
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWide band gap selenide infrared nonlinear optical materials AIIMg6Ga6Se16 with strong SHG responses and high laser-induced damage thresholds†
Linan
Wang‡
ab,
Dongdong
Chu‡
ab,
Zhihua
Yang
 ab,
Junjie
Li
*ab and
Shilie
Pan
*ab
ab,
Junjie
Li
*ab and
Shilie
Pan
*ab
aResearch Center for Crystal Materials, State Key Laboratory of Functional Materials and Devices for Special Environmental Conditions, Xinjiang Key Laboratory of Functional Crystal Materials, Xinjiang Technical Institute of Physics & Chemistry, CAS, 40-1 South Beijing Road, Urumqi 830011, China. E-mail: lijunjie@ms.xjb.ac.cn; slpan@ms.xjb.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 2nd April 2024
Abstract
Infrared (IR) nonlinear optical (NLO) materials with strong NLO response, wide band gap and high laser-induced damage threshold (LIDT) are highly expected in current laser technologies. Herein, by introducing double alkaline-earth metal (AEM) atoms, three wide band gap selenide IR NLO materials AIIMg6Ga6Se16 (AII = Ca, Sr, Ba) with excellent linear and NLO optical properties have been rationally designed and fabricated. AIIMg6Ga6Se16 (AII = Ca, Sr, Ba) are composed of unique [AIISe6] triangular prisms, [MgSe6] octahedra and [GaSe4] tetrahedra. The introduction of double AEMs effectively broadens the band gaps of selenide-based IR NLO materials. Among them, CaMg6Ga6Se16, achieving the best balance between the second-harmonic generation response (∼1.5 × AgGaS2), wide band gap (2.71 eV), high LIDT (∼9 × AgGaS2), and moderate birefringence of 0.052 @ 1064 nm, is a promising NLO candidate for high power IR laser. Theoretical calculations indicate that the NLO responses and band gaps among the three compounds are mainly determined by the NLO-active [GaSe4] units. The results enrich the chemical diversity of chalcogenides, and give some insight into the design of new functional materials based on the rare [AIISe6] prismatic units.
Introduction
Nonlinear optical (NLO) crystals are an important class of optoelectronic functional materials, which play a critical role in developing new laser light sources by the frequency conversion technology.1–8 In the ultraviolet (UV) and visible-near infrared (IR) regions, oxide-based NLO materials have achieved great progress,9–13 and a series of NLO crystals with excellent performances like LiB3O5 (LBO), BaB2O4 (β-BBO), KBe2BO3F2 (KBBF), KH2PO4 (KDP), and NH4B4O6F have been developed in recent years,14–18 while in the mid- and far-IR regions, the commercially available IR NLO materials like AgGaS2 (AGS), AgGaSe2 (AGSe) and ZnGeP2 (ZGP) suffer from their intrinsic defects, like low laser-induced damage thresholds (LIDTs) in AGS and AGSe, strong two-photon absorption around ∼1 μm and narrow IR transparent range in ZGP.19,20 Therefore, exploring new IR NLO materials with excellent performances has become an urgent need, but challenging due to the conflicts among NLO response, band gap, birefringence and IR transparent range in one material.21For an excellent IR NLO material, the following requirements should be satisfied:22 (1) noncentrosymmetric (NCS) crystal structure; (2) a large NLO response (≥0.5 × AGS, preferably ≥1.0 × AGS); (3) a wide IR transparent range that covers the two important IR atmospheric windows of 3–5 and 8–12 μm; (4) a high LIDT, which is usually related to the band gap and thermal conductivity; (5) suitable birefringence for phase-matching (PM) behavior; (6) good crystal growth habits. To achieve the large NLO response and wide IR transparent range, metal selenide has been demonstrated as a competitive system, due to the large hyperpolarizability of NLO-active [MSe4] (M = Ga, In, Ge, Sn, Zn, Cd, Hg, etc.) tetrahedral units, and red shift of the vibration peaks of M–Se compared to M–O, M–S or M–P bonding.23,24 However, for most selenides, their band gaps are narrow due to the presence of outer electrons on the 3d, 4s, 4p orbits in the Se atom.25,26
To enhance the band gap of chalcogenides, introducing an alkaline-earth metal (AEM) without d–d and f–f electron transitions into the crystal structures has been regarded as a feasible strategy.27,28 Recently, by coupling [MgQ6] (Q = S, Se) octahedral and [MQ4] (M = Al, Ga) tetrahedral units, a series of Mg-containing compounds AIB3IIC3IIIQ8VI (AI = Li, Na, Ag; BII = Mg; CIII = Al, Ga; QVI = S, Se) have been developed.29 In this work, by introducing double AEM atoms, three wide band gap selenide IR NLO materials AIIMg6Ga6Se16 (AII = Ca, Sr, Ba) have been synthesized by the high temperature solid-state reactions in sealed quartz tubes. AIIMg6Ga6Se16 (AII = Ca, Sr, Ba) crystallize in the NCS P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) space group, and are built by [AIISe6] triangular prisms, [MgSe6] octahedral and [GaSe4] tetrahedral units. The compounds show balanced optical properties including strong second-harmonic generation (SHG) responses (1.5 × AGS for CaMg6Ga6Se16, 1.0 × AGS for SrMg6Ga6Se16, 1.1 × AGS for BaMg6Ga6Se16), wide band gaps (in selenides) (2.69–2.71 eV), high LIDTs (8.0–9.0 × AGS), and moderate birefringences (0.044–0.052 @ 1064 nm). Density functional theory (DFT) calculations reveal that the SHG responses and band gaps of the compounds are mainly determined by the [GaSe4] tetrahedral units.
space group, and are built by [AIISe6] triangular prisms, [MgSe6] octahedral and [GaSe4] tetrahedral units. The compounds show balanced optical properties including strong second-harmonic generation (SHG) responses (1.5 × AGS for CaMg6Ga6Se16, 1.0 × AGS for SrMg6Ga6Se16, 1.1 × AGS for BaMg6Ga6Se16), wide band gaps (in selenides) (2.69–2.71 eV), high LIDTs (8.0–9.0 × AGS), and moderate birefringences (0.044–0.052 @ 1064 nm). Density functional theory (DFT) calculations reveal that the SHG responses and band gaps of the compounds are mainly determined by the [GaSe4] tetrahedral units.
Results and discussion
The single crystals of AIIMg6Ga6Se16 (AII = Ca, Sr, Ba) were synthesized by high temperature solid-state reactions at 900 °C, and were picked under an optical microscope for structural determinations by single crystal X-ray diffractions. The energy dispersive X-ray spectroscopy (EDS) spectra and elemental maps demonstrate the presence of Ca/Sr/Ba, Mg, Ga, Se in the structures (Fig. S1†). Meanwhile, the Raman spectra confirm the chemical bonding in the compounds (Fig. S2†). The calculated bond valence sums and global instability indexes (Tables S2, S6 and S10†) verify the reasonability of the crystal structures in the compounds. The crystal data and structure refinement information including atomic coordinates and equivalent isotropic displacement parameters, bond length and angle information are provided in Tables S1–S13.†Since the three compounds show similar cell parameters and crystal structures, CaMg6Ga6Se16 is utilized as an example to illustrate their structures here. CaMg6Ga6Se16 crystallizes in the P space group with a = b = 17.5327(3) Å, c = 7.7603(2) Å. In the asymmetric unit of CaMg6Ga6Se16, there are three Ca atoms, six Mg atoms, three Ga atoms and eleven Se atoms. The Ca atom and Mg atom are coordinated with Se atoms to form [CaSe6] triangular prisms and [MgSe6] octahedral units, while Ga atoms are bonded to four Se atoms to construct [GaSe4] tetrahedra (Fig. 1a). [GaSe4] tetrahedra are connected with each other by vertex-sharing to form the [GaSe3] single-chains and [Ga2Se5] double-chains (Fig. 1a). [MgSe6] octahedra are linked with each other by edge-sharing to form [MgSe4] single-chains and [Mg2Se6] double-chains (Fig. 1b). The formed [GaSe3] and [MgSe4] single-chains are linked by vertex-sharing to build the windmill-like [Mg3Ga3Se24] channels (Fig. 1c), which are further grouped with [Ga2Se5] and [Mg2Se6] double-chains to form the channel-like [Mg9Ga9Se24] framework (Fig. 1d). The six-coordinated Ca ions are filled in the channels (Fig. 1e) to balance the charge, resulting in the final 3D structure of CaMg6Ga6Se16 (Fig. 1f). It is worth noting that, different from CaMg6Ga6Se16 and SrMg6Ga6Se16, the asymmetric unit of BaMg6Ga6Se16 contains two crystallographically independent Ba atoms at Wyckoff 1c, 1e positions, and two partially occupied Ba atoms with the site occupancy of 0.9 and 0.1 at Wyckoff 1a, 1b positions, respectively.
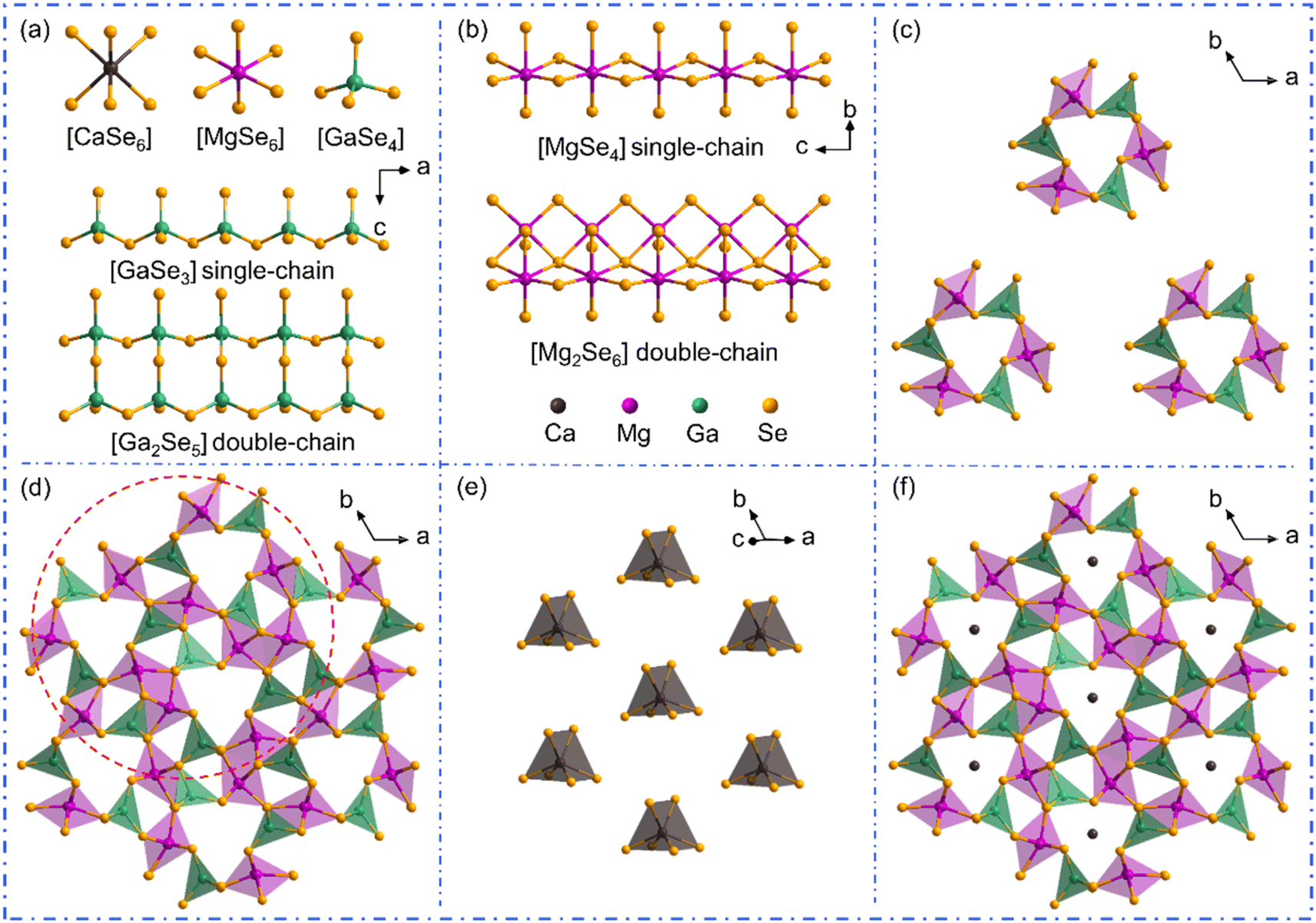 | ||
| Fig. 1 The crystal structure of CaMg6Ga6Se16. (a) The coordination environment of Ca, Mg and Ga atoms, [GaSe3] single-chain and [Ga2Se5] double-chain viewed from the b direction; (b) [MgSe4] single-chain and [Mg2Se6] double-chain viewed from the a direction; (c) windmill-like [Mg3Ga3Se24] geometries; (d) [Mg9Ga9Se24] framework; (e) [CaSe6] triangular prisms; (f) the 3D structure of CaMg6Ga6Se16 viewed from the c direction. | ||
Compared to the crystal structures of AIBII3CIII3QVI8 family compounds, the title compounds can be regarded as the atomic aliovalent substitution of monovalent AI (AI = Li, Na, Ag) by divalent AEM AII (AII = Ca, Sr, Ba). The aliovalent substitution does not cause the change of space group, but induces a doubled cell parameter c in AIIMg6Ga6Se16 compared to AIMg3Ga3Se8, which can be attributed to the formed ordered vacancy in AIIMg6Ga6Se16 (AII = Ca, Sr), or the partially occupied Ba atoms in BaMg6Ga6Se16. In addition, accompanying the fluctuation of A-site cations (Fig. 2a), the channel size fluctuates from 2.49–2.56 Å to 2.39–2.65 Å (Fig. 2b). Most of them are smaller than the AI/AII–Se bond lengths in the compounds, except in the case of AgMg3Ga3Se8 (Fig. 2b). It is worth noting that Ag is three-coordinated with three Se atoms to form a planar [AgSe3] unit in AgMg3Ga3Se8, different from the six coordinated [Li/NaSe6] triangular prism units in AIMg3Ga3Se8 (AI = Li, Na) and [Ca/Sr/BaSe6] triangular prism units in AIIMg6Ga6Se16 (AII = Ca, Sr, Ba).
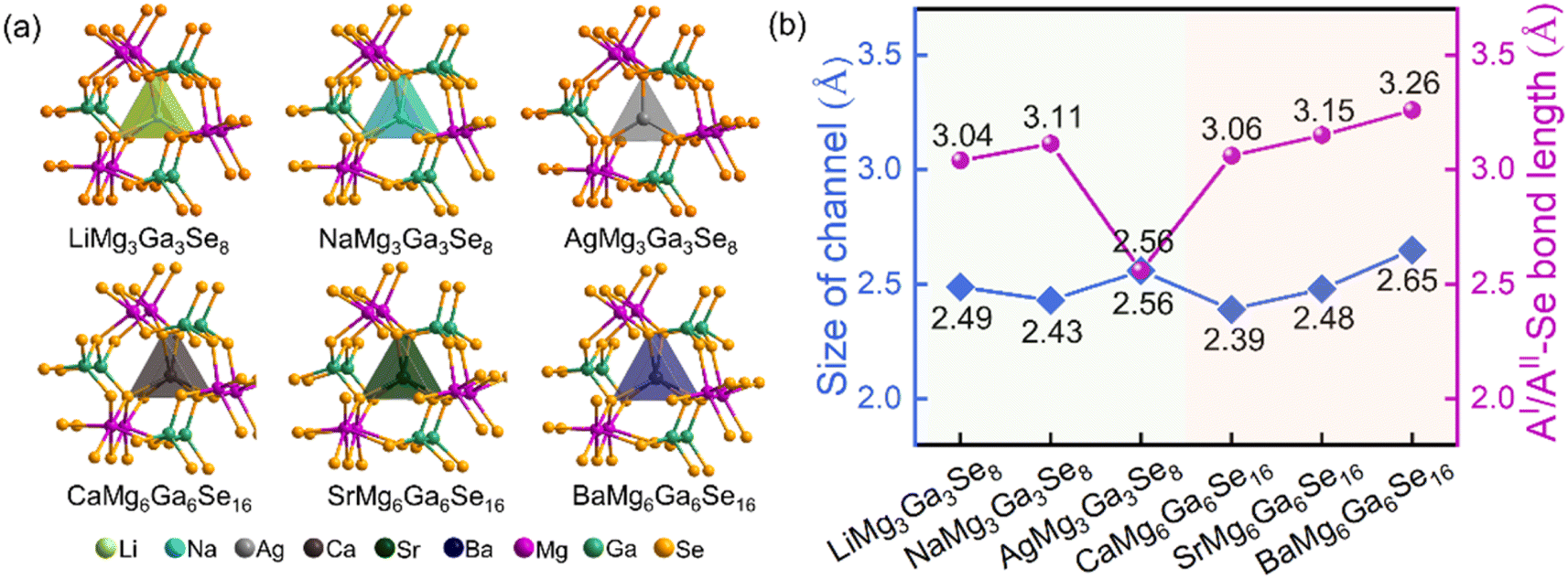 | ||
| Fig. 2 (a) The atomic modes of [Mg3Ga3Se24] channels in AIMg3Ga3Se8 (AI = Li, Na, Ag) and AIIMg6Ga6Se16 (AII = Ca, Sr, Ba); (b) the statistical analyses of the channel size and AI/AII–Se band length in the six compounds. | ||
To evaluate the optical properties, the pure-phase powder samples of the title compounds were prepared and characterized. The experimental XRD patterns are matched well with the theoretical results derived from cif files (Fig. 3a and S3†). To obtain experimental band gaps, UV-vis-NIR diffuse reflectance spectra were measured. Based on the Kubelka–Munk function,30–32 the experimental band gaps of CaMg6Ga6Se16, SrMg6Ga6Se16 and BaMg6Ga6Se16 were measured to be 2.71, 2.71 and 2.69 eV, respectively (Fig. 3b). The large band gaps inherently contribute to high resistances to laser damage.33,34 The LIDTs of the three compounds were evaluated by the single-pulse LIDT method with AGS as the reference.35–37 The measured results show that the LIDTs of CaMg6Ga6Se16, SrMg6Ga6Se16 and BaMg6Ga6Se16 powder samples were ∼9.0, ∼9.0, and ∼8.0 × AGS, respectively.
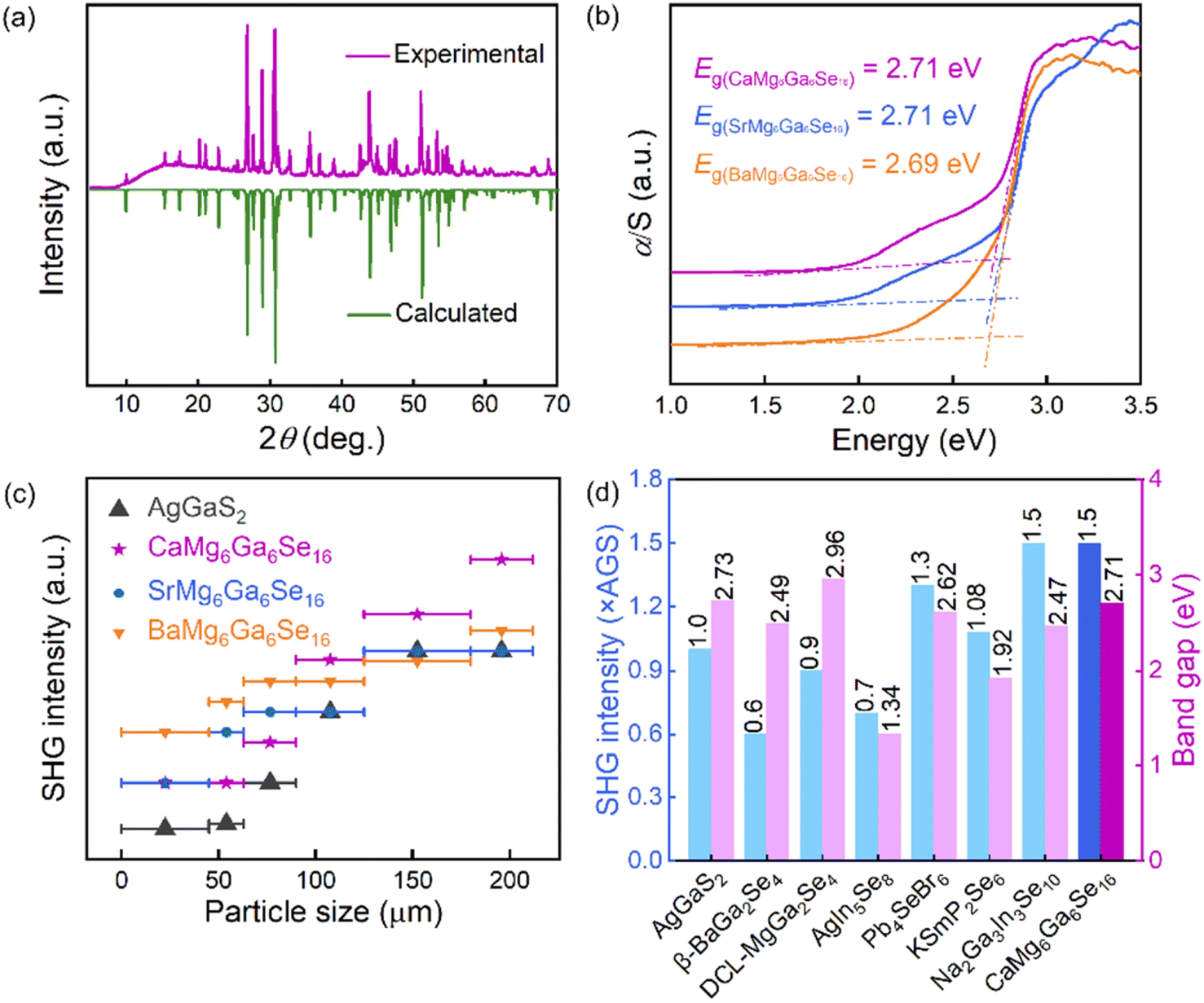 | ||
| Fig. 3 (a) The XRD patterns of CaMg6Ga6Se16; (b) the experimental band gaps of AIIMg6Ga6Se16 (A = Ca, Sr, Ba); (c) SHG intensity vs. AGS with similar particle sizes; (d) typical selenide IR NLO candidates with PM behaviours, AGS used as the references. | ||
Beyond the band gap and LIDT, SHG response is another important parameter for IR NLO materials. The SHG responses of the title compounds were evaluated by the Kurtz and Perry method with a 2.09 μm Q-switched laser.38–41 As shown in Fig. 3c, the SHG intensity of CaMg6Ga6Se16, SrMg6Ga6Se16 and BaMg6Ga6Se16 was tested to be ∼1.5, ∼1.0, and ∼1.1 × AGS, respectively, showing that the A-site AEM has an influence on the SHG responses. It can be attributed to the changes of distortion degree and hyperpolarizability of nonlinear-active [GaSe4] units induced by the different AII–Se chemical bonding (the bond length dCa–Se = 3.0609–3.0640 Å, dSr–Se = 3.1520–3.1590 Å and dBa–Se = 3.2450–3.2612 Å) in the structures. Specifically, the hyperpolarizabilities of [Ga(1–3)Se4] in CaMg6Ga6Se16 were calculated to 283.24, 640.08, and 665.56, while 253.14, 615.32, and 625.81 for SrMg6Ga6Se16, and 190.51, 575.27, and 591.85 for BaMg6Ga6Se16, thus resulting in a larger SHG response in CaMg6Ga6Se16. Moreover, the hyperpolarizabilities of [Ga(1–3)Se4] in NaMg3Ga3Se8 were calculated to be 278.91, 627.83, and 659.72, smaller than the ones in CaMg6Ga6Se16. The results indicate that the alkali metal (AM) and AEM can exert an influence on the SHG response in an indirect way by modifying the geometry of NLO tetrahedral units in the AIBII3CIII3QVI8 and AIIBII6CIII6QVI16 families. Notably, the experimental results imply that CaMg6Ga6Se16 achieves a good balance between wide band gap and large SHG response, which is comparable with the recently reported typical selenide IR NLO candidates like β-BaGa2Se4 (0.6 × AGS, 2.49 eV),25 DCL-MgGa2Se4 (0.9 × AGS, 2.96 eV),26 AgIn5Se8 (0.7 × AGS, 1.34 eV),42 Pb4SeBr6 (1.3 × AGS, 2.62 eV),43 KSmP2Se6 (1.08 × AGS, 1.92 eV)44 and Na2Ga3In3Se10 (1.5 × AGS, 2.47 eV)45 (Fig. 3d).
To understand the structure–performance relationship, the band structures, total and partial density of states (TDOS/PDOS) were computed by DFT calculations.46–48 As shown in Fig. 4a, S4a and c,† the title compounds could be direct band gap compounds with calculated GGA band gaps of 1.851 eV (CaMg6Ga6Se16), 1.866 eV (SrMg6Ga6Se16) and 1.853 eV (BaMg6Ga6Se16). Considering the underestimation of band gaps in the standard DFT calculations with GGA because of the discontinuity of the exchange–correlation energy functional,49,50 the HSE06 band gaps of the compounds were calculated to be 2.568 eV (CaMg6Ga6Se16), 2.564 eV for (SrMg6Ga6Se16) and 2.556 eV (BaMg6Ga6Se16) (Table S14†), which are close to the experimental results in Fig. 3b. From the results of TDOS and PDOS (Fig. 4b, S4b and d†), it can be seen that the tops of the valence band maximum (VBM) near the Fermi level are determined by Se-4p orbitals, and the bottoms of the conduction band minimum (CBM) are mainly occupied by Se-4s4p and Ga-4s orbitals, indicating that the band gaps of the three compounds are mainly determined by Ga–Se bonding in [GaSe4] tetrahedral units. Moreover, the calculated birefringence values are 0.052 for CaMg6Ga6Se16, 0.048 for SrMg6Ga6Se16, and 0.044 for BaMg6Ga6Se16 at 1064 nm (Fig. 4c), matched with the PM behaviours in Fig. 3c. Meanwhile, the bonding electron density difference (Δρb) and contributions (Δω) indicate that [MgSe6] and [GaSe4] units make the main contribution to the optical anisotropy in the compounds (Fig. 4d and S5†).51
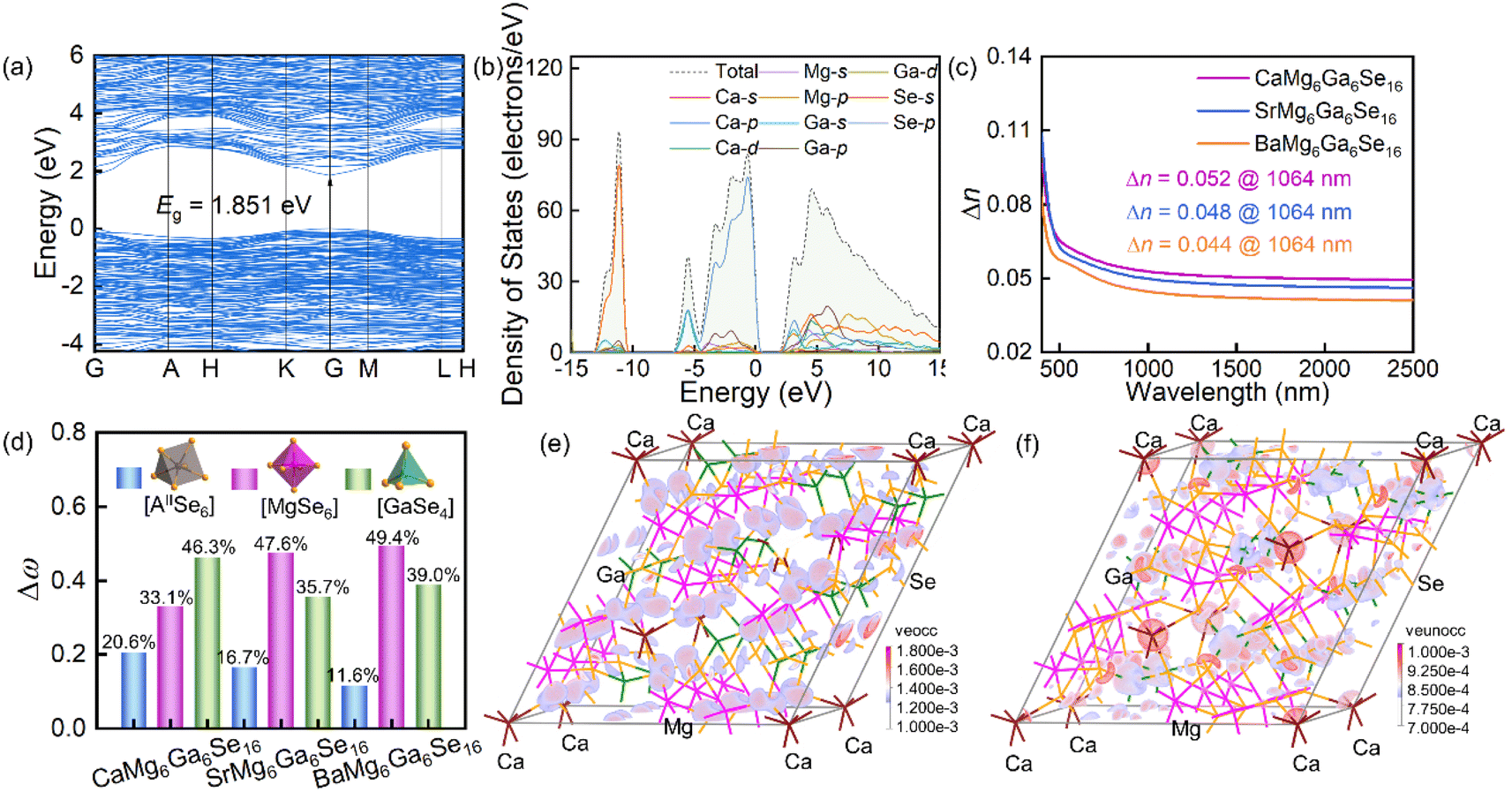 | ||
| Fig. 4 (a) The band structure and (b) total and partial density of states of CaMg6Ga6Se16; (c) the calculated birefringences of the title compounds; (d) the contributions (Δω) of [AIISe6] triangular prisms, [MgSe6] octahedra and [GaSe4] tetrahedra in AIIMg6Ga6Se16 calculated by the response electron distribution anisotropy (REDA) method; (e and f) the SHG density maps of occupied (e) and unoccupied (f) orbitals in the VE process of CaMg6Ga6Se16. | ||
To detect the origin of the NLO effect, the NLO coefficients and SHG density maps of the title compounds were investigated by DFT calculations.52–56 As shown in Table S14,† the calculated NLO coefficients are d11 = −11.05 and d22 = 3.92 pm V−1 for CaMg6Ga6Se16, d11 = −10.67 and d22 = 3.93 pm V−1 for SrMg6Ga6Se16, d11 = −10.58 and d22 = 3.71 pm V−1 for BaMg6Ga6Se16, which match the experimental results in Fig. 3c. Meanwhile, the SHG density maps of occupied and unoccupied orbitals in the Virtual-Electron (VE) process which occupied 93.74% in CaMg6Ga6Se16, 95.25% in SrMg6Ga6Se16, and 95.82% in BaMg6Ga6Se16 (the Virtual-Hole (VH) process is neglected here because it accounts for a small quantity of contributions) indicate that the NLO responses of AIIMg6Ga6Se16 (A = Ca, Sr, Ba) also mainly originate from the [GaSe4] tetrahedral units (Fig. 4e, f and S6†), matched with the anionic group theory promoted by Chen et al.57,58
Conclusions
In summary, by introducing double AEM atoms into the structure, three wide band gap selenides CaMg6Ga6Se16, SrMg6Ga6Se16 and BaMg6Ga6Se16 have been developed successfully. The compounds show strong phase-matching SHG responses (1.0–1.5 × AGS), wide band gaps (2.69–2.71 eV), high LIDTs (8.0–9.0 × AGS) and suitable birefringences (0.044–0.052 @ 1064 nm). Among them, CaMg6Ga6Se16, achieving the best balance among strong SHG response (∼1.5 × AGS), wide band gap (∼2.71 eV) and high LIDT (∼9.0 × AGS), is a promising IR NLO candidate for high power IR laser. The theoretical calculations reveal that the SHG responses and band gaps of the series of double AEM compounds originate from or are determined by the NLO-active [GaSe4] units. The results indicate that grouping 6-coordinated AEM units (triangular prisms and octahedra) and 4-coordinated NLO-active tetrahedra is a feasible way for the design of new IR NLO materials with excellent properties.Data availability
Data available in the ESI† includes experimental sections, crystallographic data, EDS and Raman spectra, XRD patterns, band structures, the calculated bonding electron density differences and SHG density maps of the title compounds.Author contributions
Linan Wang: investigation, methodology, writing – original draft; Dongdong Chu: DFT calculation; Zhihua Yang: software, validation; Junjie Li and Shilie Pan: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the High-level Talent Project of Xinjiang Uygur Autonomous Region (2020000039), National Natural Science Foundation of China (22335007, 52002398, 61835014, 51972336), Xinjiang Key Laboratory of Electronic Information Materials and Devices (2017D04029), Xinjiang Tianchi Talents Project (2023000043), and Xinjiang Tianshan Talent Program (2022TSYCTD0005).Notes and references
- Z. Y. Bai and K. M. Ok, Coord. Chem. Rev., 2023, 490, 215212 CrossRef CAS.
- H. X. Fan, N. Ye and M. Luo, Acc. Chem. Res., 2023, 56, 3099–3109 CrossRef CAS PubMed.
- Y. W. Kang and Q. Wu, Coord. Chem. Rev., 2024, 498, 215458 CrossRef CAS.
- J. J. Li and F. L. Deepak, Chem. Rev., 2022, 122, 16911–16982 CrossRef CAS.
- Y. Q. Li, J. H. Luo and S. G. Zhao, Acc. Chem. Res., 2022, 55, 3460–3469 CrossRef CAS.
- M. Mutailipu, J. Han, Z. Li, F. M. Li, J. J. Li, F. F. Zhang, X. F. Long, Z. H. Yang and S. L. Pan, Nat. Photonics, 2023, 17, 694–701 CrossRef CAS.
- L. A. Wang, Q. Sun and J. J. Li, Chin. J. Struct. Chem., 2023, 42, 100013 CrossRef CAS.
- J. J. Li, J. C. Chen, H. Wang, N. Chen, Z. C. Wang, L. Guo and F. L. Deepak, Adv. Sci., 2018, 5, 1700992 CrossRef.
- X. H. Dong, L. Huang, H. M. Zeng, Z. E. Lin, K. M. Ok and G. H. Zou, Angew. Chem., Int. Ed., 2022, 61, e202116790 CrossRef CAS PubMed.
- M. Mutailipu, K. R. Poeppelmeier and S. L. Pan, Chem. Rev., 2021, 121, 1130–1202 CrossRef CAS PubMed.
- X. L. Chen and K. M. Ok, Chem. Sci., 2022, 13, 3942–3956 RSC.
- J. Chen, C. L. Hu, Y. L. Lin, Y. Chen, Q. Q. Chen and J. G. Mao, Chem. Sci., 2022, 13, 454–460 RSC.
- J. J. Li, Z. C. Wang and F. L. Deepak, ACS Nano, 2017, 11, 5590–5597 CrossRef CAS PubMed.
- G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS PubMed.
- H. N. Liu, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, J. Am. Chem. Soc., 2023, 145, 12691–12700 CrossRef CAS PubMed.
- C. Wu, C. B. Jiang, G. F. Wei, X. X. Jiang, Z. J. Wang, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2023, 145, 3040–3046 CrossRef CAS.
- L. L. Wu, C. S. Lin, H. T. Tian, Y. Q. Zhou, H. X. Fan, S. Da Yang, N. Ye and M. Luo, Angew. Chem., Int. Ed., 2023, e202315647 Search PubMed.
- X. F. Wang, X. D. Leng, Y. Kuk, J. Lee, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2024, 63, e202315434 CrossRef CAS.
- W. F. Chen, X. M. Jiang, S. M. Pei, M. S. Zhang, B. W. Liu and G. C. Guo, Sci. China Mater., 2023, 66, 740–747 CrossRef CAS.
- M. R. Sun, C. X. Li, J. L. Shi, M. H. Lee and J. Y. Yao, Mater. Today Phys., 2023, 36, 101166 CrossRef CAS.
- D. J. Mei, W. Z. Cao, N. Z. Wang, X. X. Jiang, J. Zhao, W. K. Wang, J. H. Dang, S. Y. Zhang, Y. D. Wu, P. H. Rao and Z. S. Lin, Mater. Horiz., 2021, 8, 2330–2334 RSC.
- M. Y. Ran, A. Y. Wang, W. B. Wei, X. T. Wu, H. Lin and Q. L. Zhu, Coord. Chem. Rev., 2023, 481, 215059 CrossRef CAS.
- W. K. Wang, D. J. Mei, F. Liang, J. Zhao, Y. D. Wu and Z. S. Lin, Coord. Chem. Rev., 2020, 421, 213444 CrossRef CAS.
- J. D. Chen, H. X. Chen, F. Xu, L. L. Cao, X. T. Jiang, S. Da Yang, Y. S. Sun, X. Zhao, C. S. Lin and N. Ye, J. Am. Chem. Soc., 2021, 143, 10309–10316 CrossRef CAS PubMed.
- Y. J. Zhang, Q. Bian, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115374 CrossRef CAS.
- P. Wang, Y. Chu, A. Tudi, C. W. Xie, Z. H. Yang, S. L. Pan and J. J. Li, Adv. Sci., 2022, 9, 2106120 CrossRef CAS.
- A. Abudurusuli, J. B. Huang, P. Wang, Z. H. Yang, S. L. Pan and J. J. Li, Angew. Chem., Int. Ed., 2021, 60, 24131–24136 CrossRef CAS PubMed.
- J. L. Chen, Y. J. Zhang, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, Adv. Opt. Mater., 2023, 11, 2202147 CrossRef CAS.
- L. Luo, L. A. Wang, J. B. Chen, J. Z. Zhou, Z. H. Yang, S. L. Pan and J. J. Li, J. Am. Chem. Soc., 2022, 144, 21916–21925 CrossRef CAS PubMed.
- Y. Chu, H. S. Wang, T. Abutukadi, Z. Li, M. Mutailipu, X. Su, Z. H. Yang, J. J. Li and S. L. Pan, Small, 2023, 19, 2305074 CrossRef CAS PubMed.
- M. Mutailipu, M. Zhang, B. B. Zhang, L. Y. Wang, Z. H. Yang, X. Zhou and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 6095–6099 CrossRef CAS PubMed.
- L. A. Wang, C. C. Tu, H. B. Gao, J. Z. Zhou, H. S. Wang, Z. H. Yang, S. L. Pan and J. J. Li, Sci. China: Chem., 2023, 66, 1086–1093 CrossRef CAS.
- J. Z. Zhou, K. T. Hou, Y. Chu, Z. H. Yang, J. J. Li and S. L. Pan, Small, 2023 DOI:10.1002/smll.202308806.
- L. A. Wang, C. C. Tu, J. Z. Zhou, Y. Chu, Z. H. Yang, S. L. Pan and J. J. Li, Adv. Opt. Mater., 2024, 12, 2301634 CrossRef CAS.
- J. Z. Zhou, Z. X. Fan, K. W. Zhang, Z. H. Yang, S. L. Pan and J. J. Li, Mater. Horiz., 2023, 10, 619–624 RSC.
- J. Z. Zhou, L. A. Wang, Y. Chu, H. S. Wang, S. L. Pan and J. J. Li, Adv. Opt. Mater., 2023, 11, 2300736 CrossRef CAS.
- J. Z. Zhou, L. Luo, Y. Chu, P. Wang, Z. Y. Guo, X. Su and J. J. Li, J. Alloys Compd., 2022, 899, 163366 CrossRef CAS.
- Y. X. Han, C. L. Hu and J. G. Mao, Small, 2024, 20, 2305828 CrossRef CAS.
- W. F. Chen, B. W. Liu, S. M. Pei, X. M. Jiang and G. C. Guo, Adv. Sci., 2023, 10, 2207630 CrossRef CAS PubMed.
- X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS PubMed.
- H. S. Wang, Y. Chu, X. T. Pan, Z. H. Yang, S. L. Pan and J. J. Li, Mater. Today Phys., 2023, 38, 101243 CrossRef CAS.
- L. F. Dong, S. Z. Zhang, P. F. Gong, F. Liang and Z. S. Lin, Inorg. Chem. Front., 2023, 10, 3248–3254 RSC.
- J. K. Wang, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Adv. Opt. Mater., 2022, 10, 2102673 CrossRef CAS.
- Z. X. Chen, C. Y. Zhao, X. H. Li, W. D. Yao, W. L. Liu and S. P. Guo, Small, 2023, 19, 2206910 CrossRef CAS.
- J. Li, W. D. Yao, J. N. Li, X. H. Li, W. L. Liu and S. P. Guo, Mater. Today Phys., 2023, 32, 101007 CrossRef CAS.
- J. J. Li, Z. Lian, Q. Li, Z. C. Wang, L. F. Liu, F. L. Deepak, Y. P. Liu, B. Li, J. Y. Xu and Z. X. Chen, Nano Res., 2022, 15, 5933–5939 CrossRef CAS.
- Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 2150–2154 CrossRef CAS PubMed.
- M. Yan, R. L. Tang, W. D. Yao, W. L. Liu and S. P. Guo, Chem. Sci., 2024, 15, 2883–2888 RSC.
- H. S. Wang, X. T. Pan, W. Zhao, Y. Chu and J. J. Li, Inorg. Chem. Front., 2023, 10, 6253–6261 RSC.
- B. B. Zhang, G. Q. Shi, Z. H. Yang, F. F. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS.
- B. H. Lei, Z. H. Yang, H. W. Yu, C. Cao, Z. Li, C. Hu, K. R. Poeppelmeier and S. L. Pan, J. Am. Chem. Soc., 2018, 140, 10726–10733 CrossRef CAS PubMed.
- Y. Chu, H. S. Wang, Q. Chen, X. Su, Z. X. Chen, Z. H. Yang, J. J. Li and S. L. Pan, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202314933.
- Y. Chu, P. Wang, H. Zeng, S. C. Cheng, X. Su, Z. H. Yang, J. J. Li and S. L. Pan, Chem. Mater., 2021, 33, 6514–6521 CrossRef CAS.
- L. A. Wang, D. D. Chu, D. Q. Yin, C. W. Xie, Z. H. Yang, J. J. Li and S. L. Pan, Mater. Today Phys., 2023, 38, 101245 CrossRef CAS.
- X. F. Wang, J. J. Li, Z. J. Zhao, S. M. Huang and W. H. Xie, J. Appl. Phys., 2012, 112, 023701 CrossRef.
- M. Y. Zhang, N. W. Liang, D. Hao, Z. X. Chen, F. Zhang, J. Yin, Y. H. Yang and L. S. Yang, Nano Research Energy, 2023, 2, e9120077 CrossRef.
- C. T. Chen, Acta Phys. Sin., 1977, 26, 486–499 CrossRef CAS.
- H. T. Qiu, F. M. Li, C. C. Jin, Z. H. Yang, J. J. Li, S. L. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2024, 63, e202316194 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2311758–2311760. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00334a |
| ‡ L. A. Wang and D. D. Chu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |