
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePb2(SeO3)(SiF6): the first selenite fluorosilicate with a wide bandgap and large birefringence achieved through perfluorinated group modification†
Peng-Fei
Li
 ab,
Chun-Li
Hu
ab,
Jiang-Gao
Mao
ab and
Fang
Kong
*ab
ab,
Chun-Li
Hu
ab,
Jiang-Gao
Mao
ab and
Fang
Kong
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: kongfang@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 5th April 2024
Abstract
Birefringent crystals serve as the core elements of polarizing optical devices. However, the inherent challenge of balancing bandgap and birefringence poses a significant hurdle in designing crystals with excellent overall performance. In this study, we propose a novel approach, namely modification with perfluorinated groups, to achieve dual enhancement of the bandgap and birefringence of selenite materials. We have successfully synthesized the first selenite fluorosilicate, namely, Pb2(SeO3)(SiF6). This compound exhibits a three-dimensional structure composed of two-dimensional lead selenite layers bridged by SiF6 octahedrons. Notably, by introducing a perfluorinated SiF6 group, the bandgap of the lead selenite compound has been expanded to 4.4 eV. Furthermore, Pb2(SeO3)(SiF6) demonstrates a large birefringence (0.161 @ 546 nm), surpassing most of the selenite compounds with a bandgap larger than 4.2 eV. Theoretical calculations suggest that the large birefringence of Pb2(SeO3)(SiF6) can be attributed to the synergistic effects of SeO3, PbO4 and PbO3F4 polyhedrons. Our research not only pioneers a new system for selenite materials, enriching the diversity of selenite structures, but also provides a design methodology for obtaining wide bandgap birefringent selenite.
Introduction
Birefringent crystals play a pivotal role as optical functional components in commercial, agricultural, industrial, and military sectors.1–4 Some crystals have been commercialized, such as α-BaB2O4,5 CaCO3,6 YVO4,7 MgF2,8 TiO2,9etc. However, these commercially available birefringent crystals have inherent limitations. For example, the transparency of YVO4 is notably low below 400 nm, and the birefringence of MgF2 is comparatively small, which fail to meet the demands of modern industry.10 In recent years, with the rapid advancement of optoelectronic technology, the requirement for miniaturization of short-wave devices has been escalating. The miniaturization of such devices necessitates crystals with large birefringence and a wide bandgap.11,12 However, the inherent contradiction between the bandgap and birefringence makes wide bandgap birefringent crystals extremely rare. Consequently, addressing these contradictions and achieving a balance between these properties is an urgent issue that requires immediate attention.13–17Metal selenites, containing stereo-chemically active lone pair electrons, have shown promise as potential candidates for birefringent materials due to their significant anisotropy.18,19 Scientists have primarily explored the following avenues to enhance the birefringent performance of selenite crystals: (i) incorporation of stereo-chemically active lone pair cations, such as Pb2Bi(SeO3)2Cl3 (0.186 @ 1064 nm, 3.45 eV),20 Rb2Bi2(SeO3)3F2 (0.105 @ 546 nm, 3.72 eV),21 and Bi4TeO4F2(TeO3)(SeO3)2 (0.20 @ 1064 nm, 2.74 eV);22 (ii) incorporation of d0 transition metals susceptible to second-order Jahn–Teller (SOJT) distortion, for example, Pb2(V2O4F)(VO2)(SeO3)3 (0.105 @ 1064 nm, 2.35 eV),23 Bi4TiO2F4(SeO3)4 (0.19 @ 1064 nm, 3.58 eV)22 and Gd2F2(OH2)(MoO3)2(SeO3)2 (0.143 @ 1064 nm, 3.15 eV);24 (iii) incorporation of highly polarizable and deformable d10 transition metals, for instance, Rb2Hg2(SeO3)3 (0.055 @ 546 nm, 3.60 eV),25 Pb2Cd(SeO3)2Cl2 (0.093 @ 1064 nm, 4.1 eV)26 and Pb2Cd(SeO3)2Br2 (0.116 @ 1064 nm, 3.9 eV).26 However, the relatively low transparency below 400 nm limits their application in the ultraviolet region. To address this, researchers have introduced ions conducive to widening the bandgap in the selenite system, such as alkali metals, alkaline earth metals, etc.27 The successfully synthesized wide bandgap selenites include NaLu(SeO3)2 (0.153 @ 546 nm, 5.3 eV),28 Ga2(SeO3)3(H2O)3 (0.082 @ 532 nm, 5.3 eV),29 and Y(HSeO3)(SeO3)(H2O)·(H2O) (0.044 @ 532 nm, 5.5 eV).30 Although their bandgaps have been improved to 5.0 eV or above, their birefringence decreased obviously, except for NaLu(SeO3)2. This is mainly attributed to their limited contribution to the anisotropy of polarizability. Consequently, there is an urgent need to develop a new method to achieve enhancement of the bandgap and birefringence in selenite materials.
Our research group has done many studies for the design and synthesis of selenite materials with a wide bandgap and large birefringence, and we have discovered that the bandgap of these materials can be influenced by the type of anionic group. Currently, we are pursuing two approaches to simultaneously improve the bandgap and birefringence of selenites. The first approach involves introducing SO4 tetrahedral groups with a wide transmittance window,31,32 exemplified by compounds like Hg2(SeO3)(SO4) (0.133 @ 532 nm, 3.58 eV)33 and Hg3(SeO3)2(SO4) (0.118 @ 546 nm, 4.70 eV).34 Another approach involves the use of partially fluorinated metal oxide polyhedrons.35,36 For instance, CaYF(SeO3)2 exhibits a wide bandgap of 5.06 eV and a large birefringence of 0.138 @ 532 nm.37 It is noteworthy that there is still upside potential in enhancing the bandgap and birefringence of selenite compounds.
Based on our literature research, Si4+ in the IVA group exhibits a large bandgap and strong ultraviolet transmittance capability.38,39 Additionally, F− in the VIIA group has high electronegativity, strong ultraviolet transmittance capability, and chemical scissor action.40–42 Combining these elements to form SiF6 groups can leverage their individual advantages to positively influence the bandgap of compounds.43,44 Moreover, SiF6 groups can act as a chemical scissor, regulating the arrangement of metal cations and selenite ions, thereby enhancing the birefringence performance of the compounds. To achieve compounds with large birefringence, Pb2+ was chosen as the balancing cation. Aside from possessing stereo-chemically active lone pair electrons, Pb2+ readily forms highly anisotropic PbOxFy polyhedron when partially fluorinated.45–47 The synergistic effect of the SiF6 group, SeO3 group, and PbOxFy polyhedron holds the potential for developing new selenite materials with a wide bandgap and large birefringence (Fig. 1). Consequently, we have focused our attention on the Pb2+–Se4+–Si4+–F−–O2− system for the first time. As of now, no compounds have been discovered in this system. Through our exploration, a novel compound, Pb2(SeO3)(SiF6), has been obtained. Pb2(SeO3)(SiF6) stands as the initial example of a fluorosilicate selenite system. Theoretical calculations and experimental measurements indicate that the selenite fluorosilicate exhibits a large birefringence and a wide bandgap. The synergistic effect of the SeO3 group, PbO4 and PbO3F4 polyhedrons is the primary contributor to its large birefringence. This paper will report on the design, synthesis, structure, and optical properties of Pb2(SeO3)(SiF6).
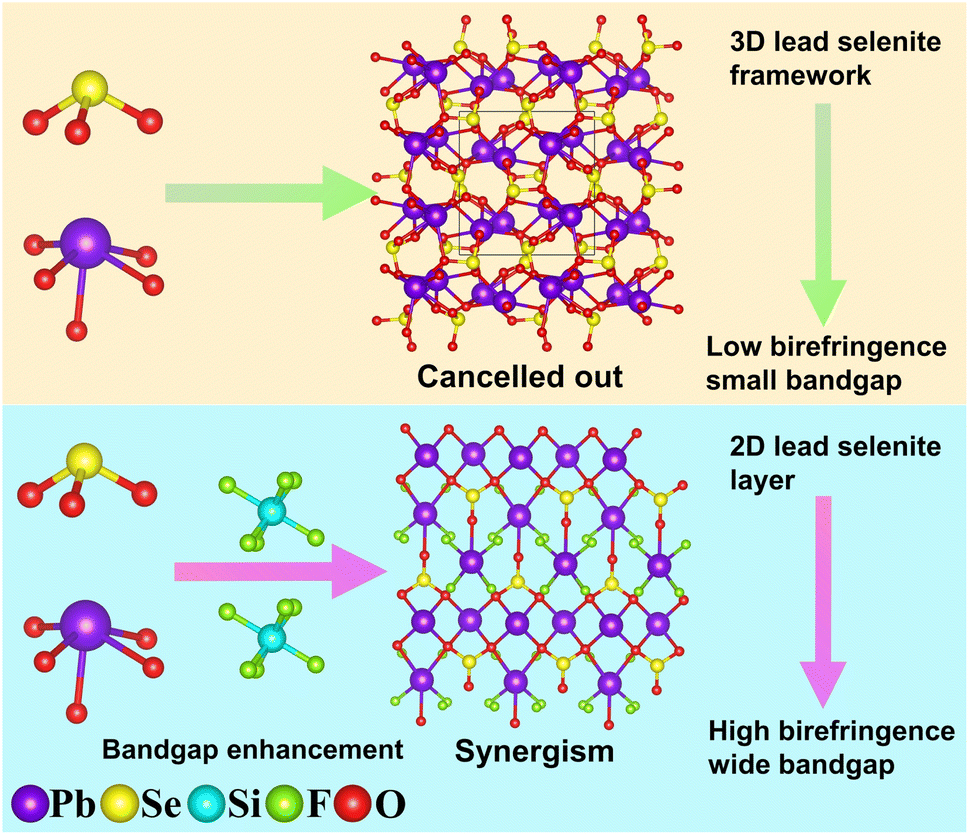 | ||
| Fig. 1 From low birefringence, small bandgap to high birefringence wide bandgap regulated by SiF6 groups. | ||
Results and discussion
Pb2(SeO3)(SiF6) was synthesized using SeO2, H2SiF6, and Pb(BF4)2 through a low-temperature (105 °C) hydrothermal method. The detailed synthesis procedures are outlined in the ESI† under the section titled “Syntheses”. The elemental distribution map confirmed the presence of fluorine (F) element (Fig. 2), and quantitative elemental analysis results revealed the stoichiometry of F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb:Si:Se as 6:2:1:1 in the compound (Fig. S1a†). The crystal purity was verified using X-ray diffraction (XRD), as depicted in Fig. S1b.† Crystallographic details of Pb2(SeO3)(SiF6) can be found in Table S1.†
:Pb:Si:Se as 6:2:1:1 in the compound (Fig. S1a†). The crystal purity was verified using X-ray diffraction (XRD), as depicted in Fig. S1b.† Crystallographic details of Pb2(SeO3)(SiF6) can be found in Table S1.†
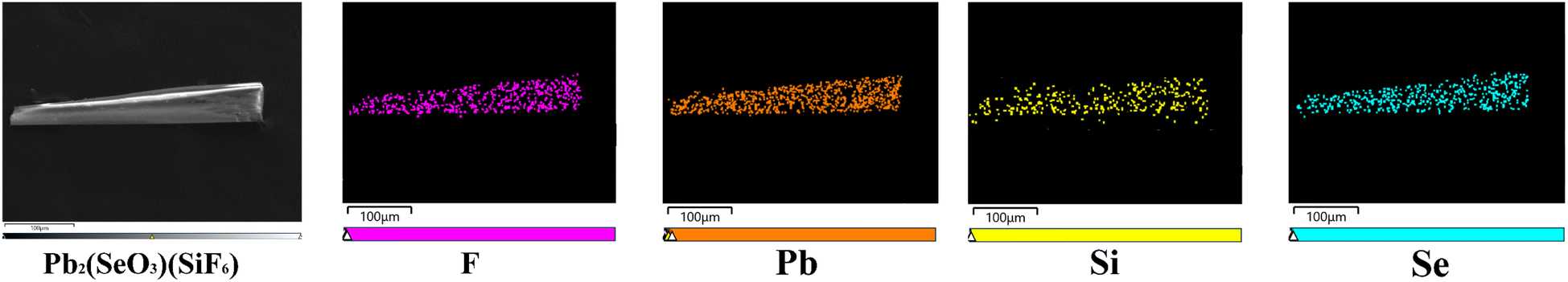 | ||
| Fig. 2 SEM images of Pb2(SeO3)(SiF6) and its elemental distribution maps. | ||
The first example of the selenite fluorosilicate compound, Pb2(SeO3)(SiF6), crystallizes in the orthorhombic space group Pnma (no. 62). The lattice parameters are a = 13.8153(4) Å, b = 5.4470(2) Å, c = 9.6098(3) Å, α = β = γ = 90°, and V = 723.16(4) Å3. Its asymmetric unit contains two lead atoms, one selenium atom, one silicon atom, four fluorine atoms, and two oxygen atoms, totaling ten atoms. Among them, F(1), F(4), and O(1) are located at general positions, while the remaining atoms occupy special positions. The selenium atom coordinates with three oxygen atoms, forming a trigonal pyramid with Se–O bond lengths ranging from 1.674(7) to 1.727(4) Å. The silicon atom bonds with six fluorine atoms, resulting in an octahedral SiF6 configuration, with Si–F bond lengths ranging from 1.674(6) to 1.699(6) Å. Within the range of 2.5 to 2.8 Å, Pb(1) coordinates with four oxygen atoms, while Pb(2) coordinates with three oxygen and four fluorine atoms. Bond valence calculations indicate that the oxidation state of Se(1) is 3.958. The oxidation state of Si(1) is 4.513, which is relatively elevated and commonly observed in hexafluoro silicate compounds, such as compounds KLiSiF6 (Si: 4.496),48 Tl2SiF6 (Si: 4.506)49 and Li2SiF6 (Si: 4.518).50 Given that it is not an isolated case, we believe the high bond valence may be caused by the overestimated bond valence parameter (1.58) for the Si4+–F bond. If the parameter was decreased to 1.55, the oxidation state of Si in Pb2(SeO3)(SiF6) will decrease to 4.16 and the same phenomenon is observed in KLiSiF6 (Si: 4.15), Tl2SiF6 (Si: 4.15) and Li2SiF6 (Si: 4.16).51 The calculated oxidation state for Pb(1) and Pb(2) is 1.374 and 1.443 respectively. The calculated oxidation state for Pb(1) and Pb(2) is 1.374 and 1.443 respectively. When considering the longer Pb–O and Pb–F bonds in the range of 2.80 to 2.97 Å, the oxidation states of Pb(1) and Pb(2) can be increased to 1.868 and 1.755, respectively, approaching their standard oxidation states (Table S2†).
Pb2(SeO3)(SiF6) exhibits a novel three-dimensional (3D) structure composed of two-dimensional (2D) lead selenite layers bridged by SiF6 groups (Fig. 3). Initially, several Pb(1)O4 polyhedrons are linked through O(1)⋯O(1) edge-sharing, forming one-dimensional (1D) lead oxide chains. These chains are reinforced by Pb(2)O3F4 polyhedrons via O(1) atoms (Fig. 3a). The Se(1)O3 groups serve as hexadentate ligands, coordinating bidentately with one Pb(1) atom and bridging to two Pb(1) and three Pb(2) atoms (Fig. 3b). These Se(1)O3 groups further connect the 1D lead oxide chains to create a 2D lead selenite layer on the bc plane (Fig. 3d). Ultimately, the SiF6 groups bridge these 2D layers, resulting in the formation of a novel 3D Pb2(SeO3)(SiF6) network structure (Fig. 3e). Notably, the SiF6 groups act as tridentate anionic ligands, coordinating bidentately with one Pb(2) atom and linking to two Pb(2) atoms (Fig. 3c). This complex configuration underscores the unique structural characteristics of the compound.
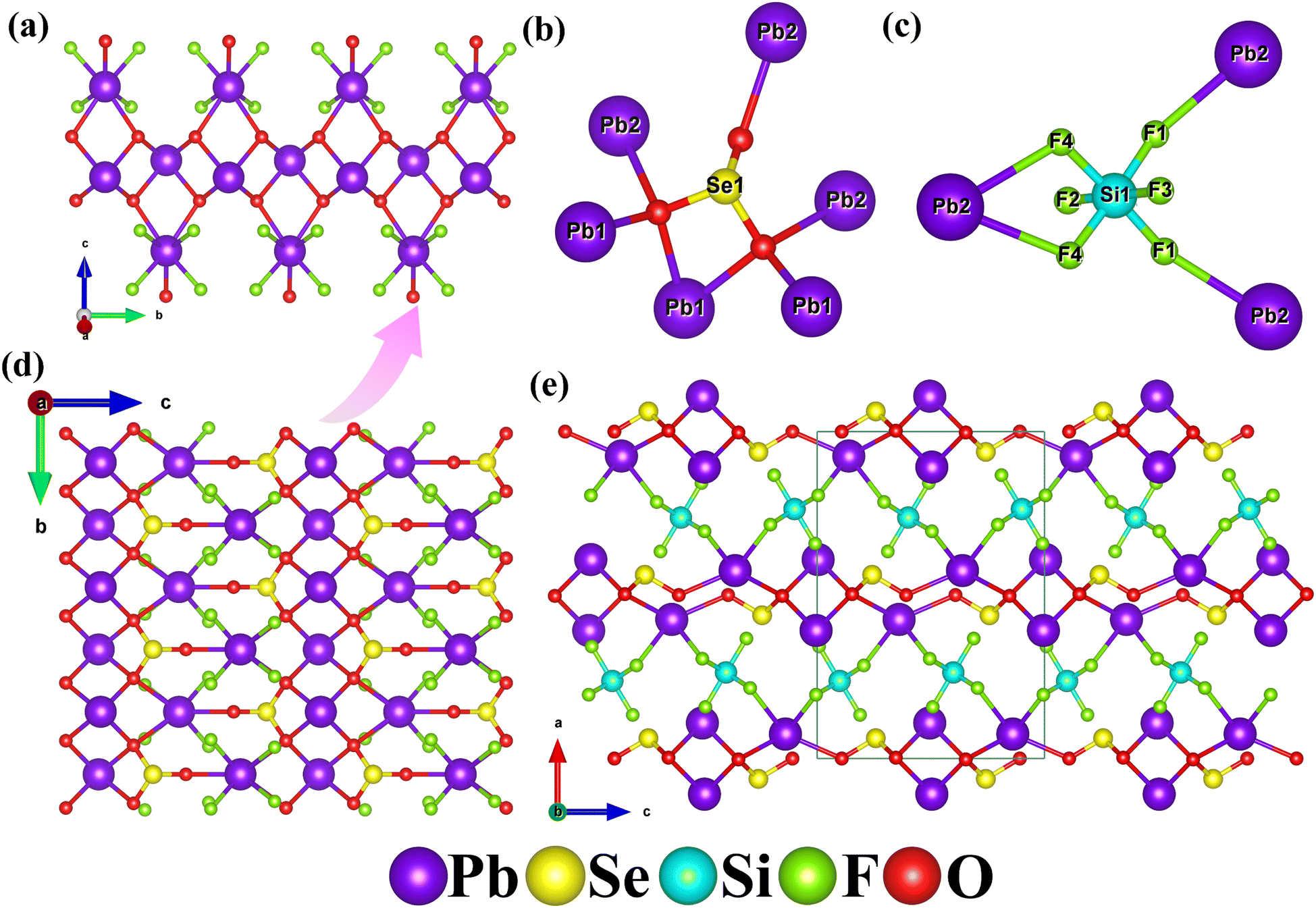 | ||
| Fig. 3 One-dimensional lead oxide chains enhanced with lead oxyfluoride modification (a), the coordination environments of the SeO3 group (b) and SiF6 group (c), two-dimensional lead selenite layer structure (d) and the 3D structure of (e) Pb2(SeO3)(SiF6). | ||
Thermogravimetric analysis (TGA) of the compound Pb2(SeO3)(SiF6) was performed within a temperature range of 20 to 1200 °C. It is noteworthy that the compound remains stable before reaching 220 °C. In the temperature range of 220 to 500 °C, the weight loss of the compound is primarily attributed to the decomposition of one molecule of SeO2, with an experimental value of 15.8%, closely approximating the theoretical value of 16.2%. Beyond 500 °C, the weight loss of the compound should be associated with the incomplete decomposition of lead and fluorine components (Fig. S2†).43 The incomplete weight loss of lead and fluorine components has been previously observed in the TGA of compounds Pb2Al3F3(Te6F2O16) and Pb2Ga3F3(Te6F2O16), as reported by our group.52 This further strengthens the reliability of our observations. In addition, Pb2(SeO3)(SiF6) also exhibits good environmental stability. The samples can still be stable after being exposed to room temperature for three months.
The infrared spectrum (IR) of Pb2(SeO3)(SiF6) was obtained at room temperature in the wavenumber range of 4000–400 cm−1. The compound exhibits good transparency in the range of 4000–800 cm−1. The characteristic peak observed around 638 cm−1 is attributed to the absorption of fluorine ions. Strong peaks in the regions of 420–510 cm−1 and 700–800 cm−1 are attributed to the bending and stretching vibrations of the Se–O bonds (Fig. S3†).
The UV-vis-NIR diffuse reflectance spectra indicate that the Pb2(SeO3)(SiF6) compound exhibits a UV cutoff edge at 247 nm, accompanied by a bandgap of 4.4 eV (Fig. S4†). Notably, this bandgap is significantly higher than those observed in previously reported lead-based compounds such as Pb3SeO5 (3.3 eV),53 CdPb8(SeO3)4Br10 (3.32 eV),45 Pb2Bi(SeO3)2Cl3 (3.45 eV),20 Pb2NbO2(SeO3)2Br (3.17 eV),54 and Pb3(SeO3)Br4 (3.35 eV).55 This significant bandgap can be primarily attributed to the contribution of the perfluorinated group SiF6. Combined with the IR spectrum, the transparency range for Pb2(SeO3)(SiF6) powder samples is from 0.25 to 12.6 μm (Fig. 4). However, considering the potential deviation in infrared cutoff edge due to multi-phonon absorption in powder and crystal measurements, a 50% correction was applied.56 Therefore, the estimated transparency range for Pb2(SeO3)(SiF6) should be from 0.25 μm to 6.3 μm, covering the ultraviolet, visible, and mid-infrared regions. This suggests that Pb2(SeO3)(SiF6) serves as a promising optical functional material.
 | ||
| Fig. 4 UV-vis-NIR diffuse reflectance and IR transmittance spectra of Pb2(SeO3)(SiF6). | ||
A large bandgap is advantageous for enhancing the laser-induced damage threshold (LIDT) of compounds. Therefore, we measured the LIDT of Pb2(SeO3)(SiF6) within the particle size range of 150 to 210 nm. The experiments revealed that the LIDT value for Pb2(SeO3)(SiF6) is 50.8 MW cm−2, approximately 19.5 times higher than that of AgGaS2 (2.6 MW cm−2). This value is slightly higher than those previously reported for the compounds Hg3Se(SeO3)(SO4) (23.35 MW cm−2)57 and Pb3(TeO3)Br4 (21.5 MW cm−2).45
The birefringence of Pb2(SeO3)(SiF6) at 546 nm was measured using a Zeiss Axio Scope A1 polarizing microscope. Under positive polarization, Pb2(SeO3)(SiF6) exhibited complete extinction. The optical path difference was determined to be 1.365 μm, with a certain crystal plane at a thickness of 9.31 μm (Fig. S5†). Utilizing the formula R = Δn × T,58 the experimental birefringence of Pb2(SeO3)(SiF6) at 546 nm was determined to be 0.147 (Fig. 5). This result aligns with the theoretical birefringence value calculated in the subsequent text.
 | ||
| Fig. 5 Experimental birefringence at 546 nm for Pb2(SeO3)(SiF6). | ||
We have conducted an investigation into the electronic structure and optical properties of Pb2(SeO3)(SiF6) utilizing density functional theory.67 The results show that Pb2(SeO3)(SiF6) is a direct bandgap compound, exhibiting a bandgap of 4.17 eV, which aligns closely with the experimental value of 4.4 eV (Fig. S6†). To ensure the precision of subsequent optical property calculations, we used a scissor operator of 0.23 eV. The partial and total density of states of Pb2(SeO3)(SiF6) are shown in Fig. 6a. Notably, the valence band top predominantly consists of O-2p and Se-4s4p orbitals, while the conduction band bottom is primarily composed of Se-4p and Pb-6p orbitals. This indicates that the bandgap of Pb2(SeO3)(SiF6) is mainly determined by Se, Pb, and O atoms.
 | ||
| Fig. 6 The total and partial density of states (a) and the calculated refractive indices and birefringence values (b) of Pb2(SeO3)(SiF6). | ||
To elucidate the theoretical birefringence of Pb2(SeO3)(SiF6), we calculated its birefringence using the formula n2(ω) = ε(ω).68 As a biaxial crystal, Pb2(SeO3)(SiF6) exhibits refractive index order: n001 > n010 > n100. The theoretical birefringence values for Pb2(SeO3)(SiF6) are 0.161 at 546 nm, 0.143 at 1064 nm, and 0.139 at 2000 nm, showing a close agreement with the measured value of 0.147 at 546 nm (Fig. 6b). Fig. 7 illustrates the pronounced asymmetry of lobes on Se(IV), indicating of the high stereo–chemical activity of lone pairs, which is much larger than those of Pd(II). From the electron density difference (EDD) map, we can also find that the Se(IV) and Pb(II) groups are almost arranged in straight lines, which leads to the large birefringence of Pb2(SeO3)(SiF6). So, the large birefringence of Pb2(SeO3)(SiF6) was contributed by SeO3, PbO4 and PbO3F4 polyhedrons primarily. Although the SiF6 groups contribute little to the birefringence of Pb2(SeO3)(SiF6) directly, the scissor effect of SiF6 octahedrons make an important role in regulation of the arrangement of lone pair containing polyhedrons.
 | ||
| Fig. 7 The electron density difference (EDD) map of Pb2(SeO3)(SiF6). | ||
Table 1 provides a comprehensive summary of the birefringence of selenite compounds with bandgaps exceeding 4.2 eV reported to date.18 It can be observed that Pb2(SeO3)(SiF6) exhibits a large birefringence in these selenites, second only to KCl·(H2SeO3)2 (0.17 @ 532 nm).64 However, Pb2(SeO3)(SiF6) demonstrates the largest birefringence among the selenites without hydrogen, and it is the sole example of lead selenite with a large birefringence (Δn ≥ 0.1) and wide bandgap (Eg ≥ 4.2 eV). This result highlights the effectiveness of perfluorinated group modification in achieving dual enhancement of the bandgap and birefringence in selenite systems.
| Compound | Space group | E g (eV) | Birefringence | Ref. |
|---|---|---|---|---|
| K4(H2SeO3)(HSO4)2(SO4) | P-1 | — | Cal.: 0.017 @ 1064 nm | 59 |
| Pb2Ga3F6(SeO3)2Cl3·2H2O | R32 | 4.59 | Cal.: 0.02 @ 1064 nm | 60 |
| NaGa3(HSeO3)6(SeO3)2 | P21/n | 5.2 | Cal.: 0.03 @ 1064 nm | 29 |
| Y3F(SeO3)4 | P63 | 5.03 | Cal.: 0.036 @ 1064 nm | 37 |
| Ga2(Se2O5)2(HSeO3)(H2SeO3)F | Pna21 | 4.71 | Cal.: 0.037 @ 1064 nm | 61 |
| Na8(SeO3)(SO4)3 | Pna21 | 5.69 | Cal.: 0.038 @ 1064 nm | 59 |
| Y(HSeO3)(SeO3)(H2O)·(H2O) | P212121 | 5.5 | Cal.: 0.041 @ 1064 nm | 30 |
| Cd5Ga4(HSeO3)2(SeO3)10 | P-1 | 4.7 | Cal.: 0.044 @ 1064 nm | 29 |
| Al(B(SeO3)3)H2O | Pbca | 4.86 | Cal.: 0.063 @ 1064 nm | 62 |
| Ga(B(SeO3)3)H2O | Pbca | 4.79 | Cal.: 0.064 @ 1064 nm | 62 |
| Ga2(SeO3)3(H2O)3 | R3c | 5.3 | Cal.: 0.079 @ 1064 nm | 29 |
| Ga(Se2O5)(HSeO3) | P21/c | 4.34 | Cal.: 0.079 @ 1064 nm | 61 |
| Na2(H2SeO3)(SO4) | P21/c | 5.04 | Cal.: 0.082 @ 1064 nm | 59 |
| Al2(SeO3)3(H2O)3 | R3c | 5.5 | Cal.: 0.084 @ 1064 nm | 29 |
| Hf(SeO3)2 | P21/c | 5.2 | Cal.: 0.096 @ 1064 nm | 63 |
| CsCl·(H2SeO3)2 | P-1 | 5.46 | Cal.: 0.1 @ 532 nm | 64 |
| Zr(SeO3)2 | P21/c | 5.1 | Cal.: 0.1025 @ 1064 nm | 63 |
| Hg3(SeO3)2(SO4) | P21 | 4.7 | Cal.: 0.103 @ 1064 nm | 34 |
| Sc(HSeO3)3 | Cc | 5.28 | Cal.: 0.105 @ 1064 nm | 19 |
| Gd(NO3)(Se2O5)·3H2O | P212121 | 5.53 | Cal.: 0.109 @ 1064 nm | 65 |
| CaYF(SeO3)2 | Pbcn | 5.06 | Cal.: 0.127 @ 1064 nm | 37 |
| NaLu(SeO3)2 | Pna21 | 5.3 | Cal.: 0.128 @ 1064 nm | 28 |
| CsCl H2SeO3 | P21/c | 5.23 | Cal.: 0.13 @ 532 nm | 64 |
| RbCl·(H2SeO3)2 | P-1 | 5.39 | Cal.: 0.14 @ 589 nm | 66 |
| CsBr·(H2SeO3)2 | P21/m | 5.19 | Cal.: 0.14 @ 532 nm | 64 |
| Pb 2 (SeO 3 )(SiF 6 ) | Pnma | 4.4 | Cal.: 0.161 @ 546 nm | This work |
| KCl·(H2SeO3)2 | P-1 | 5.44 | Cal.: 0.17 @ 532 nm | 64 |
Conclusions
In summary, the first selenite fluorosilicate, Pb2(SeO3)(SiF6), has been successfully obtained through modification using perfluorinated groups. The compound exhibits a unique 3D structure composed of 2D lead selenite layers interconnected by SiF6 groups. Excitingly, Pb2(SeO3)(SiF6) achieves a balance between a wide bandgap and large birefringence. It represents a new birefringent crystal with large birefringence (0.161 @ 546 nm), a short UV cut-off edge (247 nm), a broad transmittance window (0.25–6.3 μm), a high laser-induced damage threshold (50.8 MW cm−2) and excellent environmental stability. Notably, its birefringence is the largest among non-hydrogen selenite materials with bandgap ≥ 4.2 eV and stands as the only case of a lead selenite material with bandgap ≥4.2 eV and birefringence ≥ 0.1. Theoretical calculations proved that the large birefringence of Pb2(SeO3)(SiF6) primarily originates from SeO3 groups, PbO4 and PbO3F4 polyhedrons. Our work not only pioneers a new selenite system, but also more importantly demonstrates the significance of perfluorinated groups modification in the development of novel birefringent materials with a large bandgap.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Peng-Fei Li: investigation, formal analysis, and writing – original draft. Chun-Li Hu: theoretical calculations. Jiang-Gao Mao: supervision, resources, and funding acquisition. Fang Kong: conceptualization, project administration, and writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22031009, 22375201, 21921001 and 91963105) and the NSF of Fujian Province (Grant No. 2023J01216).Notes and references
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Nat. Photonics, 2023, 17, 694–701 CrossRef CAS.
- H. Y. Wu, C. L. Hu, M. B. Xu, Q. Q. Chen, N. Ma, X. Y. Huang, K. Du and J. Chen, Chem. Sci., 2023, 14, 9533–9542 RSC.
- S. J. Han, A. Tudi, W. B. Zhang, X. L. Hou, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2023, 62, e202302025 CrossRef CAS PubMed.
- Z. Fang, W. H. Ma, Q. Y. Chen, X. T. Zhu, X. M. Zeng, P. B. Li, Q. F. Zhou, T. T. Song and M. H. Duan, Inorg. Chem. Front., 2024, 11, 1775–1780 RSC.
- G. Zhou, J. Xu, X. Chen, H. Zhong, S. Wang, X. Ke, P. Deng and F. Gan, J. Cryst. Growth, 1998, 191, 517–519 CrossRef CAS.
- G. Ghosh, Opt. Commun., 1999, 163, 95–102 CrossRef CAS.
- M. J. Cohen, H. Luo, E. L. Dereniak, T. Tkaczyk, R. Sampson and E. L. Dereniak, presented in part at the Semiconductor Photodetectors III, 2006 Search PubMed.
- M. J. Dodge, Appl. Opt., 1984, 23, 1980–1985 CrossRef CAS.
- J. R. DeVore, J. Opt. Soc. Am., 1951, 41, 416–419 CrossRef CAS.
- M. Cheng, W. Q. Jin, Z. H. Yang and S. L. Pan, Chem. Sci., 2022, 13, 13482–13488 RSC.
- J. Guo, A. Tudi, S. Han, Z. Yang and S. Pan, Angew. Chem., Int. Ed., 2021, 60, 24901–24904 CrossRef CAS PubMed.
- Y. Lan, J. Ren, P. Zhang, X. Dong, L. Huang, L. Cao, D. Gao and G. Zou, Chin. Chem. Lett., 2024, 35, 108652 CrossRef CAS.
- M. Mutailipu, M. Zhang, Z. H. Yang and S. L. Pan, Acc. Chem. Res., 2019, 52, 791–801 CrossRef CAS PubMed.
- W. Zhou and S. P. Guo, Acc. Chem. Res., 2024, 57, 648–660 CAS.
- H. Sha, D. Yang, Y. Shang, Z. Wang, R. Su, C. He, X. Yang and X. Long, Chin. Chem. Lett., 2024, 109730 CrossRef.
- M. Y. Ran, A. Y. Wang, W. B. Wei, X. T. Wu, H. Lin and Q. L. Zhu, Coord. Chem. Rev., 2023, 481, 215059 CrossRef CAS.
- J. Chen, C. Lin, X. Jiang, G. Yang, M. Luo, X. Zhao, B. X. Li, G. Peng, N. Ye, Z. Hu, J. Wang and Y. Wu, Mater. Horiz., 2023, 10, 2876–2882 RSC.
- P. F. Li, J. G. Mao and F. Kong, Mater. Today Phys., 2023, 37, 101197 CrossRef CAS.
- Z. Bai, J. Lee, H. Kim, Y. Kuk, M. H. Choi, C. L. Hu and K. M. Ok, Small, 2023, 19, 2207709 CrossRef CAS PubMed.
- Y. J. Jia, X. Y. Zhang, Y. G. Chen, X. X. Jiang, J. N. Song, Z. S. Lin and X. M. Zhang, Inorg. Chem., 2022, 61, 15368–15376 CrossRef CAS.
- S. S. Shi, C. S. Lin, G. S. Yang, L. L. Cao, B. X. Li, T. Yan, M. Luo and N. Ye, Chem. Mater., 2020, 32, 7958–7964 CrossRef CAS.
- Y. P. Gong, C. L. Hu, F. Kong and J. G. Mao, Chem.–Eur. J., 2019, 25, 3685–3694 CrossRef CAS PubMed.
- L. Lin, X. X. Jiang, C. Wu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Dalton Trans., 2021, 50, 7238–7245 RSC.
- Y. X. Ma, P. F. Li, C. L. Hu, J. G. Mao and F. Kong, Adv. Sci., 2023, 10, 2304463 CrossRef CAS PubMed.
- J. Ren, H. Cui, L. Cheng, Y. Zhou, X. Dong, D. Gao, L. Huang, L. Cao and N. Ye, Inorg. Chem., 2023, 62, 21173–21180 CrossRef CAS PubMed.
- Y. P. Gong, C. L. Hu, Y. X. Ma, J. G. Mao and F. Kong, Inorg. Chem. Front., 2019, 6, 3133–3139 RSC.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Inorg. Chem., 2023, 62, 8494–8499 CrossRef CAS PubMed.
- P. F. Li, C. L. Hu, J. G. Mao and F. Kong, Mater. Horiz., 2024, 11, 1704–1709 RSC.
- P. F. Li, Y. P. Gong, C. L. Hu, B. Zhang, J. G. Mao and F. Kong, Adv. Opt. Mater., 2024, 12, 2301426 CrossRef CAS.
- P. F. Li, C. L. Hu, B. X. Li, F. Kong and J. G. Mao, J. Alloys Compd., 2023, 959, 170570 CrossRef CAS.
- P. Zhang, X. Mao, X. Dong, L. Huang, L. Cao, D. Gao and G. Zou, Chin. Chem. Lett., 2023, 109235, DOI:10.1016/j.cclet.2023.
- P. F. Li, C. L. Hu, Y. F. Li, J. G. Mao and F. Kong, J. Am. Chem. Soc., 2024, 146, 7868–7874 CrossRef CAS PubMed.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Mater. Chem. Front., 2022, 6, 3567–3576 RSC.
- J. Ren, Y. Chen, L. Ren, Y. Zhou, X. Dong, D. Gao, L. Huang, L. Cao and N. Ye, Inorg. Chem., 2023, 62, 9130–9138 CrossRef CAS PubMed.
- H. Qiu, S. Pan and M. Mutailipu, Fundam. Res., 2023 DOI:10.1016/j.fmre.2023.08.017.
- C. Shen, F. Zhang, T. Sasaki, C. Eerdun, M. Hayashi, H.-w. Wang, K. Tominaga, M. Mutailipu and S. Pan, Angew. Chem., Int. Ed., 2024, 63, e202319121 CrossRef CAS.
- P. F. Li, C. L. Hu, F. Kong and J. G. Mao, Angew. Chem., Int. Ed., 2023, 62, e202301420 CrossRef CAS.
- Y. Yang, Y. Xiao, B. Li, Y. G. Chen, P. Guo, B. Zhang and X. M. Zhang, J. Am. Chem. Soc., 2023, 145, 22577–22583 CrossRef CAS PubMed.
- G. Xu, H. Li, J. Han, X. Hou, Z. Yang and S. Pan, Inorg. Chem., 2023, 63, 852–859 CrossRef PubMed.
- H. Qiu, F. Li, C. Jin, Z. Yang, J. Li, S. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2023, 63, e202316194 CrossRef PubMed.
- Y. Hu, X. Jiang, T. Wu, Y. Xue, C. Wu, Z. Huang, Z. Lin, J. Xu, M. G. Humphrey and C. Zhang, Chem. Sci., 2022, 13, 10260–10266 RSC.
- M. Yan, R. L. Tang, W. D. Yao, W. Liu and S. P. Guo, Chem. Sci., 2024, 15, 2883–2888 RSC.
- R. L. Tang, X. Lian, W. D. Yao, W. Liu and S. P. Guo, Dalton Trans., 2021, 50, 16562–16567 RSC.
- Y. Shen, Y. Zhou, X. Xue, H. Yu, S. Zhao and J. Luo, Inorg. Chem. Front., 2022, 9, 5226–5230 RSC.
- P. F. Li, C. L. Hu, B. X. Li, J. G. Mao and F. Kong, Inorg. Chem. Front., 2023, 10, 7343–7350 RSC.
- C. Bai, Y. Chu, J. Zhou, L. Wang, L. Luo, S. Pan and J. Li, Inorg. Chem. Front., 2022, 9, 1023–1030 RSC.
- Y. Y. Yang, Y. Guo, B. B. Zhang, T. Wang, Y. G. Chen, X. H. Hao, X. X. Yu and X. M. Zhang, Inorg. Chem., 2022, 61, 1538–1545 CrossRef CAS PubMed.
- C. Stoll, M. Seibald, D. Baumann, J. Bandemehr, F. Kraus and H. Huppertz, Eur. J. Inorg. Chem., 2021, 2021, 62–70 CrossRef CAS.
- J. Rienmüller, J. Bandemehr and F. Kraus, Z. Naturforsch., B: J. Chem. Sci., 2021, 76, 559–565 CrossRef.
- W. Hinteregger, H. Niederwieser and Z. Huppertz, Kristallogr, 2014, 229, 77–82 Search PubMed.
- B. N. E. Brese and M. O'Keeffe, Acta Crystallogr., 1991, B47, 192–197 Search PubMed.
- P. F. Li, F. Kong and J. G. Mao, J. Solid State Chem., 2020, 286, 121288 CrossRef CAS.
- S. H. Kim, J. Yeon and P. S. Halasyamani, Chem. Mater., 2009, 21, 5335–5342 CrossRef CAS.
- H. Zhao, P. Gong, X. Zhang, Z. Lin, Z. Hu and Y. Wu, Dalton Trans., 2020, 49, 14046–14051 RSC.
- X. X. Wang, X. X. Jiang, H. M. Liu, L. Yang, Z. S. Lin, Z. G. Hu, X. G. Meng, X. G. Chen and J. G. Qin, Dalton Trans., 2018, 47, 1911–1917 RSC.
- Z. Yang, C. Hu, M. Mutailipu, Y. Sun, K. Wu, M. Zhang and S. Pan, J. Mater. Chem. C, 2018, 6, 2435–2442 RSC.
- P. F. Li, C. L. Hu, B. X. Li, J. G. Mao and F. Kong, Inorg. Chem., 2024, 63, 4011–4016 CrossRef CAS PubMed.
- H. Y. Sha, Y. R. Shang, Z. J. Wang, R. B. Su, C. He, X. M. Yang and X. F. Long, Small, 2023, e2309776, DOI:10.1002/smll.202309776.
- X. W. Zhang, Z. X. Wang, C. L. Hu, Y. F. Li, J. G. Mao and F. Kong, Inorg. Chem., 2024, 63, 6067–6074 CrossRef CAS PubMed.
- L. Liu, J. Wang, B. Xiao, X. Li, Y. Chu, T. Sun and P. S. Halasyamani, J. Mater. Chem. C, 2024, 12, 4986–4994 RSC.
- J. Zhang, H. Yu, Z. Hu, J. Wang, Y. Wu and H. Wu, Mater. Today Phys., 2023, 35, 101145 CrossRef CAS.
- M. Y. Cao, C. L. Hu, F. Kong, Z. Y. Xiong and J. G. Mao, Dalton Trans., 2021, 50, 15057–15061 RSC.
- J. Lv, Q. Li, X. Guan, N. Lin, J. Zhang, Z. Jia and X. Tao, CrystEngComm, 2023, 25, 1675–1682 RSC.
- M. Zhu, J. Wang, L. Hou, Y. Yuan, L. Liu, Y. Chu and C. Huang, Inorg. Chem., 2024, 63, 2289–2297 CrossRef CAS PubMed.
- C. Wu, L. H. Li, L. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Dalton Trans., 2020, 49, 3253–3259 RSC.
- H. Wang, L. Liu, Z. Hu, J. Wang, M. Zhu, Y. Meng and J. Xu, Inorg. Chem., 2022, 62, 557–564 CrossRef PubMed.
- C. L. Hu, Y. X. Han, Z. Fang and J. G. Mao, Chem. Mater., 2023, 35, 2647–2654 CrossRef CAS.
- H. Chen, M. Y. Ran, S. H. Zhou, X. T. Wu, H. Lin and Q. L. Zhu, Chin. Chem. Lett., 2023, 34, 107838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, computational method, crystal data, important bond distances and bond valences, element analysis, PXRD patterns, TGA curves, IR spectrum, UV-visible-NIR diffuse reflectance spectrum and band structures. CCDC 2327037. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01376j |
| This journal is © The Royal Society of Chemistry 2024 |