
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotovoltaic-driven Ni(II)/Ni(III) redox mediator for the valorization of PET plastic waste with hydrogen production†
Jianying
Wang‡
ac,
Xin
Li‡
a,
Ting
Zhang
a,
Xinyu
Chai
a,
Mingze
Xu
c,
Menglei
Feng
a,
Chengcheng
Cai
a,
Zuofeng
Chen
 *c,
Xufang
Qian
*a and
Yixin
Zhao
*ab
*c,
Xufang
Qian
*a and
Yixin
Zhao
*ab
aSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Rd., Shanghai 200240, China. E-mail: qianxufang@sjtu.edu.cn; yixin.zhao@sjtu.edu.cn
bState Key Lab of Metal Matrix Composite, Shanghai Jiao Tong University, 800 Dongchuan Rd., Shanghai 200240, China
cSchool of Chemical Science and Engineering, Tongji University, 1239 Siping Rd., Shanghai 200092, China. E-mail: zfchen@tongji.edu.cn
First published on 22nd April 2024
Abstract
Electrocatalytic valorization of PET plastic waste provides an appealing route by converting intermittent renewable energy into valuable chemicals and high-energy fuels. Normally, anodic PET hydrolysate oxidation and cathodic water reduction reactions occur simultaneously in the same time and space, which increases the challenges for product separation and operational conditions. Although these problems can be addressed by utilizing membranes or diaphragms, the parasitic cell resistance and high overall cost severely restrict their future application. Herein, we introduce a Ni(II)/Ni(III) redox mediator to decouple these reactions into two independent processes: an electrochemical process for water reduction to produce hydrogen fuel assisted by the oxidation of the Ni(OH)2 electrode into the NiOOH counterpart, followed subsequently by a spontaneous chemical process for the valorization of PET hydrolysate to produce formic acid with a high faradaic efficiency of ∼96% by the oxidized NiOOH electrode. This decoupling strategy enables the electrochemical valorization of PET plastic waste in a membrane-free system to produce high-value formic acid and high-purity hydrogen production. This study provides an appealing route to facilitate the transformation process of PET plastic waste into high-value products with high efficiency, low cost and high purity.
Introduction
Since the first synthetic polymer (phenolic resin) synthesized in 1907 by Leo Baekeland, the low-cost, lightweight, durable and versatile properties of polymer plastics have led to their widespread application in our daily lives.1 Global plastic production is expected to reach more than 500 million metric tons by 2050.2 However, with rapidly increasing production, a mass of disposable polymer waste has been discarded and accumulated in aquatic and terrestrial ecosystems.3 Improper management of these plastic wastes has not only induced a series of environmental pollution problems but also resulted in wasting of carbon resources.4 It is urgent to develop efficient recycling strategies for the valorization of plastic waste and to satisfy the demands of a circular economy.5,6Notably, electrocatalysis driven by renewable energy provides a promising way for environmental governance and sustainable energy conversion.7,8 Recently, some researchers have investigated and reported the conversion of polyethylene terephthalate (PET) plastic waste into high-value chemicals and hydrogen fuels by electrocatalysis or photo-electrocatalysis.9–13 Compared with traditional water splitting, the oxidation of PET hydrolysate, replacing sluggish water oxidation, can efficiently decrease the energy consumption needed for the hydrogen evolution reaction (HER), thus increasing the value of anodic products and avoiding the H2/O2 mixing problem9 and shows appealing prospects for the upcycling of PET plastic wastes, as shown in Fig. S1.† The oxygen evolution reaction (OER) is replaced by the PET hydrolysate oxidation, although an ion-exchange membrane still needs to be employed (indispensable) to separate anodic and cathodic chambers to avoid diffusion of liquid products.14,15 The low/poor mechanical strength and high resistance of this expensive membrane increases the cost and energy input of the electrolysis system. To develop a membrane-free electrolytic strategy is important for efficient upcycling of plastic waste into high-valued chemicals and fuels.
In 2013, Cronin et al. proposed a membrane-free decoupling strategy for water splitting using a Mo-based redox mediator, which can separate anodic and cathodic half-reactions (HER and OER) in time and space.16 Since then, a series of redox mediators (polyoxometallic acid,17 small organic molecules,18 conductive polymers19,20 and nickel hydroxides21) with high proton and electron storage/release capacity have been employed for decoupling the HER and OER half-reactions. Among these redox mediators, the earth-abundant Ni(OH)2 material shows attractive features for green hydrogen production by alkaline electrocatalysis technology without the assistance of a membrane.21–23 In the traditional decoupling process, while the decoupled HER process can be realized with the assistance of Ni(OH)2 oxidation into NiOOH, the high energy consumption and low-value OER process still needs to be conducted to refresh the Ni-based redox mediator electrode by electrochemical21,22 or chemical23 methods. It is very important to develop highly efficient and valued oxidation reactions to replace the water oxidation reaction during the decoupling process.
Recently, a series of studies have reported that oxidized Ni-based materials exhibit higher catalytic activity for the transformation of organic small-molecule alcohols into aldehydes or carboxylic acids than for water oxidation.24–26 Given that the PET hydrolysate solution contains ethylene glycol (EG) molecules, these Ni-based materials may be introduced to the valorization system for PET plastic hydrolysate. Under standard conditions, Gibbs free energy changes for the conversion of EG to formic acid, and of NiOOH to Ni(OH)2, are −404.9 kJ mol−1 and −50.2 kJ mol−1, respectively, indicating that the chemical reaction between ethylene glycol and NiOOH can occur spontaneously at normal temperature and pressure without additional energy input.27,28 Based on the above analysis, introducing the decoupling strategy for the valorization of PET plastic waste can not only decrease the cost and energy input for hydrogen production but also facilitate the conversion process of PET plastic waste. In addition, photovoltaic (PV) power generation is experiencing a surge in popularity as a sustainable electricity source and offers numerous advantages, including environmental sustainability.29 However, one significant challenge for practical application lies in the intermittent nature of PV power generation. Therefore, it is crucial to develop effective energy conversion and utilization systems capable of seamlessly addressing this intermittency.
As shown in Scheme 1, in our proof-of-concept strategy, the valorization process of PET plastic waste involves an electrochemical process (step 1) for hydrogen production and a spontaneous chemical process (step 2) for formic acid production, which is similar to the photosynthesis process and can be matched with PV equipment. For step 1, hydrogen production is achieved by reduction of water on the cathode, and the Ni(OH)2 anode is simultaneously oxidized into NiOOH without the oxygen evolution reaction occurring. For step 2, the conversion of PET hydrolysate is carried out by a spontaneous chemical oxidation process by the oxidized NiOOH electrode. The reduced Ni(OH)2 material can be sequentially employed as the anodic electrode for hydrogen production. By continuously alternating steps 1 and 2, electrochemical and spontaneous chemical processes can be integrated using the auxiliary Ni-based hydroxide electrode for the valorization of PET plastic waste and production of hydrogen in a membrane-free system, as shown in Fig. S1.†
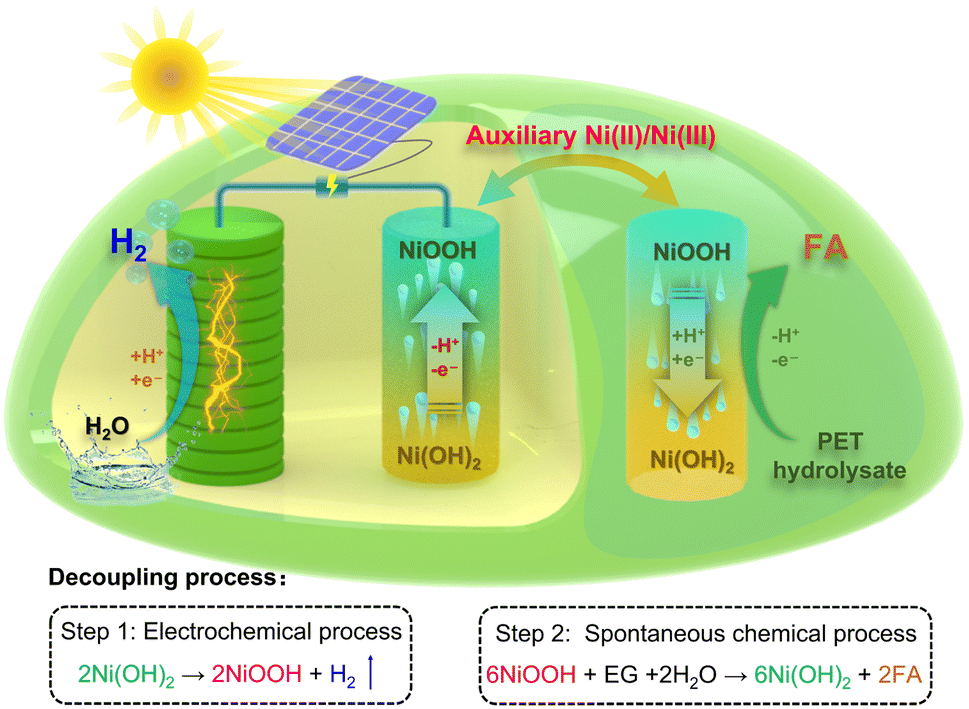 | ||
| Scheme 1 Schematic illustration of the proposed decoupling strategy with the assistance of a Ni(II)/Ni(III) redox mediator for the valorization of PET plastic waste to produce valued hydrogen and formic acid, which contains two independent electrochemical and chemical processes. | ||
Herein, we developed a decoupling strategy to produce hydrogen with high purity and formic acid, which was constituted by a two-step electrochemical–chemical process. In this strategy, hydrogen production was performed with an electrochemical process, which involved cathodic water reduction and anodic Ni(OH)2 oxidation. The subsequent PET hydrolysate oxidation was conducted with a spontaneous chemical process, which oxidizes PET hydrolysate into formic acid by reducing oxidized NiOOH back to Ni(OH)2. This strategy can separate the hydrogen production and PET hydrolysate conversion processes in space and time, thus avoiding the use of a high-cost membrane and increasing the operational flexibility for upcycling PET plastic waste. In addition, the spontaneous chemical process shows that the Ni(II)/Ni(III) redox mediator can selectively oxidize PET hydrolysate into formic acid with a high faradaic efficiency of ∼96%. Compared with traditional one-step electrolysis, the decoupled two-step strategy provides an appealing approach for upcycling PET plastic into high-value chemical and green hydrogen in a membrane-free system.
Results and discussion
In this study, Ni(OH)2, which is a conventional electrode material for rechargeable batteries and supercapacitors, was selected and employed as the auxiliary redox mediator. Ni(OH)2 nanosheets grown on nickel foam (NF) with three-dimensional structure were fabricated by an in situ anodic oxidation electrodeposition method.30 The morphology of the structure of as-prepared Ni(OH)2 was characterized by scanning electron microscopy (SEM). As shown in Fig. S2,† SEM images show that Ni(OH)2 nanosheet arrays were uniformly covered on the surface of the NF substrate. X-Ray diffraction (XRD) analysis was further conducted to investigate the as-prepared Ni(OH)2/NF electrode (Fig. S3†). The XRD pattern of the electrodeposited electrode shows three peaks attributed to the nickel substrate, with no other peaks found, indicating the amorphous features of the as-deposited nickel hydroxide material. A photograph of the as-prepared electrode is also displayed in Fig. S4.† The Ni-based electrode exhibits different colors in different oxidation states.To verify the spontaneous chemical reaction between ethylene glycol and NiOOH, a series of electrochemical measurements were carried out in a three-electrode system. As shown in Fig. 1A, the cycle voltammetry (CV) curve of the Ni(OH)2 electrode displays a characteristic Ni(II)/Ni(III) redox peak in 1 M KOH solution in the absence of PET hydrolysate. Notably, when the electrolyte contains PET hydrolysate, redox peaks assigned to the Ni(II)/Ni(III) species disappeared, and the reverse scan process exhibited a larger reaction current than the forward scan process, indicating that the high-valence-state nickel species may be reduced in situ to its initial state by PET hydrolysate. In addition, a potentiostatic electrolysis measurement was conducted at a potential of 1.40 V, where the oxygen evolution reaction will not occur, as shown in Fig. 1B. During the initial 100 s, the current derived from the oxidation of Ni(OH)2 gradually decreased from 45 mA to 0 mA, indicating that the nickel-based material has been totally oxidized to its high-valence state. It is worth noting that the reaction current sharply increased to 70 mA after injecting PET hydrolysate into the electrolyte, which is due to oxidized nickel species being reduced to Ni(OH)2 in a low-valence state. These results show that PET hydrolysate can refresh the oxidized nickel species to its reduced state.
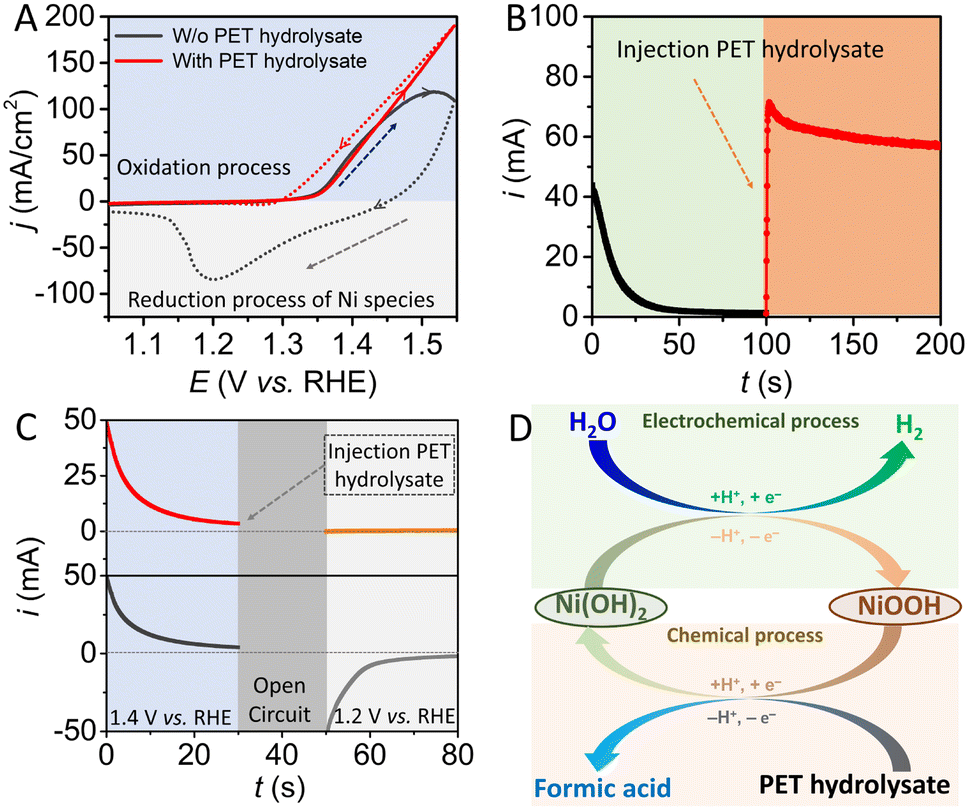 | ||
| Fig. 1 (A) Cycle voltammetry curves of the Ni(OH)2 electrode in 1 M KOH solution with and without the addition of 0.1 M PET hydrolysate; scan rate: 10 mV s−1. (B) Controlled-potential electrolysis curve of the Ni(OH)2 electrode in 1 M KOH solution at a potential of 1.40 V vs. RHE. (C) Controlled-potential electrolysis curves of the Ni(OH)2 electrode in 1 M KOH solution at potentials of 1.40 V and 1.20 V, which are separated by an open-circuit period. (D) Schematic illustration of the oxidation reaction of PET hydrolysate and HER decoupled by the Ni(OH)2 electrode. | ||
A further potentiostatic electrolysis experiment at different potentials was also carried out, as shown in Fig. 1C. First, the electrolysis process was conducted in 1 M KOH solution without the addition of PET hydrolysate at a potential of 1.40 V, to thoroughly oxidize the nickel species to the high-valence state. During the period of quiet time, PET hydrolysate was injected into the selected electrolyte. It is expected that the Ni-based electrode in the electrolyte without the addition of PET hydrolysate will display an obvious reduction current at a potential of 1.2 V, which is derived from the reduction of nickel species in the high-valence state. In contrast, the Ni-based electrode exhibited a negligible current in the electrolyte with the presence of PET hydrolysate. All above electrochemical results demonstrate that spontaneous chemical reaction can occur at the surface of the NiOOH electrode in the PET hydrolysate solution. Ethylene glycol derived from PET hydrolysate can rapidly transform the electro-oxidized NiOOH species into its initial state, as Ni(OH)2. Given that the Ni-based electrode can be oxidized by an electrochemical process, with the production of hydrogen, it is feasible to combine electrochemical and spontaneous chemical processes via the auxiliary Ni(OH)2 electrode for the production of hydrogen and valorization of PET plastic by two independent steps in a membrane-free system, as shown Fig. 1D.
To further validate the surface state of the Ni-based redox electrode, in situ Raman spectroscopy was conducted in a three-electrode electrochemical system, as shown in Fig. S5.† The corresponding spectra were collected under 785 nm laser excitation. The as-prepared Ni(OH)2 electrode was employed as the working electrode to gather Raman signals under different potentials. As shown in Fig. 2A, in the absence of PET hydrolysate in the electrolyte, two obvious scattering peaks at 474.3 cm−1 and 560 cm−1, assigned to the depolarized Eg mode and polarized A1g mode of NiOOH, appeared when the potential was above 1.36 V, and the intensity of the peaks gradually increased with increased potential.31,32 The peak intensity at 560 cm−1 is stronger than that at 474.3 cm−1, indicating the high structural disorder of the NiOOH material.33 During electrolysis, no bubbles were found at the electrode surface, indicating that no OER occurred when the potential was below 1.45 V. Notably, in the presence of PET hydrolysate in the solution, above-mentioned scattering peaks attributed to NiOOH disappeared under similar electrolysis potentials, as shown in Fig. S6.† The results indicate that the NiOOH species cannot exist stably in the PET hydrolysate solution. To verify that the spontaneous chemical reaction process can proceed between NiOOH and PET hydrolysate, a controlled experiment was conducted. As shown Fig. 2B, the Raman spectrum displays two scattering peaks derived from NiOOH at a potential of 1.40 V in the absence of PET hydrolysate. After breaking the circuit, corresponding Raman scattering peaks can still be detected from the electrode surface. Subsequently, these peaks disappeared after injecting PET hydrolysate into the solution. All above results indicate that PET hydrolysate can reduce NiOOH by a spontaneous chemical process.
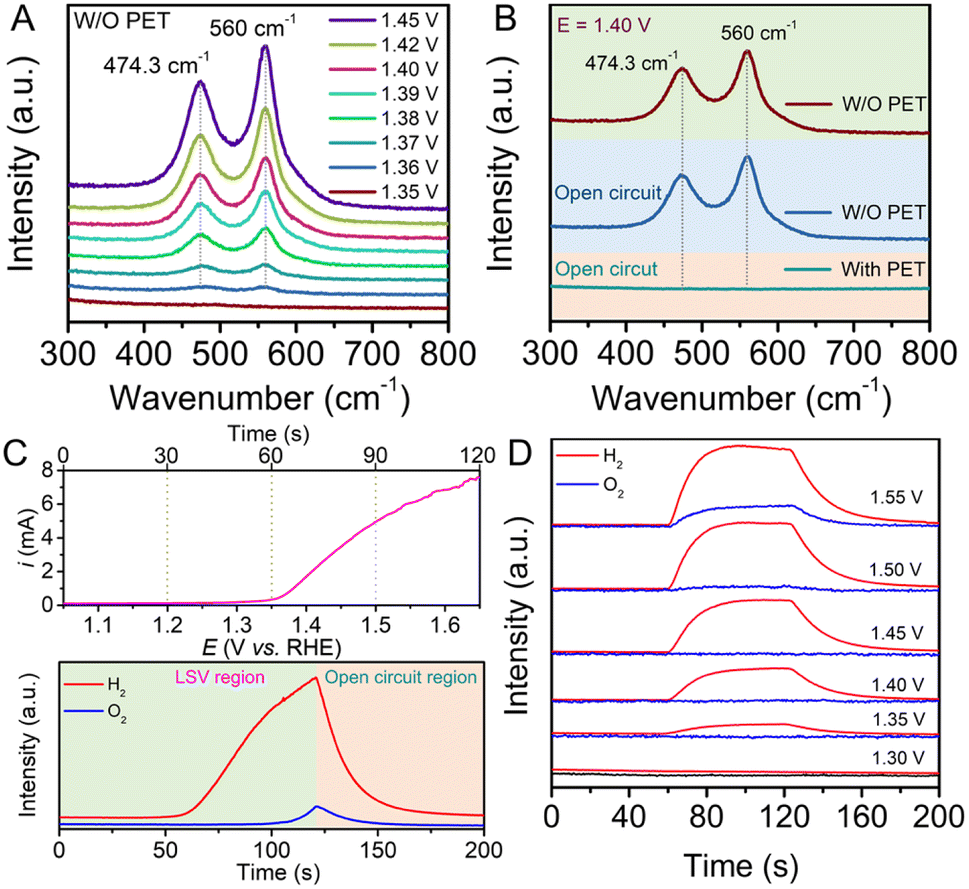 | ||
| Fig. 2 (A) In situ Raman spectra of Ni(OH)2 electrode in 1 M KOH solution without the addition of 0.1 M PET hydrolysate at different potentials. (B) Raman spectra of Ni(OH)2 electrode at the potential of 1.40 V and under open circuit in the absence/presence of 0.1 M PET hydrolysate. (C) In situ DEMS curves of H2 and O2 collected from an LSV curve for Ni(OH)2 oxidation; scan rate: 5 mV s−1. (D) In situ DEMS curves of H2 and O2 at different potentials for the oxidation of Ni(OH)2. A pure Ar gas stream was used as a purge gas during the electrolysis process. | ||
To confirm the purity of the separated hydrogen gas in the electrolysis step, we employed in situ differential electrochemical mass spectrometry (DEMS) to monitor gas evolution during the oxidation of Ni(OH)2 coupled with the HER. As depicted in Fig. 2C, the in situ DEMS curve was recorded during the Ni(OH)2 electrode oxidation using an LSV test. Production of hydrogen gas was clearly observed at the 60 second time point, corresponding to a potential of 1.35 V (vs. RHE), in the online analysis record. Furthermore, the intensity of the ion current increased progressively with higher potential. Notably, the ion current intensity of O2 remained at the background level until 100 seconds (corresponding to a potential of 1.55 V), indicating the absence of O2 generation below a potential of 1.55 V. Additionally, we collected in situ DEMS curves at various electrolysis potentials for 60 seconds, as shown in Fig. 2D. With increasing potential, the ion current intensity for H2 production began at 1.35 V and continued to increase. Similarly, the signal for O2 started to appear at a potential of 1.55 V, consistent with the results of LSV tests. The in situ DEMS measurements further substantiated that the Ni(OH)2 electrode, when used in the decoupled system, exhibited a wide potential window of approximately 200 mV without generating oxygen. Gas chromatography (GC) measurements were also performed to confirm the exclusive generation of hydrogen during the electrolysis at a potential of 1.40 V, as depicted in Fig. S7.†
According to the results of electrochemical and in situ Raman measurements, it is apparent that the Ni(OH)2 electrode can be easily oxidized into NiOOH by an electrochemical oxidation process, and the oxidized NiOOH can be rapidly reduced back to Ni(OH)2 in the presence of PET hydrolysate by a spontaneous chemical process. By combining the electrochemical step and spontaneous chemical step, pure hydrogen production and valorization of PET hydrolysate can be realized at different space and time, without the assistance of expensive membranes, as shown in Fig. 3A. For the decoupling strategy, the charge storage capacity of the redox mediator is an important factor to efficiently produce hydrogen and valorize PET plastic. In this study, the Ni(OH)2/NF electrode was systematically investigated by controlling the in situ anodic electro-deposition conditions. As shown in Fig. S8,† the peak intensity of as-prepared Ni(OH)2/NF electrodes gradually increased with increasing electrodeposition time, indicating that the loading capacity of the Ni-based materials on the NF substrate can be regulated simply by tuning the deposition time. In Fig. 3B, galvanostatic charge–discharge measurement results show that the as-prepared Ni-based redox mediator electrode exhibits a reversible capacity of 5 C cm−2, which is consistent with reported results.23 The charge/discharge performance of the prepared Ni(OH)2/NF electrode tested at higher rates is shown in Fig. S9.† To investigate the cycle stability of the Ni-based auxiliary electrode, a long-term charge–discharge test was conducted, and the electrode exhibited good maintenance of capacity for storing and releasing protons and electrons, as shown in Fig. S10.† The fabricated Ni-based electrode also exhibited a high morphological stability, without obvious change and structural cycling stability after a 20 h test, as shown in Fig. S11 and S12.†
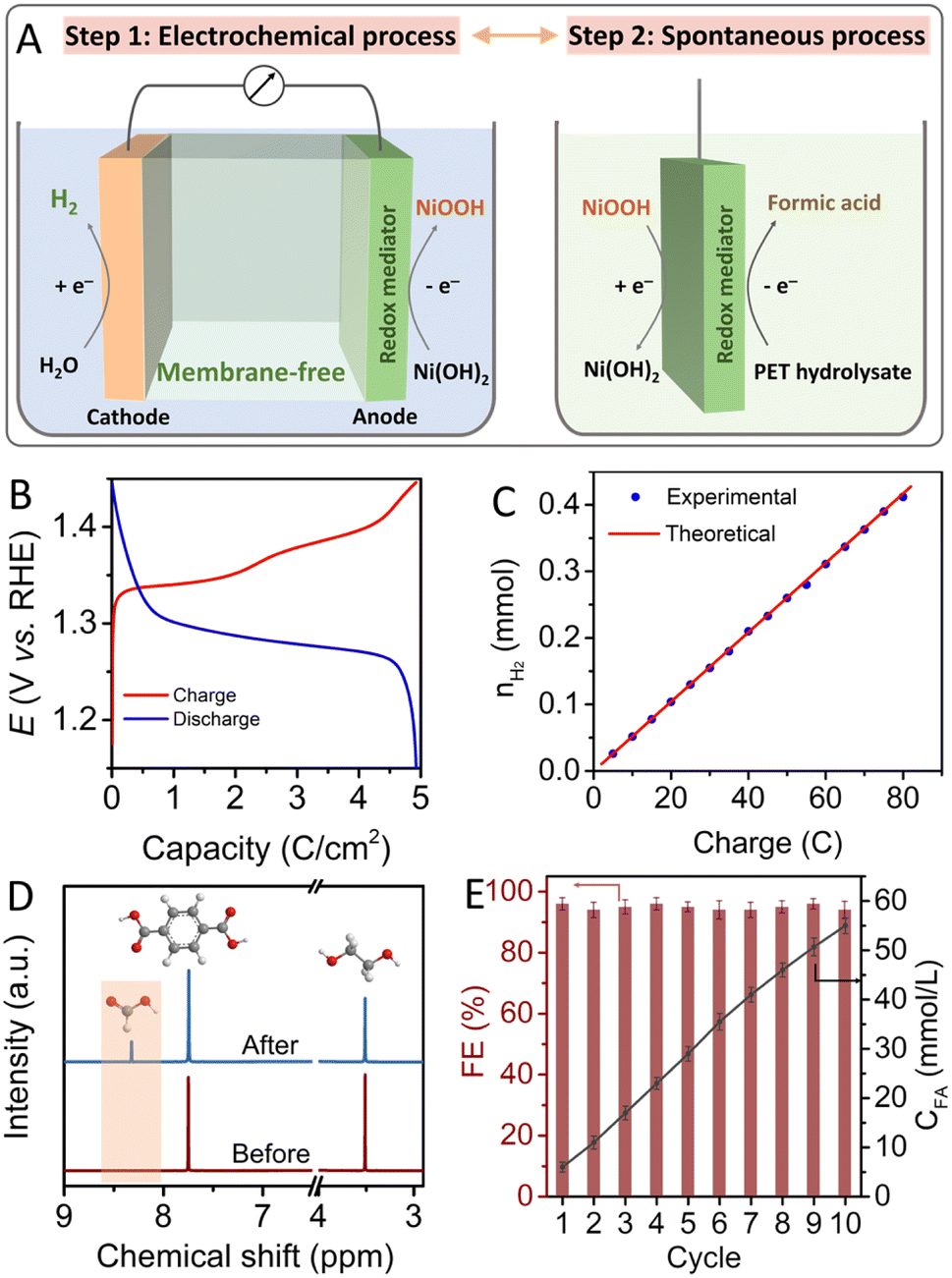 | ||
| Fig. 3 (A) Schematic illustration of the decoupling strategy for the valorization of PET plastic waste with two steps: electrochemical process and spontaneous chemical process. (B) Galvanostatic charge–discharge measurement of the Ni(OH)2/NF electrode in 1 M KOH solution at a current density of 0.2 A g−1. (C) Quantities of H2 generated with increase of passed charge in the decoupled HER process. (D) 1H NMR spectrum obtained from PET hydrolysate solution before and after immersing the oxidized NiOOH/NF electrode. (E) Faradaic efficiency of the oxidized NiOOH/NF electrode for the valorization of PET hydrolysate, and the corresponding quantities of formic acid generated for ten cycles of a spontaneous chemical process. | ||
Based on above results, we introduced Ni(OH)2/NF as an auxiliary electrode for the valorization of PET plastic waste accompanied by hydrogen production. To investigate the performance of the decoupling strategy for the valorization of PET plastic, the products of the decoupled process were detected and analyzed by gas chromatography (GC), high-performance liquid chromatography (HPLC) and nuclear magnetic resonance (NMR) methods. For the electrochemical hydrogen evolution process, Fig. S13† shows that only hydrogen gas was generated at the cathode without the generation of oxygen gas at the Ni(OH)2/NF anode. Fig. 3C shows that the faradaic efficiency for hydrogen production is approximately 100%. In Fig. 3D, the 1H NMR spectrum of as-prepared PET hydrolysate solution exhibits two apparent peaks at δ = 3.5 and 7.75 ppm assigned to ethylene glycol and terephthalate.34,35 It is worth noting that a new peak at δ = 8.3 ppm attributed to formate appeared after immersing the oxidized NiOOH/NF electrode in PET hydrolysate.36 Formate production can also be verified by 13C NMR with an apparent peak at 170.9 ppm without other products (see Fig. S14†). Combining above results, it is clear that the oxidized NiOOH can convert ethylene glycol derived from PET hydrolysate into formate, and NiOOH itself can be reduced to Ni(OH)2. Quantitative analysis of the products was further investigated using HPLC. As shown in Fig. 3E, the concentration of formic acid gradually increased to 56 mmol L−1 after immersing the Ni-based redox mediator electrode in the PET hydrolysate solution for 10 cycles of the decoupled process. Corresponding HPLC curves are shown in Fig. S15.† The faradaic efficiency of the NiOOH/NF electrode for the oxidation of PET hydrolysate is close to 96% and exhibits good stability. The structural and morphological features of the Ni(OH)2/NF electrode remain virtually unchanged after continuous recycling tests, as shown in Fig. S16.† Regarding the conversion pathway of PET hydrolysate into formic acid on the NiOOH electrode, the ethylene glycol molecules were absorbed onto the NiOOH surface and further underwent subsequent dehydrogenation by interacting with OH−, followed by C–C bond cleavage to form the target formic acid. Correspondingly, the NiOOH species were reduced back into the Ni(OH)2 species, which can be further utilized for the electrochemical hydrogen evolution process.37–39 The actual processes of the decoupled PET valorization and HER are also provided in the ESI as movie S1 and S2.† All results show that the decoupling process proceeded according to the proposed reaction route.
For comparison, conventional electrocatalytic processes with or without the assistance of anion exchange membranes (AEMs) were also conducted to investigate the performance for the conversion of PET hydrolysate by the Ni(OH)2/NF electrode. CV curves were collected to investigate the influence on the mass transfer of the AEM for the electrochemical process. As shown in Fig. 4A, the oxidation peak of the Ni(OH)2 electrode in the presence of AEM displays a lower intensity than that in the absence of AEM. In addition, the AEM electrolyzer needed a higher potential to achieve the same current density. Given that NiOOH is the active species, the conversion efficiency for the valorization of PET hydrolysate decreases in the presence of the AEM, as shown in Fig. 4B. Therefore, the construction of a membrane-free electrolyzer is beneficial to enhance mass transfer and reduce energy consumption.
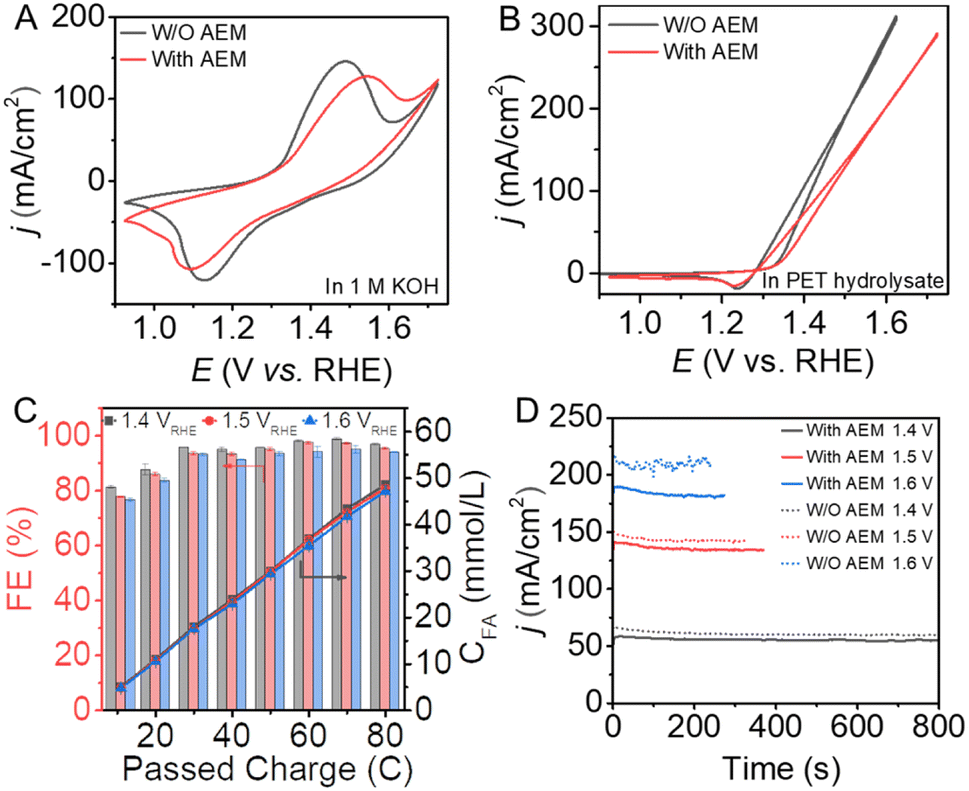 | ||
| Fig. 4 CV curves of Ni(OH)2/NF in (A) 1 M KOH solution and (B) 0.1 M PET hydrolysate solution in the presence and absence of AEM in the electrolyzer. (C) Faradaic efficiency and quantity curves for formic acid production by the Ni(OH)2/NF electrode at different potentials in the presence of AEM in 0.1 M PET hydrolysate. (D) CPE curves for the Ni(OH)2/NF electrode for the conversion of PET hydrolysate at different potentials in the presence and absence of the AEM in the electrolyzer. | ||
To investigate the efficiency and selectivity of the conventional electrolyzer with and without the assistance of the AEM, quantitative analysis measurements were conducted using HPLC. As shown in Fig. 4C, the faradaic efficiency for the formic acid production is slightly decreased as the potential increases. In the conventional electrolysis system, although the membrane-free electrolysis system can achieve a higher current density for the conversion of PET hydrolysate, as shown in Fig. 4D, there are also several drawbacks: (1) generated products may diffuse between the anode and cathode and re-oxidize or re-reduce to influence the efficiency of the system, as shown in Fig. S17;† (2) during the continuous electrolysis process, oxygen may be generated at the anode as a by-product, which can form an explosive gas with hydrogen and cause safety problems; (3) the complex PET hydrolysate can also diffuse to the cathode and poison the electrocatalyst activity. Therefore, it is crucial to develop flexible and facile electrochemical conversion technology for the efficient valorization of PET plastic waste.
Based on above results, our proof-of-concept experiment indicates the potential of the decoupling process for the valorization of PET plastic waste to produce valued formic acid and hydrogen with versatile operational conditions and without a membrane. To the best of our knowledge, the high conversion efficiency without the use of membranes represents a comparable performance with previous reports for electrocatalytic upcycling of PET hydrolysate into formic acid over Ni-based catalysts (Table S1†). Notably, unlike the decoupling strategy in water splitting, which requires input of heat or electricity to refresh the NiOOH back to Ni(OH)2, the use of PET hydrolysate in this work as a reducing reactant not only effectively reduces NiOOH into Ni(OH)2 but also generates valuable formic acid. In our proposed decoupling strategy, the spontaneous chemical process between NiOOH and PET hydrolysate proceeded at normal temperature and pressure without any other energy input.
Moreover, the integration of photovoltaics (PV) and electrolysis represents a promising approach for harnessing intermittent solar energy sources.40,41 In our previous study, our solar-powered PV-electrochemical system exhibited an impressive solar-to-chemical (STC) efficiency when upcycling PET plastic waste.42 Building upon these investigations, we now illustrate the construction of a solar-powered PET upcycling system by implementing a decoupling strategy, as depicted in Fig. 5A. This innovative approach separates the HER process, which can be powered by the PV module to produce hydrogen using the Ni(OH)2 mediator. Notably, although the HER step is endothermic, some incidental heat is generated due to physical resistance and electrochemical polarization. The excess heat can be captured and utilized for the hydrolysis of PET plastic waste, facilitating the production of desired hydrolysate through careful heat management. Subsequently, valuable formic acid can be obtained from PET plastic waste through a spontaneous chemical process. This system allows the PV-electro-chemical setup to be driven by solar energy, enabling the efficient valorization of PET plastic waste.
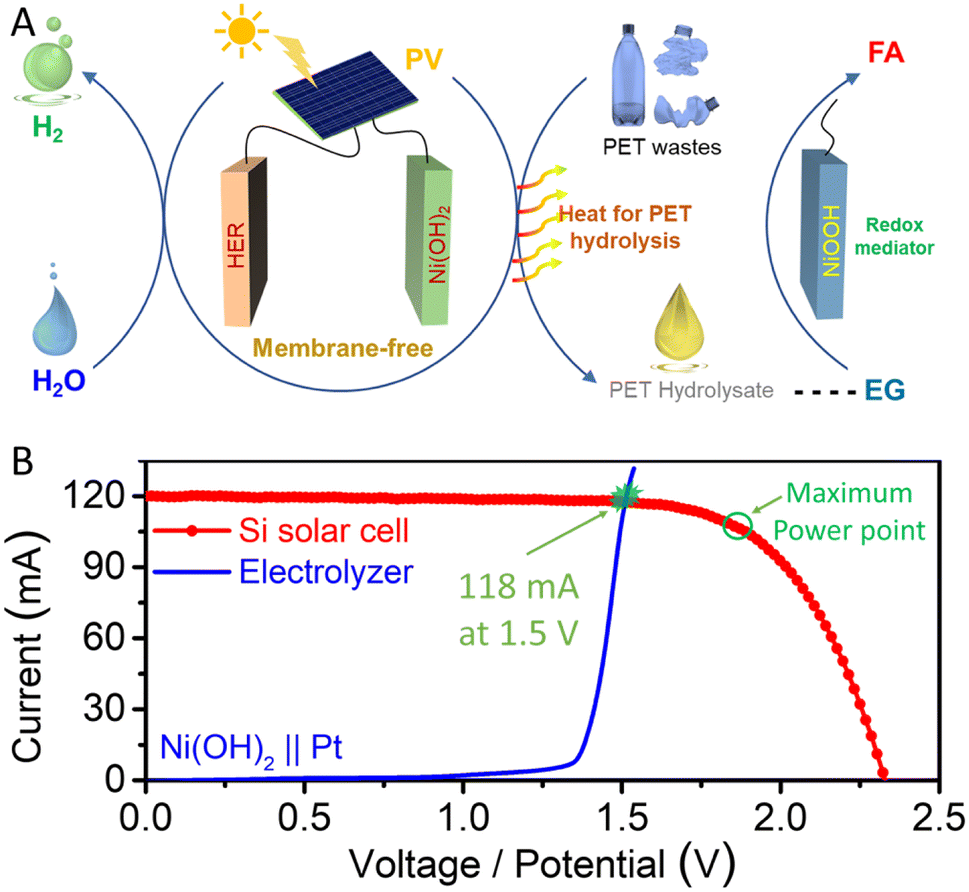 | ||
| Fig. 5 (A) Schematic diagram of the decoupling strategy for solar-powered PV-electrochemical upgrade of PET plastic waste with the assistance of the Ni-based redox mediator. (B) Current–potential (voltage) characteristic curves of Ni(OH)2‖Pt electrolyzer and a Si-based photovoltaic cell. | ||
We predicted the operating current for hydrogen production in the PV-electrolysis process by analyzing current–voltage/potential (I–V) curves, as presented in Fig. 5B. The I–V characteristics of the commercial Si-based solar cell and Ni(OH)2‖Pt electrolyzer were measured. The short-circuit current (Jsc) of the Si-based solar cell is 120 mA (∼7.5 mA cm−2), and its open-circuit potential (Voc) is 2.30 V. The crossover points in the I–V curve between the HER electrolysis process and Si-based solar cell occurred at 118 mA and 1.5 V, approximating the operating point of the entire system. The entire system achieved a solar to chemical efficiency of ∼11.03% with a current density of ∼7.5 mA cm−2 under a simulated solar irradiation intensity of 100 mW cm−2.42,43 Hence, exploration of the decoupling strategy, aided by the Ni(II)/Ni(III) redox mediator, offers an appealing avenue for the valorization of PET plastic waste, leading to the production of valuable chemicals within a solar-driven, membrane-free system.
Conclusions
In this study, a Ni(II)/Ni(III) redox-mediated decoupling strategy was introduced for the valorization of PET plastic waste to produce high-value formic acid and green hydrogen fuel in a membrane-free system. The decoupled process included an electrochemical process for hydrogen production and a spontaneous chemical process for the valorization of PET plastic waste. By employing this strategy, the decoupled HER process can be driven at a relatively low potential with assitance of Ni(OH)2 redox mediator electrodes. The spontaneous chemical process between the oxidized NiOOH and PET hydrolysate exhibited a high faradaic efficiency to produce formic acid. Compared with traditional water splitting, this decoupling strategy introduced to valorize PET plastic waste for high-value formic acid production can simultaneously reduce the energy consumption for hydrogen production. Moreover, membrane-free characteristics can reduce assembly costs, device complexity and operational difficulty of photovoltaic-driven electrochemical conversion systems for the valorization of PET plastic waste. This decoupling strategy provides a potential route for the upcycling of PET plastic waste combined with the HER and CO2 reduction reaction by utilizing appropriate redox materials as proton-electron mediators under facile conditions.Data availability
All data are available in the manuscript and in the ESI.†Author contributions
Jianying Wang, Xin Li and Yixin Zhao conceived this work. Jianying Wang and Xin Li performed this work. Ting Zhang, Xinyu Chai, Mingze Xu, Chengcheng Cai, Menglei Feng contributed to the discussion of this work. Jianying Wang, Xin Li, Xufang Qian, Zuofeng Chen and Yixin Zhao wrote and revised the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22276123 and 22025505), Natural Science Foundation of Shanghai (Grant No. 23ZR1464800) and China Postdoctoral Science Foundation (Grant No. 2021M702120).Notes and references
- A. Stubbins, K. L. Law, S. E. Munoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51–55 CrossRef CAS PubMed.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- A. Chen, M.-Q. Yang, S. Wang and Q. Qian, Front. Nanotech., 2021, 3, 723120 CrossRef.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed. Engl., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- Y. Yan, H. Zhou, S. M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622–627 CrossRef CAS PubMed.
- X. Li, J. Wang, M. Sun, X. Qian and Y. Zhao, J. Energy Chem., 2023, 78, 487–496 CrossRef CAS.
- X. Li, J. Wang, T. Zhang, T. Wang and Y. Zhao, ACS Sustain. Chem. Eng., 2022, 10, 9546–9552 CrossRef CAS.
- Q. Shi and H. Duan, Chem Catal., 2022, 2, 3471–3496 CrossRef CAS.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571–6577 RSC.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2022, 16, 4625–4633 CrossRef.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403–409 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed.
- Y. Ma, X. Dong, Y. Wang and Y. Xia, Angew. Chem., Int. Ed. Engl., 2018, 57, 2904–2908 CrossRef CAS PubMed.
- J. Wang, L. Ji, X. Teng, Y. Liu, L. Guo and Z. Chen, J. Mater. Chem. A, 2019, 7, 13149–13153 RSC.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 11741 CrossRef CAS PubMed.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- H. Dotan, A. Landman, S. W. Sheehan, K. D. Malviya, G. E. Shter, D. A. Grave, Z. Arzi, N. Yehudai, M. Halabi, N. Gal, N. Hadari, C. Cohen, A. Rothschild and G. S. Grader, Nat. Energy, 2019, 4, 786–795 CrossRef CAS.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed. Engl., 2022, 61, e202200211 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhofer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed. Engl., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- H. R. Corti and E. R. Gonzalez, Direct Alcohol Fuel Cells, 2014 Search PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- Y. Wang, M. I. Dar, L. K. Ono, T. Zhang, M. Kan, Y. Li, L. Zhang, X. Wang, Y. Yang, X. Gao, Y. Qi, M. Gratzel and Y. Zhao, Science, 2019, 365, 591–595 CrossRef CAS PubMed.
- J. Wang, L. Ji and Z. Chen, ACS Catal., 2016, 6, 6987–6992 CrossRef CAS.
- S. Lee, L. Bai and X. Hu, Angew. Chem., Int. Ed. Engl., 2020, 59, 8072–8077 CrossRef CAS PubMed.
- Y. L. Lo and B. J. Hwang, Langmuir, 1998, 14, 944–950 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- S. Song, J. Zhang, G. Gozaydin and N. Yan, Angew. Chem., Int. Ed. Engl., 2019, 58, 4934–4937 CrossRef CAS PubMed.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, e2300841 CrossRef PubMed.
- X. Liu, X. He, D. Xiong, G. Wang, Z. Tu, D. Wu, J. Wang, J. Gu and Z. Chen, ACS Catal., 2024, 14, 5366–5376 CrossRef CAS.
- H. Kang, D. He, X. Yan, B. Dao, N. B. Williams, G. I. Elliott, D. Streater, J. Nyakuchena, J. Huang, X. Pan, X. Xiao and J. Gu, ACS Catal., 2024, 14, 5314–5325 CrossRef CAS PubMed.
- F. Lv, Z. Qin, J. Wu, L. Pan, L. Liu, Y. Chen and Y. Zhao, ChemSusChem, 2023, 16, e202201689 CrossRef CAS PubMed.
- J. Kim, J. Jang, T. Hilberath, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 776–786 CrossRef.
- T. Zhang, X. Li, J. Wang, Y. Miao, T. Wang, X. Qian and Y. Zhao, J. Hazard. Mater., 2023, 450, 131054 CrossRef CAS PubMed.
- G. Gurudayal, J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional information as noted in the text. See DOI: https://doi.org/10.1039/d4sc01613k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |