
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonding bolstered head-to-tail ligation of functional chromophores in a 0D SbF3·glycine adduct for a short-wave ultraviolet nonlinear optical material†
Zhiyong
Bai
a,
Jihyun
Lee
a,
Chun-Li
Hu
*b,
Guohong
Zou
 *c and
Kang Min
Ok
*a
*c and
Kang Min
Ok
*a
aDepartment of Chemistry, Sogang University, Seoul, 04107, Republic of Korea. E-mail: kmok@sogang.ac.kr
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China
cCollege of Chemistry, Sichuan University, Chengdu, 610065, P. R. China
First published on 1st April 2024
Abstract
The key properties of nonlinear optical (NLO) materials highly rely on the quality of functional chromophores (FCs) and their optimized interarrangement in the lattice. Despite the screening of various FCs, significant challenges persist in optimizing their arrangement within specific structures. Generally, FC alignment is achieved by designing negatively charged 2D layers or 3D frameworks, further regulated by templating cations. In this study, a novel 0D adduct NLO material, SbF3·glycine, is reported. Neutrally charged 0D [SbF3C2H5NO2] FCs, comprising [SbF3] pyramids and zwitterionic glycine, are well-aligned in the structure. The alignment is facilitated by the hydrogen bonding, reinforcing a ‘head-to-tail’ ligation of [SbF3C2H5NO2] FCs. Consequently, the title compound exhibits favorable NLO properties, including a large second-harmonic generation efficiency (3.6 × KDP) and suitable birefringence (cal. 0.057 @ 1064 nm). Additionally, its short absorption cut-off edge (231 nm) positions it as a promising short-wave ultraviolet NLO material. Importantly, the binary SbF3–amino acid system is expected to serve as a new resource for exploring ultraviolet NLO crystals, owing to the abundance of the amino acid family.
Nonlinear optical (NLO) materials have the capability to efficiently generate coherent light with photon energies in the short-wavelength range, such as ultraviolet (UV, 200–400 nm) and deep-UV (below 200 nm), through a second-harmonic generation (SHG) process. Consequently, they find wide applications in various laser-driven technologies, including laser photolithography, quantum entanglement, medical treatment, and laser micromachining.1–4 From a materials perspective, the ongoing challenge lies in discovering new UV NLO materials that exhibit a large SHG coefficient, possesses significant birefringence, and have a short absorption cut-off edge (a wide band gap). However, achieving these properties poses a challenge attributable to the contradictory relationship among them.5 Over the past few decades, the exploration of promising UV NLO crystals has primarily focused on borates. This is because planar [BO3]3− and ring-type [B3O6]3− functional chromophores (FCs) in borates enable them to exhibit large SHG efficiency and birefringence.6–14 Subsequently, the exploration of UV NLO materials has extended to carbonates, nitrates, and cyanurates.15–22
In addition to π-conjugated anions, stereochemically active lone pair (SCALP) cations such as Pb2+, Sn2+, Bi3+, and Sb3+ are also considered to be good FCs for designing high-performance NLO materials.11,23–36 This is because these SCALP ions are commonly susceptible to forming highly distorted polyhedra, thereby giving rise to large SHG efficiency and birefringence. However, Pb2+ is toxic, while Sn2+ and Bi3+ usually cause red-shifted cutoff edges. In recent years, Zou and coworkers discovered that SCALP Sb3+ could be a good UV NLO FC because it can endow materials with short cutoff edges (commonly below 266 nm, the fourth harmonic generation of a 1064 nm Nd:YAG laser) while retaining a large SHG effect and birefringence. This has been demonstrated by a few antimony sulfates and phosphates, such as CsSbF2SO4,37 RbSbSO4Cl2,38 and ASbP2O7F (A = K and Rb), among others.39,40 To date, the development of Sb3+-based UV NLO materials has been mainly limited to sulfate and phosphate salts, with most of these materials exhibiting relatively lower SHG intensity attributable to the unfavorable arrangement of antimony polyhedra and the lesser contribution of SO42− or PO43− units to SHG.35,41,42 Given this context, achieving a uniformly arranged lattice of Sb3+-based polyhedra and concurrently introducing other types of FCs has become important.43 However, the desired uniform alignment of Sb3+-based polyhedra is only realized in a few crystals, such as 3D fresnoite-type ASbP2O7F (A = K and Rb). In this study, we endeavor to expand the frontiers of Sb3+-based UV NLO materials by combining Sb3+ with amino acids, a rarely explored approach. The amino acids are expected to serve not only as additional FCs but also to optimize the arrangement of Sb3+-based polyhedra through hydrogen bonding. Herein, we report a novel 0D adduct NLO material, SbF3·glycine, composed of stoichiometric antimony(III) trifluoride and the amino acid glycine. The antimony polyhedra and glycine molecules interact with each other through hydrogen bonding, resulting in a uniform ‘head-to-tail’ arrangement of [SbF3C2H5NO2] clusters, which gives rise to excellent NLO properties.
SbF3·Gly crystals were synthesized using an aqueous solution evaporation technique at room temperature (see details in the Experimental section in the ESI†). SbF3·Gly is somewhat hygroscopic in wet air; therefore, it should be stored in a dry environment, such as a desiccator. The powder X-ray diffraction patterns (XRD) recorded on a Miniflex 600 diffractometer matched well with the simulated ones (Fig. S1†), thus confirming phase purity. Elemental analysis based on an energy-dispersive spectrometer (EDS) revealed the presence of Sb, O, F, C, and N (Fig. S2†). Thermogravimetric (TG) testing showed that SbF3·Gly is thermally stable up to ca. 105 °C, beyond which it undergoes slow decomposition (Fig. S3†).
The single-crystal XRD analysis reveals a noncentrosymmetric polar space group, Pc (no. 7) for SbF3·Gly, with well-refined R values (Table S1†). It belongs to an adduct compound composed of stoichiometric SbF3 and zwitterionic glycine (Fig. 1a), which are further connected to form 0D [SbF3C2H5NO2] clusters (Fig. 1b) by sharing the O(2) atom. Antimony is four-coordinated, producing a one-sided [SbOF3] seesaw, with an Sb–O bond length of 2.243(5) Å and Sb–F interatomic distances ranging from 1.914(4) to 2.036(5) Å (Fig. 1b). The bond valence calculations reveal a bond valence sum value of +2.906 for the cationic ion Sb3+, consistent with its expected oxidation state of +3 (Table S7†). In the structure, the discrete [SbF3C2H5NO2] clusters repeat with a ‘head-to-tail’ ligation model, with SbF3 serving as the ‘head’ and glycine as the ‘tail’. It should be pointed out that the ‘head’ and ‘tail’ are connected and bolstered by N–H⋯F hydrogen bonding with H⋯F bond lengths ranging from 2.750(7) to 2.924(6) Å (Fig. 1c and Table S6†) to form the entire structure, highlighting the crucial roles of hydrogen bonding in the formation and support of ‘head-to-tail’ linkage of FCs in SbF3·Gly. Furthermore, it is worth noting that such ‘head-to-tail’ ligation of 0D [SbF3C2H5NO2] clusters aligns them with each other, resulting in a uniform arrangement in the lattice, with the lone pair of Sb3+ ions oriented along the rough [111] direction (Fig. 1d). The ‘head-to-tail’ connection gives rise to constructively added net dipole moments of [SbOF3] polyhedra along the [100] and [001] directions (Table S8†), which is favourable for stimulating large SHG for SbF3·Gly.
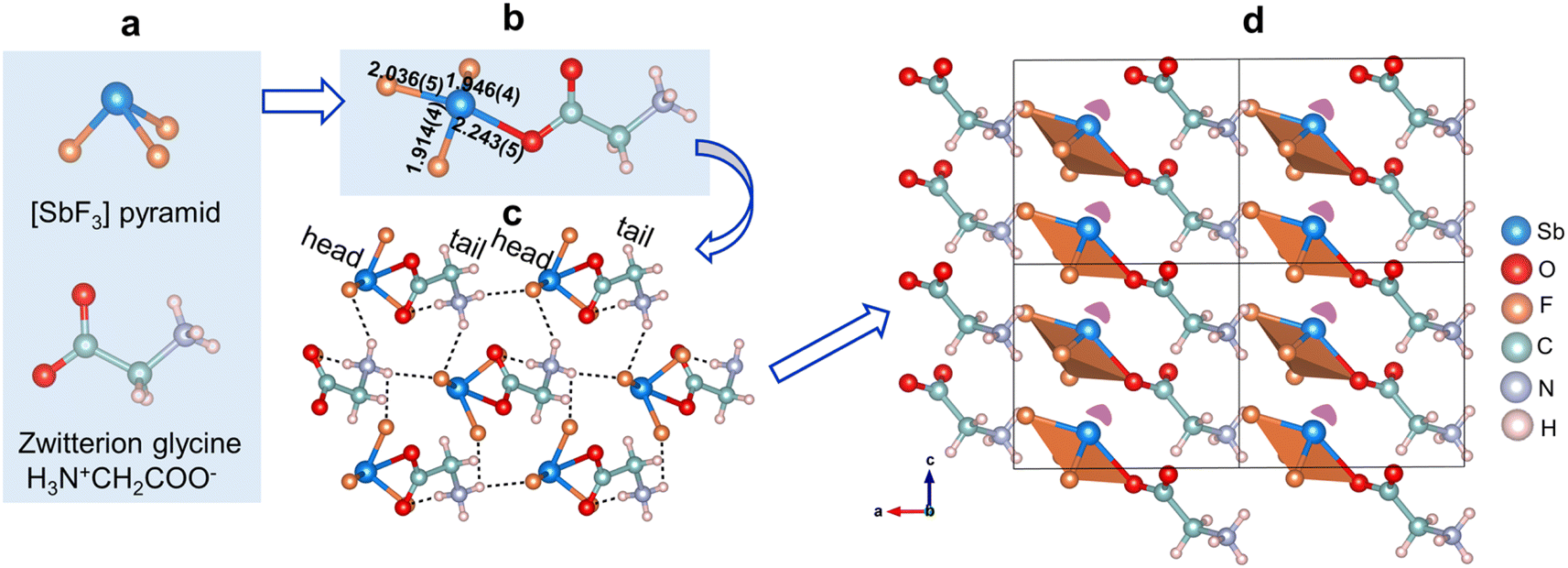 | ||
| Fig. 1 The crystal structure of the molecular SbF3·Gly compound. The SbF3 and zwitterion glycine primitives (a) combine to form 0D [SbF3C2H5NO2] clusters (b) by sharing the O(2) atom, which repeats periodically in the structure with head-to-tail ligation, reinforced by hydrogen bonding (c). The alignment of [SbF3C2H5NO2] clusters in the double unit cell (d). The purple lobe represents the lone pair of Sb3+. | ||
The UV-vis diffuse reflectance spectrum (Fig. 2a) suggests that SbF3·Gly is transparent in the UV region with a short UV absorption cut-off (λcuf-off) edge of ca. 231 nm, falling within the solar-blind UV region (210–280 nm). This value is one of the shortest observed among Sb3+-based UV NLO crystals,37,39,44 indicating that SbF3·Gly is suitable for short-wave UV applications. Furthermore, using the Kubelka–Munk function, its optical band gap is determined to be 4.63 eV (Fig. 2b).
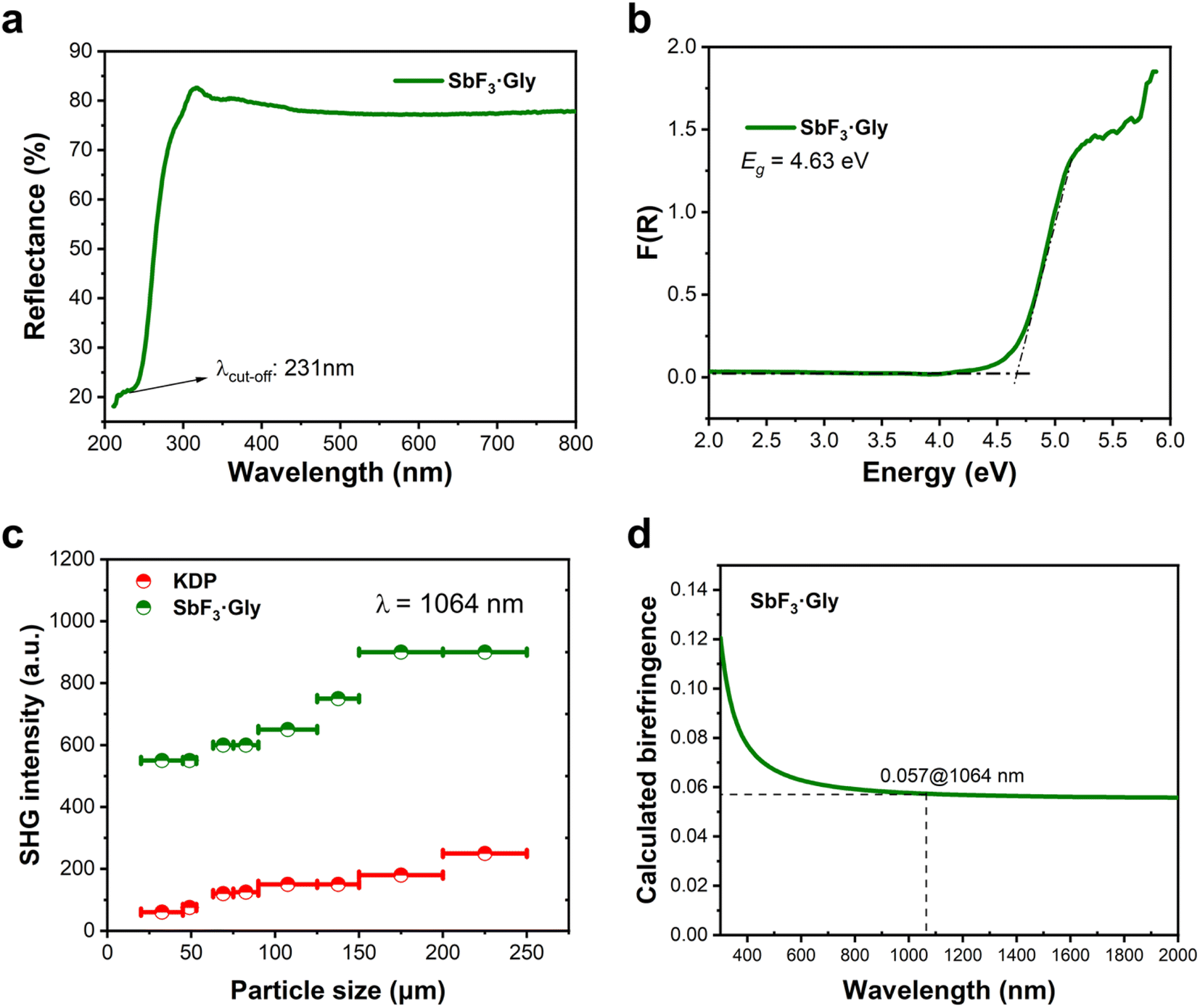 | ||
| Fig. 2 The linear and nonlinear optical properties of SbF3·Gly. (a) The UV-vis diffuse reflectance spectrum and (b) optical band gap (Eg) determined using the Kubelka–Munk equation. (c) Particle size-dependent powder SHG efficiency. (d) Calculated birefringence. | ||
As the title compound crystallizes in an acentric space group, its powder SHG efficiency was measured based on the Kurtz–Perry method under 1064 nm laser irradiation.45 The standard KH2PO4 (KDP) crystal was used as a reference. As shown in Fig. 2c, the SHG intensities increase with rising particle size before reaching maximum values independent of particle size, indicating that SbF3·Gly is phase-matchable in the visible region. The phase-matching (PM) ability of SbF3·Gly is also confirmed by its moderate birefringence, as discussed later. Furthermore, SbF3·Gly exhibits a large powder SHG efficiency, ca. 3.6 times that of KDP for the same particle size range of 200–250 μm. It should be noted that the Flack parameter for SbF3·Gly is 0.20(4) (Table S1†), indicating that the crystal is slightly twinned. Thus, SHG produced by one twin may be slightly weakened by the other. In other words, the real SHG efficiency is probably larger than 3.6 × KDP if the crystal is not twinned. The SHG ability of the SbF3·Gly adduct is comparable to that of numerous potential short-wave UV NLO crystals, including Sb-based SbB3O6 (3.5 × KDP),46 K2Sb(P2O7)F (4.0 × KDP),39 CsSbF2SO4 (3.0 × KDP),37 ASbSO4Cl2 (2.7 × KDP for A = Rb and 1.7 × KDP for A = NH4),38,41 Rb2SbF3(NO3)2 (2.7 × KDP);47 borates such as CaZn2(BO3)2 (3.8 × KDP)48 and Ca2B3O6X (1.5 × KDP for X = Cl and 2.0 × KDP for X = Br);49 carbonates such as AMCO3F (A = alkali, M = alkaline earth, 1.11–4.0 × KDP),15 NaZnCO3(OH) (5.2 × KDP),18 and LiZnCO3(OH) (3.2 × KDP),17 sulfates including MF2SO4 (3.2 × KDP for M = Zr and 2.5 × KDP for M = Hf)50 and sulfamates such as Ba(NH2SO3)2 (2.7 × KDP),51 SO2(NH2)2 (4.0 × KDP)52 and Cs2Mg(NH2SO3)4·4H2O (2.3 × KDP);53 selenites such as Sc(HSeO3)3 (5.0 × KDP)54 and Y3F(SeO3)4 (5.5 × KDP);55 and a few hybrid materials such as KLi(HC3N3O3)·2H2O (5.3 × KDP),21 Rb[PO2(NH3)(CO)2]·0.5H2O (4.2 × KDP),56 and 2(C3H7N6)+·2Cl·2H2O (4.3 × KDP),57 among others.
To better understand the structure–property relationship of SbF3·Gly, first principles calculations were carried out using the plane-wave pseudopotential method implemented in the CASTEP package, based on density functional theory (see details in the ESI†). The calculated band gap of SbF3·Gly under the GGA-PBE functional is 4.23 eV (Fig. S4†), which is slightly less than the experimental value (4.63 eV). Therefore, a scissor operator of 0.40 eV was applied when assessing the optical properties. The total and partial densities of states for SbF3·Gly are plotted in Fig. S5.† Specifically, the upper part of the valence band (−3.2 to 0.0 eV) is dominated by O 2p and Sb 5s5p electronic states, while the bands ranging from −10.0 to −3.2 eV are mainly from F 2p, O 2p, C 2p and N 2p electronic states, mixed with a small amount of H 1s and Sb 5s5p states. The conduction band primarily comprises unoccupied Sb 5p, C 2p, O 2p, and H 1s orbitals, as well as a small number of N 2p orbitals. It is worth noting that the valence band maximum is contributed by the nonbonding states of O 2p and Sb 5s5p, while the conduction band minimum originates from the π*-antibonding states of C–O bonds in glycine. Thus, the band gap of SbF3·Gly is determined by the Sb3+-based polyhedra and glycine molecules.
Fig. 2d displays the calculated birefringence (Δn) curve, from which we can observe that the birefringence of SbF3·Gly at 1064 nm is about 0.057. This value falls within a desirable range for a UV NLO material, categorized as ‘moderate’, which ensures good PM capability in the SHG process. We further examined its PM capacity based on the calculated refractive indices (Fig. S6†). The results reveal that its shortest PM wavelength reaches 366 nm, confirming its PM ability in the visible region and suggesting potential applications for UV laser output through the SHG process. It is worth noting that the shortest PM wavelength of SbF3·Gly is 135 nm longer than its λcut-off edge, resulting in a 135 nm loss of PM. This discrepancy is likely owing to the severe refractive dispersion effect (the curves become suddenly steep) below the 500 nm energy region, which prevents the PM from further approaching shorter wavelengths.
Considering structural symmetry (point group: m) and Kleinman symmetry, SbF3·Gly exhibits 10 nonvanishing SHG tensors, namely
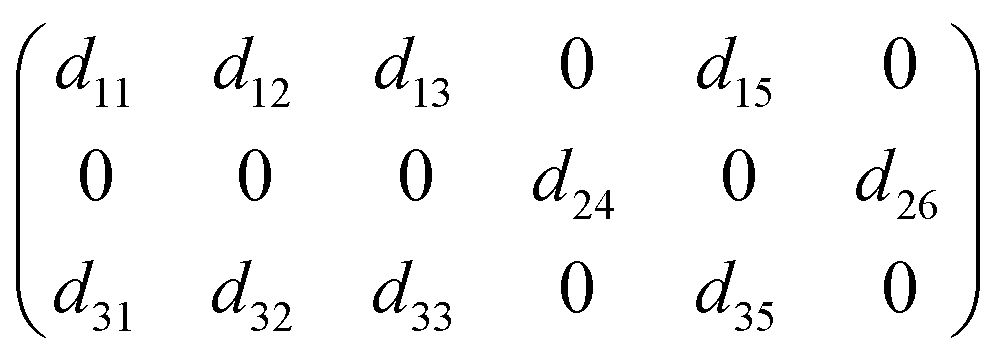
Among these, d11, d12 = d26, d13 = d35, d15 = d31, d24 = d32, and d33. The static values of these tensors were calculated and are listed in Table S9.† Notably, d32 exhibits the largest tensor, 1.48 pm V−1, which is approximately 3.8 times that of KDP (d36 = 0.39 pm V−1), aligning well with the powder SHG efficiency (3.6 × KDP). To further uncover the underlying origin of the strong SHG effect in SbF3·Gly, SHG-weighted electron density analyses for d32 were performed (Fig. 3). It was found that in the valence band (VB) (Fig. 3a), the O 2p nonbonding states contribute significantly to the SHG, with additional contributions from F 2p and C 2p states. In the conduction band (CB) (Fig. 3b), the SHG effect primarily originates from the π*-antibonding states of C–O bonds and Sb 5p orbitals, along with minor contributions from F 2p orbitals. Based on the SHG-density data, the group contributions can be delineated as 42.16% for [SbOF3] and 57.84% for glycine, indicating that the collaborative effect of [SbOF3] and glycine results in the SHG response of SbF3·Gly.
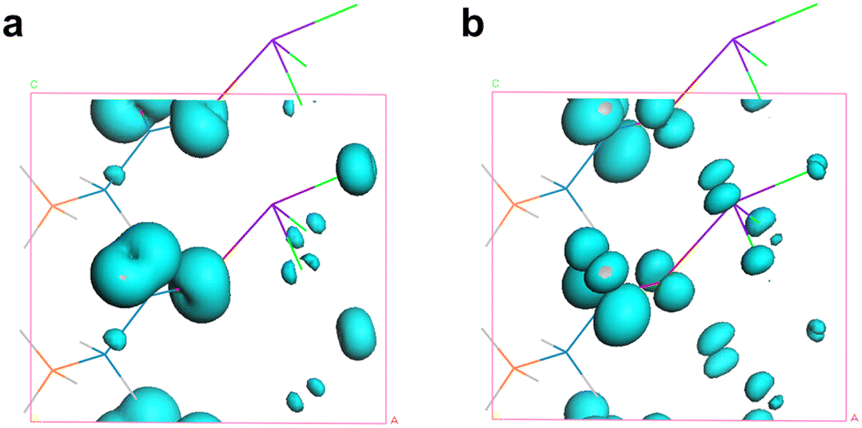 | ||
| Fig. 3 The SHG-weighted electron density in (a) the VB and (b) CB for the d32 coefficient. | ||
Conclusions
In summary, we present a new UV NLO material, SbF3·Gly, based on Sb3+. This compound comprises 0D [SbF3C2H5NO2] FCs formed by [SbF3] pyramids and zwitterionic glycine, sharing the oxygen corner. These [SbF3C2H5NO2] FCs adopt a head-to-tail arrangement sustained by hydrogen bonding, resulting in a large SHG response, 3.6 times that of KDP, and a moderate birefringence (Δn(cal.) = 0.057) at 1064 nm, which imposes a PM limit of 366 nm. In addition, the material also exhibits a short absorption edge of 231 nm, one of the smallest values among Sb3+-based NLO materials. These attributes position SbF3·Gly as a potential short-wavelength UV NLO material.Data availability
Crystallographic data for SbF3·Gly have been deposited with the CCDC under deposition number 22049585 and can be obtained from https://www.ccdc.cam.ac.uk/. Other data are available from the authors upon request.Author contributions
Z. Bai synthesized and characterized the compound, J. Lee carried out the SHG measurement, C.-L. Hu performed the theoretical calculations, G. Zou analyzed the data, and K. M. Ok devised and supervised the experiments. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (nos. 2018R1A5A1025208 and 2019R1A2C3005530).Notes and references
- M. Mutailipu, M. Zhang, Z. Yang and S. Pan, Acc. Chem. Res., 2019, 52, 791–801 CrossRef CAS.
- M. Mutailipu, K. R. Poeppelmeier and S. Pan, Chem. Rev., 2021, 121, 1130–1202 CrossRef CAS.
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Nat. Photonics, 2023, 17, 694–701 CrossRef CAS.
- Y. Li, J. Luo and S. Zhao, Acc. Chem. Res., 2022, 55, 3460–3469 CrossRef CAS PubMed.
- M. Mutailipu, Z. Yang and S. Pan, Acc. Mater. Res., 2021, 2, 282–291 CrossRef CAS.
- C. Chen, Y. Wu, A. Jiang, B. Wu, G. You, R. Li and S. Lin, J. Opt. Soc. Am. B, 1989, 6, 616–621 CrossRef CAS.
- C. Chen, B. Wu, A. Jiang and G. You, Sci. Sin., Ser. B, 1985, 28, 235–243 Search PubMed.
- S. Zhao, P. Gong, L. Bai, X. Xu, S. Zhang, Z. Sun, Z. Lin, M. Hong, C. Chen and J. Luo, Nat. Commun., 2014, 5, 4019 CrossRef CAS PubMed.
- H. Wu, S. Pan, K. R. Poeppelmeier, H. Li, D. Jia, Z. Chen, X. Fan, Y. Yang, J. M. Rondinelli and H. Luo, J. Am. Chem. Soc., 2011, 133, 7786–7790 CrossRef CAS.
- Y. Yang, S. Huang and S. Pan, J. Mater. Chem. C, 2022, 10, 11232–11238 RSC.
- G. Zou, C. Lin, H. Jo, G. Nam, T. S. You and K. M. Ok, Angew. Chem., Int. Ed., 2016, 55, 12078–12082 CrossRef CAS.
- X. Wang, Y. Wang, B. Zhang, F. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2017, 129, 14307–14311 CrossRef.
- H. Yu, H. Wu, S. Pan, Z. Yang, X. Hou, X. Su, Q. Jing, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2014, 136, 1264–1267 CrossRef CAS PubMed.
- M. Mutailipu, M. Zhang, H. Wu, Z. Yang, Y. Shen, J. Sun and S. Pan, Nat. Commun., 2018, 9, 3089 CrossRef PubMed.
- G. Zou, N. Ye, L. Huang and X. Lin, J. Am. Chem. Soc., 2011, 133, 20001–20007 CrossRef CAS PubMed.
- G. Zou, L. Huang, N. Ye, C. Lin, W. Cheng and H. Huang, J. Am. Chem. Soc., 2013, 135, 18560–18566 CrossRef CAS.
- X. Liu, L. Kang, P. Gong and Z. Lin, Angew. Chem., Int. Ed., 2021, 60, 13574–13578 CrossRef CAS.
- G. Peng, C. Lin and N. Ye, J. Am. Chem. Soc., 2020, 142, 20542–20546 CrossRef CAS.
- J. Wang, Y. Cheng, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Angew. Chem., Int. Ed., 2022, 61, e202201616 CrossRef CAS PubMed.
- X. Dong, L. Huang, Q. Liu, H. Zeng, Z. Lin, D. Xu and G. Zou, Chem. Commun., 2018, 54, 5792–5795 RSC.
- D. Lin, M. Luo, C. Lin, F. Xu and N. Ye, J. Am. Chem. Soc., 2019, 141, 3390–3394 CrossRef CAS PubMed.
- J. Lu, Y. K. Lian, L. Xiong, Q. R. Wu, M. Zhao, K. X. Shi and L. M. Wu, J. Am. Chem. Soc., 2019, 141, 16151–16159 CrossRef CAS.
- L. Qi, Z. Chen, X. Shi, X. Zhang, Q. Jing, N. Li and M. H. Lee, Chem. Mater., 2020, 32, 8713–8723 CrossRef CAS.
- M. Abudoureheman, S. Han, B. H. Lei, Z. Yang, X. Long and S. Pan, J. Mater. Chem. C, 2016, 4, 10630–10637 RSC.
- S.-H. Kim, J. Yeon and P. S. Halasyamani, Chem. Mater., 2009, 21, 5335–5342 CrossRef CAS.
- Y. Z. Huang, L. M. Wu, X. T. Wu, L. H. Li, L. Chen and Y. F. Zhang, J. Am. Chem. Soc., 2010, 132, 12788–12789 CrossRef CAS PubMed.
- M. Luo, Y. Song, F. Liang, N. Ye and Z. Lin, Inorg. Chem. Front., 2018, 5, 916–921 RSC.
- F. You, F. Liang, Q. Huang, Z. Hu, Y. Wu and Z. Lin, J. Am. Chem. Soc., 2019, 141, 748–752 CrossRef CAS.
- H. Zhang, M. Zhang, S. Pan, X. Dong, Z. Yang, X. Hou, Z. Wang, K. B. Chang and K. R. Poeppelmeier, J. Am. Chem. Soc., 2015, 137, 8360–8363 CrossRef CAS PubMed.
- X. Chen, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2020, 59, 20323–20327 CrossRef CAS.
- H. Yu, N. Z. Koocher, J. M. Rondinelli and P. S. Halasyamani, Angew. Chem., Int. Ed., 2018, 57, 6100–6103 CrossRef CAS PubMed.
- Y. J. Jia, X. Zhang, Y. G. Chen, X. Jiang, J. N. Song, Z. Lin and X. M. Zhang, Inorg. Chem., 2022, 61, 15368–15376 CrossRef CAS PubMed.
- J. Guo, S. Cheng, S. Han, Z. Yang and S. Pan, Adv. Opt. Mater., 2020, 9, 2001734 CrossRef.
- K. Chen, Y. Yang, G. Peng, S. Yang, T. Yan, H. Fan, Z. Lin and N. Ye, J. Mater. Chem. C, 2019, 7, 9900–9907 RSC.
- Q. Wei, C. He, K. Wang, X. F. Duan, X. T. An, J. H. Li and G. M. Wang, Chem.–Eur. J., 2021, 27, 5880–5884 CrossRef CAS.
- Q. Wei, K. Wang, C. He, L. Wei, X. F. Li, S. Zhang, X. T. An, J. H. Li and G. M. Wang, Inorg. Chem., 2021, 60, 11648–11654 CrossRef CAS.
- X. Dong, L. Huang, C. Hu, H. Zeng, Z. Lin, X. Wang, K. M. Ok and G. Zou, Angew. Chem., Int. Ed., 2019, 58, 6528–6534 CrossRef CAS PubMed.
- F. He, Y. Deng, X. Zhao, L. Huang, D. Gao, J. Bi, X. Wang and G. Zou, J. Mater. Chem. C, 2019, 7, 5748–5754 RSC.
- Y. Deng, L. Huang, X. Dong, L. Wang, K. M. Ok, H. Zeng, Z. Lin and G. Zou, Angew. Chem., Int. Ed., 2020, 59, 21151–21156 CrossRef CAS.
- X. Dong, H. Huang, L. Huang, Y. Zhou, B. Zhang, H. Zeng, Z. Lin and G. Zou, Angew. Chem., Int. Ed., 2024, e202318976, DOI:10.1002/anie.202318976.
- F. He, Q. Wang, C. Hu, W. He, X. Luo, L. Huang, D. Gao, J. Bi, X. Wang and G. Zou, Cryst. Growth Des., 2018, 18, 6239–6247 CrossRef CAS.
- F. Yang, L. Wang, Y. Ge, L. Huang, D. Gao, J. Bi and G. Zou, J. Alloys Compd., 2020, 834 Search PubMed.
- K. M. Ok, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef CAS.
- S. Han, A. Tudi, W. Zhang, X. Hou, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2023, 62, e202302025 CrossRef CAS.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- Y. Liu, X. Liu, S. Liu, Q. Ding, Y. Li, L. Li, S. Zhao, Z. Lin, J. Luo and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 7793–7796 CrossRef CAS PubMed.
- L. Wang, H. Wang, D. Zhang, D. Gao, J. Bi, L. Huang and G. Zou, Inorg. Chem. Front., 2021, 8, 3317–3324 RSC.
- M. Mutailipu, F. Li, C. Jin, Z. Yang, K. R. Poeppelmeier and S. Pan, Angew. Chem., Int. Ed., 2022, 61, e202202096 CrossRef CAS PubMed.
- H. Qiu, F. Li, Z. Li, Z. Yang, S. Pan and M. Mutailipu, J. Am. Chem. Soc., 2023, 145, 24401–24407 CrossRef CAS PubMed.
- C. Wu, C. Jiang, G. Wei, X. Jiang, Z. Wang, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2023, 145, 3040–3046 CrossRef CAS PubMed.
- X. Hao, M. Luo, C. Lin, G. Peng, F. Xu and N. Ye, Angew. Chem., Int. Ed., 2021, 60, 7621–7625 CrossRef CAS PubMed.
- H. Tian, N. Ye and M. Luo, Angew. Chem., Int. Ed., 2022, 61, e202200395 CrossRef CAS PubMed.
- X. Wang, X. Leng, Y. Kuk, J. Lee, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2024, 63, e202315434 CrossRef CAS.
- Z. Bai, J. Lee, H. Kim, Y. Kuk, M. H. Choi, C. L. Hu and K. M. Ok, Small, 2023, 2207709 CrossRef CAS.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Angew. Chem., Int. Ed., 2023, 62, e202301420 CrossRef CAS PubMed.
- X. Song, Z. Du, B. Ahmed, Y. Li, Y. Zhou, Y. Song, W. Huang, J. Zheng, J. Luo and S. Zhao, Inorg. Chem. Front., 2023, 10, 5462–5467 RSC.
- L. Liu, C. L. Hu, Z. Bai, F. Yuan, Y. Huang, L. Zhang and Z. Lin, Chem. Commun., 2020, 56, 14657–14660 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of crystal data and structure refinement, fractional atomic coordinates, anisotropic displacement parameters, selected bond lengths and bond angles, hydrogen bonds, dipole moments, and second-order nonlinear optical coefficients, figures of PXRD, EDS, TG, band structures, PDOS, and simulated phase-matching wavelength. CCDC 2049585. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01353k |
| This journal is © The Royal Society of Chemistry 2024 |