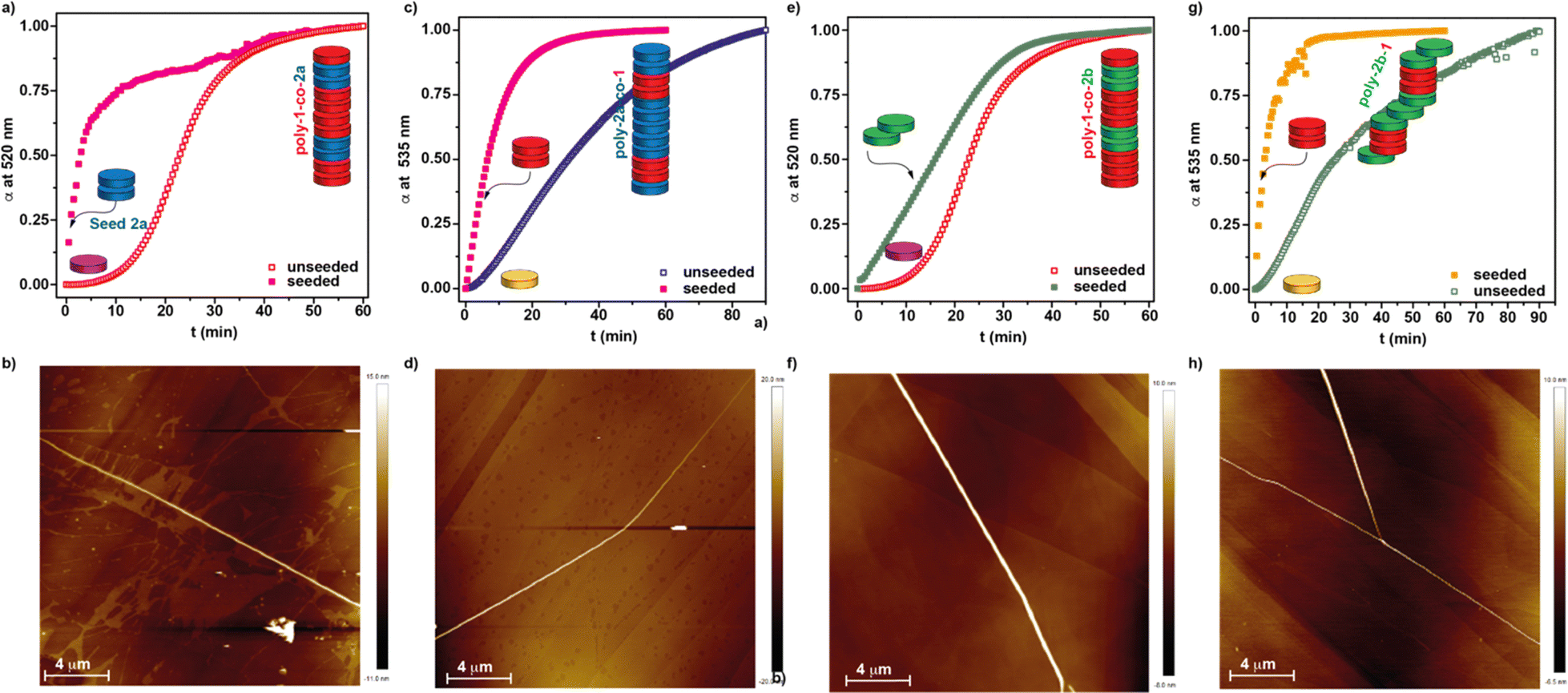 | ||
| Fig. 5 Kinetic profiles (a, c, e and g) and AFM images (b, d, f and h) of kinetically controlled SSP to attain the heteropolymers poly-1-co-2a (a), poly-2a-co-1 (c), poly-1-co-2b (e) and poly-2b-co-1 (g). Panels (b, d, f and h) display the AFM images of the heteropolymers poly-1-co-2a (b), poly-2a-co-1 (d), poly-1-co-2b (f) and poly-2b-co-1 (h), respectively. The unseeded experiments in panels (a)–(d) corresponds to the polymerization of pristine homopolymers poly-1 (red squares), poly-2a (blue squares) and poly-2b (green squares). | ||
The UV-Vis spectrum of the final poly-1-co-2a presents a similar pattern to that registered for pristine poly-1. Taking into account the stability of the fragments of poly-2a, acting as seeds, we can assume that the microstructure of the heteropolymer poly-1-co-2a would be a blocky arrangement of units with short segments of poly-2a, stemming from the seeds, and longer fragments of poly-1, corresponding to the elongated monomeric units of 1 growing from both sides of the seeds (Fig. 1c). The kinetics of both the SSP yielding the homopolymer poly-1 and the heteropolymer poly-1-co-2a indicate that the rate of the conversion is not affected by the nature of the seed (Fig. 4b and 5a). The formation of the supramolecular block copolymers poly-1-co-2a exerts a strong influence on the morphology of the aggregates. Unlike the strongly intertwined fibrillar structures visualized for pristine poly-1 (Fig. 3c and d), the AFM images of the heteropolymer poly-1-co-2a shows isolated, rod-like fibres of several micrometres in length (Fig. 5b, S9a and b†). The isolated fibres of heteropolymer poly-1-co-2a present uniform heights of ∼12 nm all along the fibre due to the similar molecular dimensions of both BQQDI 1 and PDI 2a (Fig. S9c†). This uniform height contrasts with the previous heteropolymers formed by planar PDI 2a and contorted bay-substituted PDIs reported by Würthner and coworkers, in which the height of the fibre depends on the constitutive block.20b After attaining poly-1-co-2a, we have also carried out the preparation of poly-2a-co-1 by making use of the formation of the 2M* species and by following a similar strategy to that utilized in the previous heteropolymer. Therefore, we have heated up a solution of 2a to 80 °C and cooled it down rapidly to 48 °C to generate the corresponding 2M* species. After reaching this temperature, we added the seeds of 1 prepared by sonication for 5 min of a 10 μM solution of this BQQDI in a 3% ratio. The heteropolymerization yielding the H-type SBCP poly-2a-co-1 was completed in ∼30 min (Fig. 5b and S8b†). Interestingly, the SSP affording poly-2a-co-1 is slower than the corresponding SSP homopolymerization yielding poly-2a which implies that the kinetics of the conversion is affected by the nature of the seed (Fig. 4c and 5c). As in the previous copolymer, the AFM images of poly-2a-co-1 also shows the formation of long and isolated, rod-like fibres with uniform heights of ∼15 nm (Fig. 5d and S9d–f†).
The previous copolymers poly-1-co-2a and poly-2a-co-1 are constituted by monomeric units affording H-type aggregates upon self-assembly. In the last part of this study, we have investigated the formation of SBCPs by combining BQQDI 1 and PDI 2b that self-assemble into H- and J-type aggregates, respectively. Heteropolymer poly-1-co-2b has been obtained from the metastable monomeric units 1M*, generated upon heating up a 10 μM solution of 1 in MCH/DCE 7/3 at 80 °C and rapidly cooled it down to 30 °C, and adding fragments of poly-2b acting as the seeds for the coassembly (Fig. 1c and 5e). The addition of the seeds of 2b to 1M* eliminates the lag phase observed in the kinetics of pristine 1 but slightly shortens the time at which the total conversion is completed, thus indicating the low ability of 2b to seed (Fig. 5e). Similarly to the above studied SBCPs, the AFM images of poly-1-co-2b show long fibrillar aggregates with uniform heights of ∼15 nm (Fig. 5f and S9g–i†). Noteworthy, the chiral nature of the seeds of 2b is not sufficient to induce any amplification of asymmetry effect yielding an enantioenriched helical supramolecular copolymer as demonstrates the lack of appreciable dichroic response in the CD spectrum (Fig. S10†). In addition, the UV-Vis spectrum of the final heteropolymer poly-1-co-2b presents the same pattern than pristine poly-1 corresponding to the formation of H-type aggregates (Fig. S10†). Finally, we have also performed the seeded heteropolymerization of 2b, as major component, and seeds of BQQDI 1 (Fig. 1c and 5g) by following the same strategy to that utilized for the previous heteropolymers. Thus, we prepared the metastable 2bM* species at 39 °C and, shortly after that, we added the achiral seeds of BQQDI 1. The formation of poly-2b-co-1 proceeds rapidly and completed within ∼15 min (Fig. 5g). In good agreement with that observed for the seeded supramolecular polymerization yielding poly-1-co-2a and poly-2a-co-1, in which seeds stemming from H-type aggregates are added, the kinetics are remarkably faster than the formation of the homopolymer poly-2b. Importantly, the SSP yielding poly-2a-co-1 provokes remarkable changes in the CD spectra of this copolymer. Whilst the previously reported homopolymer poly-2b presents a monosignated CD signal at λ = 635 nm, diagnostic of an off-resonance coupling of transition dipoles,26 the heteropolymer poly-2b-co-1 exhibits a bisignated CD signal at λ = 635 nm characteristic of an on-resonance coupling of the transition dipoles characteristic of H-type aggregates (Fig. S11a†).26 However, and despite the changes observed in the CD spectra, the absorption pattern of both pristine poly-2b and poly-2b-co-1 are identical and ascribable to J-type aggregates (Fig. S11b†). These (chir)optical features can be justified by considering that the heteropolymer growth around the seeds takes place yielding columnar fragments within the whole J-type aggregate giving rise to supramolecular copolymers with a blocky microstructure (Fig. 1c).
Conclusions
Herein we report on the synthesis and self-assembling features of the 3,4,9,10-benzo[d,e]isoquinolino[1,8-g,h]quinoline-tetracarboxylic diimide (BQQDI) 1 endowed with peripheral trialkoxybenzamide fragments that favors its kinetically controlled supramolecular polymerization by the formation of intramolecularly H-bonded, seven-membered pseudocycles. The electron-withdrawing character of 1, together with the similar geometry, makes BQQDI 1 an optimal candidate to perform seeded supramolecular copolymerization with the previously reported PDIs 2. Thus, we have developed the synthesis of four different supramolecular block copolymers by mixing BQQDI 1 and PDIs 2. Heteropolymers poly-1-co-2a and poly-2a-co-1 are formed between two different species that separately give rise to H-type aggregates and in combination also form H-type co-assemblies. More interesting is the case of the heteropolymers formed upon mixing achiral 1 with chiral 2b which, in turn, upon self-assembly, affords J-type aggregates. Heteropolymer poly-1-co-2b, formed by seeding the metastable monomers of 1 with fragments of 2b, presents the characteristics of H-type aggregates. However, poly-2b-co-1, prepared by seeding the chiral, metastable monomers of 2b with achiral seeds of 1, produces chiral, J-type aggregates. Interestingly, the original monosignated CD signal of pristine poly-2b changes to a bisignated CD signal most probably due to the formation of columnar domains around the seeds of 1. These findings indicate the blocky nature of the supramolecular copolymers formed. The studies presented allows shedding relevant information about the formation of kinetically controlled supramolecular heteropolymers with a controlled microstructure.Data availability
The data that supports the findings of this study have been included in the main text and ESI† and are available from the corresponding author upon reasonable request.Author contributions
A. J. Schwalb (first author): investigation, data curation, formal analysis, visualization and revising the original draft; Dr F. García: investigation, revising the original draft, funding acquisition; Dr L. Sánchez: (corresponding author): conceptualization, supervision, validation, writing – review & editing and funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIN/AEI of Spain (CNS2022-136058, PID2020-113512GB-I00 and TED2021-130285B-I00) and Comunidad de Madrid (P27/21-008) are acknowledged.Notes and references
- (a) G. Van Meer, D. R. Voelker and G. V. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112 CrossRef CAS PubMed; (b) D. Lingwood and K. Simons, Science, 2010, 327, 4 CrossRef PubMed; (c) X. Cheng and J. C. Smith, Chem. Rev., 2019, 119, 5849 CrossRef CAS PubMed.
- (a) F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS; (b) C. Sinturel, F. S. Bates, M. A. Hillmyer and N. High, ACS Macro Lett., 2015, 4, 1044 CrossRef CAS PubMed; (c) H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y.-S. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. B. Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122, 14471 CrossRef CAS PubMed.
- (a) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; (b) M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38 CrossRef CAS.
- Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848 CrossRef PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, K. J. H. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS PubMed.
- O. Dumele, L. Ðordević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 3127 CrossRef CAS PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72 CrossRef CAS PubMed.
- (a) Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761 CrossRef CAS PubMed; (b) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. Kumar Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef PubMed.
- H. M. M. ten Eikelder and A. J. Markvoort, Acc. Chem. Res., 2019, 52, 3465 CrossRef CAS PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CrossRef CAS PubMed.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566 CrossRef CAS PubMed.
- (a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed; (b) J. Kang, D. Mijayima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS PubMed; (c) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, J. Phys. Chem. B, 2019, 123, 6627 CrossRef CAS PubMed.
- (a) B. Adelizzi, P. Chidchob, N. Tanaka, B. G. A. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681 CrossRef CAS PubMed; (b) H. Su, S. A. H. Jansen, T. Schnitzer, E. Weyandt, A. T. Rösch, J. Liu, G. Vantomme and E. W. Meijer, J. Am. Chem. Soc., 2021, 143, 17128 CrossRef CAS PubMed; (c) R. Liao, F. Wang, Y. Guo, Y. Han and F. Wang, J. Am. Chem. Soc., 2022, 144, 9775 CrossRef CAS PubMed; (d) L. López-Gandul, A. Morón-Blanco, F. García and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202308749 CrossRef PubMed.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. A. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570 CrossRef CAS PubMed.
- S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed.
- (a) D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed; (b) W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044 CrossRef CAS PubMed.
- (a) T. Okamoto, S. Kumagai, E. Fukuzaki, H. Ishii, G. Watanabe, N. Niitsu, T. Annaka, M. Yamagishi, Y. Tani, H. Sugiura, T. Watanabe, S. Watanabe and J. Takeya, Sci. Adv., 2020, 6, eaaz0632 CrossRef CAS PubMed; (b) S. Kumagai, H. Ishii, G. Watanabe, C. P. Yu, S. Watanabe, J. Takeya and T. Okamoto, Acc. Chem. Res., 2022, 55, 660 CrossRef CAS PubMed.
- (a) S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343 CrossRef CAS PubMed; (b) F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695 CrossRef; (c) W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044 CrossRef CAS PubMed.
- (a) M. Wehner, M. I. S. Röhr, M. Böhler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092 CrossRef CAS PubMed; (b) C. Naranjo, S. Adalid, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202218572 CrossRef CAS PubMed.
- (a) E. E. Greciano and L. Sánchez, Chem.–Eur. J., 2016, 22, 13724 CrossRef CAS PubMed; (b) E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517 CrossRef CAS PubMed; (c) E. E. Greciano, S. Alsina, G. Ghosh, G. Fernández and L. Sánchez, Small Methods, 2020, 4, 1900715 CrossRef CAS.
- (a) P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed; (b) L. López-Gandul, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Chem. Sci., 2022, 13, 11577 RSC.
- N. S. S. Nizar, M. Sujith, K. Swathi, C. Sissa, A. Painelli and K. G. Thomas, Chem. Soc. Rev., 2021, 50, 11208 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01322k |
| This journal is © The Royal Society of Chemistry 2024 |