
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-mobility organic mixed conductors with a low synthetic complexity index via direct arylation polymerization†
Joost
Kimpel
 *a,
Youngseok
Kim
a,
Jesika
Asatryan
b,
Jaime
Martín
b,
Renee
Kroon
cd and
Christian
Müller
*a
*a,
Youngseok
Kim
a,
Jesika
Asatryan
b,
Jaime
Martín
b,
Renee
Kroon
cd and
Christian
Müller
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Göteborg, Sweden. E-mail: kimpel@chalmers.se; christian.muller@chalmers.se
bUniversidade da Coruña, Campus Industrial de Ferrol, CITENI, Esteiro, 15403, Ferrol, Spain
cLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, Norrköping, Sweden
dWallenberg Initiative Materials Science for Sustainability, Department of Science and Technology, Linköping University, Norrköping, Sweden
First published on 19th April 2024
Abstract
Through direct arylation polymerization, a series of mixed ion-electron conducting polymers with a low synthetic complexity index is synthesized. A thieno[3,2-b]thiophene monomer with oligoether side chains is used in direct arylation polymerization together with a wide range of aryl bromides with varying electronic character from electron-donating thiophene to electron-accepting benzothiadiazole. The obtained polymers are less synthetically complex than other mixed ion–electron conducting polymers due to higher yield, fewer synthetic steps and less toxic reagents. Organic electrochemical transistors (OECTs) based on a newly synthesized copolymer comprising thieno[3,2-b]thiophene with oligoether side chains and bithiophene exhibit excellent device performance. A high charge-carrier mobility of up to μ = 1.8 cm2 V−1 s−1 was observed, obtained by dividing the figure of merit [μC*] from OECT measurements by the volumetric capacitance C* from electrochemical impedance spectroscopy, which reached a value of more than 215 F cm−3.
Introduction
Organic mixed ionic–electronic conductors (OMIECs) are a class of semiconducting materials that are widely used in the field of bioelectronics1–4 as well as for energy harvesting5–7 and storage.8,9 OMIECs can transport both ions and electrons and the charge-carrier density can be modulated through the application of an electrochemical potential, for instance when used as the channel material of organic electrochemical transistors (OECTs), the basic building blocks of more advanced electrochemical circuitry.10,11 However, many high-performance materials and devices require cumbersome synthetic protocols and fabrication methods, and hence significant improvements are needed to realize a truly sustainable technology.12Some of the most promising types of OMIECs are conjugated polymers with oligoether side chains. The conjugated backbone can conduct electronic charge while the oligoether side chains result in a high ionic mobility and facilitate the ingression of counterions from the electrolyte.13,14 As a result, many conjugated polymers with oligoether side chains feature both a high charge-carrier mobility μ as well as a high volumetric capacitance C* (see Fig. 1), two parameters that are often used to compare OMIECs.15 Among p-type materials, thieno[3,2-b]thiophene-based copolymers can exhibit outstanding electrochemical performance, e.g. a [μC*] value as large as 102 to 103 F cm−1 V−1 s−1,16 which in a few cases exceeds that of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS, see Fig. 1).17,18
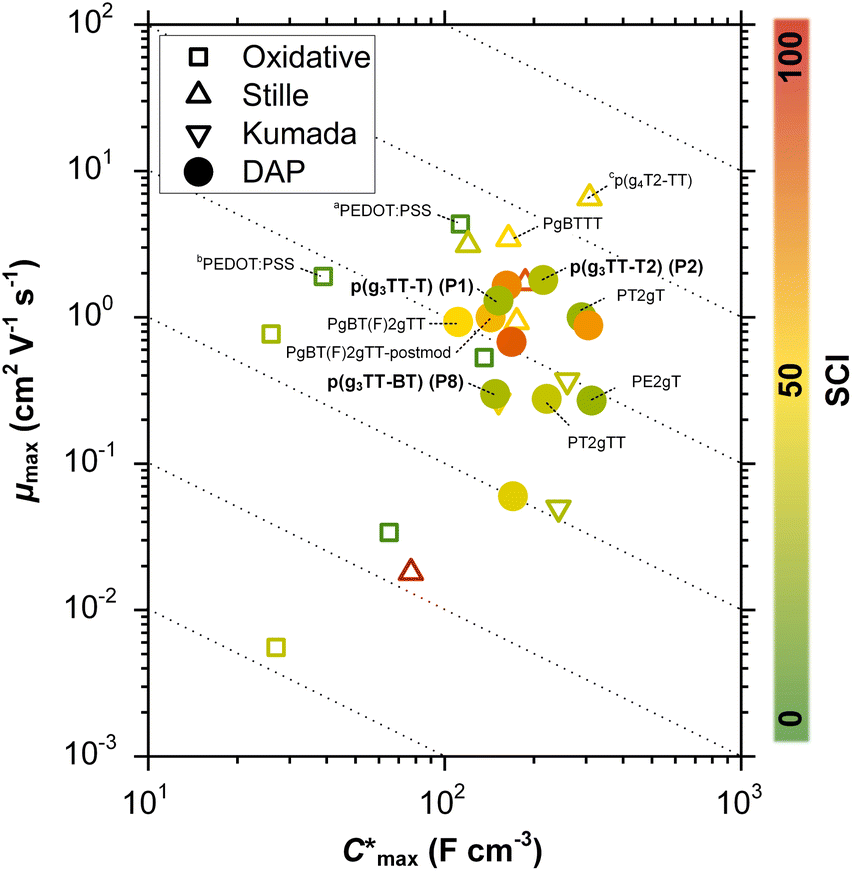 | ||
Fig. 1 Ashby plot of maximum volumetric capacitance 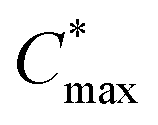 and maximum charge-carrier mobility μmax of previously reported p-type polymers,15–18,35–37,39–56 and selected polymers synthesized in this work (bold) by oxidative polymerization (squares), Stille coupling (upward triangles), Kumada coupling (downward triangles) or DAP (circles), with the synthetic complexity index (SCI) of each synthesis indicated by a red-green scale. aPoly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) post-treated with sulfuric acid,17bPEDOT:PSS post-treated with ethylene glycol,15cp(g4T2-TT) (see Fig. S1† for chemical structure) fractionated by size-exclusion chromatography (highest molecular weight fraction, catalyst removed).16 See Fig. S2† for the Ashby plot of SCI vs. [μC*]max. and maximum charge-carrier mobility μmax of previously reported p-type polymers,15–18,35–37,39–56 and selected polymers synthesized in this work (bold) by oxidative polymerization (squares), Stille coupling (upward triangles), Kumada coupling (downward triangles) or DAP (circles), with the synthetic complexity index (SCI) of each synthesis indicated by a red-green scale. aPoly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) post-treated with sulfuric acid,17bPEDOT:PSS post-treated with ethylene glycol,15cp(g4T2-TT) (see Fig. S1† for chemical structure) fractionated by size-exclusion chromatography (highest molecular weight fraction, catalyst removed).16 See Fig. S2† for the Ashby plot of SCI vs. [μC*]max. | ||
Thieno[3,2-b]thiophene-based copolymers, and more generally many state-of-the-art OMIEC materials, are synthesized via cross coupling polycondensation reactions between aryl halides and other reactive monomers. The most widely used cross coupling polycondensation reactions such as Stille coupling (organotin), Suzuki coupling (organoboron) and Kumada coupling (organomagnesium) share a common drawback: one of the monomers must be functionalized to include the reactive group that facilitates the polymerization. Besides adding a synthetic step, the functionalization makes precursors toxic (Stille), atom inefficient (Suzuki), or pyrophoric and difficult to handle (Kumada). As a result, OMIECs made by these techniques feature a relatively high synthetic complexity index (SCI). The SCI is a factor that assesses the accessibility of a compound by considering a variety of parameters including the number of synthetic steps, the yield, the number of work-up operations and the hazards involved, and should therefore be kept low.19 The SCI is a value relative to the highest SCI in the set of compounds that is compared.
An alternative synthesis route is direct arylation polymerization (DAP), a polycondensation method that uses a palladium catalyst and various additives to couple unfunctionalized aromatics and aryl halides yielding conjugated polymers.20–22 Avoidance of functionalization leads to a better atom economy as well as less toxic reactive species and side products. This all results in a lower SCI.
Despite the clear advantages of DAP, the resulting materials often underperform compared to other conventional routes. Compared to Stille-made polymers, direct arylation polymers often have a lower molecular weight23 and a greater prevalence of defects.24 Homocoupling defects are defined by subsequent monomers in the chain repeating themselves. This is caused by aryl nucleophiles (Ar–H) and aryl electrophiles (Ar–Br) in DAP being much closer in reactivity. Accordingly, the C–H bond must be sufficiently active to undergo reaction and prevent homocoupling of the dibrominated monomer – a side reaction also seen in Stille and Suzuki coupling despite highly orthogonal reactivity of the monomers in those polymerization reactions.25,26 Generally, homocoupling defects somewhat limit the synthesis of high molecular-weight polymers as per Carothers' equation. Branching defects, i.e. β-defects, are more detrimental to the device properties of the final material and are an explicit class of defects that can arise when DAP is used. Branching occurs when an unintended aryl C–H is activated on the monomer. Activation of β-protons can cause cross-conjugation in the polymer as well as poor π-stacking due to crowding of polymer chains. For instance, an increased branching content in a diketopyrrolopyrrole (DPP) based copolymer led to a significant decrease in photovoltaic device efficiency due to poor π-stacking.27 Instead, naphthalenediimide (NDI) based copolymers made with controlled defect-free DAP to minimize branching defects showed more reproducible results in field effect transistors.28,29 In some cases, β-defects led to crosslinking and thus less soluble polymers.30
One strategy to avoid coupling defects is through judicious monomer design. Homocoupling can be mostly circumvented by using aromatic units with low C–H bond dissociation energies. This can be achieved by making the aromatic unit electron-poor, in the case of 1,2,4,5-tetrafluorobenzene, or electron rich, in the case of 3,4-ethylenedioxythiophene (EDOT).31,32 Occurrence of β-defects can be completely ruled out by substituting all aryl protons except the desired reaction sites. This design principle has been employed in case of direct arylation between EDOT and 2,7-dibromo-9,9-dioctylfluorene, as well as direct arylation between EDOT and dibromo-EDOT analogues, yielding polymers without any branching defects.33,34 The possibility of defect formation shrinks the pool of monomers for use in DAP. Even so, with all these design principles in mind, a highly active monomer, which circumvents homocoupling and branching, can lead to successful DAP and high-performance polymers.
Since some of the best performing OMIEC materials comprise thieno[3,2-b]thiophene units,16 3,6-bis(triethylene glycol monomethyl ether)thieno[3,2-b]thiophene (g3TT), which only features two active C–H bonds, is an attractive monomer for DAP. The oligoether chains block positions that could have led to β-defects. Ding et al. have polymerized g3TT in combination with a fluorinated bis(thiophenyl)benzothiadiazole to obtain the polymer PgBT(F)2gTT (see Fig. S1† for chemical structure), albeit only achieving a low number-average molecular weight of Mn ≈ 4 kg mol−1 and a relatively low figure-of-merit of [μC*] = 103 F cm−1 V−1 s−1, which has recently been improved to 145 F cm−1 V−1 s−1 through post-polymerization modification (see Fig. 1 and S1†).35,36 In another recent study g3TT was paired with thiophene, resulting in a Mn ≈ 11 kg mol−1 and a low figure-of-merit of [μC*] = 61 F cm−1 V−1 s−1.37 In case of a polythiophene with oligoether side chains, prepared by Stille coupling, intermediate values of Mn ≈ 20 kg mol−1 yield an optimal OECT performance and thus it can be anticipated that higher [μC*] values can be achieved if g3TT is paired with other comonomers that enable a higher degree of polymerization.38
In this work, the versatility of g3TT monomers for DAP is explored. We demonstrate that g3TT can be combined with a wide range of common conjugated comonomers, including electron-rich, electron-deficient, and neutral comonomers such as thiophene (T), benzothiadiazole (BT), and fluorene (F), respectively. Gratifyingly, the makeup of g3TT-copolymers allows for absolute molecular weight determination through high-temperature NMR. The most promising copolymer, p(g3TT-T2), gave rise to a state-of-the-art OECT performance with [μC*] = 370 F cm−1 V−1 s−1. Importantly, the here described polymers are associated with a lower SCI than other OMIEC materials made by DAP or Stille coupling, which can be attributed to fewer synthetic steps and less toxic reagents (see Fig. 1; see Table S1, Fig. S1† for considered chemical structures, Fig. S2† for SCI vs. [μC*]max, and Fig. S3–S28† for SCI calculations).
Results and discussion
Synthesis
3,6-Bis(triethylene glycol monomethyl ether)thieno[3,2-b]thiophene (g3TT) was synthesized by Ullmann-type coupling of triethylene glycol monomethyl ether to 3,6-dibromothieno[3,2-b]thiophene (see Fig. 2). To circumvent the use of harmful solvents and to improve the yield, we attempted the reaction using triethylene glycol monomethyl ether as the reaction medium, i.e. the compound acted as both the solvent and reagent. Coupling of the glycol chain to the aromatic unit was achieved by using copper(I)-catalyzed nucleophilic aromatic substitution. Commonly, the oligoether chain is deprotonated using a strong base, e.g. potassium tert-butoxide (KOtBu), to make it more nucleophilic. We explored the use of sodium hydroxide and cesium hydroxide monohydrate instead of highly reactive and highly hygroscopic KOtBu. Ullmann couplings also benefit from the inclusion of amine ligands to stabilize the copper(I) species. Hence, alongside the solvent and base change we investigated a variety of amine additives (see Fig. S29, Table S3 and ESI Section S2.3† for synthetic details).57,58 Ultimately, the use of KOtBu as a base and glycol as the reaction medium significantly increased the final yield of g3TT to around 50%, more closely resembling the efficiency of Ullmann coupling for thiophene.46 Any changes to this protocol were either detrimental (addition of amine ligand and change of base) or did not improve this initial method (increasing the amount of KOtBu). After a full work-up including extraction, flash column chromatography and recrystallization, g3TT was obtained. The final g3TT product formed crystals –indicative of the high purity of the compound– and its structure was confirmed with NMR techniques and single crystal X-ray diffraction (see Fig. S30–S35†).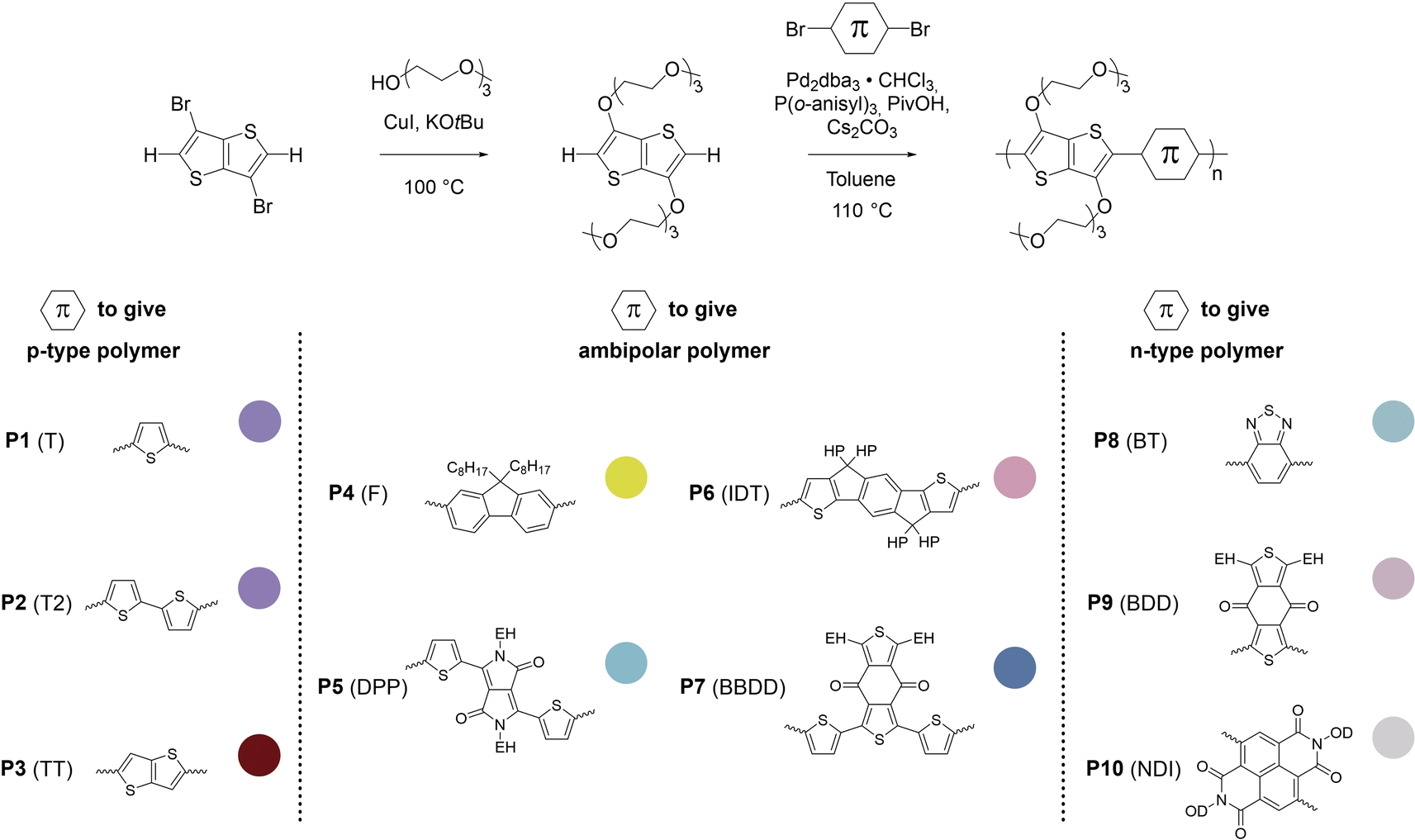 | ||
| Fig. 2 Synthesis of g3TT and direct arylation polymerization (DAP) of g3TT with comonomers that tend to give polymers with p-type, ambipolar or n-type character. The insets show colors extracted from photographs of thin films of the polymers P1–P10. EH = 2-ethylhexyl, HP = 4-hexylphenyl, OD = 2-octyldodecyl. | ||
DAP of g3TT and a variety of common brominated comonomers was performed. The chosen comonomers ranged from units that generally result in p-type polymers (thiophene (T), bithiophene (T2), and thieno[3,2-b]thiophene (TT)), to units that generally result in ambipolar polymers (fluorene (F), diketopyrrolopyrrole (DPP), indacenodithiophene (IDT), and bisthiophenylbenzodithiophenedione (BBDD)), as well as units that generally result in n-type polymers (benzothiadiazole (BT), benzodithiophenedione (BDD), and naphthalenediimide (NDI)), yielding polymers P1–P10 which featured a wide range of colors (see Fig. 2 and ESI Section S2.4† for synthetic details). Final polymers were obtained by precipitating the reaction mixtures into hexane, treating the crude with a palladium scavenging agent, and subsequently purifying the re-precipitated solids by Soxhlet extraction (Table 1).
| Polymer | Total yielda (%) | Yieldb (%) | M n,SEC (kg mol−1) | Đ | M n,NMR (kg mol−1) |
|---|---|---|---|---|---|
| a Yield of polymer before Soxhlet extraction relative to the loading of monomers and theoretical yield. b Yield of highest molecular weight fraction from Soxhlet extraction (for P1–P3 and P8, an average yield from multiple syntheses is given, see Table S5), relative to the loading of monomers and theoretical yield. c From the materials post-Soxhlet, measured by SEC at 70 °C against poly(methyl methacrylate) (PMMA) standards using dimethylformamide (DMF) with 0.1 wt% LiBr as the eluent. d From the materials post-Soxhlet, determined by end group analysis of NMR spectra recorded at 120 °C in C2D2Cl4 using a Bruker Avance NEO 600 spectrometer. Error determined by adding signal-to-noise of main chain and end-group in quadrature (Section S3.2). e Polymer did not dissolve in DMF with 0.1 wt% LiBr. f Polymers elute later than PMMA calibration suggesting oligomers, confirmed by room temperature NMR using a Bruker Avance NEO 600 spectrometer. | |||||
| P1 (T) | 87 | 48 | 14 | 6.4 | 14 ± 0.4 |
| P2 (T2) | 93 | 28 | 29 | 2.2 | 39 ± 6 |
| P3 (TT) | 77 | 38 | 5 | >10 | 3 ± 0.1 |
| P4 (F) | 97 | 31 | 34 | 1.7 | 49 ± 6 |
| P5 (DPP) | 73 | 17 | Oligomericf | n.a.f | Oligomericf |
| P6 (IDT) | 96 | 27 | 9 | 1.4 | 29 ± 3 |
| P7 (BBDD) | 84 | 50 | 15 | 6.3 | 16 ± 2 |
| P8 (BT) | 84 | 43 | n.a.e | n.a.e | 13 ± 0.2 |
| P9 (BDD) | 82 | 67 | 90 | 1.3 | 21 ± 1 |
| P10 (NDI) | 98 | 94 | oligomericf | n.a.f | Oligomericf |
By size-exclusion chromatography (SEC), we determined the number-average molecular weight Mn,SEC with a Polargel column using dimethylformamide (DMF) with 0.1 wt% LiBr as the eluent (see Fig. S36†). The polar column was necessary due to the high polarity of the oligoether side chains. A range of polymer molecular weights was obtained using DAP under the aforementioned conditions; oligomers for P5 (DPP) and P10 (NDI), Mn,SEC <10 kg mol−1 for P3 (TT) and P6 (IDT), and Mn,SEC >10 kg mol−1 for the other polymers reaching up to 91 kg mol−1 for P9 (BDD). Aggregation peaks in chromatograms of P2 (T2), P3 (TT), and P7 (BBDD) suggest that the polymers are only partially soluble. In those cases, only the molecular weight of the soluble fraction could be calculated. We suspect that P9 also aggregates, though its aggregates passed through the filter, which would result in an overestimate of the molecular weight and thus a high Mn,SEC. In case of P8 (BT) the chromatogram could not be recorded due to the insolubility of the polymer in DMF with 0.1 wt% LiBr. Note that the reported chromatograms were recorded with relative calibration using PMMA as a standard and thus the here quoted values must not be taken as absolute.
In some specific cases, end groups of polymers can confidently be assigned by high-temperature NMR.59,60 Then, by comparing the end-group signal with the main-chain signal, the absolute number-average molecular weight can be determined by NMR, Mn,NMR. High-temperature NMR spectra were recorded by dissolving the polymers in tetrachloroethane-d2 at 120 °C using a 600 MHz spectrometer (see Fig. S37–S44†). All polymers display two unique signals indicative of g3TT end groups, i.e. a peak assigned to the aromatic C–H (ca. 6.40 ppm, purple in Fig. 3) and a peak assigned to CH2 from the glycol chains closest to the thieno[3,2-b]thiophene unit (ca. 4.30 ppm, blue in Fig. 3). The aromatic C–H is a clear end-group signal owing to g3TT only possessing two aromatic C–H, which are consumed during the cross-coupling reaction. The oligoether signal is assigned to the end group since the ratio between the integrals of the aromatic C–H and the oligoether CH2 end group signal lies between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.99 and 1:2.15 (around 1:2) for all polymers. For P7–P9, the other oligoether CH2 peak from the end group is also visible (ca. 4.55 ppm in Fig. S42–S44,† turquoise in Fig. 3). In all other cases, this end-group peak has merged with the oligoether CH2 peak assigned to the polymer repeat unit. By comparing the CH2 oligoether end-group signals (ca. 4.30 ppm, blue in Fig. 3) to the CH2 oligoether main-chain signal (ca. 4.60–4.70 ppm, red in Fig. 3), Mn,NMR can be assigned. The end group glycol signal is more shielded than the main-chain glycol signal, also observed for the alkyl chain end group and main chain comparison in case of extensively studied poly(3-hexylthiophene) (P3HT).59,61 MALDI-ToF of P2 confirmed debromination of bithiophene end groups (Fig. S45†). All bromines were ultimately replaced with a hydrogen, commonly observed in case of direct arylation polymerization.62 Since the synthesis of all polymers is comparable, we argue that none of the polymers P1–4 and P6–P9 possess any residual bromine end groups. Assuming a statistical mixture of end groups of the two monomers, this allowed for the determination of Mn,NMR according to:
:1.99 and 1:2.15 (around 1:2) for all polymers. For P7–P9, the other oligoether CH2 peak from the end group is also visible (ca. 4.55 ppm in Fig. S42–S44,† turquoise in Fig. 3). In all other cases, this end-group peak has merged with the oligoether CH2 peak assigned to the polymer repeat unit. By comparing the CH2 oligoether end-group signals (ca. 4.30 ppm, blue in Fig. 3) to the CH2 oligoether main-chain signal (ca. 4.60–4.70 ppm, red in Fig. 3), Mn,NMR can be assigned. The end group glycol signal is more shielded than the main-chain glycol signal, also observed for the alkyl chain end group and main chain comparison in case of extensively studied poly(3-hexylthiophene) (P3HT).59,61 MALDI-ToF of P2 confirmed debromination of bithiophene end groups (Fig. S45†). All bromines were ultimately replaced with a hydrogen, commonly observed in case of direct arylation polymerization.62 Since the synthesis of all polymers is comparable, we argue that none of the polymers P1–4 and P6–P9 possess any residual bromine end groups. Assuming a statistical mixture of end groups of the two monomers, this allowed for the determination of Mn,NMR according to:
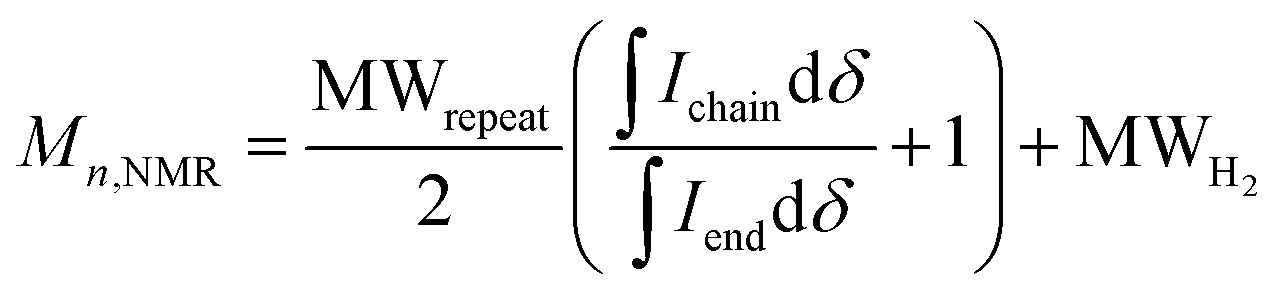 | (1) |
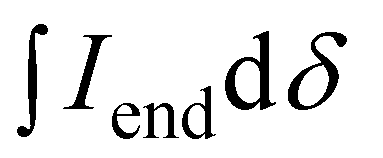 is the integral of the peak assigned to the oligoether CH2 of the end-group side chain that is oriented to the end of the polymer chain (ca. 4.30 ppm),
is the integral of the peak assigned to the oligoether CH2 of the end-group side chain that is oriented to the end of the polymer chain (ca. 4.30 ppm), 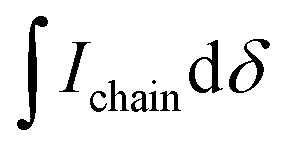 is the integral of the main-chain oligoether CH2 signal assigned to the g3TT repeat unit (ca. 4.60–4.70 ppm), MWrepeat is the molecular weight of the repeat unit, and MWH2 is the molecular weight of the hydrogens at the ends of the polymer chain (see ESI Section S3.1† for derivation of eqn (1)).
is the integral of the main-chain oligoether CH2 signal assigned to the g3TT repeat unit (ca. 4.60–4.70 ppm), MWrepeat is the molecular weight of the repeat unit, and MWH2 is the molecular weight of the hydrogens at the ends of the polymer chain (see ESI Section S3.1† for derivation of eqn (1)).
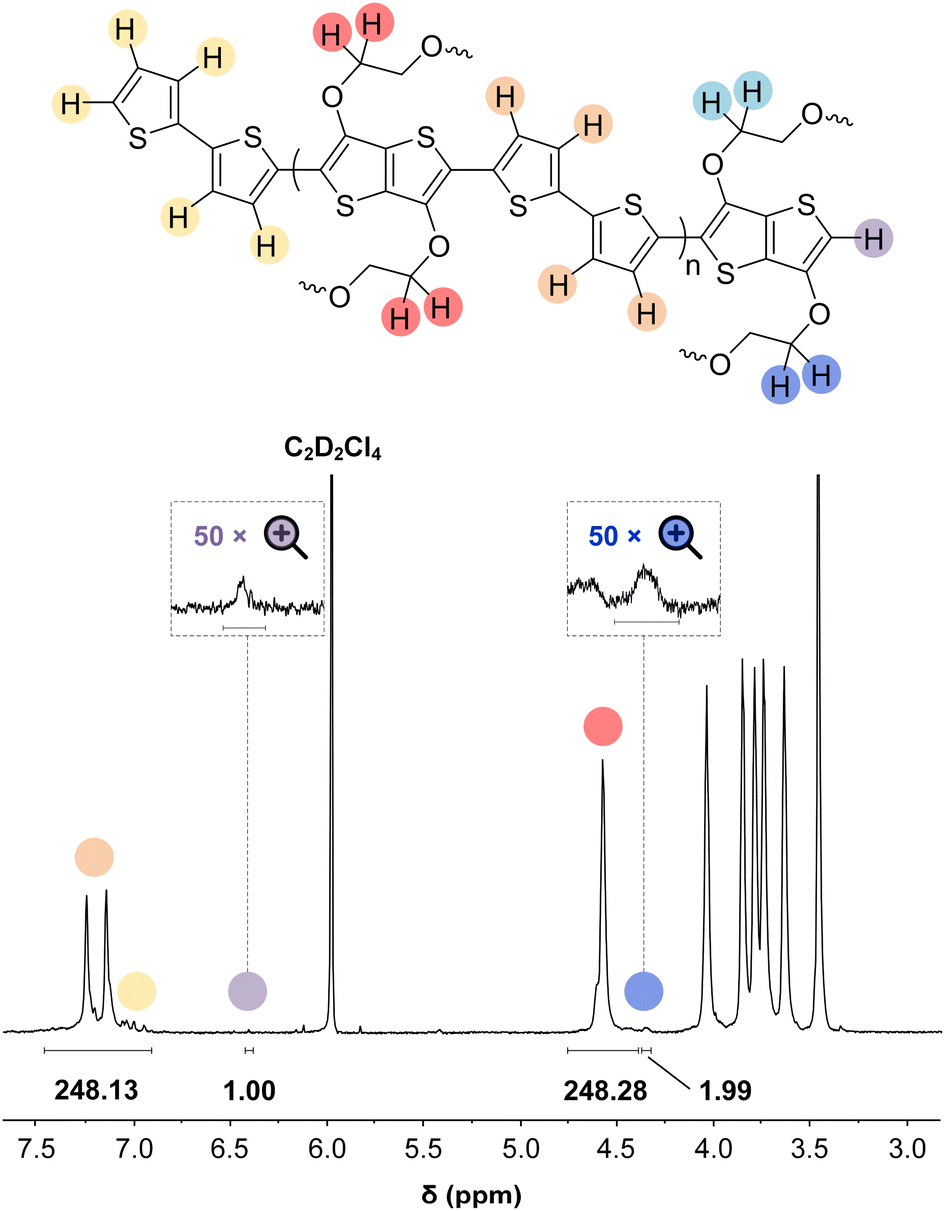 | ||
| Fig. 3 NMR spectrum of p(g3TT-T2) (P2 (T2)) recorded in tetrachloroethane-d2 at 120 °C, allowing end-group analysis by comparing the end-group oligoether CH2 (blue) and the main chain oligoether CH2 (red). The signals at circa 6.40 ppm and circa 4.30 are assigned to the aromatic C–H of g3TT (purple) and the end-group oligoether CH2 (blue), respectively, because the ratio of the integrated areas is 1:2. | ||
By comparing the aromatic proton signals of the comonomer (>7.00 ppm) and the methylene proton signal of the glycol chain closest to the aromatic system of g3TT in the main chain (Ar–O–CH2–, 4.25–4.70 ppm), the relative incorporation of comonomer and g3TT could be determined. The well-matching integrals of the peaks suggest that the comonomers and g3TT are incorporated by the same amount. A maximum deviation of 3% in repeat unit integral is observed, favoring additional g3TT incorporation compared to the comonomer. From this, we expect that the homocoupling amount is minimal.
No clear trend in molecular weight could be linked to the electronic character of the comonomers. The lower molecular weights can be explained in some cases: (i) P3 (TT) is highly planar and one-dimensional causing strong aggregation, which possibly causes precipitation during the polymerization, as observed by blockage in the pipette upon dilution with chloroform, (ii) for P5 (DPP) the brominated DPP comonomer may suffer from activated protons leading to β-defects, as observed in an experimental and computational study on DPP reactivity by Bura et al.,63 which would then cause a monomer imbalance (excess of C–H), and (iii) P10 (NDI) can be expected to experience strong transannular strain between the oligoether side chain from g3TT and the aromatic proton or the carbonyl on NDI.
The SCI of the newly synthesized polymers was compared against values for previously reported polymers (see Fig. 1 and ESI Section S1† for SCI calculations).19 A lower SCI value indicates a more benign and scalable synthesis. For the polymers in this work, the average yield was used for the SCI calculation in case the material was synthesized multiple times. When considering polymers made by polycondensation, the polymers synthesized in this work feature lower SCI values of 29 to 32 compared to previously reported g3TT based polymers made by DAP with a SCI = 35–57 (see Table S1†),35–37 thiophene-based polymers with oligoether side chains made by Stille coupling with a SCI = 36 to 93 (see Table S1†),41,46,64 and even lower than commercially available poly[2,5-bis(3-tetradecylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT) with a SCI = 36, also made by Stille coupling (see Table S1†).49 The only material with a similar SCI of 26 is a recently reported polythiophene comprising 3,4-bisoligoether thiophene made by DAP.37 Generally, higher yield, fewer synthetic steps and fewer hazardous starting materials are the main reasons for the here described low SCI. This is illustrated by calculating the SCI that would be obtained if PgBT(F)2gTT, PgBT(F)2gTT-postmod and PT2gTT (see Fig. 1 and ESI S4 and S8†) were instead prepared using g3TT synthesized as described here. The SCI is then reduced by five points.
One concern associated with an SCI analysis is the variation in reaction yields and accordingly the error in the SCI. To this end, we calculated the error in SCI based on a comparison of repeated syntheses (Table S3†). The monomer synthesis to g3TT was repeated a dozen times, and selected polymers were resynthesized several times (Tables S4 and S5†). Through this effort, a range of SCI errors was found between about 2 and 4. For instance, the SCI of P2 (T2) of 32 ± 1.5 and the SCI of P8 (BT) is 29 ± 3.8. In other words, the repeated SCI values are only slightly affected by variations in yield.
In some cases, synthetic steps can be skipped since the intermediate is commercially available (Section S1.2†). By considering commercially available intermediates as a new starting point in the synthesis, a ‘commercially available’ SCI (SCIcomm.avail.) can be calculated. This is the case for seven syntheses in the SCI range 50–93, and PBTTT with an SCI of 37. On average, this decreases the SCI by 13 points, which supports the inclusion of SCIcomm.avail. in future SCI endeavors.
Thin-film nanostructure
UV-vis absorption spectra of thin films of all polymers P1–P10 spin-coated from chloroform solutions feature strong absorption bands in the visible region (see Fig. 4a and S46; see Table S6† for optical bandgaps). Some polymers such as P1 and P2 feature vibronic peaks around 600 nm, which in case of PBTTT indicate the presence of ordered domains.65,66 Grazing-incidence wide-angle X-ray scattering (GIWAXS) was performed to examine the solid-state structure of polymer films spin-coated from chloroform solutions in more detail (see Fig. 4b and S47, Table S7†). Diffractograms of polymers P2–P4, P6 and P9 feature a broad amorphous halo at q = 14.8 to 15.0 nm−1 (d = 0.42 to 0.43 nm), while P1, P7 and P8 feature a π-stacking peak at q010 = 16.7 to 16.8 nm−1 (d010 = 0.36 to 0.38 nm), indicating the presence of ordered domains. The amorphous halo observed in case of several polymers (P2–P4 and P6) also entails a shoulder at higher q values, which suggests that there is a minor degree of π-stacking also for these polymers. For polymers P1, P2 and P7–P9 diffractograms also feature lamellar stacking peaks at q100 of 2.87 to 3.55 nm−1 (d100 = 1.8 to 2.2 nm), respectively, confirming the presence of ordered domains. A coherence length of lamellar stacking of up to 10.4 nm in case of P2 (note that a second order diffraction at q200 = 7.61 nm−1 is also observed) suggests that ordered domains can reach a significant size. Nevertheless, differential scanning calorimetry (DSC) thermograms did not reveal any endothermic melting peaks (data not shown), suggesting that only a small fraction of the polymer films is composed of ordered domains. Degradation during the DSC measurements was ruled out by thermogravimetric analysis (TGA; see Fig. S48†).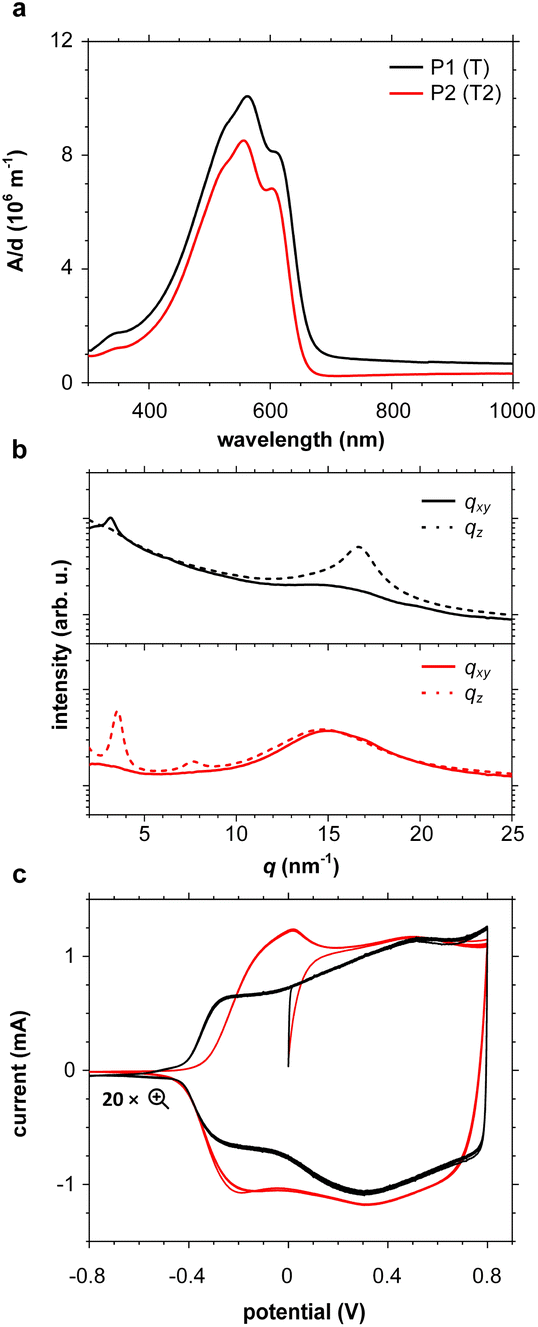 | ||
| Fig. 4 (a) Thickness-normalized UV-vis absorption spectra (absorbance A divided by film thickness d), (b) GIWAXS in-plane diffractograms (qxy; solid lines) and out-of-plane (qz; dashed lines), and (c) cyclic voltammograms measured in 0.1 M NBu4PF6 in acetonitrile of thin films of polymers P1 (black) and P2 (red). | ||
Electrochemical properties
Cyclic voltammetry (CV) was used to determine the oxidation onset potentials Eox of thin films of polymers P1–P10 spin-coated from chloroform solutions, using in all cases an acetonitrile-based electrolyte (0.1 M NBu4PF6 in acetonitrile, AcN, see Fig. 4c and S47†), and for some polymers also an aqueous electrolyte (0.1 M NaCl in H2O, see Fig. S49 and Table S6†). Polymers with small, simple comonomers, i.e.P1 (T), P2 (T2), P3 (TT) and P8 (BT), feature a low Eox of 4.7 eV to 4.8 eV vs. ferrocene/ferrocenium (Fc/Fc+) in AcN, which can be attributed to the electron-rich g3TT core. Interestingly, the electron-withdrawing character of BT does not seem to influence Eox. Larger comonomers exert a greater effect on Eox, e.g. Eox = 5.6 eV in case of P4 (F), which is similar to values reported for polyfluorene,67 and in case of P9 (BDD) the electron-withdrawing ketones in BDD increase Eox to 5.7 eV. Oxidation of polymers P1–P5 and P7–P9 was accompanied by a change in color to grey, and the appearance of the neat polymer was recovered upon subsequent reduction, in agreement with the reversibility of CV measurements (see Fig. S50†). Only the polymers P1 (T), P2 (T2), P3 (TT) and P8 (BT), which do not carry alkyl side chains on the comonomer, showed a notable response when carrying out CV with an aqueous electrolyte (see Fig. S51†). Note that all other polymers P4–7, P9 and 10 possess a mixture of oligoether and alkyl side chains. We argue that the more polar nature of polymers P1–3 and P8 facilitates the uptake of ions from an aqueous electrolyte – the oligoether side chains of P1–3 and P8 constitute over 50% of the total mass, but less than 40% in case of the other polymers.OECT device characterization
OECTs were prepared by spin-coating the active layer from chloroform solutions on pre-patterned substrates with source-drain metal electrodes. A 0.1 M NaCl aqueous electrolyte was used, and devices comprised a three-electrode configuration with an Ag/AgCl reference electrode and a Pt counter electrode (see Fig. 5a for device architecture). At first, output characteristics were obtained (see Fig. S52†) to determine the type of response (n/p-type) and device operation regime of P1–P3 and P8, which are the only polymers that respond to an aqueous electrolyte (as per CV measurements) owing to the large oligoether side-chain fraction. Devices based on polymers P1, P2 and P8 showed enhancement mode p-type operation with a near zero source-drain current IDS at a gate potential VGS = 0 V and increased IDS at negative VGS. Noteworthy is that P8 shows p-type behavior despite the n-type character of BT, which is attributed to the strong p-type character of g3TT. The response of devices based on P3 was limited (not shown), likely because of the low molecular weight of the polymer and thus the material was not studied further. The polymers P1, P2 and P8 exhibited a negligible contact resistance between the active-layer film and the source/drain Au electrodes. Moreover, OECTs based on P1, P2 and P8 could operate in the saturation regime with a uniform IDS at a VDS < −0.5 V, and hence devices based on these three polymers were chosen for an in-depth analysis.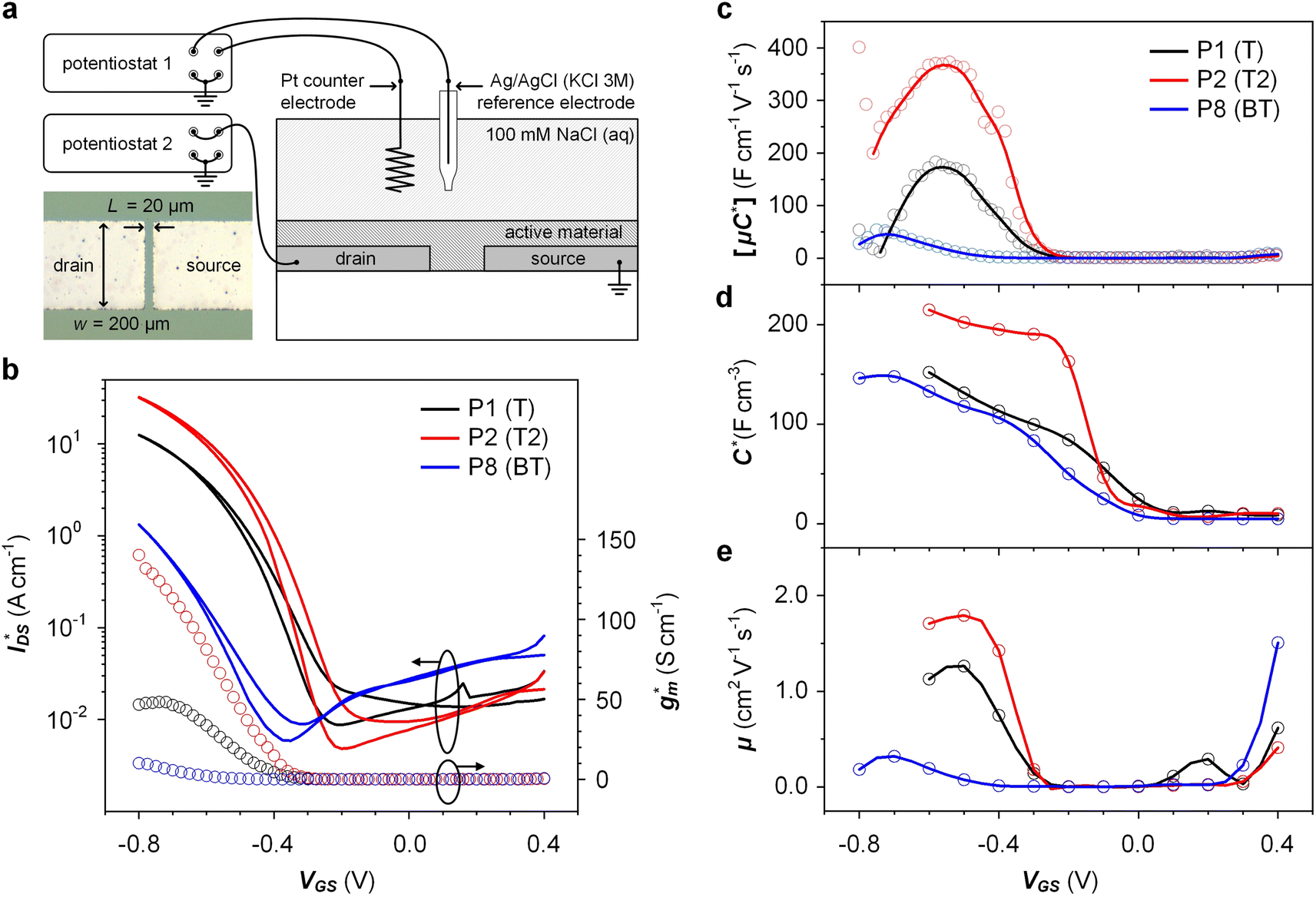 | ||
Fig. 5 Ionic/electronic properties of conjugated polymers. (a) Schematics of OECT device characterizations. (b) Transfer curves and corresponding normalized transconductance, 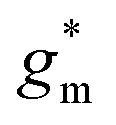 , as a function of gate potential, VGS. Drain current, IDS, and gm were normalized by the channel dimensions (i.e., , as a function of gate potential, VGS. Drain current, IDS, and gm were normalized by the channel dimensions (i.e., 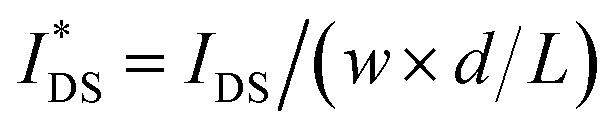 and and 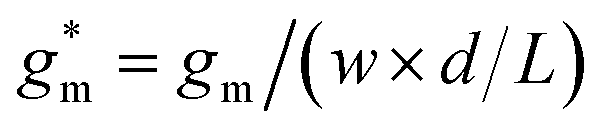 ). The characterization was conducted with a backward scan of VGS from −0.8 to +0.4 V at a constant VDS of −0.6 V. (c–e) Plots of electrical/electrochemical properties as a function of gate potential; (c) product of carrier mobility and volumetric capacitance [μC*] obtained from transfer curves. (d) Volumetric capacitance C* from EIS. (e) Carrier mobility μ calculated by dividing [μC*] by C*. Trend lines (solid line) estimated using spline interpolation of measured values (open symbols). ). The characterization was conducted with a backward scan of VGS from −0.8 to +0.4 V at a constant VDS of −0.6 V. (c–e) Plots of electrical/electrochemical properties as a function of gate potential; (c) product of carrier mobility and volumetric capacitance [μC*] obtained from transfer curves. (d) Volumetric capacitance C* from EIS. (e) Carrier mobility μ calculated by dividing [μC*] by C*. Trend lines (solid line) estimated using spline interpolation of measured values (open symbols). | ||
Devices based on P2 feature the highest dimension-normalized drain current 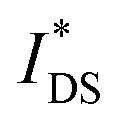 and dimension-normalized transconductance
and dimension-normalized transconductance 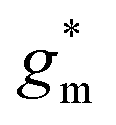 of 32 A cm−1 and 140 S cm−1, followed by P1 and P8 with
of 32 A cm−1 and 140 S cm−1, followed by P1 and P8 with 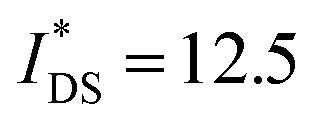 and 1.3 A cm−1, and
and 1.3 A cm−1, and 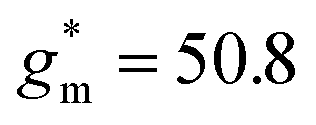 and 10.3 S cm−1, respectively. For devices based on polymers P1 and P2 we observed a similar threshold voltage Vth of −0.38 V and −0.35 V respectively, while devices based on P8 had a lower Vth of −0.53 V (see Table 2). The [μC*] values (see Fig. 5c) were extracted according to:
and 10.3 S cm−1, respectively. For devices based on polymers P1 and P2 we observed a similar threshold voltage Vth of −0.38 V and −0.35 V respectively, while devices based on P8 had a lower Vth of −0.53 V (see Table 2). The [μC*] values (see Fig. 5c) were extracted according to:
| ∂gm/∂VGS = [μC*]·(wd/L) | (2) |
 determined through EIS; maximum hole mobility μmax obtained by dividing [μC*] by C*. Values within brackets denote the corresponding gate potential for each value; the mean and min–max error of the values extracted from two devices are given
determined through EIS; maximum hole mobility μmax obtained by dividing [μC*] by C*. Values within brackets denote the corresponding gate potential for each value; the mean and min–max error of the values extracted from two devices are given
| Polymer | E ox,H2O (eV) | E ox,AcN (eV) | V th (V) | [μC*]max (F cm−1 V−1 s−1) |
|
μ max (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| P1 (T) | 4.35 | 4.69 | −0.38 ± 0.02 | 168 ± 17 (−0.55 V) | >152 ± 17 (−0.6 V) | 1.3 ± 0.2 (−0.5 V) |
| P2 (T2) | 4.52 | 4.77 | −0.35 ± 0.01 | 368 ± 9 (−0.55 V) | >215 ± 24 (−0.6 V) | 1.8 ± 0.2 (−0.5 V) |
| P8 (BT) | 4.48 | 4.77 | −0.53 ± 0.01 | 50 ± 6 (−0.75 V) | 148 ± 16 (−0.7 V) | 0.3 ± 0.1 (−0.7 V) |
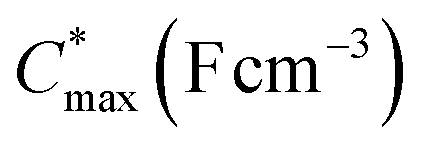
Electrochemical impedance spectroscopy (EIS, see Fig. S53†) was used to determine the volumetric capacitance C* (see Fig. 5d). The C* values gradually increased with decreasing VGS (=−offset potential E vs. Ag/AgCl) up to 152, 215 and 148 F cm−3 for P1 and P2 (at VGS = −0.6 V) and P8 (at VGS = −0.7 V), respectively (see Table 2). P1, P2, and P8 polymers exhibited comparable onset potential values for C* at VGS = −0.1 to 0 V, attributed to their similar Eox values (see Table 2). Also, it is notable that the electron-withdrawing character of BT did not significantly influence the onset and maximum value of the electrochemical capacitance. We divided the [μC*] values obtained from OECT characterization by C* from EIS to obtain μ values for P1, P2 and P8. The polymers P1 and P2 feature a high μmax = 1.3 and 1.8 cm2 V−1 s−1 (see Table 2). Thin films of P1 and P2 comprise ordered domains with a face-on and edge-on texture, respectively (see Fig. 4b), with the latter being beneficial for in-plane charge transport, which may explain the somewhat higher μmax value for P2. However, it can be expected that both the texture and degree of order are affected by electrochemical oxidation, as recently reported for other polythiophenes and thieno[3,2-b]thiophene–thiophene copolymers with oligoether side chains.68,69
Besides the Vth, the onset and peak potentials for [μC*] and μ of P8 are also shifted to more negative values compared with P1 and P2. The higher magnitude of Vth of P8 based OECTs is tentatively assigned to a more localized highest-occupied molecular orbital (HOMO) on the g3TT unit in case of P8 (see inset Table S8†), which may increase the energy barrier for charge conduction along the polymer backbone, and thus a higher oxidation level is required for hopping of charges to occur in case of P8 compared to P1 and P2. Accordingly, P8 featured a lower μmax = 0.3 cm2 V−1 s−1 despite having similar electrochemical properties as P1 and P2 (see Table 2). An alternative explanation of the lower μmax is the lower order of P8 compared to P1 and P2 as per the UV-vis absorption spectra and GIWAXS diffractograms (see Fig. S46 and S47†). Comparison of the μmax and  values obtained for P1, P2 and P8 with those reported for other OMIECs, including other polymers with oligoether side chains as well as PEDOT:PSS, reveals that the here synthesized materials feature an electrochemical response that is comparable to state-of-the-art materials (see Fig. 1).
values obtained for P1, P2 and P8 with those reported for other OMIECs, including other polymers with oligoether side chains as well as PEDOT:PSS, reveals that the here synthesized materials feature an electrochemical response that is comparable to state-of-the-art materials (see Fig. 1).
Under a pulsed gate potential, P2-based OECTs showed a promising degree of operational stability up to 200 cycles (see Fig. S54†). The on-current values at VGS = −0.6 V decreased by only 6% from that of the initial cycle (Ion = 130 μA), while the off-current value at VGS = +0.4 V gradually increased from 40 nA to 600 nA, which we assign to the gradual reduction of the polymer film by oxygen.70 Even under excessive electrochemical stress during the cyclic measurement (i.e., VGS = −0.6 and +0.4 V with VDS = −0.6 V for on- and off-state), P2 shows a comparable operational stability as the state-of-the-art polymer p(g2T-TT),41 which was characterized with a smaller potential window (VGS was cycled between −0.4 and +0.0 V with VDS = −0.4 V).
Conclusions
A variety of potential OMIEC materials was prepared by direct arylation polymerization (DAP) of a thieno[3,2-b]thiophene monomer with oligoether side chains (g3TT) with a variety of electron-rich, electron-deficient and neutral comonomers. In case of polymers that combined the monomer g3TT with the small comonomers thiophene, bithiophene or benzothiadiazole, the materials were active as p-type OMIECs in OECTs operating with an aqueous electrolyte. In particular the copolymer p(g3TT-T2) (P2) gave rise to a state-of-the-art OECT performance with a [μC*]max value of 370 F cm−1 V−1 s−1, facilitated by a high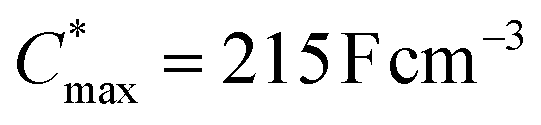 and μmax = 1.8 cm2 V−1 s−1. All synthesized polymers feature a lower synthetic complexity index (SCI) compared to many other polymers made through other polycondensation methods, which considerably enhances the scalability of conjugated polymers and is an important step toward the development of sustainable organic electronics.12
and μmax = 1.8 cm2 V−1 s−1. All synthesized polymers feature a lower synthetic complexity index (SCI) compared to many other polymers made through other polycondensation methods, which considerably enhances the scalability of conjugated polymers and is an important step toward the development of sustainable organic electronics.12
Data availability
Data are available from the authors upon reasonable request.Author contributions
JK conceived the study and wrote the manuscript together with CM, and carried out the synthesis, single crystal X-ray diffraction, polymer characterization, UV-vis spectroscopy, cyclic voltammetry, and DFT calculations. YK fabricated and characterized OECT devices and carried out EIS. JA carried out GIWAXS measurements and processed data together with YK; JM supervised the GIWAXS analysis. RK and CM supervised the study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge funding from the European Union's Horizon 2020 research and innovation programme through the Marie Skłodowska-Curie grant agreement no. 955837 (HORATES) and the Knut and Alice Wallenberg Foundation through a Wallenberg Scholar grant. J. M. thanks MICINN/FEDER for grant Ref. PID2021-126243NB-I00. We thank Lars Öhrström for providing access to single X-ray crystallography, Ergang Wang for providing several commercially available monomers, Diego Ropero Hinojosa and Michael Sommer for providing access to high temperature NMR, as well as Rukiya Matsidik and Michael Sommer for performing MALDI-ToF measurements. DFT calculations were enabled by resources provided by Chalmers e-Commons. The project was in part performed at the Chalmers Materials Analysis Laboratory (CMAL).References
- A. Giovannitti, C. B. Nielsen, D.-T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 13066 CrossRef CAS PubMed
.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS PubMed
- S. T. M. Tan, A. Giovannitti, A. Marks, M. Moser, T. J. Quill, I. McCulloch, A. Salleo and G. E. Bonacchini, Adv. Mater., 2022, 34, 2202994 CrossRef CAS
- Y. He, N. A. Kukhta, A. Marks and C. K. Luscombe, J. Mater. Chem. C, 2022, 10, 2314–2332 RSC
- J. Brebels, J. V. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2017, 5, 24037–24050 RSC
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed
- J. Liu, G. Ye, H. G. O. Potgieser, M. Koopmans, S. Sami, M. I. Nugraha, D. R. Villalva, H. Sun, J. Dong, X. Yang, X. Qiu, C. Yao, G. Portale, S. Fabiano, T. D. Anthopoulos, D. Baran, R. W. A. Havenith, R. C. Chiechi and L. J. A. Koster, Adv. Mater., 2021, 33, 2006694 CrossRef CAS PubMed
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC
- A. V. Volkov, H. Sun, R. Kroon, T.-P. Ruoko, C. Che, J. Edberg, C. Müller, S. Fabiano and X. Crispin, ACS Appl. Energy Mater., 2019, 2, 5350–5355 CrossRef CAS
- H. Sun, M. Vagin, S. Wang, X. Crispin, R. Forchheimer, M. Berggren and S. Fabiano, Adv. Mater., 2018, 30, 1704916 CrossRef PubMed
- M. Massetti, S. Zhang, P. C. Harikesh, B. Burtscher, C. Diacci, D. T. Simon, X. Liu, M. Fahlman, D. Tu, M. Berggren and S. Fabiano, npj Flexible Electron., 2023, 7, 11 CrossRef CAS
- I. McCulloch, M. Chabinyc, C. Brabec, C. B. Nielsen and S. E. Watkins, Nat. Mater., 2023, 22, 1304–1310 CrossRef CAS PubMed
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS PubMed
- N. A. Kukhta, A. Marks and C. K. Luscombe, Chem. Rev., 2022, 122, 4325–4355 CrossRef CAS PubMed
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed
- S. Griggs, A. Marks, D. Meli, G. Rebetez, O. Bardagot, B. D. Paulsen, H. Chen, K. Weaver, M. I. Nugraha, E. A. Schafer, J. Tropp, C. M. Aitchison, T. D. Anthopoulos, N. Banerji, J. Rivnay and I. McCulloch, Nat. Commun., 2022, 13, 7964 CrossRef CAS
- S.-M. Kim, C.-H. Kim, Y. Kim, N. Kim, W.-J. Lee, E.-H. Lee, D. Kim, S. Park, K. Lee, J. Rivnay and M.-H. Yoon, Nat. Commun., 2018, 9, 3858 CrossRef PubMed
- R. K. Hallani, B. D. Paulsen, A. J. Petty II, R. Sheelamanthula, M. Moser, K. J. Thorley, W. Sohn, R. B. Rashid, A. Savva, S. Moro, J. P. Parker, O. Drury, M. Alsufyani, M. Neophytou, J. Kosco, S. Inal, G. Costantini, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2021, 143, 11007–11018 CrossRef CAS PubMed
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, Macromolecules, 2015, 48, 453–461 CrossRef CAS
- M. Sévignon, J. Papillon, E. Schulz and M. Lemaire, Tetrahedron Lett., 1999, 40, 5873–5876 CrossRef
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS PubMed
- P.-O. Morin, T. Bura, B. Sun, S. I. Gorelsky, Y. Li and M. Leclerc, ACS Macro Lett., 2015, 4, 21–24 CrossRef CAS PubMed
- A. L. Mayhugh, P. Yadav and C. K. Luscombe, J. Am. Chem. Soc., 2022, 144, 6123–6135 CrossRef CAS PubMed
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135–147 CrossRef CAS
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed
- T. J. Aldrich, A. S. Dudnik, N. D. Eastham, E. F. Manley, L. X. Chen, R. P. H. Chang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Macromolecules, 2018, 51, 9140–9155 CrossRef CAS
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705–6711 CrossRef CAS PubMed
- A. S. Dudnik, T. J. Aldrich, N. D. Eastham, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 15699–15709 CrossRef CAS PubMed
- K. Okamoto, J. B. Housekeeper, F. E. Michael and C. K. Luscombe, Polym. Chem., 2013, 4, 3499–3506 RSC
- W. Elsawy, M. Son, J. Jang, M. J. Kim, Y. Ji, T.-W. Kim, H. C. Ko, A. Elbarbary, M.-H. Ham and J.-S. Lee, ACS Macro Lett., 2015, 4, 322–326 CrossRef CAS PubMed
- F. Livi, N. S. Gobalasingham, E. Bundgaard and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2598–2605 CrossRef CAS
- K. Yamazaki, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2013, 34, 69–73 CrossRef CAS PubMed
- J. F. Ponder Jr., S. A. Gregory, A. Atassi, A. A. Advincula, J. M. Rinehart, G. Freychet, G. M. Su, S. K. Yee and J. R. Reynolds, Angew. Chem., Int. Ed., 2023, 62, e202211600 CrossRef PubMed
- B. Ding, G. Kim, Y. Kim, F. D. Eisner, E. Gutiérrez-Fernández, J. Martín, M.-H. Yoon and M. Heeney, Angew. Chem., Int. Ed., 2021, 60, 19679–19684 CrossRef CAS PubMed
- B. Ding, I.-Y. Jo, H. Yu, J. H. Kim, A. V. Marsh, E. Gutiérrez-Fernández, N. Ramos, C. L. Rapley, M. Rimmele, Q. He, J. Martín, N. Gasparini, J. Nelson, M.-H. Yoon and M. Heeney, Chem. Mater., 2023, 35, 3290–3299 CrossRef CAS PubMed
- B. Ding, V. Le, H. Yu, G. Wu, A. V. Marsh, E. Gutiérrez-Fernández, N. Ramos, M. Rimmele, J. Martín, J. Nelson, A. F. Paterson and M. Heeney, Adv. Electron. Mater., 2024, 2300580 CrossRef CAS
- J. Tropp, D. Meli, R. Wu, B. Xu, S. B. Hunt, J. D. Azoulay, B. D. Paulsen and J. Rivnay, ACS Mater. Lett., 2023, 5, 1367–1375 CrossRef CAS
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, J. Am. Chem. Soc., 2019, 141, 4345–4354 CrossRef CAS PubMed
- A. Giovannitti, C. B. Nielsen, J. Rivnay, M. Kirkus, D. J. Harkin, A. J. P. White, H. Sirringhaus, G. G. Malliaras and I. McCulloch, Adv. Funct. Mater., 2016, 26, 514–523 CrossRef CAS
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 12017–12022 CrossRef CAS PubMed
- R. Halaksa, J. H. Kim, K. J. Thorley, P. A. Gilhooly-Finn, H. Ahn, A. Savva, M.-H. Yoon and C. B. Nielsen, Angew. Chem., Int. Ed., 2023, 62, e202304390 CrossRef CAS PubMed
- D. G. Harman, R. Gorkin, L. Stevens, B. Thompson, K. Wagner, B. Weng, J. H. Y. Chung, M. in het Panhuis and G. G. Wallace, Acta Biomater., 2015, 14, 33–42 CrossRef CAS
- A. I. Hofmann, W. T. T. Smaal, M. Mumtaz, D. Katsigiannopoulos, C. Brochon, F. Schütze, O. R. Hild, E. Cloutet and G. Hadziioannou, Angew. Chem., Int. Ed., 2015, 54, 8506–8510 CrossRef CAS PubMed
- S. Inal, J. Rivnay, A. I. Hofmann, I. Uguz, M. Mumtaz, D. Katsigiannopoulos, C. Brochon, E. Cloutet, G. Hadziioannou and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 147–151 CrossRef CAS
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef PubMed
- L. Lindell, A. Burquel, F. L. E. Jakobsson, V. Lemaur, M. Berggren, R. Lazzaroni, J. Cornil, W. R. Salaneck and X. Crispin, Chem. Mater., 2006, 18, 4246–4252 CrossRef CAS
- X. Luo, H. Shen, K. Perera, D. T. Tran, B. W. Boudouris and J. Mei, ACS Macro Lett., 2021, 10, 1061–1067 CrossRef CAS PubMed
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS PubMed
- J. W. Onorato, Z. Wang, Y. Sun, C. Nowak, L. Q. Flagg, R. Li, B. X. Dong, L. J. Richter, F. A. Escobedo, P. F. Nealey, S. N. Patel and C. K. Luscombe, J. Mater. Chem. A, 2021, 9, 21410–21423 RSC
- L. Porcarelli, A. S. Shaplov, M. Salsamendi, J. R. Nair, Y. S. Vygodskii, D. Mecerreyes and C. Gerbaldi, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CrossRef CAS PubMed
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed
- A. S. Shaplov, P. S. Vlasov, M. Armand, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, I. A. Godovikov and Y. S. Vygodskii, Polym. Chem., 2011, 2, 2609–2618 RSC
- C. K. Song, B. J. Eckstein, T. L. D. Tam, L. Trahey and T. J. Marks, ACS Appl. Mater. Interfaces, 2014, 6, 19347–19354 CrossRef CAS PubMed
- Q. Cai and W. Zhou, Chin. J. Chem., 2020, 38, 879–893 CrossRef CAS
- R. Lv, Y. Wang, C. Zhou, L. Li and R. Wang, ChemCatChem, 2013, 5, 2978–2982 CrossRef CAS
- D. S. Dissanayake, E. Sheina, M. C. Biewer, R. D. McCullough and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 79–82 CrossRef CAS
- J. W. Wackerly and J. F. Dunne, J. Chem. Educ., 2017, 94, 1790–1793 CrossRef CAS
- N. Yaghoobi Nia, M. Bonomo, M. Zendehdel, E. Lamanna, M. M. H. Desoky, B. Paci, F. Zurlo, A. Generosi, C. Barolo, G. Viscardi, P. Quagliotto and A. Di Carlo, ACS Sustain. Chem. Eng., 2021, 9, 5061–5073 CrossRef CAS
- S. Broll, F. Nübling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481–7488 CrossRef CAS
- T. Bura, S. Beaupré, M.-A. Légaré, J. Quinn, E. Rochette, J. T. Blaskovits, F.-G. Fontaine, A. Pron, Y. Li and M. Leclerc, Chem. Sci., 2017, 8, 3913–3925 RSC
- M. Moser, L. R. Savagian, A. Savva, M. Matta, J. F. Ponder Jr, T. C. Hidalgo, D. Ohayon, R. Hallani, M. Reisjalali, A. Troisi, A. Wadsworth, J. R. Reynolds, S. Inal and I. McCulloch, Chem. Mater., 2020, 32, 6618–6628 CrossRef CAS
- L. Biniek, N. Leclerc, T. Heiser, R. Bechara and M. Brinkmann, Macromolecules, 2013, 46, 4014–4023 CrossRef CAS
- J. E. Cochran, M. J. N. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS
- N. Takeda, S. Asaoka and J. R. Miller, J. Am. Chem. Soc., 2006, 128, 16073–16082 CrossRef CAS PubMed
- N. Siemons, D. Pearce, C. Cendra, H. Yu, S. M. Tuladhar, R. K. Hallani, R. Sheelamanthula, G. S. LeCroy, L. Siemons, A. J. P. White, I. McCulloch, A. Salleo, J. M. Frost, A. Giovannitti and J. Nelson, Adv. Mater., 2022, 34, 2204258 CrossRef CAS PubMed
- R. Wu, M. Matta, B. D. Paulsen and J. Rivnay, Chem. Rev., 2022, 122, 4493–4551 CrossRef CAS PubMed
- S. Zhang, P. Ding, T.-P. Ruoko, R. Wu, M.-A. Stoeckel, M. Massetti, T. Liu, M. Vagin, D. Meli, R. Kroon, J. Rivnay and S. Fabiano, Adv. Funct. Mater., 2023, 33, 2302249 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Description of synthetic procedures and experimental details; derivation and calculation of molecular weight determination of polymers by high temperature NMR; definition, derivation, and synthetic routes considered for the synthetic complexity index (SCI) calculations; single crystal X-ray structure, NMR spectra, size-exclusion chromatograms, TGA thermograms, DSC thermograms, UV-vis spectra, CV curves, GIWAXS patterns, EIS spectra, OECT output characteristics, OECT cycling stability curves, and computational results. CCDC 2336289. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01430h |
| This journal is © The Royal Society of Chemistry 2024 |