
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single hydrogen bond that tunes flavin redox reactivity and activates it for modification†
Debarati
Das
 and
Anne-Frances
Miller
*
and
Anne-Frances
Miller
*
Department of Chemistry, University of Kentucky, Lexington, Kentucky, USA. E-mail: Debarati.Das@uky.edu; Afmill3r2@gmail.com
First published on 24th April 2024
Abstract
Electron bifurcation produces high-energy products based on less energetic reagents. This feat enables biological systems to exploit abundant mediocre fuel to drive vital but demanding reactions, including nitrogen fixation and CO2 capture. Thus, there is great interest in understanding principles that can be portable to man-made devices. Bifurcating electron transfer flavoproteins (Bf ETFs) employ two flavins with contrasting reactivities to acquire pairs of electrons from a modest reductant, NADH. The bifurcating flavin then dispatches the electrons individually to a high and a low reduction midpoint potential (E°) acceptor, the latter of which captures most of the energy. Maximum efficiency requires that only one electron accesses the exergonic path that will ‘pay for’ the production of the low-E° product. It is therefore critical that one of the flavins, the ‘electron transfer’ (ET) flavin, is tuned to execute single-electron (1e−) chemistry only. To learn how, and extract fundamental principles, we systematically altered interactions with the ET-flavin O2 position. Removal of a single hydrogen bond (H-bond) disfavored the formation of the flavin anionic semiquinone (ASQ) relative to the oxidized (OX) state, lowering 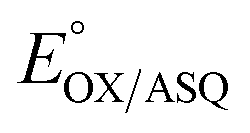 by 150 mV and retuning the flavin's tendency for 1e−vs. 2e− reactivity. This was achieved by replacing conserved His 290 with Phe, while also replacing the supporting Tyr 279 with Ile. Although this variant binds oxidized FADs at 90% the WT level, the ASQ state of the ET-flavin is not stable in the absence of H290's H-bond, and dissociates, in contrast to the WT. Removal of this H-bond also altered the ET-flavin's covalent chemistry. While the WT ETF accumulates modified flavins whose formation is believed to rely on an anionic paraquinone methide intermediate, the FADs of the H-bond lacking variant remain unchanged over weeks. Hence the variant that destabilizes the anionic semiquinone also suppresses the anionic intermediate in flavin modification, verifying electronic similarities between these two species. These correlations suggest that the H-bond that stabilizes the crucial flavin ASQ also promotes flavin modification. The two effects may indeed be inseparable, as a Jekyll and Hydrogen bond.
by 150 mV and retuning the flavin's tendency for 1e−vs. 2e− reactivity. This was achieved by replacing conserved His 290 with Phe, while also replacing the supporting Tyr 279 with Ile. Although this variant binds oxidized FADs at 90% the WT level, the ASQ state of the ET-flavin is not stable in the absence of H290's H-bond, and dissociates, in contrast to the WT. Removal of this H-bond also altered the ET-flavin's covalent chemistry. While the WT ETF accumulates modified flavins whose formation is believed to rely on an anionic paraquinone methide intermediate, the FADs of the H-bond lacking variant remain unchanged over weeks. Hence the variant that destabilizes the anionic semiquinone also suppresses the anionic intermediate in flavin modification, verifying electronic similarities between these two species. These correlations suggest that the H-bond that stabilizes the crucial flavin ASQ also promotes flavin modification. The two effects may indeed be inseparable, as a Jekyll and Hydrogen bond.
Introduction
Electron bifurcation generates high-energy reductants required for demanding reactions such as fixation of CO2 or N2, based on less potent reductants.1,2 Tight coupling between a pair of electron transfers enables a thermodynamically favourable transfer to ‘pay for’ an unfavourable one, effectively concentrating the energy of both electrons on one of them.3,4 Although electron bifurcation was first recognized in the cytochrome bc1 complex of respiratory electron transfer, it is exemplified in a more accessible form by the so-called Bifurcating Electron Transfer Flavoproteins (Bf ETFs), in which the relative simplicity and air tolerance of the system facilitate experimentation.5The best-characterized Bf ETFs are heterodimers with two subunits called EtfA and EtfB, in which subunit A is composed of two domains, domain I and domain II, whereas subunit B makes up domain III (Fig. 1).6,7 Domain II bears a non-covalently bound FAD known as the Electron Transfer FAD (ET-FAD), which is also present in the non-bifurcating ‘canonical’ ETFs of mitochondria.7 Bf ETFs carry a second FAD bound between domains I and III, which canonical ETFs lack (they have an AMP instead).8,9 This second FAD called the bifurcating FAD (Bf-FAD) is the site of bifurcation, at which two electrons acquired as H− from NADH are dispatched individually via two separate but tightly coupled transfers: to a higher E° acceptor (exergonic pathway) and to a lower E° acceptor (endergonic pathway, E° = reduction midpoint potential).
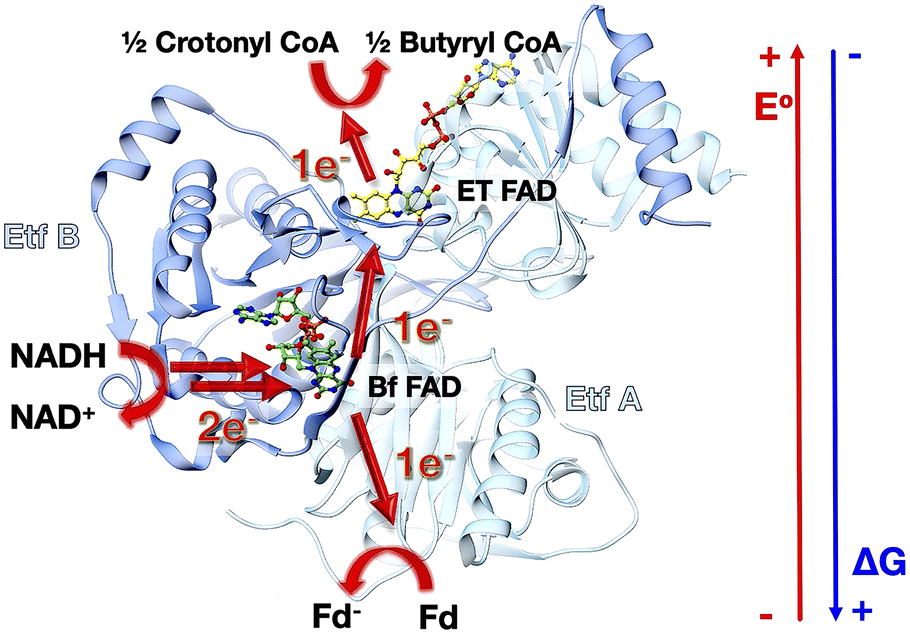 | ||
| Fig. 1 Structure and function of the electron transfer flavoprotein from Acidaminococcus fermentans (Afe). AfeETF is a heterodimer composed of a larger subunit EtfA (light teal) and a smaller subunit EtfB (cornflower blue). EtfA consists of two domains: domain I and domain II, whereas EtfB contains domain III. Domain I and domain III form the base of the ETF, and domain II sits on top of the base and undergoes almost 90° rotation relative to the base in the course of turnover. Bf-FAD (green) is positioned at the interface between domains I and III whereas the ET-FAD (yellow) is situated in domain II. | ||
Flavin based electron bifurcation (FBEB) was discovered in 2008.2 While bifurcation in the bc1 complex occurs at a quinone with E° in the range of +90 mV to −90 mV, the lower −207 mV E° of the flavin enables FBEB to capture more energy from NADH (E° ≈ −320 mV) and to produce electrons at potentials as low as −500 mV (all potentials are vs. the NHE).10 Besides Bf ETFs, FBEB is executed by NADPH dependent ferredoxin-NADH reductase, heterodisulfide reductase, Bf hydrogenase and NADH dehydrogenase in conjunction with [FeFe] hydrogenase.11 In FBEB, NADH donates a pair of electrons to Bf-FAD; one electron from the resulting reduced flavin hydroquinone (HQ) flows to the high E° ET-FAD leaving an unstable semiquinone (SQ) state of the Bf-FAD (Fig. 1). This reduces a low E° carrier: ferredoxin (Fd) or flavodoxin (Fld) semiquinone, thus producing very low E° reducing equivalents.4 Bifurcating activity is natural for a flavin, since unstable SQ states are the rule in organic redox compounds such as quinones and other dyes. However, the ET-FAD's pattern of two sequential one-electron (1e−) reactions is unusual. This requires that the 1e− reduced SQ state be stable over a substantial potential range. Free flavins populate SQ states at only a 1% level when [OX] = [HQ].12 However in proteins, the relative stability of flavin SQ states can be tuned over hundreds of mV.13 This, in turn, determines whether the flavin will display sequential 1e− redox reactions involving a SQ intermediate 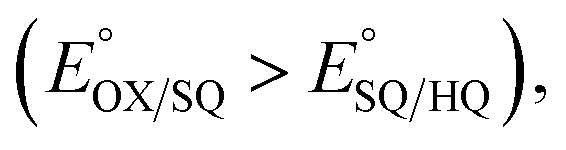 or 2e− reactivity characterized by a single 2e− potential
or 2e− reactivity characterized by a single 2e− potential 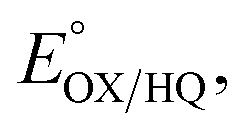 under equilibrium conditions. Thus, to favour this unusual 1e− transfer, the ETF protein must stabilize the ET-FAD's SQ state, which is an anionic semiquinone (ASQ) throughout the physiological range.14,15
under equilibrium conditions. Thus, to favour this unusual 1e− transfer, the ETF protein must stabilize the ET-FAD's SQ state, which is an anionic semiquinone (ASQ) throughout the physiological range.14,15
ET-FAD's unusually high 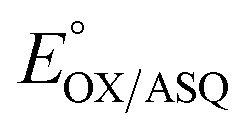 should also favour ASQ as the resting state of ET-FAD in vivo.15,16 Thus, one more electron would suffice to completely reduce the ET-FAD, leaving the second electron no alternative to the uphill ET path to Fd/Fld.17 This mechanism of electron gating would be an automatic consequence of the remarkable stabilization of the ASQ state of ET-FAD. An additional mechanism proposed to gate electron transfer is conformational dynamics, wherein rotation of domain II either moves the ET-flavin closer (≤18 Å) to the Bf-flavin facilitating electron transfer, or removes it to >35 Å away, essentially severing the exergonic ET path and thereby increasing the branching ratio in favour of the endergonic alternative.18 Conformational dynamics can complement redox tuning, and indeed the different conformations of the ETF may have distinct redox properties.19
should also favour ASQ as the resting state of ET-FAD in vivo.15,16 Thus, one more electron would suffice to completely reduce the ET-FAD, leaving the second electron no alternative to the uphill ET path to Fd/Fld.17 This mechanism of electron gating would be an automatic consequence of the remarkable stabilization of the ASQ state of ET-FAD. An additional mechanism proposed to gate electron transfer is conformational dynamics, wherein rotation of domain II either moves the ET-flavin closer (≤18 Å) to the Bf-flavin facilitating electron transfer, or removes it to >35 Å away, essentially severing the exergonic ET path and thereby increasing the branching ratio in favour of the endergonic alternative.18 Conformational dynamics can complement redox tuning, and indeed the different conformations of the ETF may have distinct redox properties.19
Proteins exploit a variety of mechanisms to tune flavin E°s, including control over the local dielectric, distortion of flavin geometry, hydrogen bonds (H-bonds) and for the anionic states (ASQ and AHQ), net electrostatics.14,20–24 Studies of canonical and Bf ETFs showed that the unusually high 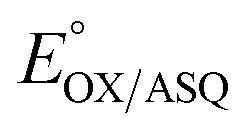 of ET-FAD can be attributed in part to a 99% conserved Arg253 and a 100% conserved H-bond to N5 from Ser or Thr270 (Fig. 2 for flavin position numbering, amino acids' numbering is from AfeETF, and the specified residues all derive from the A subunit).16,23–28 However, these interactions do not suffice to explain the stability of ET-FAD's ASQ, as ASQ accumulates even when these residues are replaced.14,26 Moreover, the positive charge of the Arg would stabilize ET-FAD's AHQ state as well as its ASQ, and the H-bond from Ser/Thr is weak based on computations.28
of ET-FAD can be attributed in part to a 99% conserved Arg253 and a 100% conserved H-bond to N5 from Ser or Thr270 (Fig. 2 for flavin position numbering, amino acids' numbering is from AfeETF, and the specified residues all derive from the A subunit).16,23–28 However, these interactions do not suffice to explain the stability of ET-FAD's ASQ, as ASQ accumulates even when these residues are replaced.14,26 Moreover, the positive charge of the Arg would stabilize ET-FAD's AHQ state as well as its ASQ, and the H-bond from Ser/Thr is weak based on computations.28
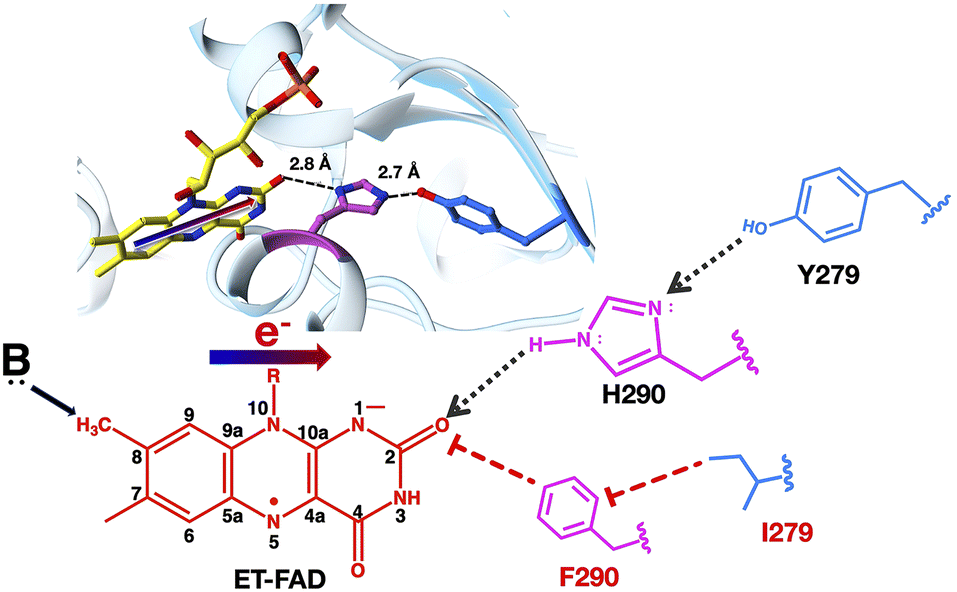 | ||
Fig. 2 Role of H290 in modulating the 1e− reactivity of ET-FAD. The δNH of H290 is a H-bond donor to O2, which stabilizes excess electron density of the ASQ in the N1C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2 locus, thereby favouring ET-FAD ASQ (ETASQ). An analogous shift of electron density makes the C8m methyl group acidic in the ET-FAD's OX state (ETOX) and is proposed to enable population of the paraquinoid methide intermediate invoked in flavin modification at C8m. Therefore, we hypothesize that H-bonding from H290 to flavin O2 activates the flavin for modification at C8m. O2 locus, thereby favouring ET-FAD ASQ (ETASQ). An analogous shift of electron density makes the C8m methyl group acidic in the ET-FAD's OX state (ETOX) and is proposed to enable population of the paraquinoid methide intermediate invoked in flavin modification at C8m. Therefore, we hypothesize that H-bonding from H290 to flavin O2 activates the flavin for modification at C8m. | ||
We hypothesize that a 90% conserved histidine (His) residue close to the ET-flavin O2 donates an important H-bond, thereby stabilizing negative charge over the N1–C2O2 locus in the ET-FAD's ASQ state. This is predicted to stabilize ASQ specifically, because the AHQ state tends to concentrate excess electron density near N5 instead, in consequence of N5's protonation.28 Thus, we propose that His290 (H290) of AfeETF is crucial for its stable ET-FAD ASQ state (ETASQ) and 1e− reactivity.
Another intriguing anomaly of the ET-FAD is its unusual propensity for chemical modification.29–31 The proposed mechanisms of modification invoke deprotonation of the C8 methyl (C8m) or nucleophilic attack at C6 and a paraquinone methide-type transition state (reviewed in the ESI† of Mohamed Raseek,32 and see ref. 23 and 33–36). Like the ASQ, the proposed methide state is anionic and excess electron density can be distributed over the flavin π system to the electronegative N1–C2O2 locus.36,37 Therefore, we postulate that the H-bond from H290 to O2 could also underlie ET-flavin's unusually high susceptibility to modification.38 If so, replacement of the H290 residue in AfeETF should diminish both the stability of ETASQ and ET-FAD modification. Indeed, our characterization of the doubly substituted H290F and Y279I (H290FY279I) variant of AfeETF documents both the anticipated results. Thus, we suggest a chemical explanation for the unusually high tendency of ETFs to carry modified flavins, unexplained since the 1970s.39 We propose that evolution achieved the remarkably stable ASQ of ET-flavin at the cost of promoting flavin modification.
Experimental section
Site – directed mutagenesis
The plasmid bearing the genes for EtfB and EtfA with a C terminal His tag and carbenicillin resistance were the kind gift of R. Hille with consent from N. P. Chowdhury and W. Buckel.6 Mutations were introduced in Acfer_0555 at the H290 codon using the polymerase chain reaction (Q5 high fidelity DNA polymerase, New England Biolabs, M0492S). Six different variants of EtfA were designed: H290A, H290E, H290N, H290K, H290T and a doubly substituted variant H290FY279I. The forward and reverse primers were designed using a NEB base changer and were ordered from Eurofins genomics. The primers, the melting temperatures and the annealing temperatures used during the reaction are provided in Table S1.† Desired mutations were confirmed and undesired changes were excluded, by sequencing of the resulting plasmids. The mutated constructs were transformed into an Escherichia coli expression host Nico21 (DE3).Expression of the wild type and variant AfeETFs
The expression levels and solubilities of the variants were initially assessed via SDS-PAGE of the supernatant and pellet resulting from centrifugation of lysed cells expressing variant ETFs (lysed using BugBuster), (MilliporeSigma™ 705844).For larger-scale expression, 10 mL of liquid culture of transformed Nico21 cells were grown overnight in an incubator shaker (Innova 4330) at 37 °C at 220 rpm and used to inoculate 1 L Terrific Broth augmented with carbenicillin (100 μg mL−1). This was shaken at 180 rpm and 37 °C while OD600 was monitored at regular intervals. At an OD600 of 0.5, the temperature was lowered to 21 °C and ETF gene expression was induced with anhydrotetracycline (0.2 μg mL−1). The cells were shaken at 180 rpm at 21 °C overnight (12–18 hours). Cells were then harvested by centrifugation (Thermo Scientific 75004521) at 4500 rpm and 4 °C for 30 min, followed by a wash with phosphate-buffered saline (pH 7.4) before storage at −80 °C.
Purification of the wild type and the variants
Harvested cells (11–12 g from 1 L growth) were thawed at 4 °C and resuspended in a lysis buffer (1 mL g−1 pellet, buffer compositions are in the ESI†). The resuspended cells were sonicated at 50% amplitude in pulses of 10 s interspersed with 10 s off for 30 min. The lysed cells were centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. The supernatant was mixed with 2 mL nickel-nitrilotriacetic acid resin pre-equilibrated with 5 column volumes (CVs) of a buffer composed of 50 mM HEPES pH 7.5 and 10 mM imidazole, and stirred for 1 h at 4 °C. The slurry was loaded into a column. The resin was washed with 15 CVs of the wash buffer (as above but containing 15 mM imidazole). Finally, the protein was eluted with 4 CVs of the elution buffer (as above but containing 150 mM imidazole). The eluted protein was concentrated to 3 mL in a centrifugal filter cell (Amicon UFC801024) and transferred to phosphate buffer (50 mM KPO4 pH 7.2) by gel filtration on a pre-equilibrated DG 10.
000g for 30 min. The supernatant was mixed with 2 mL nickel-nitrilotriacetic acid resin pre-equilibrated with 5 column volumes (CVs) of a buffer composed of 50 mM HEPES pH 7.5 and 10 mM imidazole, and stirred for 1 h at 4 °C. The slurry was loaded into a column. The resin was washed with 15 CVs of the wash buffer (as above but containing 15 mM imidazole). Finally, the protein was eluted with 4 CVs of the elution buffer (as above but containing 150 mM imidazole). The eluted protein was concentrated to 3 mL in a centrifugal filter cell (Amicon UFC801024) and transferred to phosphate buffer (50 mM KPO4 pH 7.2) by gel filtration on a pre-equilibrated DG 10.
To restore FAD lost during purification, the protein was incubated within 1 mM FAD overnight. Excess FAD was then removed by gel filtration over a DG 10 column pre-equilibrated with 50 mM KPO4 at pH 7.0, our default buffer. The resultant protein was concentrated using centrifugal filters and stored at −80 °C in aliquots of 50 μL.
Quantification of protein and FAD
Protein was quantified using Pierce's A660 assay vs. a standard curve of Bovine Serum Albumin (BSA). The molecular mass of ETF from AfeETF is 61 kDa.6 For each FAD quantification, 400 μL of 20 μM sample was denatured at 100 °C for 10 minutes in darkness to release FAD, and then cooled to room temperature. The solution was cleared of protein by centrifugation at a high speed for 10 minutes. The supernatant was collected, and the concentration of FAD was determined from the UV visible spectrum based on the absorbance at 450 nm, A450, and the corresponding extinction coefficient (ε450) of 11300 M−1 cm−1.8
Melting temperature of wild type and H290FY279I variants
Differential scanning fluorimetry was used to estimate the melting midpoint temperatures of WT and H290FY279I ETF. 30 μL samples of 1 mg mL−1 WT- or H290FY279I in default buffer were diluted with 30 μL default buffer and augmented with 15 μL of 50× SYPRO orange stock solution in 1.5 mL centrifuge tubes, yielding triplicate samples of 25 μL each. Samples were loaded into wells of a 96 well plate and then heated from 10 °C to 95 °C while fluorescence was recorded at 0.5 °C intervals. The exposure of the protein's hydrophobic residues upon melting of the core is reflected by a change in dye fluorescence.40Preparation of Ti2+ citrate (pH 7.0 100 mM)
All the solutions used were equilibrated vs. an inert atmosphere for 2–3 days in an antechamber before being brought into the main anaerobic chamber. To an 80 mL solution of 0.2 M Na citrate, 5.3 mL of commercially available 30% (w/v) Ti–chloride (TiCl3 in HCl, FW: 154.2 is 1.9 M) was added. 20 mL of 8% (w/v) Na2CO3 was added to the above mixture. The pH of the solution was checked with pH paper and adjusted to pH 7.0 with Na2CO3. The bottle was wrapped with aluminium foil and stored shielded from light inside an anaerobic chamber.Reductive titrations
Anaerobic reductive titration was carried out in an inert atmosphere as provided by a glove box (Belle Technology, UK). Samples were ≥20 μM in concentration and 400 μL in volume. For the reductant NADH, 2 equivalents were sufficient to completely reduce the ETF, and the NADH concentration of stock solutions was confirmed via NADH's A340 and ε340 = 6220 M−1 cm−1.41 Titrations were carried out in self-masked cuvettes (Fireflysci, type30BM, 700 μL, 5 mm pathlength) in a temperature-controlled sample holder (Quantum-Northwest) monitored optically using a HP 8452A spectrophotometer refurbished and modernized by OLIS. Reductive titrations with dithionite (ε315 = 8.02 mM−1 cm−1) were carried out in a similar fashion using a fresh stock solution of dithionite prepared in 0.02 M KOH solution (pH 11) inside the glove box and diluted as needed using 50 mM KPO4 at pH 7.0 immediately before use.42 Titanium citrate (ε340 = 0.74 mM−1 cm−1) was also used to reduce the ETF.43 A 100 mM stock of titanium citrate solution was prepared as described above. Titanium citrate is a 1e− donor so 4 stoichiometric equivalents were needed to completely reduce the ETF.Determination of reduction midpoint potentials
Reduction midpoint potentials were determined using xanthine/xanthine oxidase and xanthine as the reducing system.44 Catalytic amounts of methyl viologen were included as a mediator and a reference dye was chosen such that the potential of the dye would be within ±60 mV of the potential describing the phase under study. The goal is for the ETF and the dye to undergo simultaneous or at least overlapping reduction as the reductant is added, at equilibrium with one another. Initial selections of reference dyes were made based on reported potentials, but extensive trials were required to identify dyes that actually co-reduced with the ETF and equilibrated on the time scale of the reduction. Each reaction involved 400 μM xanthine, 20 μM ETF, 1 μM methyl viologen, and 1–5 μL of the reference dye, as required to obtain an optical signal for the dye of sufficient magnitude to be quantified with precision, but with minimal obstruction of the signals of ETF's flavins. Reactions were initiated by addition of xanthine oxidase and monitored optically at 30 s intervals. The concentration of xanthine oxidase provided ranged from 10 nM–100 nM with larger values used to attain lower potentials, but with values kept small enough to ensure that reduction was slow enough that the system remained at equilibrium. E° values that did not vary upon alteration of the rate of reduction were taken as confirmation of equilibration between the ETF and the dye.Optical signatures were used to identify and quantify each state of ETF consumed or generated by the reduction. Different dyes were chosen for each of the reactions, on the basis of numerous trials to identify those whose conversion from 90% oxidized to 90% reduced overlapped best with the analogous interval for the ETF. For H290FY279I, 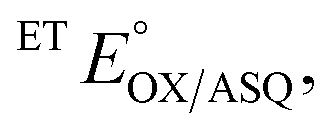
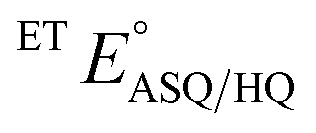 and
and 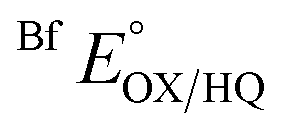 were measured using new methylene blue (E° = −35.75 mV), Nile blue (E° = −130.75 mV), and safranin O (E° = −303.75) at pH 7.5, respectively, calculated from published E°′ values (E°′ = −21 mV for new methylene blue; E°′ = −116 mV for Nile blue; E°′ = −289 mV for safranine O, respectively).45 The buffer used for the reactions was KPO4 pH 7.0; however, since xanthine is dissolved in 10 N NaOH, the pH of the system increased to pH 7.5. Phase 1's
were measured using new methylene blue (E° = −35.75 mV), Nile blue (E° = −130.75 mV), and safranin O (E° = −303.75) at pH 7.5, respectively, calculated from published E°′ values (E°′ = −21 mV for new methylene blue; E°′ = −116 mV for Nile blue; E°′ = −289 mV for safranine O, respectively).45 The buffer used for the reactions was KPO4 pH 7.0; however, since xanthine is dissolved in 10 N NaOH, the pH of the system increased to pH 7.5. Phase 1's 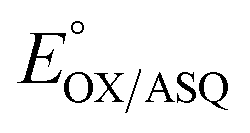 was monitored on the basis of ET-FAD ASQ formation as indicated by absorbance near 374 nm. The reaction was deemed complete when maximal absorbance at 374 nm was attained in conjunction with decreased A454, based on plots of A374vs. A454 (figure insets). Oxidized new methylene blue's λmax of 590 nm and ETFOX's λmax of 454 nm were used to monitor the progress of their reductions. Using absorbances at these wavelengths, the total oxidized population and the total reduced population (=total − OX) were calculated for the ETF and the dye. The ratios of oxidized to reduced populations for the ETF and dye were calculated at each step, [FADox]/[FADASQ] and [Dyeox]/[DyeRed], and analysed using the Nernst equation (eqn (1)).
was monitored on the basis of ET-FAD ASQ formation as indicated by absorbance near 374 nm. The reaction was deemed complete when maximal absorbance at 374 nm was attained in conjunction with decreased A454, based on plots of A374vs. A454 (figure insets). Oxidized new methylene blue's λmax of 590 nm and ETFOX's λmax of 454 nm were used to monitor the progress of their reductions. Using absorbances at these wavelengths, the total oxidized population and the total reduced population (=total − OX) were calculated for the ETF and the dye. The ratios of oxidized to reduced populations for the ETF and dye were calculated at each step, [FADox]/[FADASQ] and [Dyeox]/[DyeRed], and analysed using the Nernst equation (eqn (1)).
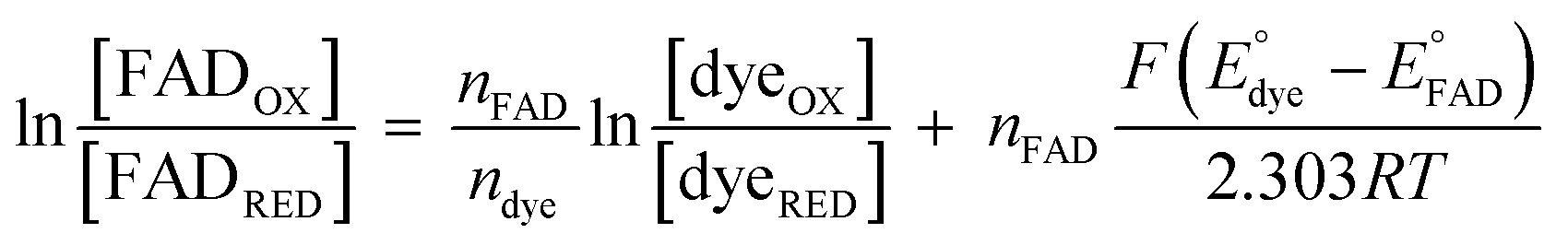 | (1) |
To do so, we plotted  vs.
vs.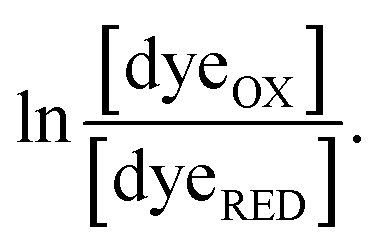 The resulting slope is equal to
The resulting slope is equal to 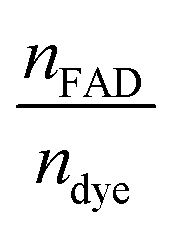 and since the ndye = 2, the nFAD was obtained. The determined nFAD was then used with the known
and since the ndye = 2, the nFAD was obtained. The determined nFAD was then used with the known 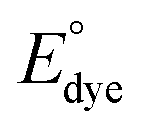 to solve for
to solve for  by equating the intercept to
by equating the intercept to 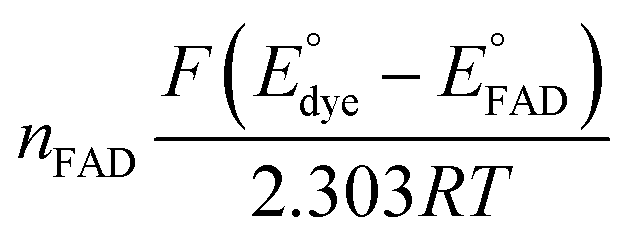 for the reaction under study.
for the reaction under study.
Because of the number of electrons acquired by the flavin, the slopes for phases 1, 2 and 3 are expected to be 0.5, 0.5 and 1 respectively because the dyes used all undergo 2e− reduction. The slopes for individual phases are provided in Table S2.†
Mass spectrometry
Samples of flavin released from 10 μM and 15 μM Afe WT and variant H290FY279I, respectively, in bis tris propane (pH 9.0), were sent to the University of Texas Southwestern's Proteomics core facility for intact mass analysis using a Sciex X500B QTOF mass spectrometer.Results
Stability and integrity of variants relative to the wild type
ET-FAD's unique 1e− reactivity is crucial to Bf ETF's bifurcating activity, but requires a stable ASQ state. Thus, specialized redox tuning imposed by the protein environment plays an essential role in bifurcation. Previous efforts demonstrate the importance of electrostatics and H-bonds to N5.14,16,25,26 However these interactions have analogues in the Bf site, which does not stabilize ASQ. Instead, the current efforts take aim at an interaction that is unique to the ET site.To probe the role of H290 in tuning the reactivity of ET-FAD, we engineered AfeETF variants where H290 is replaced with A, N, E, K or T. Unfortunately, any consequences for redox tuning were confounded by the poor stability of these AfeETF variants. We succeeded in eliminating H290's H-bonding functionality by using a double replacement found in nature, wherein the H290F substitution is enabled by simultaneous substitution of nearby Y279 with I (Fig. 2). The doubly substituted H290FY279I-AfeETF (H290FY279I-ETF or ‘H290FY279I’ hereafter) retained acceptable stability, based on its Tm of 55.5 °C measured using differential scanning fluorimetry (vs. 65.5 °C for WT-AfeETF, WT-ETF or ‘WT’ hereafter, Fig. 3). The variant's significantly depressed Tm verifies H290's influence on the stability of AfeETF and suggests why no prior work has been published. However, H290FY279I retained the full FAD complement and a band II excitation maximum close to that of the WT, in contrast to variants in which H290 alone was replaced (Table 1).
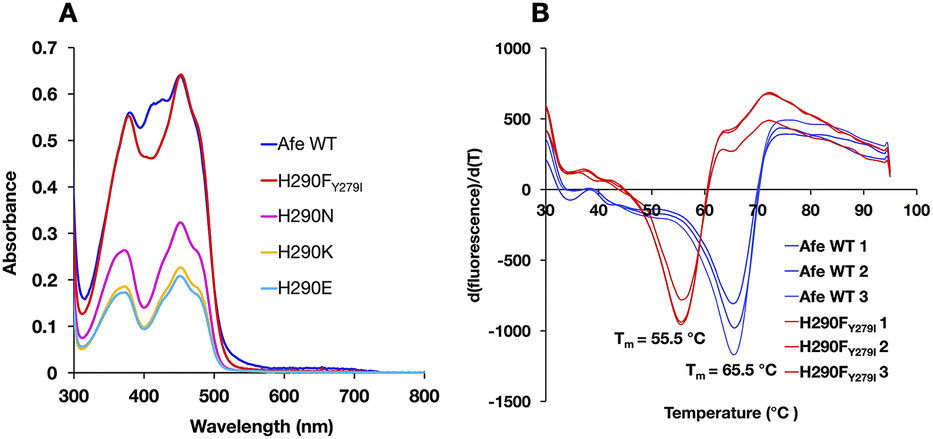 | ||
| Fig. 3 Amino acid substitution at position 290 perturbs the electronic spectrum of bound flavin and diminishes the stability of AfeETF. (A) UV/vis spectra of different AfeETF variants produced by amino acid substitution at position 290 of chain A, normalized to a concentration of 20 μM. (B) Differential scanning fluorescence comparing triplicate samples of H290FY279I with triplicates of WT-AfeETF. | ||
Because the H290FY279I variant best preserved the overall integrity of the system, it was further characterized to elucidate the specific contributions of H290 in tuning the chemical reactivity of ET-FAD.
Flavins in H290FY279I ETF lack the modification found in WT-ETF
The optical signatures of the ETF's flavins are influenced by both the binding site environments and the natures of the flavins. The optical spectrum of H290FY279I retained the characteristic band I at 454 nm and the band II at 374 nm, as in the WT. However, the variant's spectrum had a more pronounced dip between band I and band II and lacked a shoulder near 415 nm that is seen in the published spectra of most Bf ETFs.26 The absent features have been attributed to 8-formyl flavin (8fF) in the ET site by Augustin et al.46To test for the presence of a modified flavin, the spectra of flavins released from our ETF variants were compared. FAD released from WT-AfeETF displayed a pronounced blue shift and amplitude gain for band II indicating that some of the FAD of WT-AfeETF is 8fF, as found for human ETF.46 Because human ETF lacks the Bf-flavin, the modified flavin can be attributed specifically to the ET site. The spectra of flavins bound in ETF differ from those of released flavins by the much shallower dip between the two bands and the shoulders near 415 nm (Fig. 4A) in WT-AfeETF. These features must therefore reflect the consequences of the WT protein environment. Indeed, they match the signature of the ASQ of 8fF which would be populated due to the WT ET site's strong stabilization of that state.30 This is corroborated by a discernible long-wavelength signature of flavin SQ peaking near 650 nm in WT-AfeETF but not the variant spectrum. Thus, it appears that the ET-FAD of WT-AfeETF is prone to conversion to 8fF ASQ at pH 7.0, as also documented for the Bf ETF from R. palustris at pH 9.32
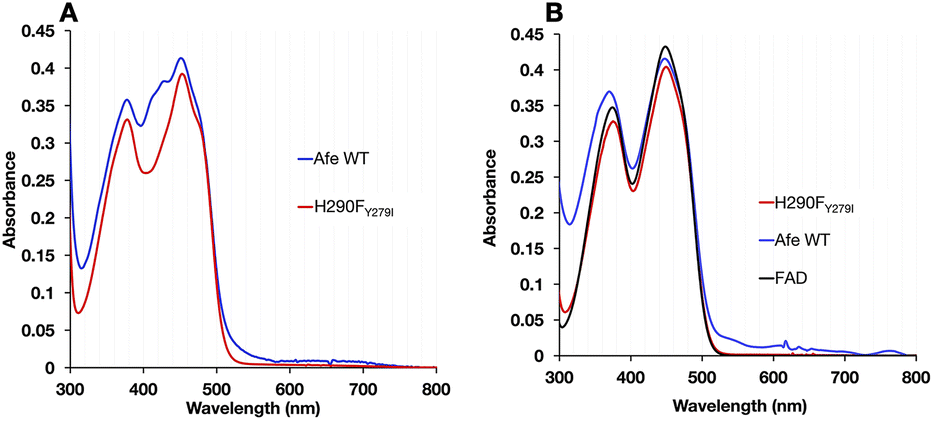 | ||
| Fig. 4 Spectral signatures of flavin modification are absent from H290FY279I. (A) Spectra of the WT (blue) and H290FY279I (red) ETF; (B) spectra of flavins released from the WT (blue) and H290FY279I (red). Visible spectra of 20 μM WT and variant H290FY279I in 50 mM PO4 buffer pH 7 and their released flavins along with free FAD were recorded. In A, the ETF spectra of the WT and H290FY279I are distinguished by the absence from the latter of the features near 415 nm attributed to 8-formyl flavin (8fF). In (B) the spectrum of the flavin released from the WT deviates from the authentic FAD in having a stronger and blue-shifted band II, near 370 nm, whereas flavin from the variant resembles authentic FAD. Equally strong band I and band II and a blue shifted band II are characteristic of 8fF. Since AfeETF contains two FADs, the signals of both contribute to all spectra, and changes affecting the ET-FAD are proportionately less significant due to admixture of the unchanged spectrum of Bf-FAD. | ||
In contrast, the optical spectrum of H290FY279I-ETF lacked the signatures of both 8fF and its spontaneous reduction to ASQ, and the spectrum of released FAD resembled that of authentic FAD instead. Both distinctions confirm the absence of flavin modification in H290FY279I-ETF.
Phases of stepwise reduction are less distinct in H290FY279I-ETF, for reduction by NADH
To test for retention of biochemical activity by both of H290FY279I's flavins we assessed the acceptance of the hydride from NADH by the Bf-flavin, and subsequent acquisition of single reducing equivalents by the ET-FAD.47,48 NADH is the natural substrate of Bf ETFs.2 For WT-AfeETF, stepwise reduction with sub-stoichiometric aliquots of NADH occurred in three phases associated with distinct spectral changes (Fig. 5A).6,19,47,49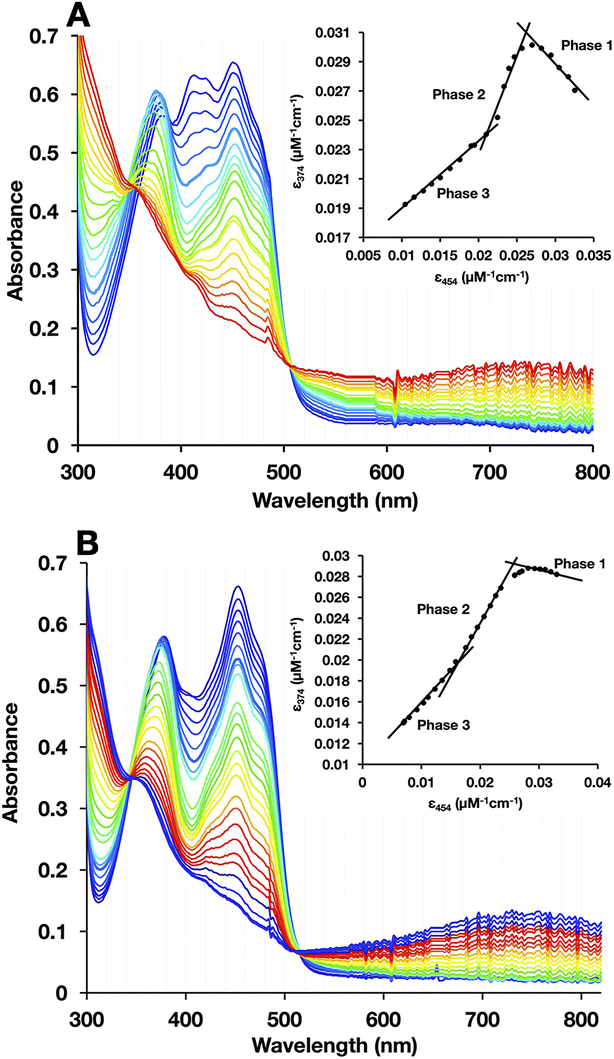 | ||
| Fig. 5 Reductive titrations with NADH reveal the sequence of events that are separated in WT but overlap in H290FY279I. (A) WT- and (B) H290FY279I-ETF. 20 μM WT or 20 μM H290FY279I in 50 mM PO4, pH 7, was reduced using stepwise additions of NADH, and spectra were collected after each addition. (Insets) Plots of A374vs. A454 that document the changing natures of the chemical events in the course of titration, and related differences between the WT and the variant. In the WT, the three phases are characterized by obviously different slopes, whereas phases 1 and 2 are much less distinct in the variant. In the insets, the first data point collected is on the right and subsequence points fall to its left. | ||
Phase 1 was marked by a decrease in A454 that occurred without net loss of absorbance near 374 nm, indicating conversion of oxidized ET-FAD (ETOX) to the ASQ state (ETASQ).50 This assignment is also supported by the loss of the modified ET-FAD's features between 400 and 430 nm in the first phase of titration. Phase 2 was associated with decreases in A374 greater than the accompanying decreases on the red edge of band I, characteristic of reduction of ETASQ to ETAHQ. Phase 3 also involved simultaneous decreases in absorbance bands I and II but of comparable magnitudes, more strongly affecting the centre of band I and accompanied by formation of a robust charge transfer (CT) band from 600 to >820 nm. These are consistent with the comparable strengths of OX FAD's bands I and II, the shape of the OX signal being replaced by that of HQ, and formation of a NAD+-flavin HQ complex. The latter is only expected in the Bf site where NAD(H) binds, confirming the reduction of BfOX to BfHQ.17,49 The CT bands formed were comparable, indicating that amino acid substitution in the ET site has relatively minor long-range effects on the Bf site.
As is common in titrations with NADH, a large population undergoes 2e− reduction directly to the HQ because NADH is intrinsically a 2e− donor and our reactions lack the natural electron acceptor Fld or Fd that would allow the two electrons to access separate acceptors. Our use of a lower pH (pH 7) further favours full reduction of the flavins.8,49 Thus, accumulation of ASQ between phases 1 and 2 is not quantitative. Nevertheless, the formation of ETASQ in WT agrees with the 1e− reactivity of ET-FAD in contrast with Bf-FAD's 2e− reactivity seen in phase 3. The Bf site does not stabilize the Bf FAD SQ, consistent with the crossed reduction potentials of Bf FAD that are crucial for electron bifurcation.4,49
Three phases were also seen in the titration of the H290FY279I variant (Fig. 5B); however, they were less distinct (compare insets). Difference spectra at different points in the titration confirmed that ASQ is formed, but there is much less of it, even at its maximum (Fig. 6). For the WT, the plot of A374vs. A454 in the inset shows a steep rise for phase 1 accompanied by an isosbestic point near 390 nm, confirming the dominance of a single conversion. However, an isosbestic point is not evident in the variant, indicating that 1e− reduction to ASQ occurs in conjunction with a second reaction, identified as 2e− reduction to HQ by the nature of the spectral changes. The overlapping occurrence of the two reductions suggests similar driving forces for them and thus diminished separation of the ET-FAD's E°s in the variant. Both the WT and H290FY279I display isosbestic points associated with phase 3, near 350 nm, consistent with phase 3's assignment to 2e− reduction of Bf-flavin. The fact that this phase is unchanged is consistent with the location of the amino acid substitutions, far from the Bf-flavin, in a separate domain. Thus, although the variant retains the reactions seen in the WT, the ET-flavin's 1e− equilibria are not separate and sequential, so we observe conversion of more ET sites directly from OX to HQ in the variant.
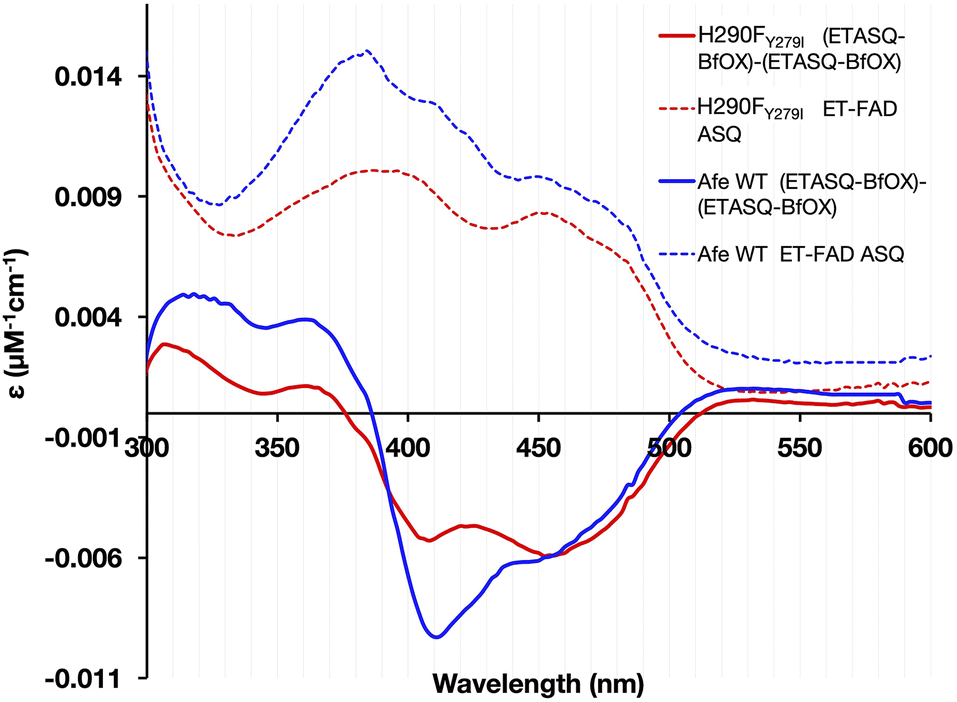 | ||
| Fig. 6 Degree to which ASQ forms upon addition of 0.5 equivalents of NADH. The solid blue and red difference spectra depict the maximal accumulation of ETASQ seen at the expense of ETOX based on the spectrum in which maximal ASQ is present (end of phase 1, ETASQ–BfOX) minus the starting spectrum (ETOX–BfOX), in reductive titration with NADH. Maximal presence of ETASQ was identified on the basis of maximal absorbance at 374 nm and occurred when half an equivalent of NADH per ETF had been added. As is evident from the difference spectra, this gain at 374 nm as well as 525 nm was accompanied by decreased absorbance at 454 nm. The presence of 8fF in WT-ETF explains the stronger negative contribution at 410 nm than at 454 nm in that case. For reference, the corresponding spectra of ETOX are also shown for each case (dotted lines), after deconvolution to remove spectral contributions from the Bf-FAD. The ETOX spectrum of WT-ETF also displays the signatures of 8fF. The smaller amplitude of the difference spectrum for the H290FY279I variant and the smaller gain at 370 nm demonstrate that significantly less ETASQ accumulates in the variant. | ||
H290FY279I-ETF accumulates less ASQ than the WT upon reaction with NADH
The magnitude of maximal ASQ formation was assessed upon reduction of the ETFs by half an equivalent of NADH. This carries one electron equivalent per ETF as required to produce the theoretical maximal yield of ETASQ, possible only if no ET-FAD undergoes 2e− reduction to AHQ. The greater relative amplitude of band II in the WT confirms that a larger amount of ASQ accumulated in the WT than in the H290FY279I variant (Fig. 6). This suggests that H290 and/or Y279 aids in stabilizing ASQ vs. OX and AHQ.Reductive titration with dithionite reveals a second ASQ species in the variant
The low ASQ yields of H290FY279I could reflect relief of a kinetic block of ETASQ reduction or thermodynamics.16 Additionally, maximal formation of ETASQ based on 2e− reduction of Bf-FAD requires a single reducing equivalent to end up on ET-FAD based on intra-ETF electron transfer but transfer of the other electron to a second ETF's ET-FAD (inter-ETF transfer). The H290FY279I substitutions could modulate these processes and thereby alter the relative rates of formation/consumption of the two reduced states of ET-FAD.As a control for mechanistic effects, we compared with the results of stepwise reduction by a 1e− donor, dithionite. This choice also avoids conformational consequences of substrate binding/product release that may be coupled to reaction with the natural substrate.
In WT-AfeETF, we could again identify three phases of reduction confirming the contrasting reactivities of Bf-FAD and ET-FAD. However, the H290FY279I variant displayed novel behaviour, producing a second ASQ species identified by increased absorbance at 374 nm and decreased absorbance at 454 nm, but distinguished by a sharp feature at 400 nm (best seen in the inset to panel 7B, spring green curve). This formed as part of the later population of ASQ and persisted until the last stages of full reduction (Fig. 7B). Indeed, it has been assigned to BfASQ in ETF lacking ET-FAD.47,51
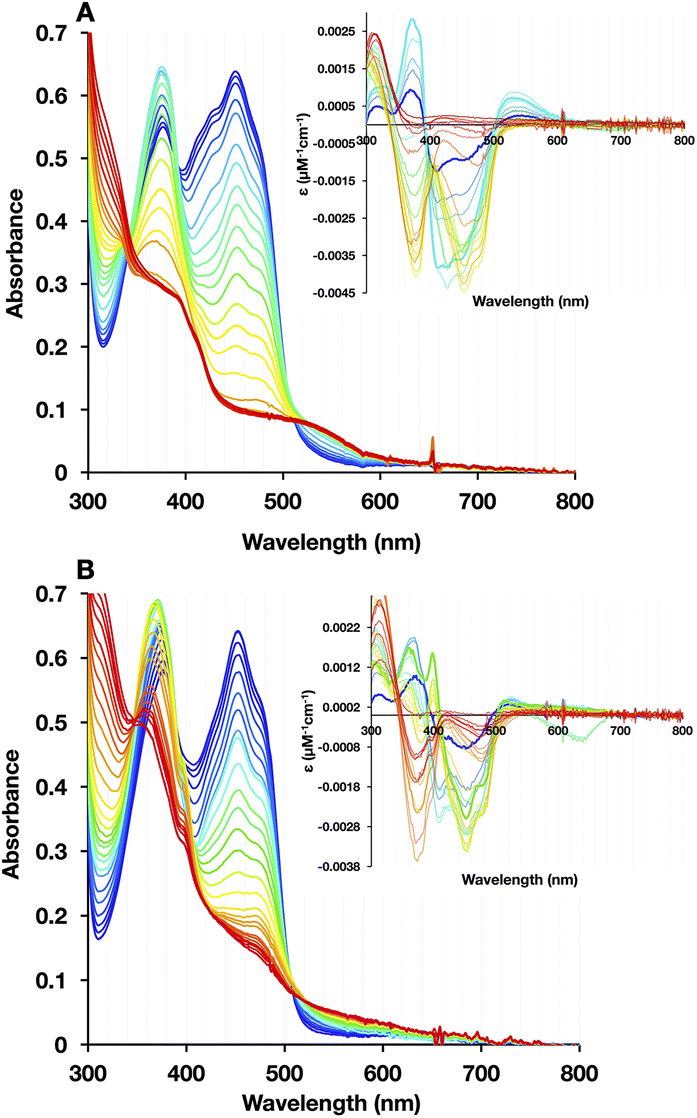 | ||
| Fig. 7 Reductive titrations with dithionite revealing the sequence of events and flavin reactivities. (A) WT and (B) H290FY279I. 20 μM WT or H290FY279I in 50 mM PO4 pH 7.0 was reduced by stepwise addition of dithionite. The visible spectra recorded during reduction of the WT change in the familiar three phases. However in the case of the variant, an additional spectral feature at 400 nm appears in the second half of the reduction before disappearing at the end. The presence of this sharp hook-like feature at 400 nm has been attributed to BfASQ in Bf ETF lacking ET-FAD. | ||
The formation of BfASQ is a distinction from the WT, so we tested the generality of the result with another 1e− donor: Ti2+ citrate. Because the formation of BfASQ has been attributed to ETFs depleted of ET-FAD, the integrity of the reduced H290FY279I variant was assessed after full reduction. The reduced ETF was passed through centrifugal filters to remove any unbound FAD from the sample. The resulting ETF had a diminished FAD:ETF stoichiometry, as low as 1 (see Table S3†), consistent with a much weaker OX spectrum for the recovered H290FY279I (Fig. 8B). The retained flavin is inferred to be Bf-FAD based on the shape of the spectrum, with its broader dip between band I and band II and the vibrational structure on band I.26 Thus, our data show that ET-FAD dissociated upon reduction, so the observed ASQ of Bf-FAD derives from ETF retaining only Bf-FAD.
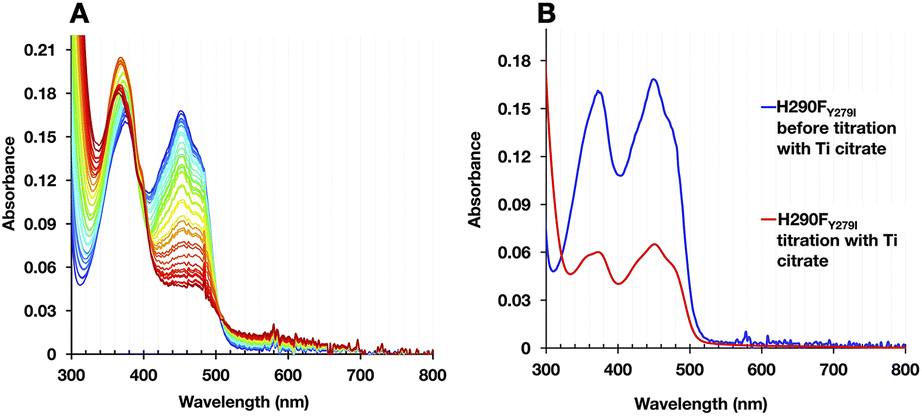 | ||
| Fig. 8 Reductive titration of H290FY279I with Ti2+ citrate. (A) Accumulation of Bf-FAD ASQ during reductive titration with Ti2+ citrate and (B) evidence of flavin depletion in the course of reductive titration. 10.7 μM H290FY279I ETF in 50 mM PO4 pH 7.0 was reduced by stepwise addition of Ti2+ citrate. Optical spectra recorded during the reductive titration revealed formation of Bf-FAD ASQ characterized by a sharp feature (shoulder) at 400 nm. Spectra of the variant ETF demonstrated overall depletion of FAD during reduction based on the amplitude, but retention of Bf-FAD, as demonstrated by retention of vibrational features in band I attributable to Bf-FAD in spectra recorded after separation of the ETF from the released FAD followed by reoxidation. | ||
These observations concur with dissociation of ET-FAD's ASQ state in particular, since flavin loss is not significant when the reductant is NADH, which was shown to produce less ETASQ. Moreover, the loss of ET-FAD from the variant but not the WT ties it to the absence of H290. Thus, the H-bond from H290 seems required to stabilize ASQ in the ET site. Although this finding warns that caution is needed in conducting and interpreting E° determinations, the dissociation of ET-FAD upon reduction is fully consistent with a role for His290 in stabilizing the reduced state(s) of this flavin.
Quantitative assessment of reactivity: E° values
The suppression of ET-flavin modification and the formation of ASQ in H290FY279I could both be explained by diminished stabilization of anionic states of the ET-flavin. However, the emergence of 1e− reactivity for the distant Bf-flavin indicates that our amino acid substitutions or at least the resulting ET-FAD dissociation may also have long-range effects, including on Bf ETF's conformational equilibrium. To guide the formulation of hypotheses, we assessed the magnitudes of energies involved. We quantified the destabilization of ASQ by measuring the E°s of the ET- and Bf-flavin of H290FY279I,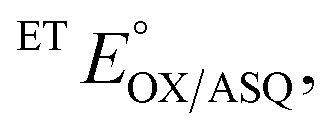
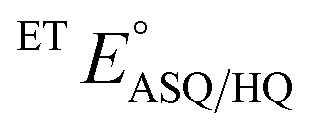 and
and 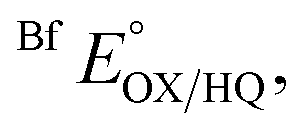 for comparison with those of the WT. Each of
for comparison with those of the WT. Each of 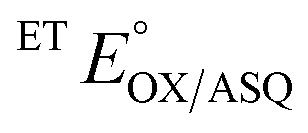 and
and  reports the stability of one state relative to the other, and the maximum population of the ASQ state (vs. the total population) can be calculated from these values as well (see the Discussion).
reports the stability of one state relative to the other, and the maximum population of the ASQ state (vs. the total population) can be calculated from these values as well (see the Discussion).
E°s were determined using the xanthine oxidase system to deliver reducing equivalents slowly and continuously, in the presence of a mediator to hasten equilibration between the ETF and a dye used to report on the ambient potential.44 Because the variant did not display the signature of BfASQ in the course of titration, the data could be interpreted in terms of simple reduction of the flavins without consideration of flavin dissociation. For the ET-flavin, the WT- and H290FY279I ETFs displayed E°s so different that different dyes were required (see the Experimental section). The resulting E°s as determined at pH 7.5 are presented in Table 2 (also see ESI Fig. S1†).
| H290FY279I pH 7.5a (mV) | Literature WTb, adj. to pH 7.5 (mV) | |
|---|---|---|
a All values vs. NHE (normal hydrogen electrode).
b Calculated based on E°'s reported for pH 7.0. For reduction by 1e− only for 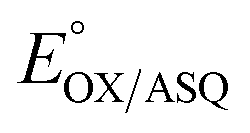 (no change), for 1H+/1e− for (no change), for 1H+/1e− for 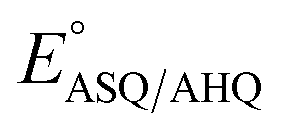 (−60 mV per pH unit), or for 1H+/2e− for (−60 mV per pH unit), or for 1H+/2e− for 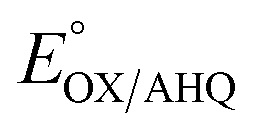 (−30 mV per pH unit).
c All measurements were made in duplicate when results were within 5 mV and in triplicate when a larger scatter was obtained.
d (−30 mV per pH unit).
c All measurements were made in duplicate when results were within 5 mV and in triplicate when a larger scatter was obtained.
d

|
||
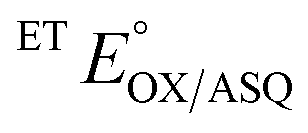
|
−22 ± 0.8c | +134 ± 5 |
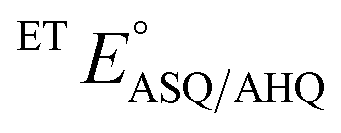
|
−138 ± 2.7 | −66 ± 9 |
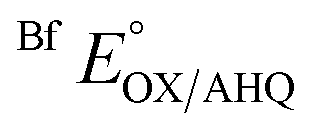
|
−301 ± 0.03 | −264 ± 5 |
| ETΔΕ°d | 116 | 200 |
The  of the variant was 70 mV lower than the corresponding WT value (Table 2) and the variant's
of the variant was 70 mV lower than the corresponding WT value (Table 2) and the variant's 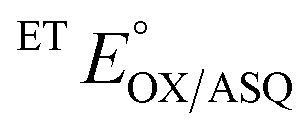 was 150 mV lower than the literature value for the WT, adjusted to pH 7.5.19 Thus, both anionic states are less favourable in H290FY279I than in the WT. Moreover, we can attribute these effects to a single H-bond from H290 to the flavin, since the largest distinction between His and Phe is H-bonding capacity. A 150 mV change in E° corresponds to a H-bond strength of 15 kJ mol−1, which is stronger than average, although not extreme.52 The separation between
was 150 mV lower than the literature value for the WT, adjusted to pH 7.5.19 Thus, both anionic states are less favourable in H290FY279I than in the WT. Moreover, we can attribute these effects to a single H-bond from H290 to the flavin, since the largest distinction between His and Phe is H-bonding capacity. A 150 mV change in E° corresponds to a H-bond strength of 15 kJ mol−1, which is stronger than average, although not extreme.52 The separation between 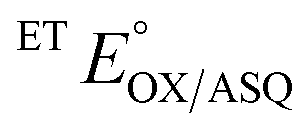 and
and  is 84 mV smaller in the variant than in the WT, resulting in a diminished population of the ASQ state even at the optimal potential, intermediate between
is 84 mV smaller in the variant than in the WT, resulting in a diminished population of the ASQ state even at the optimal potential, intermediate between 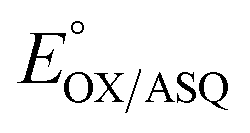 and
and  This is consistent with the greater co-occurrence of 1e− and 2e− reactivity seen for the variant in Fig. 5.
This is consistent with the greater co-occurrence of 1e− and 2e− reactivity seen for the variant in Fig. 5.
Removal of H290 had a much smaller effect on 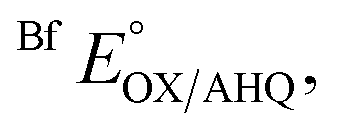 consistent with the locations of the substitutions in the ET site, far from the site of Bf-FAD, and indicating retention of the basic integrity of the ETF over the titration, despite the amino acid substitutions.
consistent with the locations of the substitutions in the ET site, far from the site of Bf-FAD, and indicating retention of the basic integrity of the ETF over the titration, despite the amino acid substitutions.
Suppression of ET-flavin modification upon replacement of H290 and Y279
While oxidized WT-AfeETF's optical spectrum displayed features indicative of 8fF, they were virtually absent from the H290FY279I variant (Fig. 4). This circumstantial evidence suggested that removal of the H-bond from H290 destabilizes the anionic paraquinone methide invoked as an intermediate in 8fF formation since 1959.33–37,53,54 The methide intermediate is deemed to be favoured by displacement of electron density into the N1C2O2 locus, making the 8-methyl protons acidic.37 Meanwhile, the H290 near O2 is positioned to favour such a charge migration by donating a H-bond to the flavin O2.6,28 Therefore, we hypothesized that if a methide intermediate is essential, then H290FY279I should not undergo modification. This was confirmed by comparison of flavin modification yields in WT and H290FY279I.
To maximize the ETF's tendency to form 8fF, samples were held at pH 9.0. To suppress other reactions, they were kept anaerobic in darkness at 4 °C. Aliquots were withdrawn at intervals and diluted in pH 9.0 buffer. Fig. 9 compares the UV/visible spectra collected over regular time intervals, for H290FY279I and the WT.
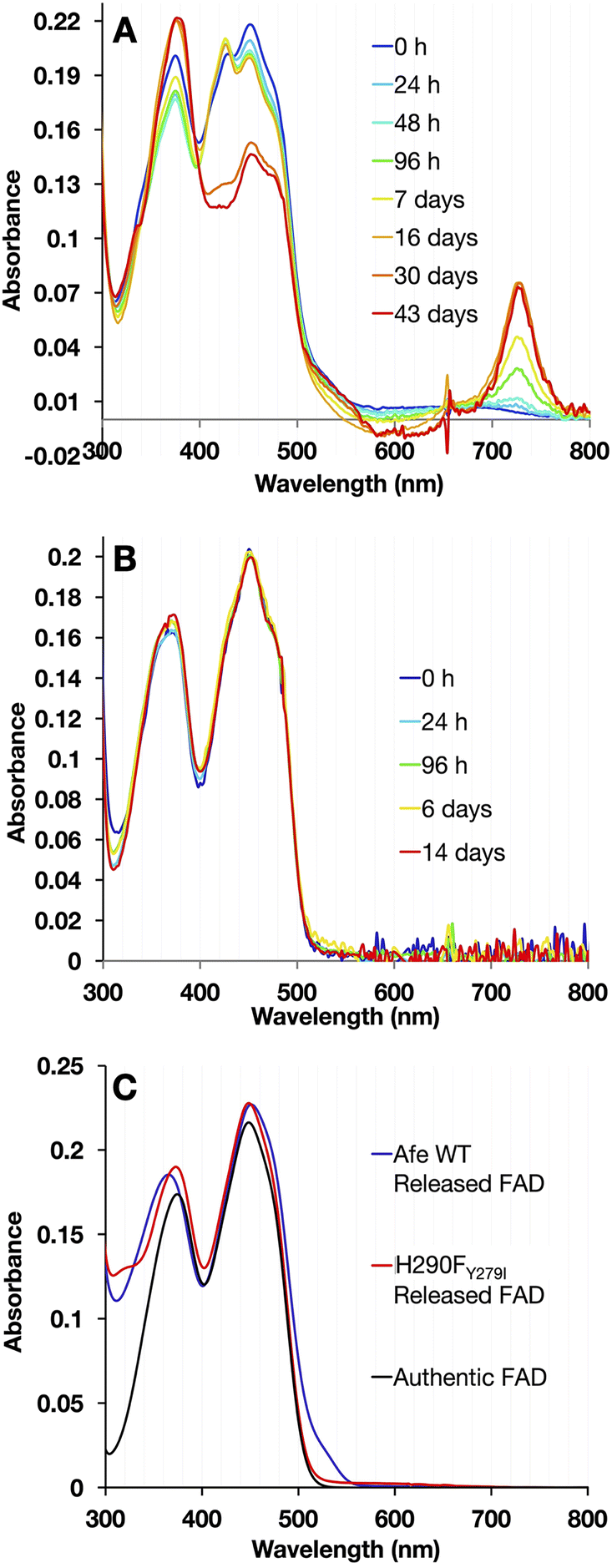 | ||
| Fig. 9 8-Formyl flavin formation at pH 9 in WT- vs. H290FY279I-ETF. Visible spectra of (A) WT-ETF and (B) H290FY279I-ETF at pH 9 were recorded at intervals over a period of 144 h. Visible spectra of the WT demonstrated an increase in absorbance at 424 nm characteristic of 8fF and the presence of NSQ which decreased over time while bands near 530 and 730 nm increased. Meanwhile, the H290FY279I variant remained unmodified and fully oxidized at pH 9. The spectra collected over 384 h for WT were normalized using the isosbestic point at 406 nm and the spectra collected after 30 days and 43 days resembled 8fF ASQ and were normalized using 660 nm. (C) Comparison flavins released from WT- and H290FY279I-ETF after full reaction at pH 9. Flavin released from WT-ETF deviated from authentic FAD in a blue-shifted band II and a prominent shoulder near 530 nm. | ||
ET-FAD in WT-AfeETF changed to 8fF based on the growth in absorbance at 424 nm characteristic of 8fF ASQ, which replaced a weak wide feature from 600–700 nm characteristic of NSQ in the initial spectrum. Eventually, a small amount of 8-amino flavin formed, based on the appearance of a shoulder at 530 nm and a charge transfer band near 730 nm (Fig. 9A).32 A striking drop in the amplitude of band I also signalled reduction of the ET-flavin.32 Meanwhile, the variant remained unmodified and fully oxidized over the same period of time (Fig. 9B).
To distinguish between alterations in the protein environment vs. modification of the flavins themselves, the spectra of released flavins were also compared. Flavins released from WT-ETF after full reaction at pH 9 deviated spectroscopically from authentic FAD as revealed by the blue shift of band II to λmax = 365 nm, whereas the FAD released from H290FY279I resembled authentic FAD (λmax = 374 nm). A short λmax for band II has been assigned to 8fF by Augustin et al.46 The formation of 8fF by WT-ETF was also confirmed by mass spectrometry but was not detected in flavins released from the H290FY279I variant (ESI Fig. S2†). Flavin released from WT-ETF additionally displayed a small long-wavelength shoulder near 530 nm, which has been assigned to 8 amino flavin.32,55
Thus, replacement of H290 by a non-H-bonding side chain not only destabilizes ETASQ, but also impedes flavin modification, consistent with anionic character shared between intermediate(s) in flavin modification and the ASQ state.
Discussion
Contributions of H290 and Y279 to stability and FAD binding
While the possible significance of H290 has been noted before, substitutions at this position have not previously been published, to our knowledge.7,56 We succeeded in replacing it by simultaneously replacing an additional participant in the H-bond chain. The H290F substitution appears in several naturally occurring ETFs, and in them we noted the almost complete conservation of I in the position of the tyrosine (Y279). Thus, it appears that these two residues function together. Indeed, H290FY279I was the one variant studied that retained good FAD binding.To assess the structural and stability roles of H290, we tried replacing it with smaller polar as well as non-polar residues. At our pH of 7.0, we expected that H290 would be neutral, especially as the recipient of an H-bond from nearby Y279 (Fig. 1). However, we tested the possibility that H290 might bear positive charge by replacing it with K, in parallel with substituents that are neutral or negatively charged: A, T, N and E. The H290A and H290T variants showed no expression in the soluble phase, whereas variants H290E, H290K and H290N expressed soluble protein, suggesting a requirement for a polar residue, in the presence of Y279. Even these soluble variants failed to incorporate the ET-FAD, suggesting that H290's H-bond to O2 is significant even in the FAD's OX state. Glutamate (E) could disfavour ET-FAD binding due to repulsion between its expected negative charge and the partial negative charge expected at O2. Lysine (K) should be better able to bind flavin based on its expected positive charge; however, its larger size compared to His could interfere with ET-FAD binding. Asparagine (N) replicates His' polarity in-plane and H-bonding, but Phe (F) replicates His' aromatic ring. Thus, the greater stability and flavin occupancy of ETF containing Phe, vs. Asn, indicate that the special electronic and geometric properties of a π system are more important than H-bonding per se, in this position. Aromatic pi systems' interactions depend on the orientation of the ring, with excess negative charge above and below but electron depletion in the plane of the ring, so the edge of the Phe ring could possibly interact favourably with excess negative charge on O2. The Phe side chain also occupies a similar shape of the void to that of His, raising the importance of packing.
Contributions of H290 to redox tuning and the nature of ET-flavin reactivity
Non-covalent interactions between flavins and their protein environments play crucial roles in tuning flavin reactivities.14,20,23,24,27,46,57 Thus they are critical determinants of the energetics of electron transfer. Moreover, the complementary reactivities of the Bf-FAD and ET-FAD are crucial for electron bifurcation. For the ET-FAD to serve as a 1e− gate, allowing only one electron per NADH to exploit the exergonic path, it must have an unusually stable SQ state. Indeed, the ET-FAD of the ETF of Methylophilus methylotrophus (MmeETF) holds the record for the highest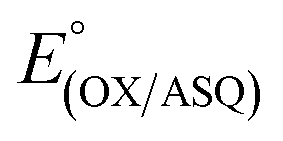 of any flavin, at 150 mV.16 The large gap between it and MmeETF's
of any flavin, at 150 mV.16 The large gap between it and MmeETF's 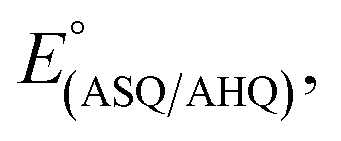
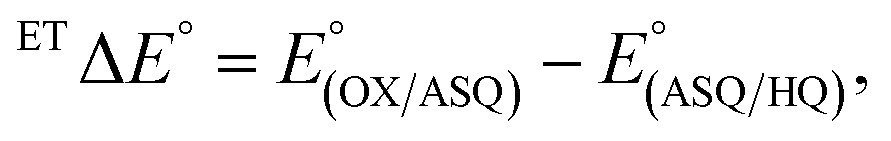 produces a wide potential range in which the ASQ is the most stable state of this flavin.
produces a wide potential range in which the ASQ is the most stable state of this flavin.
In the case of WT-AfeETF,  (at pH 7, Table 3), so the formation constant of ASQ, KSQ,WT = [ASQ]2/[OX][HQ], is 760 (eqn (2a)), which results in maximal fractional accumulation of SQ to fSQ,max = 93% of the population at pH 7 (eqn (3)). Thus, the ET-flavin acts as a 1e− carrier for some 93% of reductions based on thermodynamic tuning alone. In contrast, the ASQ state of Bf-FAD is not significantly populated in our flavin-replete AfeETF preparations. A population <1% corresponds to a KSQ of 0.0004 and BfΔE° more negative than −200 mV, embodying the greater energy required to form SQ than to reduce it further.
(at pH 7, Table 3), so the formation constant of ASQ, KSQ,WT = [ASQ]2/[OX][HQ], is 760 (eqn (2a)), which results in maximal fractional accumulation of SQ to fSQ,max = 93% of the population at pH 7 (eqn (3)). Thus, the ET-flavin acts as a 1e− carrier for some 93% of reductions based on thermodynamic tuning alone. In contrast, the ASQ state of Bf-FAD is not significantly populated in our flavin-replete AfeETF preparations. A population <1% corresponds to a KSQ of 0.0004 and BfΔE° more negative than −200 mV, embodying the greater energy required to form SQ than to reduce it further.
 | (2a) |
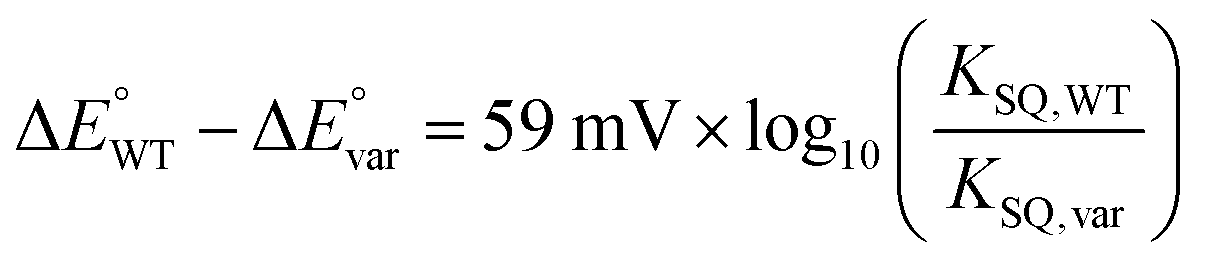 | (2b) |
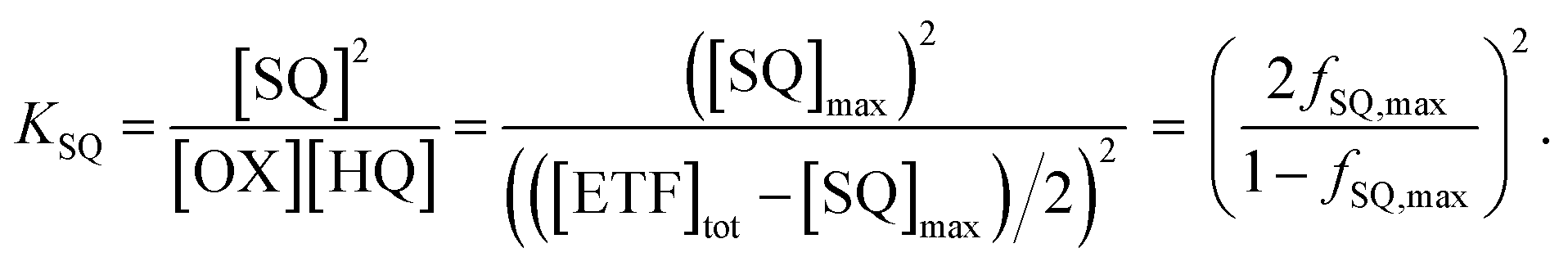 | (3) |
| H290FY279I (mV, pH 7.0)b | WTd (mV, pH 7.0) | Free flavin | |||
|---|---|---|---|---|---|
| ET | Bf | ET | Bf | ||
a All values vs. NHE.
b Calculated for pH 7.0 based on 1e− only for 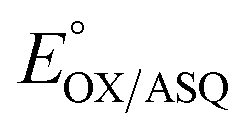 (no change), 1H+/1e− for (no change), 1H+/1e− for  (−60 mV per pH unit), or 1H+/2e− for (−60 mV per pH unit), or 1H+/2e− for  (−30 mV per pH unit).
c All measurements were made in duplicate when results were within 5 mV and in triplicate when a larger scatter was obtained.
d Ref. 19.
e (−30 mV per pH unit).
c All measurements were made in duplicate when results were within 5 mV and in triplicate when a larger scatter was obtained.
d Ref. 19.
e
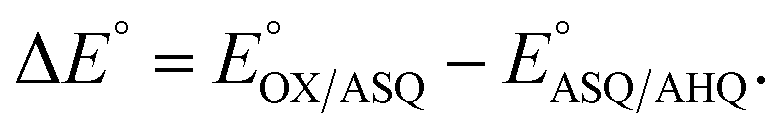
|
|||||

|
−22 ± 0.8c | +134 ± 5 | |||
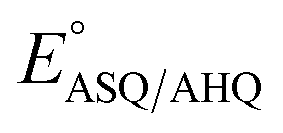
|
−108 | −36 ± 9 | |||
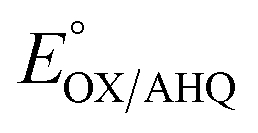
|
−286 | −249 ± 5 | −207 | ||
| ΔE°e | 86 | <−200 | 170 | ≤−200 | −200 |
| K SQ | 29 | 0.0004 | 760 | <0.0004 | 0.0004 |
| Max popASQ | 72% | <1% | 93% | <1% | 1% |
For the H290FY279I variant at pH 7.0, our E° values yield KSQ = 28.6 and maximal fractional accumulation of SQ to 72% of the population. Consideration that 28% of the population of ET-FAD undergoes reduction directly from OX to AHQ, instead of OX to ASQ, it is consistent with the less steep slope of the A370vs. A450 line for the variant (Fig. 5B inset).
If the WT slope of early points in the reduction is taken for OX → ASQ (for ET-FAD) and the slope towards the end of reduction is taken for OX → AHQ (the Bf-FAD in this case), we can calculate their linear combination that yields the observed slope for phase 1 of H290FY279I reduction (note that the early points in reductive titration are those on the right side in the insets of Fig. 5 and the order in which data are acquired is right to left). This exercise suggests that only 40–50% of sites undergo 1e− reduction. Thus, the E° changes are also associated with a significant change in the nature of ET-FAD's reactivity from a 1e− carrier to a 2e− carrier.
The modest shifts in E° moreover confirm the importance of additional interactions. If we assume that H-bond donation from S270 to N5 has a comparable effect on ΔE° and combine that with the 80 mV effect of R253, a site lacking those three interactions would have KSQ = 0.04 and a maximal SQ population on the order of only 10%, providing little electron gating, compared to the 93% observed with all three interactions present.14,26 Thus, we argue that the ET site achieves its unusual 1e− activity by cumulative effects of several weak interactions rather than any one alone.
The use of multiple interactions may imbue the protein with more nuanced control over reactivity. While the elevated E°s of the ET-FAD have been attributed in part to electrostatics between the flavin and nearby R253, this would tend to stabilize the anionic reduced states in general vs. the neutral OX state.16,25,26 Indeed, replacement of the Arg with neutral residues lowered 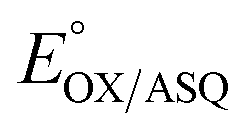 but not
but not 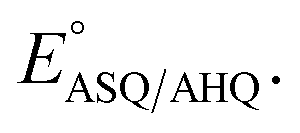 In contrast, H-bonds are more position-specific since they incorporate up to 10% covalent character.58,59 H290's H-bond to O2 has different effects on the reduction of OX to ASQ than it does on reduction of ASQ to HQ. In the former, the flavin accommodates an additional electron and the H-bond to O2 draws excess electron density into the N1–C2O2 locus where electronegative atoms and resonance stabilization are beneficial. However, as part of reduction of ASQ to AHQ a proton is acquired at N5, over-riding the effect of H-bonding at O2 and concentrating excess electron density at N5.28 This is consistent with the smaller shift in
In contrast, H-bonds are more position-specific since they incorporate up to 10% covalent character.58,59 H290's H-bond to O2 has different effects on the reduction of OX to ASQ than it does on reduction of ASQ to HQ. In the former, the flavin accommodates an additional electron and the H-bond to O2 draws excess electron density into the N1–C2O2 locus where electronegative atoms and resonance stabilization are beneficial. However, as part of reduction of ASQ to AHQ a proton is acquired at N5, over-riding the effect of H-bonding at O2 and concentrating excess electron density at N5.28 This is consistent with the smaller shift in 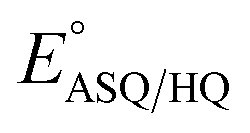 upon replacement of H290.
upon replacement of H290.
Conclusions: a unifying explanation for the stable ASQ and modification at C8m of ET-FAD
We hypothesized that if H290 acts by stabilizing excess electron density in N1–C2O2, then variants lacking H290 should not accumulate chemically modified flavins if the latter form via a paraquinoid methide intermediate. This is because the methide exploits a paraquinoid resonance structure that benefits from stabilization of negative charge at N1–C2O2, like ASQ. This hearkens back to the ideas of Hemmerich and Massey.60 Indeed, our H290FY279I variant does not display the features of 8fF that are evident in the WT at pH 7.0 (Fig. 4). We sought to maximize modification by long incubation at pH 9.0 since the proposed iminoquinone methide intermediate should be favoured at high pH.32,33,35–37,53 Indeed, the spectral features acquired by WT ETF at pH 9.0 are attributable to 8fF while the variant retains the characteristic spectral features of FAD. The flavins released from WT-ETF after two weeks included 8fF, but flavins released from H290FY279I did not, as confirmed by UV-vis spectrometry (Fig. 9C) and mass spectrometry (ESI Fig. S2†). Our spectra further indicate that oxidation of C8m involved migration of electron density into the π system of the flavin, since the HQ state of 8fF forms spontaneously. This too is consistent with the proposed modification via methide (see Raseek et al. and its ESI†).32 However, unlike the WT, the variant H290FY279I did not develop any significant spectral changes over two weeks.
Thus, our data provide evidence for 8fF formation via reductive nucleophilic attack by OH− on C8m in the paraquinoid methide state of flavin that concentrates electron density in N1–C2O2 and depletes the xylene ring, especially C8m. They also explain why some of our ETFs gradually become partially reduced during storage in our glove box, even though we avoid use of H2 in the atmosphere. Iminoquinone methide formation involves abstraction of an acidic proton from C8m by a base. This unusual acidity of a methyl proton is made possible by extensive delocalization of the resulting negative charge towards the flavin's pyrimidine ring. The presence of H290 close to O2 of the N1C2O2 locus thereby makes the C8m proton more acidic. Thus, the same interaction that stabilizes ASQ also favours iminoquinone methide, causing the ET-FAD's 1e− reactivity and modification at C8m to go hand in hand.28 We propose that the constraints of flavin electronics may have required nature to tolerate flavin modification in order to stabilize ASQ and thereby deploy a flavin in the capacity of a 1e− gate, in ETFs.
Data availability
All data required to support the claims made are provided, in the manuscript and its ESI.†Author contributions
DD: conceptualization, methodology, investigation, validation, visualization, writing – original draft, and writing – review & editing. AFM: conceptualization, methodology, investigation, validation, writing – review & editing, supervision, resources, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Debarati Das and Anne-Frances Miller gratefully acknowledge the support of NSF under award CHE 2108134; Dr Kee-Yuen Martin Chow, Department of Molecular and Cellular Biochemistry, University of Kentucky for differential scanning fluorimetry support; Dr Andrew Lemoff, Department of Biochemistry, UT Southwestern Medical Center for mass spectrometry support; Arthur Sebesta, Department of Chemistry and Todd Stone, Physical Plant Division, University of Kentucky, for infrastructural support; Melissa Cowan, University of Kentucky for administrative support; Chandendu Arteev for support with illustrations.References
- J. L. Yuly, C. E. Lubner, P. Zhang, D. N. Beratan and J. W. Peters, Electron bifurcation: progress and grand challenges, Chem. Commun., 2019, 55(79), 11823–11832 RSC.
- G. Herrmann, E. Jayamani, G. Mai and W. Buckel, Energy conservation via electron-transferring flavoprotein in anaerobic bacteria, J. Bacteriol., 2008, 190, 784–791 CrossRef CAS PubMed.
- J. W. Peters, A.-F. Miller, A. K. Jones, P. W. King and M. W. W. Adams, Electron bifurcation, Curr. Opin. Chem. Biol., 2016, 31, 146–152 CrossRef CAS PubMed.
- W. Nitschke and M. J. Russell, Redox bifurcations: Mechanisms and importance to life now, and at its origin, BioEssays, 2012, 34, 106–109 CrossRef CAS PubMed.
- P. Mitchell, The protonmotive Q cycle: a general formulation, FEBS Lett., 1975, 59, 137–139 CrossRef CAS PubMed.
- N. P. Chowdhury, A. M. Mowafy, J. K. Demmer, V. Upadhyay, S. Koelzer, E. Jayamani, J. Kahnt, M. Hornung, U. Demmer, U. Ermler and W. Buckel, Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans, J. Biol. Chem., 2014, 289, 5145–5157 CrossRef CAS PubMed.
- D. L. Roberts, F. E. Frerman and J. J. Kim, Three-dimensional structure of human electron transfer flavoprotein to 2.1-A resolution, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14355–14360 CrossRef CAS PubMed.
- K. Sato, Y. Nishina and K. Shiga, Interaction between NADH and electron-transferring flavoprotein from Megasphaera elsdenii, J. Biochem., 2013, 153, 565–572 CrossRef CAS PubMed.
- E. R. DuPlessis, R. J. Rohlfs, R. Hille and C. Thorpe, Electron-transferring flavoprotein from pig and the methylotrophic bacterium W3A1 contains AMP as well as FAD, Biochem. Mol. Biol. Int., 1994, 32, 195–199 CAS.
- N. P. Chowdhury, J. Kahnt and W. Buckel, Reduction of ferredoxin or oxygen by flavin-based electron bifurcation in Megasphaera elsdenii, FEBS J., 2015, 282, 3149–3160 CrossRef CAS PubMed.
- W. Buckel and R. K. Thauer, Flavin-Based Electron Bifurcation, A New Mechanism of Biological Energy Coupling, Chem. Rev., 2018, 118, 3862–3886 CrossRef CAS PubMed.
- S. G. Mayhew, The Effects of pH and Semiquinone Formation on the Oxidation-Reduction Potentials of Flavin Mononucleotide: A Reappraisal, Eur. J. Biochem., 1999, 265, 698–702 CrossRef CAS PubMed.
- M. Stankovich, in Chemistry and biochemistry of flavoproteins and flavoenzymes, ed. F. Müller, CRC Press, Boca Raton, 1990, vol. 1, pp. 402–425 Search PubMed.
- K.-Y. Yang and R. P. Swenson, Modulation of the redox properties of the flavin cofactor through hydrogen-bonding interactions with the N(5) atom: Role of αSer254 in the electron-transfer flavoprotein from the methylotrophic bacterium W3A1, Biochemistry, 2007, 46, 2289–2297 CrossRef CAS PubMed.
- K. J. Griffin, T. M. Dwyer, M. C. Manning, J. D. Meyer, J. F. Carpenter and F. E. Frerman, alphaT244M mutation affects the redox, kinetic, and in vitro folding properties of Paracoccus denitrificans electron transfer flavoprotein, Biochemistry, 1997, 36, 4194–4202 CrossRef CAS PubMed.
- F. Talfournier, A. W. Munro, J. Basran, M. J. Sutcliffe, S. Daff, S. K. Chapman and N. S. Scrutton, αArg-237 in Methylophilus methylotrophus (sp. W3A1) Electron-transferring Flavoprotein Affords ∼200-Millivolt Stabilization of the FAD Anionic Semiquinone and a Kinetic Block on Full Reduction to the Dihydroquinone, J. Biol. Chem., 2001, 276, 20190–20196 CrossRef CAS PubMed.
- G. J. Schut, N. R. Mohamed-Raseek, M. Tokmina-Lukaszewska, D. E. Mulder, D. M. N. Nguyen, G. L. Lipscomb, J. P. Hoben, A. Patterson, C. E. Lubner, P. W. King, J. W. Peters, B. Bothner, A. F. Miller and M. W. W. Adams, The catalytic mechanism of electron bifurcating electron transfer flavoproteins (ETFs) involves an intermediary complex with NAD+, J. Biol. Chem., 2019, 294, 3271–3283 CrossRef CAS PubMed.
- J. K. Demmer, N. P. Chowdhury, T. Selmer, U. Ermler and W. Buckel, The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-coA dehydrogenase complex from Clostridium difficile, Nat. Commun., 2017, 8, 1577 CrossRef PubMed.
- J. Sucharitakul, S. Buttranon, T. Wongnate, N. P. Chowdhury, M. Prongjit, W. Buckel and P. Chaiyen, Modulations of the reduction potentials of flavin-based electron bifurcation complexes and semiquinone stabilities are key to control directional electron flow, FEBS J., 2021, 288, 1008–1026 CrossRef CAS PubMed.
- L. H. Bradley and R. P. Swenson, Role of Glutamate-59 Hydrogen Bonded to N(3)H of the Flavin Mononucleotide Cofactor in the Modulation of the Redox Potentials of the Clostridium beijerinckii Flavodoxin. Glutamate-59 Is Not Responsible for the pH Dependency but Contributes to the Stabilization of the Flavin Semiquinone, Biochemistry, 1999, 38, 12377–12386 CrossRef CAS PubMed.
- S. Frago, G. Goni, B. Herguedas, J. R. Peregrina, A. Serrano, I. Perez-Dorado, R. Molina, C. Gomez-Moreno, J. A. Hermoso, M. Martinez-Julvez, S. G. Mayhew and M. Medina, Tuning of the FMN binding and oxido-reduction properties by neighboring side chains in Anabaena flavodoxin, Arch. Biochem. Biophys., 2007, 467, 206–217 CrossRef CAS PubMed.
- D. M. Hoover, C. L. Drennan, A. L. Metzger, C. Osborne, C. H. Weber, K. A. Pattridge and M. L. Ludwig, Comparisons of wild-type and mutant flavodoxins from Anacystis nidulans. Structural determinants of the redox potentials, J. Mol. Biol., 1999, 294, 725–743 CrossRef CAS PubMed.
- J. D. Walsh and A.-F. Miller, Flavin reduction potential tuning by substitution and bending, J. Mol. Struct., 2003, 623, 185–195 CrossRef CAS.
- A. Lostao, C. Gomez-Moreno, S. G. Mayhew and J. Sancho, Differential stabilization of the three FMN redox forms by tyrosine 94 and tryptophan 57 in flavodoxin from Anabaena and its influence on the redox potentials, Biochemistry, 1997, 36, 14334–14344 CrossRef CAS PubMed.
- T. M. Dwyer, L. Zhang, M. Muller, F. Marrugo and F. E. Frerman, The functions of the flavin contact residues αArg249 and βTyr16, in human electron transfer flavoprotein, Biochim. Biophys. Acta, 1999, 1433(1–2), 139–152 CrossRef CAS PubMed.
- N. Mohamed-Raseek and A.-F. Miller, Contrasting roles for two conserved arginines: Stabilizing flavin semiquinone or quaternary structure, in bifurcating electron transfer flavoproteins, J. Biol. Chem., 2022, 298, 101733 CrossRef CAS PubMed.
- L. M. Schopfer, M. L. Ludwig and V. Massey, in Flavins and Flavoproteins 1990, ed. B. Curti, S. Ronchi and G. Zanetti, Walter de Güyter, Berlin, 1991, pp. 399–404 Search PubMed.
- M. González-Viegas, R. K. Kar, A. F. Miller and M. A. Mroginski, Noncovalent interactions that tune the reactivities of the flavins in bifurcating electron transferring flavoprotein, J. Biol. Chem., 2023, 299, 104762 CrossRef PubMed.
- H. O'Neill, S. G. Mayhew and G. Butler, Cloning and analysis of the genes for a novel electron-transferring flavoprotein from Megasphaera elsdenii, J. Biol. Chem., 1998, 273, 21015–21024 CrossRef PubMed.
- C. M. Byron, M. T. Stankovich, M. Husain and V. L. Davidson, Unusual Redox Properties of Electron-Transfer Flavoprotein from Methylophilus methylotrophus, Biochemistry, 1989, 28, 8582–8587 CrossRef CAS PubMed.
- T. C. Lehman and C. Thorpe, A new form of mammalian electron transfer flavoprotein, Arch. Biochem. Biophys., 1992, 292, 594–599 CrossRef CAS PubMed.
- N. Mohamed-Raseek, C. van Galen, R. Stanley and A. F. Miller, Unusual reactivity of a flavin in a bifurcating electron-transferring flavoprotein leads to flavin modification and a charge-transfer complex, J. Biol. Chem., 2022, 298, 102606 CrossRef CAS PubMed.
- V. Konjik, S. Brünle, U. Demmer, A. Vanselow, R. Sandhoff, U. Ermler and M. Mack, The Crystal Structure of RosB: Insights into the Reaction Mechanism of the First Member of a Family of Flavodoxin-like Enzymes, Angew. Chem., Int. Ed., 2017, 56, 1146–1151 CrossRef CAS PubMed.
- I. Jhulki, P. K. Chanani, S. H. Abdelwahed and T. P. Begley, A remarkable oxidative cascade that replaces the riboflavin C8 methyl with an amino group during roseoflavin biosynthesis, J. Am. Chem. Soc., 2016, 138, 8324–8327 CrossRef CAS PubMed.
- K. Yorita, T. Matsuoka, H. Masaki and V. Massey, Interaction of two arginine residues in lactate oxidase with the ezyme flavin: Conversion of FMN to 8-formyl-FMN, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13039–13044 CrossRef CAS PubMed.
- D. P. H. M. Heuts, N. S. Scrutton, W. S. McIntire and M. W. Fraaije, What's in a covalent bond?, FEBS J., 2009, 276, 3405–3427 CrossRef CAS PubMed.
- M. Mewies, J. Basran, L. C. Packman, R. Hille and N. Scrutton, S,, Involvement of a Flavin Iminoquinone Methide in the Formation of 6-Hydroxyflavin Mononucleotide in Trimethylamine Dehydrogenase: A Rationale for the Existence of 8R-Methyl and C6-Linked Covalent Flavoproteins, Biochem, 1997, 36, 7162–7168 CrossRef CAS PubMed.
- P. Trickey, M. A. Wagner, M. S. Jorns and F. S. Mathews, Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme, Structure, 1999, 7, 331–345 CrossRef CAS PubMed.
- S. Ghisla and S. G. Mayhew, Identification and properties of 8-hydroxyflavin-adenine dinucleotide in electron transfer flavoprotein from Peptostreptococcus elsdenii, Eur. J. Biochem., 1976, 63, 373–390 CrossRef CAS PubMed.
- K. Huynh and C. L. Partch, Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay, Curr. Protoc. Protein Sci., 2015, 79, 28.9.1–28.9.14, DOI:10.1002/0471140864.ps2809s79.
- E. Haid, P. Lehmann and J. Ziegenhorn, Molar absorptivities of beta-NADH and beta-NAD at 260 nm, Clin. Chem., 1975, 21, 884–887 CrossRef CAS.
- S. G. Mayhew, The Redox Potential of Dithionite and SO−2 from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen plus Hydrogenase, Eur. J. Biochem., 1978, 85, 535–547 CrossRef CAS PubMed.
- L. C. Seefeldt and S. A. Ensign, A Continuous, Spectrophotometric Activity Assay for Nitrogenase Using the Reductant Titanium(III) Citrate, Anal. Biochem., 1994, 221, 379–386 CrossRef CAS PubMed.
- V. Massey, in Flavins and flavoproteins, ed. B. Curti, S. Ronchi and G. Zanetti, Walter de Gruyer, Berlin, 1991, pp. 59–66 Search PubMed.
- A. F. Miller, H. D. Duan, T. A. Varner and N. Mohamed Raseek, Reduction midpoint potentials of bifurcating electron transfer flavoproteins, Methods Enzymol., 2019, 620, 365–398 CAS.
- P. Augustin, M. Toplak, K. Fuchs, E. C. Gerstmann, R. Prassl, A. Winkler and P. Macheroux, Oxidation of the FAD cofactor to the 8-formyl-derivative in human electron-transferring flavoprotein, J. Biol. Chem., 2018, 293, 2829–2840 CrossRef CAS PubMed.
- W. Vigil Jr, J. Tran, D. Niks, G. J. Schut, X. Ge, M. W. W. Adams and R. Hille, The reductive half-reaction of two bifurcating electron-transferring flavoproteins: Evidence for changes in flavin reduction potentials mediated by specific conformational changes, J. Biol. Chem., 2022, 298, 101927 CrossRef PubMed.
- J. Sucharitakul, W. Buckel and P. Chaiyen, Rapid kinetics reveal surprising flavin chemistry in bifurcating electron transfer flavoprotein from Acidaminococcus fermentans, J. Biol. Chem., 2021, 296, 100124 CrossRef CAS PubMed.
- H. D. Duan, C. E. Lubner, M. Tokmina-Lukaszewska, G. H. Gauss, B. Bothner, P. W. King, J. W. Peters and A. F. Miller, Distinct properties underlie flavin-based electron bifurcation in a novel electron transfer flavoprotein FixAB from Rhodopseudomonas palustris, J. Biol. Chem., 2018, 293, 4688–4701 CrossRef CAS PubMed.
- N. Mohamed-Raseek, H. D. Duan, P. Hildebrandt, M. A. Mroginski and A.-F. Miller, Spectroscopic, thermodynamic and computational evidence of the locations of the FADs in the nitrogen fixation-associated electron transfer flavoprotein, Chem. Sci., 2019, 10, 7762–7772 RSC.
- W. Vigil, D. Niks, S. Franz-Badur, N. Chowdhury, W. Buckel and R. Hille, Spectral deconvolution of redox species in the crotonyl-CoA-dependent NADH:ferredoxin oxidoreductase from Megasphaera elsdenii. A flavin-dependent bifurcating enzyme, Arch. Biochem. Biophys., 2021, 701, 108793 CrossRef CAS PubMed.
- A. Fersht, Enzyme Structure and Mechanism, Freeman & Company, London, 2nd edn, 1985 Search PubMed.
- J. M. Robbins, M. G. Souffrant, D. Hamelberg, G. Gadda and A. S. Bommarius, Enzyme-mediated conversion of flavin adenine dinucleotide (FAD) to 8-formyl FAD in formate oxidase results in a modified cofactor with enhanced catalytic properties, Biochemistry, 2017, 56, 3800–3807 CrossRef CAS PubMed.
- P. Hemmerich, B. Prijs and H. Erlenmeyer, Studien in der lumiflavin-reihe V. Spezifische reaktivität 8-ständiger substituenten am isoalloxazin-kern. Flavin-dimere, Helv. Chim. Acta, 1959, 27, 2164–2177 CrossRef.
- P.-S. Song, E. B. Walker, R. D. Vierstra and K. L. Poff, Roseoflavin as a blue light receptor analog: spectroscopic characterization, Photochem. Photobiol., 1980, 32, 393–398 CrossRef CAS.
- H. S. Toogood, D. Leys and N. S. Scrutton, Dynamics driving function − new insights from electron transferring flavoproteins and partner complexes, FEBS J., 2007, 274, 5481–5504 CrossRef CAS PubMed.
- L. H. Bradley and R. P. Swenson, Role of Hydrogen Bonding Interactions to N(3)H of the Flavin Mononucleotide Cofactor in the Modulation of the Redox Potentials of the Clostridium beijerinckii Flavodoxin, Biochemistry, 2001, 40, 8686–8695 CrossRef CAS PubMed.
- H. J. Kulik, N. Luehr, I. S. Ufimtsev and T. J. Martinez, Ab initio quantum chemistry for protein structures, J. Phys. Chem. B, 2012, 116, 12501–12509 CrossRef CAS PubMed.
- A. V. Morozov, T. Kortemme, K. Tsemekhman and D. Baker, Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6946–6951 CrossRef CAS PubMed.
- V. Massey and P. Hemmerich, Active site probes of flavoproteins, Biochem. Soc. Trans., 1980, 8, 246–257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01642d |
| This journal is © The Royal Society of Chemistry 2024 |