
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox regulation of Ni hydroxides with controllable phase composition towards biomass-derived polyol electro-refinery†
Zhuxin
Gui
a,
Yingshuai
Jia
a,
Xianping
Liao
b,
Tianlan
Yan
a,
Boxu
Gao
a,
Wenbiao
Zhang
 ab,
Li
Chen
c,
Qingsheng
Gao
*b,
Yahong
Zhang
a and
Yi
Tang
*a
ab,
Li
Chen
c,
Qingsheng
Gao
*b,
Yahong
Zhang
a and
Yi
Tang
*a
aDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials, Fudan University, Shanghai 200433, P. R. China. E-mail: yitang@fudan.edu.cn
bCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, P. R. China. E-mail: tqsgao@jnu.edu.cn
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai 200062, P. R. China
First published on 23rd April 2024
Abstract
Electrocatalytic refinery from biomass-derived glycerol (GLY) to formic acid (FA), one of the most promising candidates for green H2 carriers, has driven widespread attention for its sustainability. Herein, we fabricated a series of monolithic Ni hydroxide-based electrocatalysts by a facile and in situ electrochemical method through the manipulation of local pH near the electrode. The as-synthesized Ni(OH)2@NF-1.0 affords a low working potential of 1.36 VRHE to achieve 100% GLY conversion, 98.5% FA yield, 96.1% faradaic efficiency and ∼0.13 A cm−2 of current density. Its high efficiency on a wide range of polyol substrates further underscores the promise of sustainable electro-refinery. Through a combinatory analysis via H2 temperature-programmed reduction, cyclic voltammetry and in situ Raman spectroscopy, the precise regulation of synthetic potential was discovered to be highly essential to controlling the content, phase composition and redox properties of Ni hydroxides, which significantly determine the catalytic performance. Additionally, the ‘adsorption–activation’ mode of ortho-di-hydroxyl groups during the C–C bond cleavage of polyols was proposed based on a series of probe reactions. This work illuminates an advanced path for designing non-noble-metal-based catalysts to facilitate electrochemical biomass valorization.
Introduction
The rapidly depleted fossil resource calls for the development of renewable and greener hydrogen energy, which is well recognized as a promising candidate to facilitate the strategy of carbon neutrality as well as the revolution of the global energy landscape.1 However, the advancement of H2 fuel economy has been largely limited by the difficulties in the safe and efficient storage/transportation of energy materials, urgently requiring the development of appropriate H2 carriers.2 Formic acid (FA), as the simplest carboxylic acid, has received widespread and growing attention not only because of its common industrial applications but also for its future potential to be considered as a sustainable and environmentally benign liquid hydrogen carrier in order to achieve a low-carbon community.3–7 The industrial production of FA is mostly based on the hydrolysis of methyl formate or the acidification of formate.8 Those routes rely on the utilization of toxic CO (generated from the fossil resource) as a starting material, accompanied by strict reaction conditions (e.g., 4 MPa),8 spawning massive energy consumption, pollutant emissions and production risk, as well as violating the original intention of a low-carbon economy. Hence, it is extremely crucial to develop more efficient and greener routes towards FA production.Electrocatalytic refinery (E-refinery) from biomass to high-value-added chemicals has attracted increasing attention due to the sustainability of feedstocks and mild conditions of the electrochemical process.9 Glycerol (GLY), mainly obtained from byproducts of the biodiesel industry, is one of the leading biomass-derived platform molecules recognized by the US Department of Energy.10 Its continuously decreasing price and excessive reservation guarantee the potential and importance of chemical upgrading.11 Therefore, the electrocatalytic glycerol oxidation reaction (GOR) to produce formic acid at the anode has sparked numerous academic and industrial interests.12 The synthetic pathway from C3 polyhydric alcohol to C1 carboxylic acid caters to the demand for atom economy.13 Moreover, compared to the electrooxidation of methanol towards FA, the utilization of GLY takes advantage of its low toxicity and volatility, which ensures safety in its wide application.14,15 Furthermore, the replacement of sluggish oxygen evolution reaction (OER) with dynamically advantageous GOR can reduce the energy consumption from the anodic polarization, efficiently coupling with the electroreduction process in the cathode.16
Ni hydroxide is regarded as a high-profile candidate for organic electrooxidation because of its remarkable intrinsic activity as well as its low cost (compared to noble metal catalysts).17 Nevertheless, the onset GOR potential of the reported mono-component Ni(OH)2 electrode was limited to 1.48 VRHE, associated with high kinetic barriers.18 In order to upgrade the electrocatalytic performance of Ni(OH)2, the cation–intermediate interaction during GLY E-refinery was proposed via employing the costly LiOH electrolyte to enhance the faradaic efficiency (FE) of FA to 81.3% at a working potential of 1.50 VRHE.19 Alternatively, tremendous efforts have been devoted to further advancing the electrooxidation of GLY to FA over multi-component Ni-based electrocatalysts, such as Ni–Mo–N (97% FE, 1.35 VRHE), NiCo hydroxide (94.3% FE, 1.62 VRHE), NiVRu LDHs (97% FE, 1.40 VRHE) and NiCo2O4 (89.9% FE, 1.40 VRHE).20–23 However, the construction of multi-component catalysts possibly elevates the difficulty of clearly validating the promotion effect of each active species, while accounting for the increase in complexity and cost of catalyst synthesis. It should be noted that Ni(oxy)hydroxide (NiOOH) was broadly determined as the real active phase for many organic electrooxidations through the redox cycle of Ni3+/Ni2+ over Ni(OH)2 catalyst.24,25 To date, the catalytic effect of Ni(OH)2 structures and properties on these reactions has rarely been investigated and discussed. Our vision in this work is to explore the topmost GOR performance (high FA yield and FE at a low working potential) by rationally tuning the structures and redox properties of Ni hydroxides. More significantly, the development of a mono-component Ni-based catalyst will also benefit the elucidation of the GOR mechanism as well as boost the industrial application of GLY E-refinery and FA green production due to the well-defined structure and simplicity of preparation.
Herein, a series of monolithic Ni(OH)2 electrocatalysts were constructed via a facile electrodeposition process at varied potentials (E), which were denoted as Ni(OH)2@NFs-E. The cathodic E adopted for electrodeposition would bring about the increase in local pH near the electrode derived by the neutral hydrogen evolution reaction (HER), thus serving as an in situ manipulation of Ni(OH)2 content and phase composition (Fig. 1a). Ni hydroxides with the morphology of nanosheets directly grew on nickel foam (NF), enabling their direct attachment to the conductive substrate for efficient charge transport during electrooxidation. With the regulation of Ni(OH)2 content and phase composition by precisely controlling the deposition potential, ∼100% conversion of GLY, 98.5% yield of FA, 96.1% FE and ∼0.13 A cm−2 of current density could be accomplished over Ni(OH)2@NF-1.0 at a low working potential of 1.36 VRHE. It represents an outstanding and competitive GOR performance among the reported state-of-the-art catalysts.19–23,26–37 More importantly, the high efficiency of Ni(OH)2@NF-1.0 for the electro-synthesis of FA via C–C bond cleavage was confirmed in a wide substrate scope of various biomass-derived polyols from C2 to C6. Systematic electrochemical measurements revealed that the significantly superior GOR activity of Ni(OH)2@NF-1.0 is ascribed to the regulated redox properties of Ni(OH)2 and the enhanced catalyst dehydrogenation in anodic polarization. This phenomenon originates from the electrochemically controlled Ni hydroxide phase composition, which possesses a higher proportion of α-Ni(OH)2. Additionally, Ni(OH)2@NF-1.0, with its greater ratio of γ-NiOOH/α-Ni(OH)2 active phase, takes advantage of product desorption and inhibits the excessive oxidation of FA, endowing itself with attractive FA selectivity and FE. Furthermore, the pathway of GOR and the mechanism of C–C bond cleavage in polyol electrooxidation were attentively explored. We proposed the ‘adsorption–activation’ mode of ortho-di-hydroxyl groups in GOR over NiOOH/Ni(OH)2 through the experimental observations of probe reactions. This work sheds light on the design and development of cost-efficient and practical mono-component catalysts in the biomass-derived polyol E-refinery towards high-purity and high-value-added chemicals.
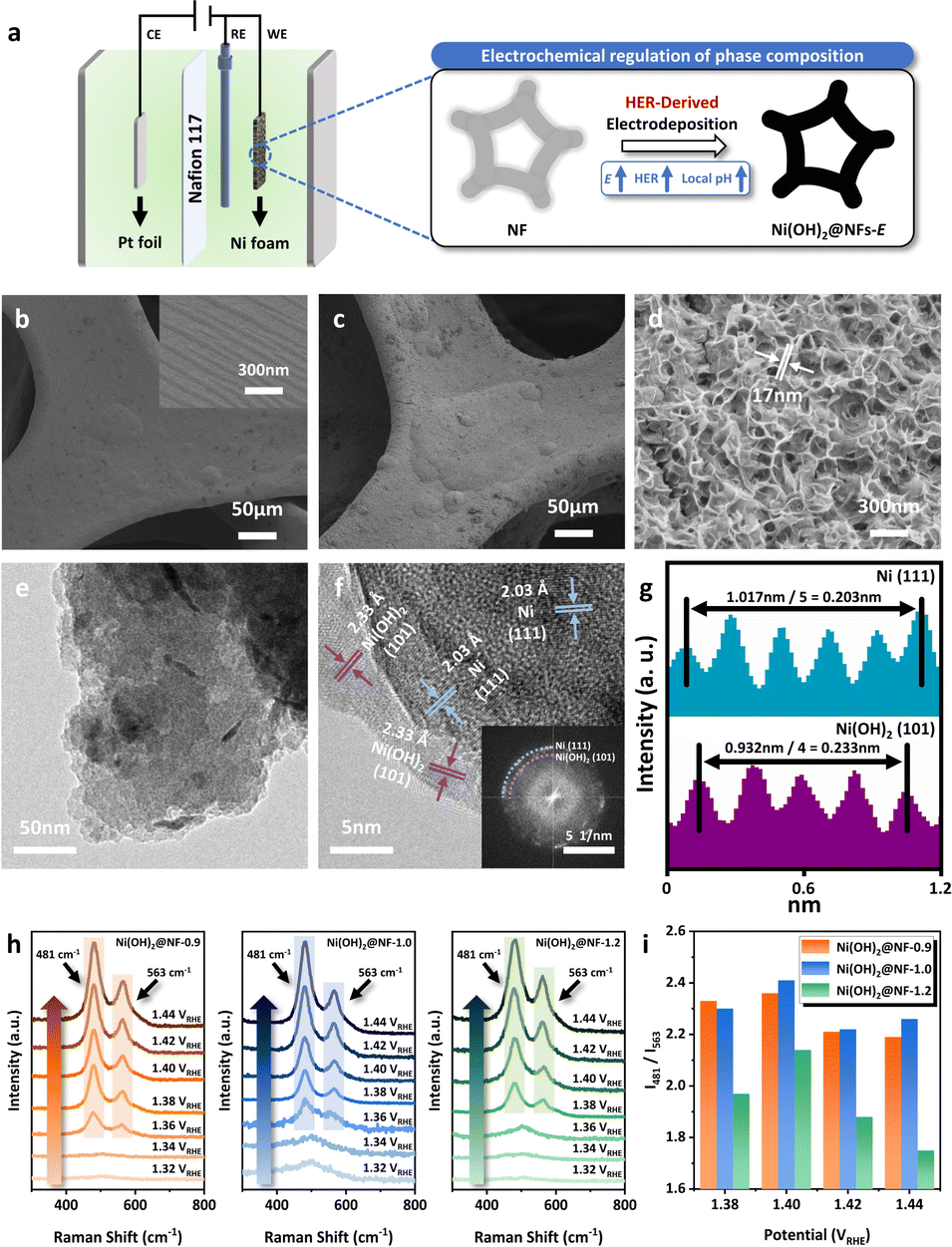 | ||
| Fig. 1 (a) Schematic illustration of the catalyst synthesis. SEM images of (b) Bare NF and (c and d) Ni(OH)2@NF-1.0. (e and f) TEM images of Ni(OH)2@NF-1.0 (inset: reduced FFT images of HR-TEM). (g) Line scan measurements of HR-TEM images. (h) In situ Raman spectra of Ni(OH)2@NFs-E at various potentials in 1 M KOH. (i) Intensity ratio of two NiIII–O peaks (I481/I563) calculated from in situ Raman characterization. | ||
Results and discussion
Preparation and characterization of catalysts
A series of monolithic Ni hydroxide-based catalysts were directly synthesized over nickel foam (NF) via an eco-friendly and facile method of electrodeposition (Fig. 1a) under different potentials. Other than the conventional electrodeposition of Ni hydroxides using nickel nitrate to generate OH− by the reduction of NO3− (NO3− + 7H2O + 8e− → NH4+ + 10OH−),38–40 the adoption of nickel sulfate as a precursor can achieve the regulation of the local pH near the electrode via the neutral HER at cathodic potentials (2H2O + 2e− → H2↑ + 2OH−), facilitating the deposition of Ni2+ in the electrolyte to generate uniformly distributed black substance over NF (Fig. S1, ESI†). The morphology of Ni(OH)2@NF-0.9, Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2 was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As presented in SEM images (Fig. 1c, d, S2 and S3, ESI†), Ni(OH)2@NFs-E exhibit the morphology of nanosheets with a thickness of 17 nm. However, the overall structure of Ni(OH)2@NF-1.2 is looser than that of Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 (Fig. 1c, S2b and S3b, ESI†), possibly because of the more significant H2 evolution during the electrodeposition resulting in the crack of the loaded substance (Fig. S3b, ESI†). Energy dispersive X-ray spectroscopy (EDS) mapping attached to SEM shows the uniform distribution of Ni and O elements for all three samples (Fig. S6, ESI†). High-resolution TEM (HR-TEM) images in Fig. 1f then reveal the composition of active species loaded by the electrochemical method, displaying the (101) crystal plane of Ni(OH)2 (d = 0.233 nm). A small amount of metallic Ni, indicated by the (111) crystal plane (d = 0.203 nm), can also be observed by HR-TEM, which might be formed by the reduction of Ni2+ during the synthetic process (Ni2+ + 2e− → Ni0).41–43 However, X-ray diffraction (XRD) can only detect the strong signal of metallic Ni as substrate (2θ = 44.5°, 51.9°, 76.4°, 92.9°, 98.4°) instead of Ni(OH)2 (Fig. S7, ESI†), which could result from the low amount or high dispersion of Ni(OH)2 over NF. A series of vibration peaks can be observed in the attenuated total reflectance–Fourier transform infrared spectroscopy (ATR-FTIR) patterns for Ni(OH)2@NFs-E (Fig. S8, ESI†).44–47 The peak centered at 3640 cm−1 should be assigned to the stretching mode of the surface hydroxyl group (–OH), while the ones at 1456 and 1346 cm−1 represent the bending mode of the lattice –OH. Vibrations of the lattice mode for Ni hydroxides can also be found at 1115, 928, 508 and 430 cm−1. A broad band at 1630 cm−1 corresponds to the –OH bending vibration from the free absorbed H2O. The peak at 621 cm−1 is attributed to the vibration of SO42− introduced by the precursor electrolyte.46,48 The above results suggest the identical chemical composition of Ni(OH)2 for all three materials synthesized by electrochemical methods.X-ray photoelectron spectroscopy (XPS) was conducted to explore the surface chemical and electronic state of Ni(OH)2@NFs-E. Ni 2p3/2 spectra (Fig. S9, ESI†) indicate the major existence of Ni2+ at the binding energy of 855.8 eV over Ni(OH)2@NFs-E with the hardly unchanged Ni0 peak (852.3 eV) and satellite peak (861.3 eV),49–52 which is in agreement with TEM and ATR-FTIR results. O 1s spectra of Ni(OH)2@NFs-E can be deconvoluted into three peaks (Fig. S9, ESI†), assigning to the Ni–O (OLat) at 529.3 eV, Ni–OH at 531.3–531.4 eV, as well as oxygen species absorbed on the structure defects (OAds) at 533.0 eV.53,54 Therefore, XPS measurements confirm that the active species loaded by electrodeposition at different potentials are mainly composed of Ni hydroxides, which also share a comparable surface Ni valence.
Furthermore, in situ Raman spectroscopy was carried out to unveil the phase composition of Ni(OH)2, which plays a decisive role in its electrochemical behavior during the anodic polarization (Fig. 1h).55 A series of Raman spectra were recorded at different working potentials in 1 M KOH. For Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0, only a broad peak around 501 cm−1 can be observed below 1.34 VRHE, attributed to Ni(OH)2 over the catalyst surface.56,57 The pair of bands at 481 and 563 cm−1 emerged at 1.36 VRHE and became more divided under higher potentials, which can be assigned to the different stretching modes of NiIII–O bond in NiOOH and represents the occurrence of Ni(OH)2 oxidation.56,57 Meanwhile, for Ni(OH)2@NF-1.2, peaks at 481 and 563 cm−1 appeared at a higher potential of 1.38 VRHE, demonstrating its more difficult transformation from Ni(OH)2 to NiOOH than that of Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0. A detailed comparison of the intensity ratio of two NiIII–O bands centered at 481 and 563 cm−1 (I481/I563) reveals the difference in Ni(OH)2 phase composition for the three materials.58–63 As is illustrated in Fig. 1i and S10, ESI,†I481/I563 of Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 were practically equivalent under 1.38 VRHE, 1.40 VRHE, 1.42 VRHE and 1.44 VRHE, while being significantly larger than that of Ni(OH)2@NF-1.2. It has been reported that the larger I481/I563 should be associated with the higher proportion of γ-NiOOH generated from the anodic oxidation process,58–63 which confirms the greater ratio of α-Ni(OH)2 within Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 than that of Ni(OH)2@NF-1.2. This phenomenon possibly originated from the higher local pH for Ni(OH)2@NF-1.2 during the HER-derived synthetic process under a more negative cathodic potential of −1.2 VAg/AgCl, which induces the aging process of α-Ni(OH)2 to produce β-Ni(OH)2.64–66 Hence, it is concluded that the phase composition of Ni hydroxides can be electrochemically regulated by the precise control of the deposition potential, which leads to the evaluation of GOR performance and further characterizations of Ni(OH)2@NFs-E.
Electrocatalytic performance of GOR
The GOR activities of Ni(OH)2@NFs-E were evaluated in a 1.0 M KOH aqueous solution with 100 mM GLY under a low working potential of 1.36 VRHE (Fig. 2a), which showed the trend of Ni(OH)2@NF-1.0 > Ni(OH)2@NF-0.9 > Ni(OH)2@NF-1.2 (FA yield 98.5% > 76.8% > 70.4%, respectively). Bare NF only displayed a low FA yield of 5.6% at the same condition (Fig. 2a), which illustrates the improved reactivity after the electrodeposition of Ni hydroxides. Especially, Ni(OH)2@NF-1.0 exhibited outstanding as well as optimum GOR performance among Ni(OH)2@NFs-E (Fig. 2a), which can achieve ∼100% conversion of GLY, 98.5% yield and 96.1% FE of FA. Electrocatalytic performance was further investigated under different working potentials for Ni(OH)2@NF-1.0 (Fig. 2b). At potentials ranging from 1.32 VRHE to 1.38 VRHE, the selectivity and FE of FA were maintained above 98.5% and 91.4%, respectively. The improvement in working potential to 1.40 VRHE led to the decay of FA selectivity and FE, which might be caused by the excessive oxidation of FA or the subtle oxygen evolution reaction (OER) under high potential. The conversion of GLY would decline with the decrease in potential below 1.36 VRHE. On the other hand, the GOR performance of Ni(OH)2@NF-1.0 was measured as a function of reaction time at 1.36 VRHE (Fig. S11, ESI†). FA yield reached its maximum after 10 h of reaction, while the selectivity of FA was kept above 95.7% during GOR (Fig. S12, ESI†). Five consecutive batch reactions (10 h in total) were carried out to evaluate the catalytic stability of Ni(OH)2@NF-1.0 (Fig. 2c). The selectivity, FE and productivity of FA were well maintained at 95.9%, 96.9% and 0.55 mmol cm−2 h−1 in average during the 5-cycle GOR, respectively (Fig. 2c). Three successive long-term electrolyses (30 h in total) were also conducted on Ni(OH)2@NF-1.0 (Fig. S13, ESI†). The conversion of GLY was kept stable above 92% during the 3-cycle GOR. FA selectivity and FE were slightly declined in the third cycle to 92.8% and 83.2%, respectively. Nickel species could barely be detected by ICP-AES in the electrolyte after long-term GOR (Table S1, ESI†), demonstrating the excellent mechanical stability of the electrode that prevents the leaching of metal species and the pollution of the final product. The above results illustrate the considerable catalytic stability of Ni(OH)2@NF-1.0. Linear sweep voltammetry (LSV) curves demonstrated that the current density was dramatically enhanced with the addition of 100 mM substrate in the anolyte (Fig. 2d). Ni(OH)2@NF-1.0 required only 1.35 VRHE to generate a current density of 100 mA cm−2 with the presence of GLY, which was significantly lower than the potential of OER at 100 mA cm−2. The favorable kinetics of the GOR (compared to that of the OER) was further confirmed by Tafel slopes calculated from LSV. As is depicted in Fig. S14a, ESI,† the Tafel slope decreased from 141.89 mV dec−1 to 67.47 mV dec−1 after adding 100 mM GLY on Ni(OH)2@NF-1.0. LSV curves (Fig. 2e) and Tafel plots (Fig. S14b, ESI†) of Ni(OH)2@NFs-E measured in 1 M KOH with 100 mM GLY also exhibited a similar trend with long-term electrolysis, which Ni(OH)2@NF-1.0 delivered an excellent and highest current density of ∼0.13 A cm−2 at 1.36 VRHE and Ni(OH)2@NF-1.2 generated only 23.67 mA cm−2 at the same potential. To sum up, Ni(OH)2@NF-1.0 showed a prominent electrocatalytic performance of GOR, which is competitive among the state-of-the-art catalysts, including non-noble-metal oxides/hydroxides or even noble-metal-added hydroxides (Fig. 2f, Table S2, ESI†).19–23,26–37 More importantly, we further expanded the range of reaction substrates to a series of biomass-derived polyols (Table 1). The electrooxidation of ethylene glycol (EG), erythritol (ERY), xylitol (XYL) and mannitol (MAN) was carried out over Ni(OH)2@NF-1.0 under the optimized reaction conditions. The delightful performance of polyol E-refinery to produce FA was accomplished by Ni(OH)2@NF-1.0, especially when the carbon number (n) of the substrate ranged from 3 to 5. It firmly proves the catalytic universality and reactivity of Ni(OH)2@NF-1.0 in the electrochemical valorization of biomass towards C1 carboxylic acid.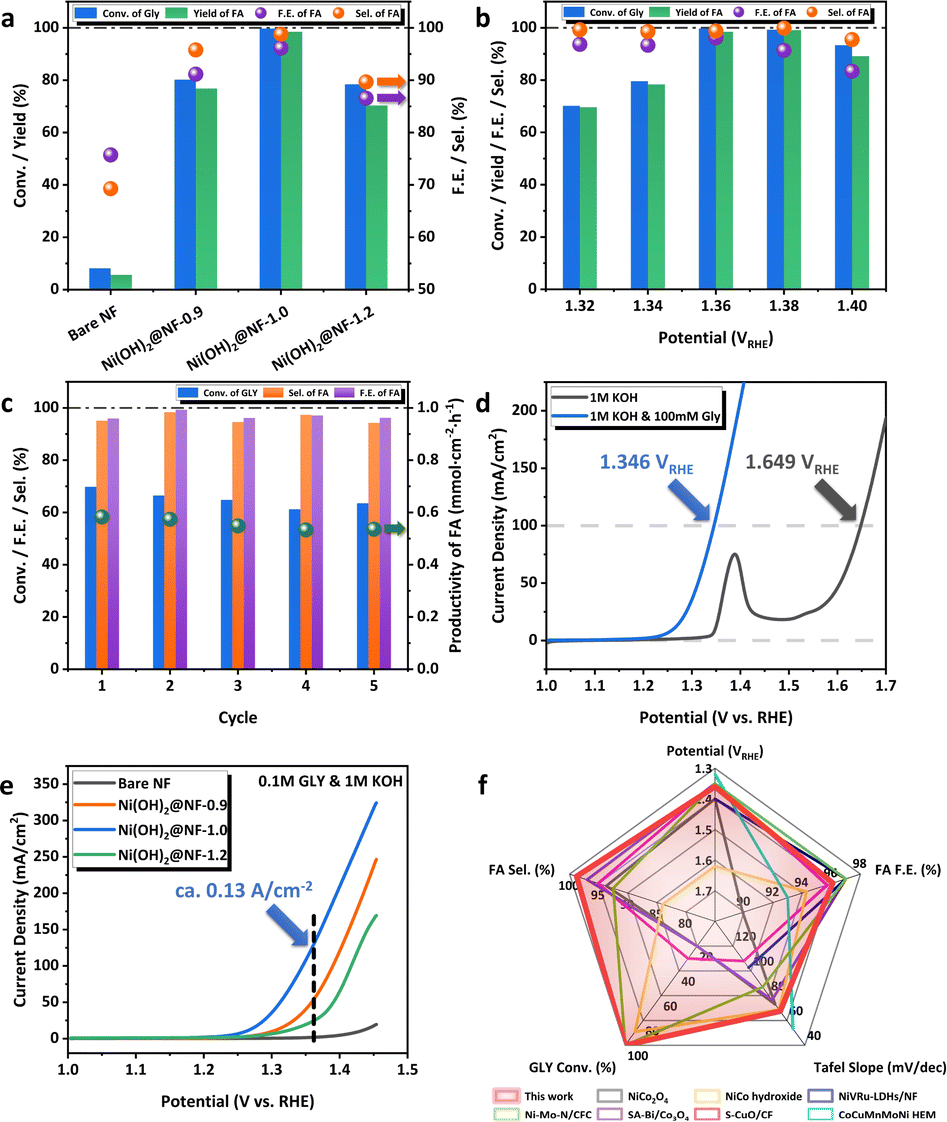 | ||
| Fig. 2 (a) Conversion of GLY, yield, FE and selectivity of FA for Ni(OH)2@NFs-E at 1.36 VRHE in 10 h. (b) GOR performance of Ni(OH)2@NF-1.0 at various potentials. (c) Five consecutive batch electrolysis (10 h in total) over Ni(OH)2@NF-1.0. (d) LSV curves of Ni(OH)2@NF-1.0 in 1 M KOH with or without 100 mM GLY at the scan rate of 5 mV s−1. (e) LSV curves of Ni(OH)2@NFs-E in 1 M KOH with 100 mM GLY at the scan rate of 5 mV s−1. (f) Comparison of GOR performance with state-of-the-art catalysts.20–23,26,27,29 | ||
|
|
|||||
|---|---|---|---|---|---|
| n | Potential (VRHE) | Conv. (%) | Yield (%) | Sel. (%) | FE (%) |
| 2 | 1.40 | 83.1 | 69.8 | 83.9 | 68.7 |
| 3 | 1.36 | 99.7 | 98.5 | 98.8 | 96.1 |
| 4 | 1.32 | >99.9 | >99.9 | >99.9 | 93.1 |
| 5 | 1.28 | 99.6 | 93.7 | 93.7 | 93.3 |
| 6 | 1.24 | 99.3 | 79.1 | 79.1 | 83.9 |
Based on the systematic evaluation of electrocatalytic activity, we achieved an appealing performance of biomass-derived polyol E-refinery with the optimization of the potential conducted in the material synthesis, while the as-synthesized Ni(OH)2 is believed to be the active site for GOR. Combined with the conclusions of catalyst characterization, the phase composition of Ni hydroxides can be regulated by the variation of electrodeposition potential, and the proportion of α-Ni(OH)2 within Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 was not only similar but also greater than that of Ni(OH)2@NF-1.2. Hence, the GOR activity difference between Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 as well as the structure–function relationship about Ni(OH)2 phase composition deserve to be carefully discussed in the following section.
Regulation of Ni(OH)2 content over NF under different deposition potentials and its effect on GOR performance
As mentioned before, the fabrication of Ni hydroxides over NF was based on the regulation of local pH by HER. Meanwhile, the rate of hydrogen evolution varied significantly under different deposition potentials, which can be indicated by LSV curves recorded under the conditions of material synthesis (Fig. S15a, ESI†). Furthermore, the deposition current as well as charge passed within the same time increased with the improvement in the deposition potential (Fig. S15B, ESI†). Therefore, the amount of electrodeposited Ni(OH)2 on NF should be different for Ni(OH)2@NFs-E, while the content of catalytic active species would significantly affect the GOR performance. H2 temperature-programmed reduction (H2-TPR) was conducted to measure the amount of active species (Fig. S16, ESI†). In accordance with our expectations, the amount as well as the loading mass of Ni hydroxides showed the trend of Ni(OH)2@NF-1.2 ≫ Ni(OH)2@NF-1.0 > Ni(OH)2@NF-0.9. Based on that, the difference between Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 in electrocatalytic activity can be explained by the inferior content of active species for Ni(OH)2@NF-0.9. Since Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 were identical in terms of morphology, electronic states and active phase structures. To reassure this point, the deposition time of Ni(OH)2@NF-0.9 was prolonged from 600 to 3000 s in order to increase the surface loading of active species. H2 consumption measured by TPR for Ni(OH)2@NF-0.9–3000 s was consistent with that of Ni(OH)2@NF-1.0 (Fig. S16, ESI†). The GOR performance of Ni(OH)2@NF-0.9 was enhanced towards a satisfactory level (FA yield 76.8% for Ni(OH)2@NF-0.9 vs. FA yield 95.7% for Ni(OH)2@NF-0.9–3000 s) with the improvement of active species content (Fig. S17, ESI†). On the other hand, with a much greater amount of Ni hydroxides, the GOR performance of Ni(OH)2@NF-1.2 was much inferior to that of Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 (Fig. S17, ESI†). The catalytic activity of Ni(OH)2@NF-1.2 further declined with the decrease in deposition time from 600 to 300 s (FA yield 70.4% for Ni(OH)2@NF-1.2 vs. FA yield 43.4% for Ni(OH)2@NF-1.2–300 s), which excludes the effect of active species content for Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2. The calculation of turnover frequency (TOF) values for Ni(OH)2@NFs-E (Table S3, ESI†) illustrates that the difference in GOR activity between Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0 is primarily induced by the variation of catalytic species content, and their TOF values are similar after the calibration of the loaded Ni(OH)2 amount. The TOF of glycerol for Ni(OH)2@NF-1.2 is much less than that of Ni(OH)2@NF-0.9 and Ni(OH)2@NF-1.0, indicating its low reactivity of chemical structures. In addition, the superior intrinsic activity of Ni(OH)2@NF-1.0 (compared to that of Ni(OH)2@NF-1.2) was also confirmed by the characterization of electrochemically active surface areas (ECSA) through the double-layer capacitance (Cdl) method (Fig. S18, ESI†). LSV curves of GOR normalized by Cdl (Fig. S18d, ESI†) clearly exhibited the improved specific current density of Ni(OH)2@NF-1.0, which indicated its advantage of intrinsic activity against that of Ni(OH)2@NF-1.2 and was in line with the comparison of geometric activities.The above investigations motivated us to consider that the difference in GOR performance between these two samples should be induced by their distinct phase composition of Ni hydroxides. Therefore, before entering the in-depth discussion, it is necessary to clarify the consistency of actual active sites for Ni(OH)2@NFs-E during GOR. As is illustrated in the XPS analysis (Fig. S19, ESI†), the binding energies of various Ni or O valence states were barely changed for these samples before and after GOR, while the ratio of Ni2+/Ni0 remained stable in the reaction (Table S4, ESI†). Hence, the surface electronic states of the as-synthesized Ni(OH)2@NFs-E were identical with the ones after GOR, thus providing a foundation for the subsequent identification of the structure–function relationship.
Elucidation of structure–function relationship of GOR over NiOOH/Ni(OH)2
To begin with, we focus on the difference in catalytic activity induced by the Ni(OH)2 phase composition. It is well known that the kernel of boosting the alkaline nucleophile electrooxidation for Ni(OH)2 is to promote the catalyst dehydrogenation process generated by electric energy, which yields the real active phase of NiOOH (Ni(OH)2 + OH− → NiOOH + H2O + e−).24,25 Cyclic voltammetry (CV) measurements of Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2 were conducted in 1 M KOH to explore the redox properties of NiOOH/Ni(OH)2. Fig. 3a clearly showed that the oxidation peak of Ni2+ → Ni3+ over Ni(OH)2@NF-1.0 shifted to the cathodic region compared to that of Ni(OH)2@NF-1.2, which is in accordance with the observation in Fig. 1h and should be the firm evidence of the enhanced catalyst dehydrogenation for the former. The regulation of NiOOH/Ni(OH)2 redox properties would be associated with its phase composition. α-Ni(OH)2 can be dehydrogenated to produce γ-NiOOH at a lower anodic potential than that of β-Ni(OH)2 to β-NiOOH.58–60 The higher proportion of α-Ni(OH)2 for Ni(OH)2@NF-1.0, which was confirmed by in situ Raman spectroscopy (Fig. 1h, i and S10, ESI†), leads to its lower dehydrogenation potential than that of Ni(OH)2@NF-1.2. The promoted Ni2+ → Ni3+ process of Ni(OH)2@NF-1.0 and the participation of NiOOH in GLY E-refinery can also be proved by in situ Raman measurements at the controlled potential of 1.36 VRHE with the addition of GLY (Fig. 3b). When applying the anodic potential, two NiIII–O bands at 481 and 563 cm−1 were clearly observed for Ni(OH)2@NF-1.0 instead of Ni(OH)2@NF-1.2, exhibiting the inadequate NiOOH species over the catalyst surface and the more difficult transformation from Ni2+ to Ni3+ for Ni(OH)2@NF-1.2. Those NiIII–O peaks then disappeared after the injection of 0.1 M GLY and could not be detected throughout the GOR process, which validates the NiOOH participation in nucleophile electrooxidation. Corresponding to a previous report, the electro-produced NiOOH will spontaneously extract proton from the organic substrate, which subsequently yields the oxidized product and re-hydrogenated Ni(OH)2.24,25 It can be illustrated from the CV curves that the reduction peak of Ni3+ → Ni2+ for Ni(OH)2@NF-1.0 also shifted to a lower potential (Fig. 3a), indicating the more difficult re-hydrogenation process of NiOOH → Ni(OH)2. It needs to be further verified whether the regulation of NiOOH/Ni(OH)2 redox properties would affect the re-hydrogenation process in GOR. We then carried out a series of multi-potential step measurements to probe this point (Fig. 3c and d).25 For both two samples, a working potential of 1.36 VRHE was applied, and a positive current could be observed in this stage (0–2 min), representing the oxidation of Ni(OH)2 to NiOOH. Then the anodic potential was removed to maintain an open-circuit state (2–4 min). If a reductive potential of 1.00 VRHE (compared to 1.36 VRHE) was applied immediately after the open-circuit potential (4–6 min), a cathodic current clearly occurred, which implies the reduction of NiOOH to Ni(OH)2. The negative current density of Ni(OH)2@NF-1.2 outweighed that of Ni(OH)2@NF-1.0 and demonstrates the easier reduction process for the former. However, when 0.1 M GLY was injected during the process of open-circuit state (2–4 min), a slight anodic current appeared in the third stage (4–6 min) instead of the cathodic current for both Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2. It explains that the spontaneous step of deprotonation from GLY would be negligibly affected by the regulated NiOOH/Ni(OH)2 redox properties. Consequently, the catalytic cycle of GOR, which is composed of the electro-generated Ni(OH)2 → NiOOH and spontaneous deprotonation from a nucleophile reagent, can be enhanced by the electrochemical regulation of Ni hydroxide phase composition and its dehydrogenation process at a low working potential (Fig. 3e).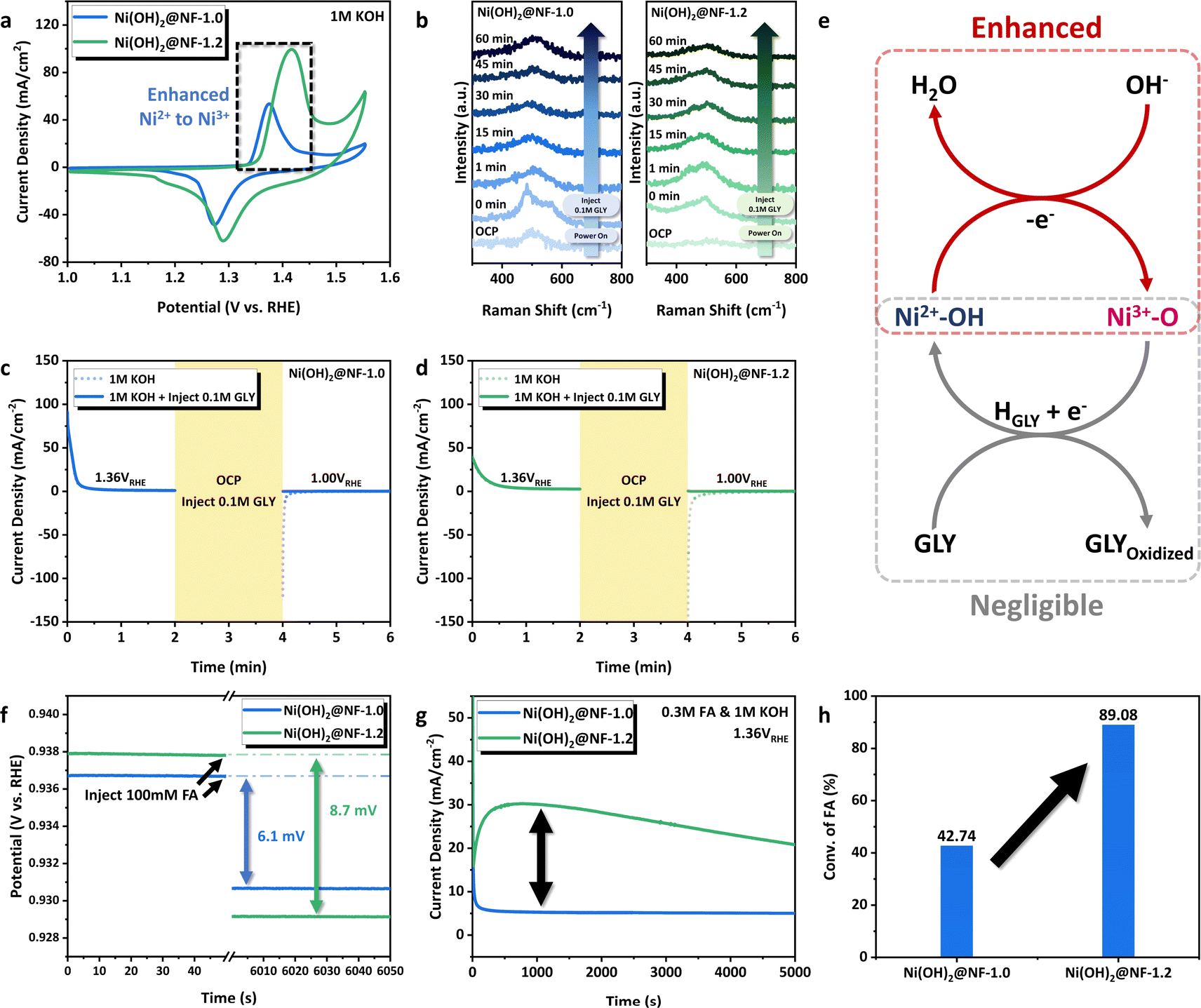 | ||
| Fig. 3 (a) CV curves of Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2 in 1 M KOH at the scan rate of 2 mV s−1. (b) In situ Raman spectroscopy of Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2 in 1 M KOH at 1.36 VRHE with the injection of 100 mM GLY. (c and d) Multi-potential step curves of Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2. (e) Schematic illustration of the catalytic cycle for GOR over NiOOH/Ni(OH)2. (f) The variation of OCP as a function of time with the injection of 100 mM FA for Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2. (g) I–t curves of FAOR conducted at 1.36 VRHE in 1 M KOH. (h) Conversion of FA after 10 h FAOR. | ||
We also notice that there is a clear gap for FA selectivity and FE between Ni(OH)2@NF-1.0 (FA selectivity/FE 98.8%/96.1%) and Ni(OH)2@NF-1.2 (FA selectivity/FE 89.7%/86.6%). The difficulty in desorption and over-oxidation of FA for Ni(OH)2@NF-1.2 are suspected as the primary reasons. To prove this, the variation of open-circuit potential (OCP) with time was first measured for Ni(OH)2@NF-1.0 and Ni(OH)2@NF-1.2 (Fig. 3f). With the injection of 100 mM FA, a more significant OCP change occurred for Ni(OH)2@NF-1.2 (8.7 mV) than that of Ni(OH)2@NF-1.0 (6.1 mV) in 1 M KOH, which demonstrates more organic adsorbate in the inner Helmholtz layer and stronger adsorption of FA for Ni(OH)2@NF-1.2.67–70 Therefore, FA produced during GOR would be more difficult to accumulate over the surface of Ni(OH)2@NF-1.0, yielding a higher selectivity and FE. On the other hand, FA oxidation reaction (FAOR) under the same conditions as GOR (1.36 VRHE, 1 M KOH) was conducted on two samples to characterize the performance of further conversion of the main product in GLY E-refinery. Surprisingly, the I–t curves in Fig. 3g suggested a greater current density for Ni(OH)2@NF-1.2 in FAOR, while the conversion of FA in a 10 h reaction (Fig. 3h) evidently showed much superior FAOR performance than that of Ni(OH)2@NF-1.0 (89.1% vs. 42.7%). In addition, Ni(OH)2@NF-1.2–300 s, with the same content of active phase compared to that of Ni(OH)2@NF-1.0, still exhibited advantageous FA adsorption (OCP changes 8.3 mV for Ni(OH)2@NF-1.2-300 s vs. 6.1 mV for Ni(OH)2@NF-1.0) and conversion performance (88.7% vs. 42.7%) after 10 h (Fig. S20, ESI†).67–70 It excludes the effect of Ni(OH)2 content on FA electrooxidation mentioned above. The promoted FAOR for Ni(OH)2@NF-1.2 is possibly linked to the favorable adsorption of organic species over metal hydroxide with reference to the reported results.27,67–70 Based on the above experimental observations, we conclude that Ni(OH)2@NF-1.0 with the greater ratio of γ-NiOOH/α-Ni(OH)2 takes advantage of product desorption and inhibits the further conversion of FA at a low working potential, thus endowing it with outstanding catalytic and electrochemical selectivity.
Reaction pathway of GOR and mechanism insight into C–C bond cleavage in ortho-hydroxyl polyol electrooxidation
In order to study the reaction pathway of GOR, high performance liquid chromatography (HPLC) elution curves of electrolyte at different reaction times were attentively investigated (Fig. 4a). Abundant and stable reaction intermediates were not present during the GOR process over Ni(OH)2@NF-1.0, while trace amounts of glyceric acid (GLA), glycolic acid (GA) and tartronic acid (TA) were detected in the middle stage of GOR (Fig. 4b). Hence, the reaction pathway of GOR is summarized in Fig. 4c. Three molecules of FA were produced in two C–C bond cleavages from GLA and GA, respectively. It was considered the major route for ‘GLY to FA’ that accounts for the high FA selectivity over Ni(OH)2@NF-1.0. On the other hand, the route that produces TA from two consecutive primary hydroxyl group oxidations of GLY was determined to be the minor one. Since oxalic acid (OA), as the by-product of C–C bond cleavage from TA, was not found in any reaction period, GLA and GA, as two major intermediates, could almost completely be converted to FA (Fig. S21, ESI†) under the same reaction condition (1.36 VRHE, 10 h), which further confirms the reaction pathway mentioned above.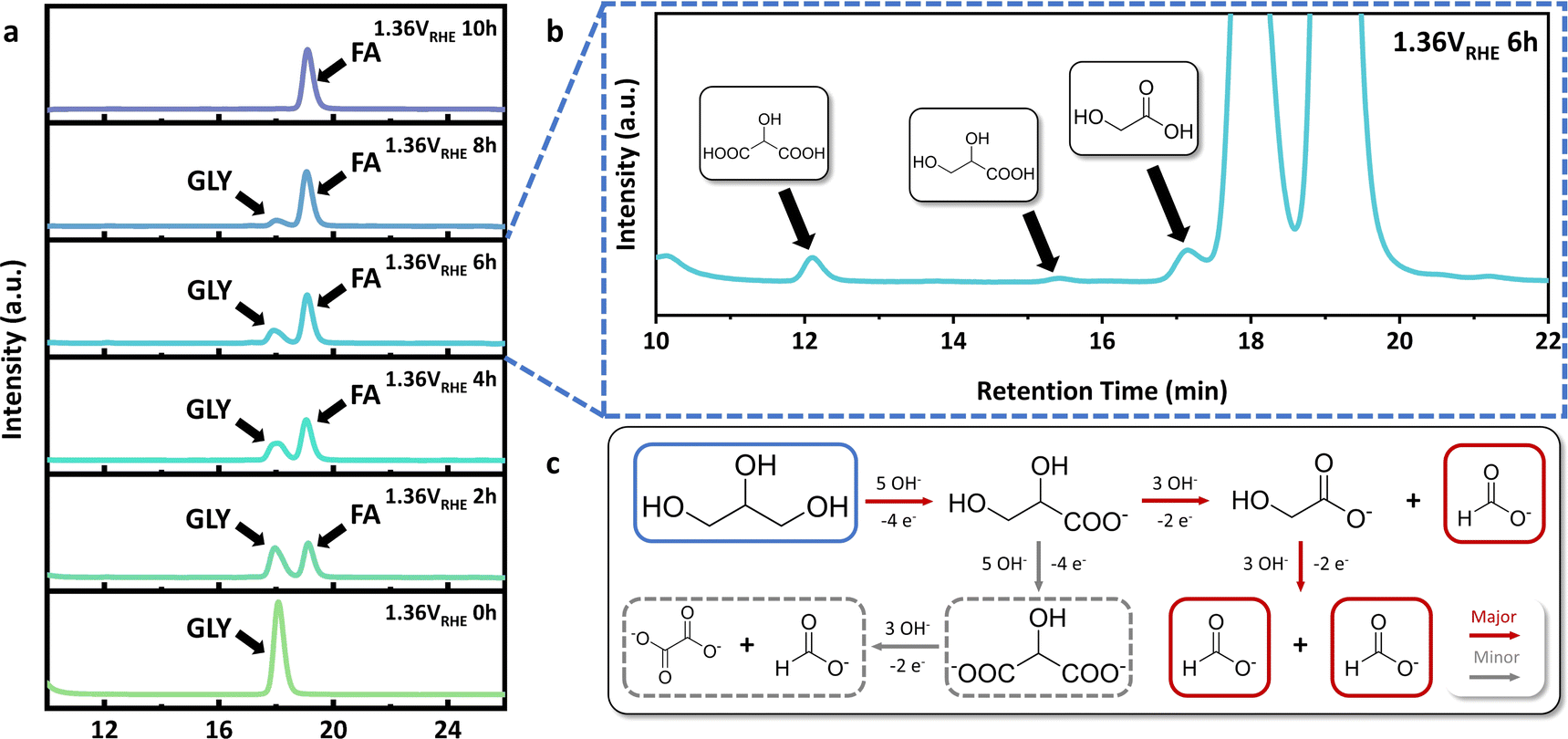 | ||
| Fig. 4 (a) HPLC elution curves of electrolyte at different times of GOR over Ni(OH)2@NF-1.0. (b) Enlarged HPLC elution curve of electrolyte at 6 h of GOR. (c) Summarized GOR pathway. | ||
Although many researchers have agreed on the identification of reaction intermediates and pathway, the mechanism of C–C bond cleavage in polyol electrooxidation was still under debate.21,27,71 We notice that the multi-dentate ‘adsorption–activation’ mode of C–C bond cleavage was proposed by Liu et al. in the thermal-catalytic conversion of sugar over tungsten trioxide-based catalyst.72 It inspired us to envision the multi-dentate interaction between NiOOH/Ni(OH)2 and –OH groups of GLY in electrocatalytic C–C bond cleavage. Combining with the GOR pathway summarized above, we suppose that the ‘adsorption–activation’ of GLY over catalyst surface during the bond breaking is possibly based on the ortho-hydroxyl groups. To validate those hypotheses, 1,2-propylene glycol (1,2-PG) and 1,3-propylene glycol (1,3-PG) were selected as the probe molecules, which the former has ortho-OH groups and the latter does not. The adsorption behavior of 1,2-PG and 1,3-PG over Ni(OH)2@NF-1.0 were characterized by the measurement of OCP variation before and after their injection (Fig. 5a). Interestingly, after 100 mM 1,2-PG was introduced, more obvious OCP change (11.5 mV) could be observed than that of 100 mM 1,3-PG (5.4 mV), which suggests the stronger adsorption for 1,2-PG.67–70 Thereby, with the equal hydroxyl groups concentration, the structure of ortho-OH groups will benefit the adsorption of polyol over NiOOH/Ni(OH)2, thus explaining the rationality of the bidentate adsorption in GOR. Moreover, probe reactions of 1,2-PG and 1,3-PG were conducted to explore the mechanism of C–C bond cleavage based on different adsorption modes. I–t curves (Fig. 5b) displayed that the electrooxidation of 1,2-PG would generate significantly larger current density than that of 1,3-PG, further indicating the different ‘adsorption–activation’ mode between ortho-hydroxyl and non-ortho-hydroxyl polyols. Reaction results of two probe molecules prove this point subsequently (Fig. S22 and S23, ESI†). During the electrooxidation of 1,3-PG, the relative content of 3-hydroxypropionic acid (3-HPA) gradually increased as the reaction proceeded (Fig. S23a, ESI†). Noticeably, 3-HPA was produced faster than malonic acid (MA), demonstrating that the oxidation of 1,3-PG mainly depends on the ‘adsorption–activation’ of single hydroxyl group. Nevertheless, equimolar acetic acid (AA) and formic acid (FA) were directly generated from the oxidation of 1,2-PG (Fig. S23b, ESI†), without the detection of lactic acid (LA) or hydroxyacetone (HA) as the product of single –OH group activation. Therefore, we deduce that the oxidation of 1,2-PG should obey the ‘adsorption–activation’ of ortho-OH groups, which leads to direct C–C bond cleavage. Based on the above experimental results, we propose the ortho-di-hydroxyl groups adsorption configuration of GLY on NiOOH/Ni(OH)2, and the corresponding mechanism of C–C bond cleavage in GOR. As is illustrated in Fig. 5d, the dehydrogenation of Ni(OH)2 to NiOOH occurs firstly under oxidative potential in alkaline solution. Then, a molecule of GLY is absorbed over catalyst surface by its interaction with primary and secondary –OH groups, followed by the spontaneous deprotonation process from GLY to yield glyceraldehyde (GLAD). GLAD is hydrated and oxidized immediately to GLA,73 which was the only C3 intermediate detected by HPLC in our observations. The first C–C bond cleavage takes place after the further dehydrogenation of GLA to produce one molecule of GA and FA respectively. Similarly, GA would be oxidized and undergo the second C–C bond cleavage, which proceeds with the bidentate ‘adsorption–activation’ mode and generates two molecules of FA. The ‘adsorption–activation’ of ortho-hydroxyl groups for GLY over NiOOH/Ni(OH)2 results in the strong interaction between active sites and intermediates, leading to the trace amount of C3 or C2 product (GLA and GA) as well as the high selectivity of FA throughout the reaction process (Fig. S12 and S25, ESI†). The present work paves the way for the development of electrocatalytic refinery from biomass towards high value-added chemicals over non-noble metal and mono-component catalysts.
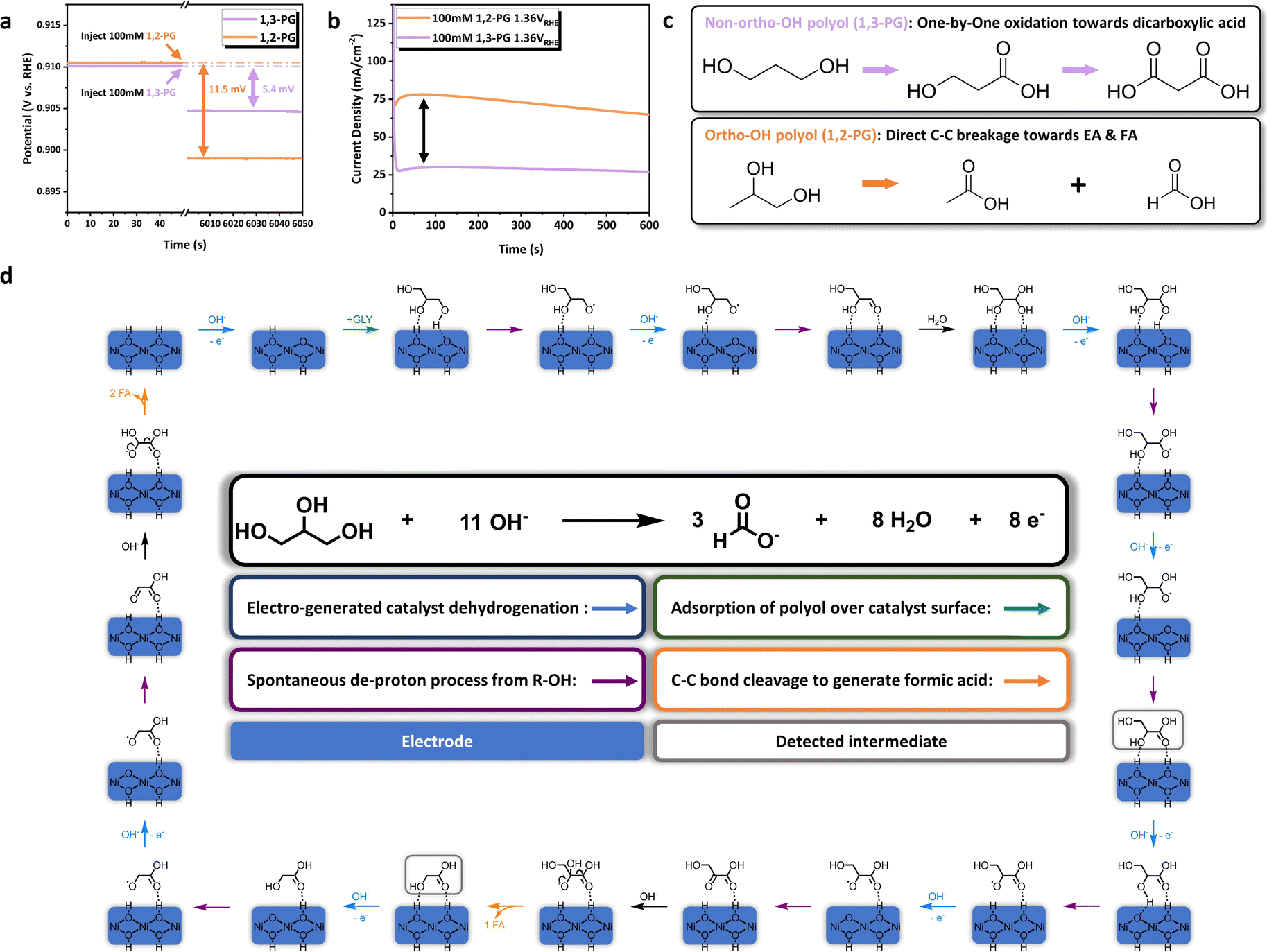 | ||
| Fig. 5 (a) The variation of OCP as a function of time with the injection of 100 mM 1,2-PG or 1,3-PG over Ni(OH)2@NF-1.0. (b) Probe reactions of 1,2-PG or 1,3-PG over Ni(OH)2@NF-1.0. (c) Reaction process of non-ortho-OH polyol and ortho-OH polyol in electrooxidation. (d) Proposed mechanism of C–C bond cleavage in GOR towards FA over NiOOH/Ni(OH)2. | ||
Conclusion
In summary, we developed a series of monolithic Ni(OH)2 catalysts via an eco-friendly and facile electrodeposition method. Ni(OH)2 nanosheets were directly constructed on NF and confirmed by SEM, TEM, XRD, ATR-FTIR and XPS. In situ Raman spectroscopy further revealed that the phase composition of Ni hydroxides can be electrochemically controlled by adjusting the deposition potential. A ∼100% GLY conversion, 98.5% FA yield, 96.1% FE and ∼0.13 A cm−2 of current density could be achieved on Ni(OH)2@NF-1.0 at a low working potential of 1.36 VRHE, thus representing a remarkable and competitive GOR performance among various state-of-the-art catalysts, including non-noble metal oxides/hydroxides and noble metal-based hydroxides. More importantly, Ni(OH)2@NF-1.0 presents the delightful performance in FA electro-synthesis from various biomass-derived polyols, which demonstrates its catalytic universality. Precise regulation of the potential conducted during the catalyst synthesis is highly relevant to the activity/selectivity of GOR. On the one hand, the increase in catalytic species can be realized by the manipulation of the synthetic potential, which results in the promotion of GOR activity. On the other hand, the electrochemically regulated phase composition of Ni(OH)2 significantly determined the redox properties of Ni3+/Ni2+. Therefore, the electro-generated dehydrogenation of the catalyst is prominently enhanced for Ni(OH)2@NF-1.0 with the greater ratio of α-Ni(OH)2, which exhibits much superior GOR activity. Ni(OH)2@NF-1.0, with a higher proportion of γ-NiOOH/α-Ni(OH)2 active phase, is also endowed with outstanding FA selectivity and FE originating from its product desorption and inhibition of excessive FA oxidation. Furthermore, the pathway of GLY E-refinery and the mechanism of C–C bond cleavage in ortho-hydroxyl polyol electrooxidation were carefully investigated. Based on the experimental observations of the probe reactions, we propose the ‘adsorption–activation’ mode of ortho-hydroxyl groups in GOR over NiOOH/Ni(OH)2. This work provides a strategy for designing high-performance Ni-based electrocatalysts that will boost the future development of sustainable biomass-derived polyol electrocatalytic valorization.Data availability
Supporting data for this work have been uploaded as the article ESI.†Author contributions
Zhuxin Gui: methodology, investigation, writing—original draft, writing—review and editing. Yingshuai Jia: methodology, investigation, writing—review and editing. Xianping Liao: investigation. Tianlan Yan: investigation. Boxu Gao: investigation. Wenbiao Zhang: investigation. Li Chen: investigation. Qingsheng Gao: conceptualization, writing—review and editing, funding acquisition, supervision. Yahong Zhang: supervision. Yi Tang: conceptualization, writing—review and editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the following funding agencies for supporting this work: the Natural Science Foundation of China (no. 22088101 to Yi Tang and 22175077 to Qingsheng Gao); the National Key R&D Program of China (no. 2023YFA1507602 and 2018YFA0209402 to Yi Tang).Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- F. Dawood, M. Anda and G. M. Shafiullah, Int. J. Hydrogen Energy, 2020, 45, 3847–3869 CrossRef CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2015, 45, 3954–3988 RSC.
- K. Sordakis, C. H. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- S. Chatterjee, I. Dutta, Y. W. Lum, Z. P. Lai and K. W. Huang, Energy Environ. Sci., 2021, 14, 1194–1246 RSC.
- I. Dutta, S. Chatterjee, H. F. Cheng, R. J. Parsapur, Z. L. Liu, Z. B. Li, E. Y. Ye, H. Kawanami, J. S. C. Low, Z. P. Lai, X. J. Loh and K. W. Huang, Adv. Energy Mater., 2022, 12, 2103799 CrossRef CAS.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, Weinheim, 2016 Search PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 2–21 CrossRef.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- J. X. Wu, X. J. Yang and M. Gong, Chin. J. Catal., 2022, 43, 2966–2986 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- Z. Y. Zhang, L. Xin, L. Qi, D. J. Chadderdon and W. Z. Li, Appl. Catal., B, 2013, 136, 29–39 CrossRef.
- K. Zakaria, M. McKay, R. Thimmappa, M. Hasan, M. Mamlouk and K. Scott, ChemElectroChem, 2019, 6, 2578–2585 CrossRef CAS.
- X. H. Wu, Y. Wang and Z. S. Wu, Chem, 2022, 8, 1–36 Search PubMed.
- M. Bender, Y. Lam, S. Hammes-Schiffer and K. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- J. L. Zhang, Y. Shen and H. Y. Li, ACS Appl. Energy Mater., 2023, 6, 5508–5518 CrossRef CAS.
- J. X. Wu, J. L. Li, Y. F. Li, X. Y. Ma, W. Y. Zhang, Y. M. Hao, W. B. Cai, Z. P. Liu and M. Gong, Angew. Chem., Int. Ed., 2022, 61, e202113362 CrossRef CAS PubMed.
- Y. Li, X. F. Wei, L. S. Chen, J. L. Shi and M. Y. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- Z. Y. He, J. W. Hwang, Z. H. Gong, M. Z. Zhou, N. Zhang, X. W. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed.
- Q. Z. Qian, X. Y. He, Z. Y. Li, Y. X. Chen, Y. F. Feng, M. Y. Cheng, H. K. Zhang, W. T. Wang, C. Xiao, G. Q. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS PubMed.
- W. S. Luo, H. Tian, Q. Li, G. Meng, Z. W. Chang, C. Chen, R. X. Shen, X. Yu, L. B. Zhu, F. T. Kong, X. Z. Cui and J. L. Shi, Adv. Funct. Mater., 2023, 2306995 Search PubMed.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Chem. Soc., Perkin Trans. 2, 1972, 10, 1396–1403 RSC.
- W. Chen, C. Xie, Y. Y. Wang, Y. Q. Zou, C. L. Dong, Y. C. Huang, Z. H. Xiao, Z. X. Wei, S. Q. Du, C. Chen, B. Zhou, J. M. Ma and S. Y. Wang, Chem, 2020, 6, 2974–2993 CAS.
- R. Y. Fan, X. J. Zhai, W. Z. Qiao, Y. S. Zhang, N. Yu, N. Xu, Q. X. Lv, Y. M. Chai and B. Dong, Nano-Micro Lett., 2023, 15, 190 CrossRef CAS PubMed.
- Y. Wang, Y. Q. Zhu, Z. H. Xie, S. M. Xu, M. Xu, Z. Z. Li, L. N. Ma, R. X. Ge, H. Zhou, Z. H. Li, X. G. Kong, L. R. Zheng, J. H. Zhou and H. H. Duan, ACS Catal., 2022, 12, 12432–42433 CrossRef CAS.
- Y. H. Pei, Z. F. Pi, H. Zhong, J. Cheng and F. M. Jin, J. Mater. Chem. A, 2022, 10, 1309–1319 RSC.
- L. F. Fan, Y. X. Ji, G. X. Wang, J. X. Chen, K. Chen, X. Liu and Z. H. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- Y. Xu, T. T. Liu, K. K. Shi, H. J. Yu, K. Deng, X. Wang, Z. Q. Wang, L. Wang and H. J. Wang, J. Mater. Chem. A, 2022, 10, 20365–20374 RSC.
- J. W. Du, Y. Qin, T. Dou, J. M. Ge, Y. P. Wang, X. H. Zhao, F. Z. Zhang and X. D. Lei, ACS Appl. Nano Mater., 2022, 5, 10174–10182 CrossRef CAS.
- L. F. Fan, Y. X. Ji, G. X. Wang, Z. F. Zhang, L. C. Yi, K. Chen, X. Liu and Z. H. Wen, J. Energy Chem., 2022, 72, 424–431 CrossRef CAS.
- H. B. Wan, C. C. Dai, L. J. Jin, S. Z. Luo, F. X. Meng, G. Chen, Y. Duan, C. T. Liu, Q. F. Xu, J. M. Lu and Z. C. Xu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS PubMed.
- S. Cychy, S. Lechler, Z. J. Huang, M. Braun, A. C. Brix, P. Blumler, C. Andronescu, F. Schmid, W. Schuhmann and M. Muhler, Chin. J. Catal., 2021, 42, 2206–2215 CrossRef CAS.
- J. L. Zhang and Y. Shen, J. Electrochem. Soc., 2021, 168, 084510 CrossRef CAS.
- X. T. Han, H. Y. Sheng, C. Yu, T. Walker, G. Huber, J. S. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- M. Houache, R. Safari, U. Nwabara, T. Rafaideen, G. Botton, P. Kenis, S. Baranton, C. Coutanceau and E. Baranova, ACS Appl. Energy Mater., 2020, 3, 8725–8738 CrossRef CAS.
- C. C. Streinz, S. Motupally and J. W. Weidner, J. Electrochem. Soc., 1995, 142, 4051–4056 CrossRef CAS.
- G. W. Yang, Y. Q. Wu, B. L. Wang, W. Y. Guo, Z. Y. Ren, Z. H. Bu, C. C. Miao and H. L. Li, J. Solid State Electrochem., 2012, 16, 3761–3767 CrossRef CAS.
- Z. P. Yao, J. K. Wang, Y. J. Wang, T. P. Xie, C. Y. Li and Z. H. Jiang, Electrochim. Acta, 2021, 382, 138342 CrossRef CAS.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solutions, Dekker, 1985 Search PubMed.
- D. M. Zhu, C. Q. Xia, Z. D. Yang, T. Yang, Q. Li and R. P. Liu, Electrochim. Acta, 2020, 334, 135591 CrossRef CAS.
- J. J. Zhang, T. A. Zhang and S. Feng, Int. J. Hydrogen Energy, 2021, 46, 41–49 CrossRef CAS.
- J. Bantignies, S. Deabate, A. Righi, S. Rols, P. Hermet, J. Sauvajol and F. Henn, J. Phys. Chem. C, 2008, 112, 2193–2201 CrossRef CAS.
- D. Hall, D. Lockwood, C. Bock and B. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- D. Hall, D. Lockwood, S. Poirier, C. Bock and B. MacDougall, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- A. Sika-Nartey, Y. Sahin, K. Ercan, Z. Kap, Y. Kocak, A. Erdali, B. Erdivan, Y. Turkmen and E. Ozensoy, ACS Appl. Nano Mater., 2022, 5, 18855–18870 CrossRef CAS.
- C. Faure and C. Delmas, J. Power Sources, 1991, 35, 279–290 CrossRef CAS.
- A. Grosvenor, M. Biesinger, R. S. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- M. Biesinger, B. P. Payne, L. W. Lau, A. Gerson and R. S. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- X. Y. Lu, K. H. Wu, B. S. Zhang, J. N. Chen, F. Li, B. J. Su, P. Q. Yan, J. M. Chen and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 14528–14535 CrossRef CAS PubMed.
- J. F. Xie, L. Gao, S. S. Cao, W. W. Liu, F. C. Lei, P. Hao, X. Y. Xia and B. Tao, J. Mater. Chem. A, 2019, 7, 13577–13584 RSC.
- A. Mansour and C. Melendres, Surf. Sci. Spectra, 1994, 3, 255–262 CrossRef CAS.
- H. L. Huang, C. Yu, X. T. Han, H. W. Huang, Q. B. Wei, W. Guo, Z. Wang and J. S. Qiu, Energy Environ. Sci., 2020, 13, 4990–4999 RSC.
- P. Oliva, J. Leonardi and J. Laurent, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- B. Trzesniewski, O. Diaz-Morales, D. Vermaas, A. Longo, W. Bras, M. Koper and W. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef CAS PubMed.
- J. W. Huang, Y. Y. Li, Y. D. Zhang, G. F. Rao, C. Y. Wu, Y. Hu, X. F. Wang, R. F. Lu, Y. R. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 CrossRef CAS PubMed.
- R. Kostecki and F. McLarnon, J. Electrochem. Soc., 1997, 144, 485–493 CrossRef CAS.
- M. Louie and A. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- B. Yeo and A. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- Q. Zhang, W. Xiao, H. C. Fu, X. L. Li, J. L. Lei, H. Q. Luo and N. B. Li, ACS Catal., 2023, 13, 14975–14986 CrossRef CAS.
- Y. P. Wei, L. Y. Yi, R. F. Wang, J. Y. Li, D. Z. Li, T. H. Li, W. Sun and W. H. Hu, Small, 2023, 19, 2301267 CrossRef CAS.
- R. Zhao, S. Xu, D. Y. Liu, L. T. Wei, S. Y. Yang, X. L. Yan, Y. B. Chen, Z. H. Zhou, J. Z. Su, L. J. Guo and C. Burda, Appl. Catal., B, 2023, 338, 123027 CrossRef CAS.
- M. Bernard, P. Bernard, M. Keddam, S. Senyarich and H. Takenouti, Electrochim. Acta, 1996, 41, 91–93 CrossRef CAS.
- M. Freitas, J. Power Sources, 2001, 93, 163–173 CrossRef CAS.
- S. Klaus, Y. Cai, M. Louie, L. Trotochaud and A. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- Y. X. Lu, T. Y. Liu, C. L. Dong, Y. C. Huang, Y. F. Li, J. Chen, Y. Q. Zou and S. Y. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- P. Zhou, X. S. Lv, S. S. Tao, J. C. Wu, H. F. Wang, X. X. Wei, T. H. Wang, B. Zhou, Y. X. Lu, T. Frauenheim, X. Z. Fu, S. Y. Wang and Y. Q. Zou, Adv. Mater., 2022, 34, 2204089 CrossRef CAS PubMed.
- Y. C. Yang, D. X. Xu, B. L. Zhang, Z. M. Xue and T. C. Mu, Chem. Eng. J., 2022, 433, 133842 CrossRef CAS.
- W. Chen, J. Q. Shi, C. Xie, W. Zhou, L. T. Xu, Y. Y. Li, Y. D. Wu, B. B. Wu, Y. C. Huang, B. Zhou, M. Yang, J. L. Liu, C. L. Dong, T. H. Wang, Y. Q. Zou and S. Y. Wang, Natl. Sci. Rev., 2023, 10, nwad099 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, C. Hao, S. Wang and H. C. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206399119 CrossRef CAS PubMed.
- W. Chen, Y. Y. Wang, B. B. Wu, J. Q. Shi, Y. Y. Li, L. T. Xu, C. Xie, W. Zhou, Y. C. Huang, T. H. Wang, S. Q. Du, M. L. Song, D. D. Wang, C. Chen, J. Y. Zheng, J. L. Liu, C. L. Dong, Y. Q. Zou, J. Chen and S. Y. Wang, Adv. Mater., 2022, 34, 2105320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental Section, supporting figures and tables. See DOI: https://doi.org/10.1039/d4sc01221f |
| This journal is © The Royal Society of Chemistry 2024 |