
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[(C5H6N2)2H](Sb4F13): a polyfluoroantimonite with a strong second harmonic generation effect†
Jia-Hang
Wu
ab,
Chun-Li
Hu
ac,
Ya-Feng
Li
 b,
Jiang-Gao
Mao
ac and
Fang
Kong
*ac
b,
Jiang-Gao
Mao
ac and
Fang
Kong
*ac
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, P. R. China. E-mail: kongfang@fjirsm.ac.cn
bCollege of Chemistry, Fuzhou University, Fuzhou 350108, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100039, P. R. China
First published on 30th April 2024
Abstract
It is of great difficulty to create a new antimonite with second-harmonic-generation (SHG) intensity larger than 6 times that of KDP. In this study, a polyfluoroantimonite strategy has been proposed to explore fluoroantimonites with large nonlinear optical (NLO) coefficients. Under the cooperation of chemical (highly asymmetric π-conjugated organic amine) and physical (viscous reaction medium ethylene glycol) methods, two novel polyfluoroantimonites, namely, (3PC)2(Sb4F14) and (3AP)2(Sb4F13), have been achieved. Interestingly, these two structures contain two new polyfluoroantimonite groups respectively, an isolated (Sb4F14)2− four-member polyhedral ring and an infinite [Sb4F13]∞− helical chain. More importantly, the polar (3AP)2(Sb4F13) displays a strong SHG intensity of 8.1 × KDP, a large birefringence of 0.258@546 nm and a high laser-induced damage threshold (LIDT) value of 149.7 MW cm−2. Theoretical calculations indicated that its strong SHG effect stems from the synergistic effect of the helical [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cation, with a contribution ratio of 48.93% and 50.77% respectively. This work provides a new approach for the design and synthesis of high-performance fluoroantimonites.
Introduction
Nonlinear optical crystals capable of widening the wavelength range of output radiations have attracted widespread attention. SHG crystals, as the most representative NLO materials, have been applied in fields such as optical communication, laser technology, biomedicine, photonics, and sensor technology.1–9 Fluoroantimonites are considered as a potential candidate for excellent SHG materials. On the one hand, the Sb3+ cation with stereochemically active-lone-pair (SCALP) electrons can undergo second-order Jahn–Teller (SOJT) distortion, resulting in high polarizability.10 On the other hand, fluorine, being the most electronegative, can enlarge the band gaps of target compounds due to the deeper F-2p orbit than O-2p.11–14 Recently, scientists have developed various design strategies to create new SHG fluoroantimonites, mainly including the following: (a) introduction of tetrahedral groups (SO42− and PO43−), which can widen the band gaps of fluoroantimonites, such as CsSbF2SO4 (3.0 × KDP, 4.76 eV) and Rb2Sb(P2O7)F (5.1 × KDP, 4.74 eV);15,16 (b) combination of Sb3+ with delocalized π-conjugated groups such as oxalate and nitrate as in RbSb2(C2O4)F5 (1.3 × KDP) and Rb2SbF3(NO3)2 (2.7 × KDP);17,18 (c) introduction of SOJT distorted d0 transition metal cations (Mo6+, W6+, Nb5+, etc.) such as Cs6Sb4Mo3O5F26 (0.7 × KDP).19 However, the development of new fluoroantimonites with high SHG intensity is still a challenge. The SHG intensities of most reported fluoroantimonites are less than 6 × KDP. Consequently, it is necessary to develop a new strategy to enhance the SHG effects of fluoroantimonites further.It is believed that the condensation of functional building blocks can enhance the density of SHG active groups, thereby impacting the structure and SHG capability of target compound.20–25 This approach has facilitated the development of numerous high-performance polyiodates, such as GdI5O14 (15 × KDP) and [o-C5H4NHOH]2[I7O18(OH)]·3H2O (8.5 × KDP).26,27 If fluoroantimonate groups can be condensed into polyfluoroantimonite groups, it is expected to obtain new NLO materials with strong SHG effects in polyfluoroantimonite system. However, research on polyfluoroantimonites is very rare, especially for the NLO material, which can be attributed to the following two reasons: (i) the synthetic conditions for polyfluoroantimonites are rather complicated and still unclear. (ii) The bond energy of the Sb–F bond is lower than that of the Sb–O bond, which makes the polymerization of fluoroantimonite groups difficult compared to oxysalts.28,29
In order to explore new fluoroantimonites with strong SHG effects, we have dedicated our efforts to polyfluoroantimonite system, and found a universal synthesis method for polyfluoroantimonites, which involves using organic amine groups (chemical method) and enhancing the viscosity of the reaction media (physical method). Highly asymmetric organic amine groups may not only facilitate the polymerization of fluoroantimonite groups during their protonation process, but also trigger the formation of noncentrosymmetric (NCS) structures. Furthermore, the viscous reaction medium can reduce the migration rate of fluoroantimonite groups, which can also promote the combination of fluoroantimonate groups to form new polyfluoroantimonite anions. Two organic amine cations, namely, 3-pyridinecarboxamide [3PC+, (C6H7N2O)+] and 3-aminopyridine [3AP+, (C5H7N2)+], were identified as the highly asymmetric NLO active groups through theoretical calculations.30–32 The 3PC+ and 3AP+ cations feature significant polarizability anisotropy (δ) and hyperpolarizability (β), surpassing most of the inorganic or organic π-conjugated groups, such as B3O63−, (C3N3O3)3−, (C5H6NO)+, and (C4H6N3)+ (Fig. 1). The selection of viscous solvents is also very important. For instance, phosphoric acid exhibits a propensity to selectively bind with organic ligands, leading to the formation of by-products during the reaction process, and highly viscous glycerol is not conducive to crystallization.33–35 Finally, ethylene glycol, a viscous and neutral organic solvent, was selected as the reaction medium.
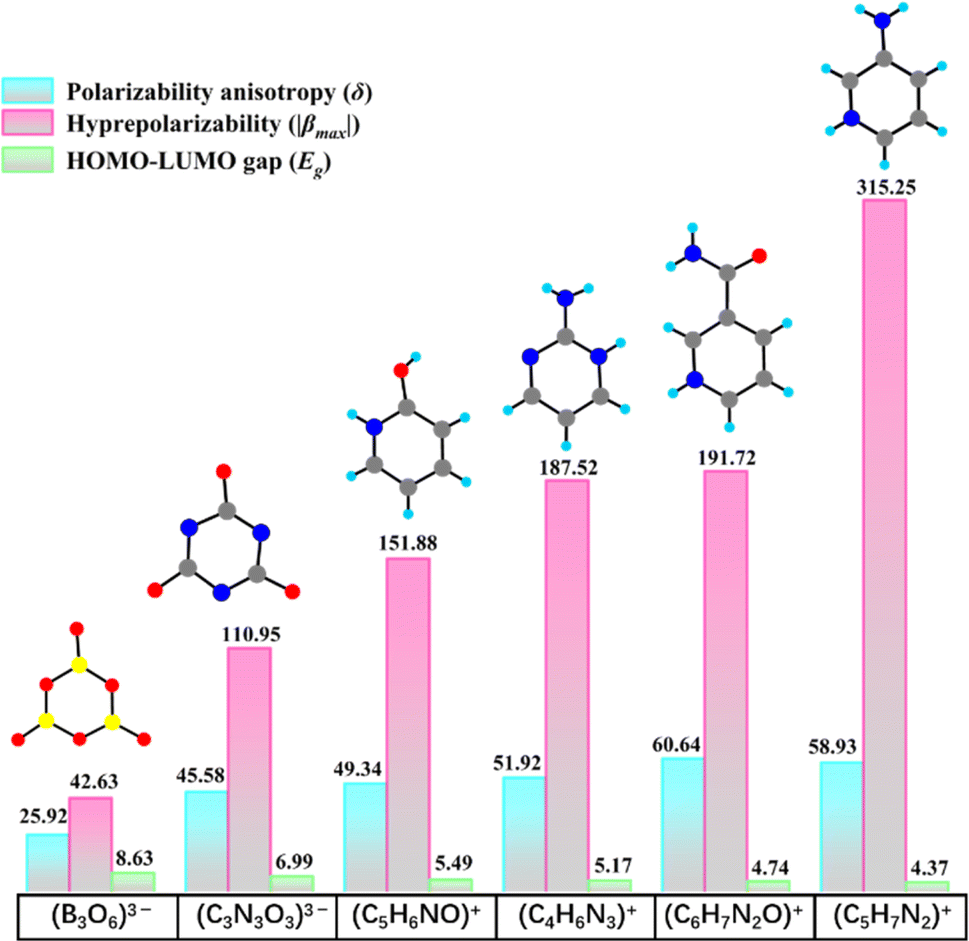 | ||
| Fig. 1 Geometry and key NLO parameters of the conventional functional groups, the 3PC+ cation and the 3AP+ cation. | ||
Guided by the above method, an appropriate amount of hydrofluoric acid was added dropwise into a mixture of organic amines (3PC and 3AP) and SbF3 to protonate the amines, and ethylene glycol was used to enhance the viscosity of the reaction medium. Two novel organic–inorganic hybrid polyfluoroantimonites, namely, (3PC)2(Sb4F14) and (3AP)2(Sb4F13), have been successfully synthesized by a facile solvent evaporation method. (3AP)2(Sb4F13) crystallizes in a NCS space group because the 3AP+ cation features a larger hyperpolarizability than the 3PC+ cation. Interestingly, (3AP)2(Sb4F13) displays a unique [Sb4F13]∞− helical chain, a strong SHG effect (8.1 × KDP) under 1064 nm laser radiation, a large birefringence (0.258 @ 546 nm) and a high LIDT value (149.7 MW cm−2). Herein, we report the syntheses, structural analyses, optical properties and first-principles studies of (3PC)2(Sb4F14) and (3AP)2(Sb4F13).
Results and discussion
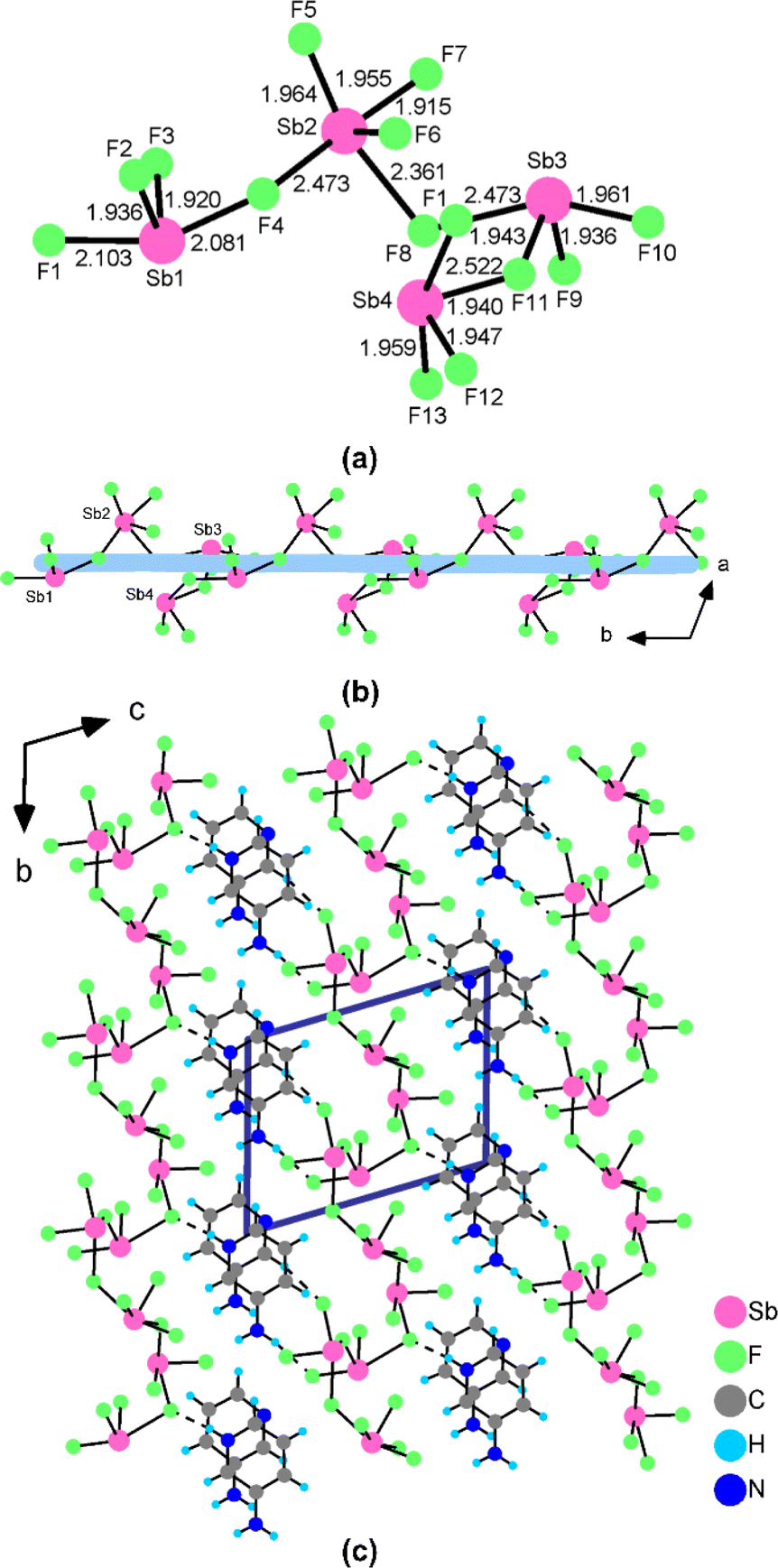
Single crystals of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) were synthesized using a facile solvent evaporation method (Fig. S1†). The detailed crystallographic data for both structures are given in Table S1.† The atomic coordinates and bond lengths are listed in Tables S2 and S3.† The presence of Sb, F, C and N elements was confirmed by field-emission scanning electron microscopy (FESEM) analyses (Fig. S2†). Powder XRD analyses confirmed the purity of the products (Fig. S3†).(3PC)2(Sb4F14) crystallizes in the triclinic crystal system with the CS space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2). There are one (Sb2F7)− anion and one 3PC+ cation in its asymmetric unit (Table S2†). Sb(1) and Sb(2) are coordinated to four and five fluorine atoms, respectively, forming the SbF4 seesaw and SbF5 square pyramid, with lone-pair electrons located in the open sites. The Sb–F bond lengths range from 1.927(3) to 2.521(3) Å (Table S3†). Two Sb(1)F4 and two Sb(2)F5 groups further linked to a (Sb4F14)2− four-member polyhedral ring (4-MPR) via corner-sharing of F(1) and F(4) atoms (Fig. 2a). The bond-valence-sum results show that the bond valences of Sb(1) and Sb(2) are 3.093 and 3.181, respectively, suggesting their oxidation states of +3. As shown in Fig. 2b, the (Sb4F14)2− anionic clusters, separated by the 3PC+ cations, were stacking linearly along the c-axis to form the structure of (3PC)2(Sb4F14). The 3PC+ cations were connected with (Sb4F14)2− anion clusters through hydrogen bonds with hydrogen bond lengths in the range of 3.108(6)–3.229(5) Å (Table S4†). The adjacent 3PC+ cationic pyrimidine rings are parallel to each other, with an inter-ring distance of 3.323 Å (Fig. S4a†).
(No. 2). There are one (Sb2F7)− anion and one 3PC+ cation in its asymmetric unit (Table S2†). Sb(1) and Sb(2) are coordinated to four and five fluorine atoms, respectively, forming the SbF4 seesaw and SbF5 square pyramid, with lone-pair electrons located in the open sites. The Sb–F bond lengths range from 1.927(3) to 2.521(3) Å (Table S3†). Two Sb(1)F4 and two Sb(2)F5 groups further linked to a (Sb4F14)2− four-member polyhedral ring (4-MPR) via corner-sharing of F(1) and F(4) atoms (Fig. 2a). The bond-valence-sum results show that the bond valences of Sb(1) and Sb(2) are 3.093 and 3.181, respectively, suggesting their oxidation states of +3. As shown in Fig. 2b, the (Sb4F14)2− anionic clusters, separated by the 3PC+ cations, were stacking linearly along the c-axis to form the structure of (3PC)2(Sb4F14). The 3PC+ cations were connected with (Sb4F14)2− anion clusters through hydrogen bonds with hydrogen bond lengths in the range of 3.108(6)–3.229(5) Å (Table S4†). The adjacent 3PC+ cationic pyrimidine rings are parallel to each other, with an inter-ring distance of 3.323 Å (Fig. S4a†).
 | ||
| Fig. 2 The configuration of the (Sb4F14)2− 4-MPR and the Sb–F bond lengths (Å) (a) and the crystal structure of (3PC)2(Sb4F14) (b). | ||
When the 3PC+ cation was replaced by the 3AP+ cation, a NCS polyfluoroantimonite, namely, (3AP)2(Sb4F13), was obtained. (3AP)2(Sb4F13) crystallizes in a chiral triclinic space group P1 (No. 1) and its asymmetric unit is composed of one 3AP+ cation, one 3AP molecule and one (Sb4F13)– anion (Table S2†). Sb(1), Sb(3) and Sb(4) adopt the SbF4 seesaw configuration, whereas Sb(2) is in the SbF5 square pyramid configuration. The Sb–F bond distances are in the range of 1.915(5)–2.522(6) Å (Table S3†). The calculated total bond valences for Sb(1), Sb(2), Sb(3) and Sb(4) are 3.045, 3.104, 2.857 and 2.818, respectively, indicating that their oxidation states are all +3. Its structure features a unique [Sb4F13]∞− helical polyfluoroantimonite chain along the b axis with 3AP+ cations and 3AP molecules located at the inter-chain space to keep charge balance (Fig. 3c). Sb(1)F4, Sb(2)F5, Sb(3)F4 and Sb(4)F4 groups are interconnected by corner-sharing [F(4), F(8) and F(11)] to form a (Sb4F14)2− tetramer (Fig. 3a). Neighbouring (Sb4F14)2− tetramers are further interconnected into a unique [Sb4F13]∞− anionic chain along the b axis via Sb(1)–F(10)–Sb(4) bridges (Fig. 3b). The 3AP+ cations are linked with the [Sb4F13]∞− helical polyfluoroantimonite chains through hydrogen bonds with the hydrogen bond lengths in the range of 2.697(12)–3.080(15) Å (Table S4†). There is also π–π interaction between neighbouring pyridine rings with a dihedral angle of 2.257° and inter-ring distances ranging from 3.837 to 4.001 Å (Fig. S4b†). The dipole moment of the Sb4F14 tetramer in (3AP)2(Sb4F13) was calculated. The local dipole moments have the values of 8.988–19.160 D for SbF4 units, and 8.719 D for SbF5 units. The local dipole moment of the Sb4F14 tetramer is 18.558 D (Table S5†), implying its polar nature.
 | ||
| Fig. 3 The configuration of the (Sb4F14)2− tetramer and the Sb–F bond lengths (Å) (a), the [Sb4F13]∞− helical polyfluoroantimonite chain (b) and the crystal structure of (3AP)2(Sb4F13) (c). | ||
Benefitting from the viscous reaction media and the organic amine groups, both (3PC)2(Sb4F14) and (3AP)2(Sb4F13) exhibit novel polyfluoroantimonite structures formed by the condensation of the SbF4 seesaw and SbF5 square pyramid (Fig. S5†). These two types of polyfluoroantimonite groups have not been reported previously. For (3PC)2(Sb4F14) and (3AP)2(Sb4F13), the Sb–F bonds of the Sb–F–Sb bridges are 2.074(3)–2.521(3) and 1.940(6)–2.522(6) Å, respectively, which are longer than their terminal Sb–F bonds [1.900(3)–1.940(3) and 1.915(5)–1.964(6) Å]. These values are comparable with those of polyfluoroantimonite crystals reported before.29
The crystal structures of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) undergo a transition from CS to NCS arrangements. To comprehend the reasons behind this transition, a detailed structural comparison has been conducted between the two compounds. Specifically, the basic building units in these two compounds are similar, including the condensed fluoroantimonite groups and highly asymmetric π-conjugated organic amine cations. The difference lies in the arrangement of organic amine cations and the polymerization modes of fluoroantimonite groups. In comparison to the antiparallel arrangement of 3PC+ cations, the 3AP+ cations and 3AP molecules exhibit a parallel pairing pattern (Fig. 4a and b), favouring the formation of NCS structures. It is worth noting that this parallel pairing pattern is exceptionally rare in organic amines. Furthermore, there have been significant changes in the polymerization modes of fluoroantimonite groups. The (Sb4F14)2− tetramer in (3PC)2(Sb4F14) appears as a 4-MPR configuration, while the tetramer in (3AP)2(Sb4F13) features an open semi-circular configuration (Fig. 4c and d). We calculated the hyperpolarizability of these two condensed Sb4F14 groups. The hyperpolarizability of the semi-circular Sb4F14 tetramer (β = 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 047) is 38 times more than that of the Sb4F14 4-MPR (β = 1778).36 Therefore, the polymerization mode of the fluoroantimonite groups is also crucial. The hyperpolarizabilities of closed cyclic clusters are often small due to the opposite arrangements of the functional groups within the rings.
047) is 38 times more than that of the Sb4F14 4-MPR (β = 1778).36 Therefore, the polymerization mode of the fluoroantimonite groups is also crucial. The hyperpolarizabilities of closed cyclic clusters are often small due to the opposite arrangements of the functional groups within the rings.
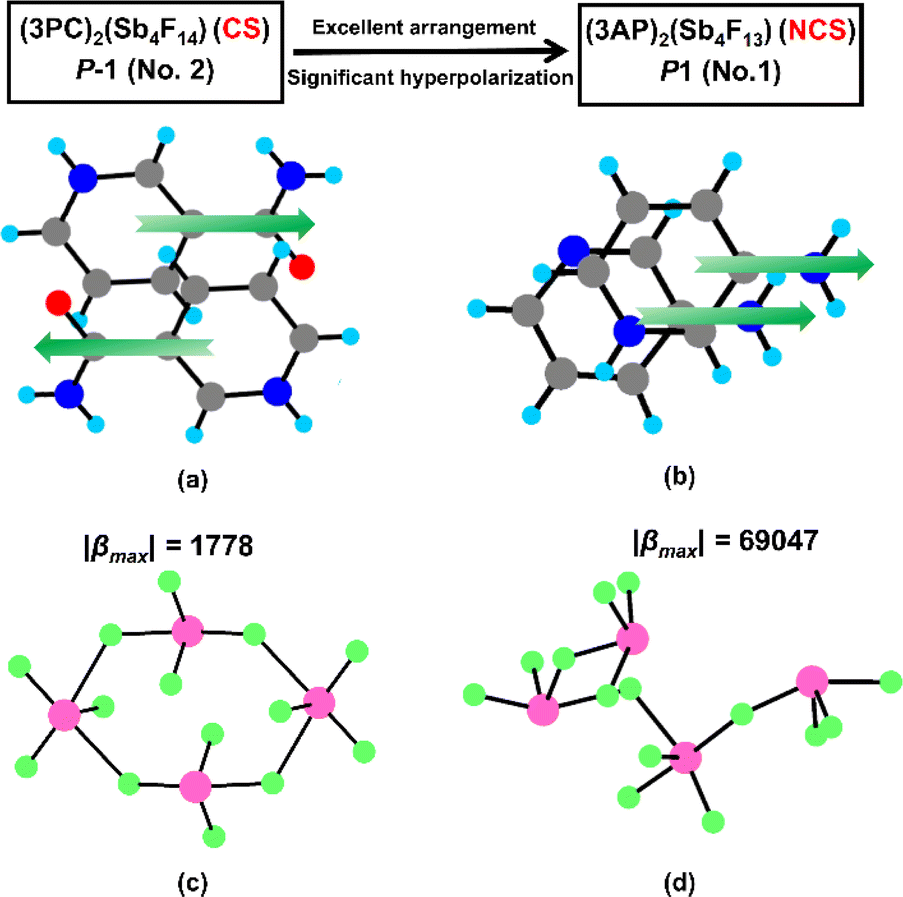 | ||
| Fig. 4 The arrangement modes of 3PC+ (a) and 3AP+ (b) cations, and the Sb4F14 tetramers in (3PC)2(Sb4F14) (c) and (3AP)2(Sb4F13) (d). | ||
Thermogravimetric analyses of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) were performed in the temperature range of 20–800 °C under a nitrogen atmosphere. (3PC)2(Sb4F14) and (3AP)2(Sb4F13) can stabilize to 134 and 153 °C, respectively (Fig. S6†). The NCS polyfluoroantimonite, (3AP)2(Sb4F13), underwent complete decomposition in the temperature range of 153 to 600 °C (Fig. S6†). Compared to other inorganic–organic hybrid NLO crystals, such as α- and β-(C4H5N2O)(IO3)·HIO3 (120 °C), (C9H14N)SbCl4 (122 °C) and KLi(HC3N3O3)·2H2O (125 °C), (3AP)2(S4F13) exhibits better thermal stability.37–39
The infrared (IR) spectra of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) were analysed at room temperature in the wavelength range of 4000–400 cm−1. Fig. S7† displays the IR spectra of the two compounds, and the corresponding assignments of the IR absorption peaks can be found in Table S6.† The absorption peaks around 3460 and 3250 cm−1 can be attributed to the N–H vibrations. The peaks observed at 3070–2850 cm−1 and 1050–650 cm−1 correspond to the C–H vibrations. The bands at 1690–1380 cm−1 and 1350–1110 cm−1 are assigned to C–C vibrations and C–N vibrations, respectively. The sharp absorption peaks in the range of 600–450 cm−1 can be attributed to the Sb–F vibrations. The above assignments are consistent with previously reported compounds.40–43
The UV-vis-NIR diffuse reflectance spectra reveal that the cutoff edge of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) is 272 and 313 nm, respectively. Their experimental optical band gaps were estimated to be 3.71 and 3.05 eV (Fig. S8†). Notably, the NCS (3AP)2(Sb4F13) possesses a larger bandgap in comparison with the 1.97 eV of the 3AP molecule (Fig. 5). This phenomenon can be attributed to the highly electronegative fluorine anions. Compared with C-2p or N-2p states, the F-2p orbitals are usually located at deeper energy levels, which induces the high level valence band to shift toward lower energy, resulting in a wide band gap.44,45 Under the excitation at 330 nm, (3AP)2(Sb4F13) exhibits a blue emission band with a peak at 400 nm (Fig. S9†), but its photoluminescence quantum yield is less than 1%. A large red shift is observed compared to the neutral 3AP, which is likely because of the protonation of the organic molecule and the influence of [Sb4F13]∞− on its highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).46,47
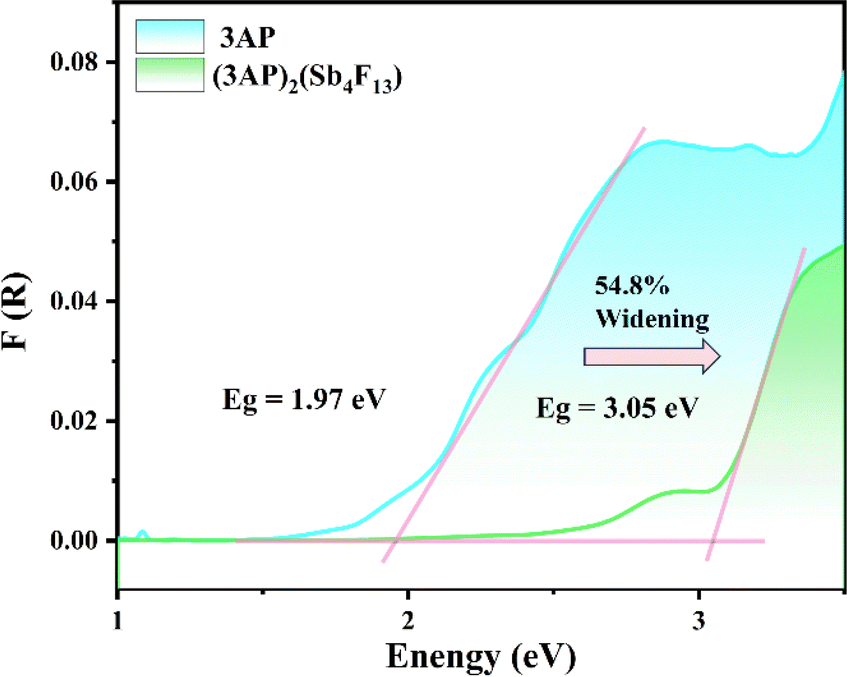 | ||
| Fig. 5 The experimental bandgap spectra of 3AP and (3AP)2(Sb4F13). | ||
The birefringence of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) was determined at λ = 546 nm using a polarized microscope (ZEISS AxioScope. A1).48 Both crystals can achieve complete extinction under ortho-polarized light (Fig. S10 and S11†). The optical path differences of the two samples were measured to be 9.591 and 7.323 μm with the thicknesses of 64.88 and 28.39 μm (Fig. S10c and S11c†), respectively. According to the formula R = |ne – no| × d = Δn × d, the experimental birefringences of (3PC)2(Sb4F14) and (3AP)2(Sb4F13) at 546 nm were measured to be 0.148 and 0.258, respectively. Due to the SCALP electrons of Sb3+ and the large anisotropy of organic amine cations, they both exhibit high birefringence. In particular, the birefringence of (3AP)2(Sb4F13) exceeds those of all inorganic fluoroantimonites, such as K2SbMoO2F7 (0.220@550 nm), (NH4)3SbF3(NO3)3 (0.164@546 nm) and K2SbP2O7F (0.157@546 nm).49–51
Under 1064 nm laser irradiation, powder SHG measurement was performed on (3AP)2(Sb4F13) using KDP (particle size 210–300 μm) as a reference. As shown in Fig. 6a, (3AP)2(Sb4F13) exhibits a strong SHG intensity of 8.1 × KDP. Furthermore, it can realize phase matching at 1064 nm (Fig. 6b). In terms of structure–property relationships, the excellent SHG property of (3AP)2(Sb4F13) can be ascribed to its polar structure, characterized by the unique [Sb4F13]∞− polyfluoroantimonite chain and the highly asymmetric organic amine groups 3AP. Note that the SHG intensity of (3AP)2(Sb4F13) is larger than those of most of the reported fluoroantimonites (Table S7†). To evaluate the comprehensive performance of (3AP)2(Sb4F13), we further compared (3AP)2Sb4F13 with other inorganic–organic hybrid Sb(III)-based compounds in terms of SHG intensity, band gap, and thermal stability (Table S8†). As shown in Fig. 7, compared with representative compounds such as (C9H14N)SbCl4, (C9H26N3)SbCl6 and L-H2his·SbBr5·H2O, (3AP)2(Sb4F13) shows great comprehensive performance.38,52,53
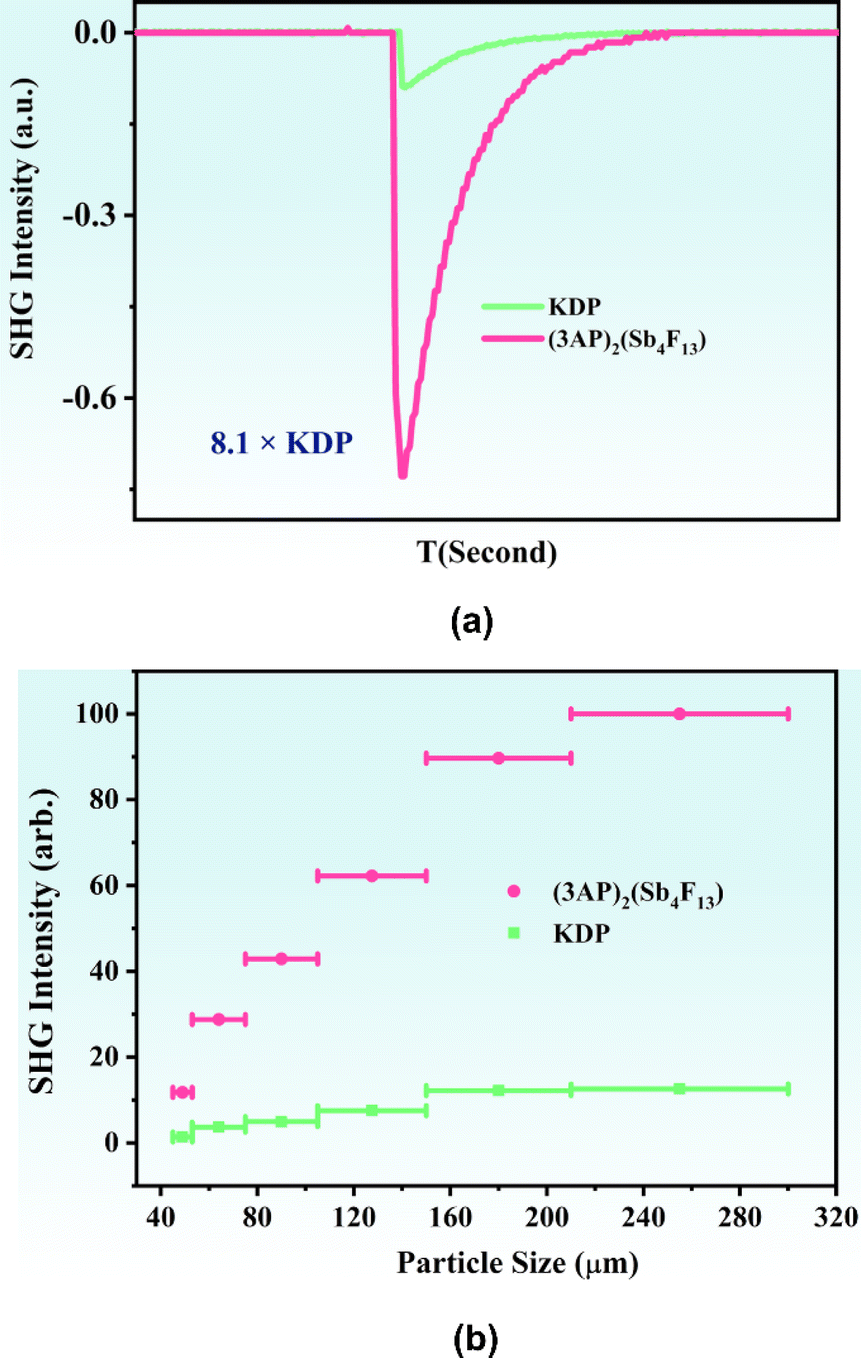 | ||
| Fig. 6 Oscilloscope traces of the SHG signals for (3AP)2(Sb4F13) (150–210 μm) (a) and the SHG intensity vs. particle size of (3AP)2(Sb4F13) under 1064 nm laser irradiation (b). | ||
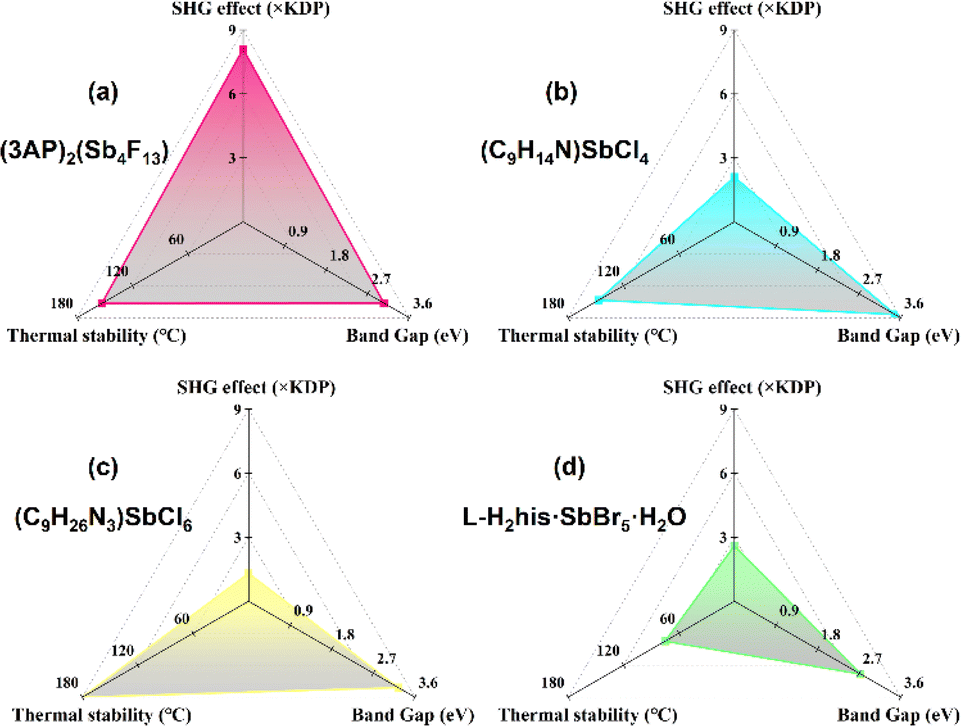 | ||
| Fig. 7 The comparison of the SHG effect, band gap and thermal stability between (3AP)2(Sb4F13) (a) and other representative inorganic–organic hybrid Sb(III)-based compounds: (b) (C9H14N)SbCl4, (c) (C9H26N3)SbCl6 and (d) L-H2his·SbBr5·H2O. | ||
Based on the reported powder method, we conducted a preliminary estimation of the LIDT value for (3AP)2(Sb4F13) using sieved crystals.54 The powder LIDT of (3AP)2(Sb4F13) and AGS was assessed with a Q-switched pulse laser featuring a flat-top laser beam distribution, yielding values of 149.7 and 4.0 MW cm−2, respectively. It suggests that the LIDT of (3AP)2(Sb4F13) is approximately 37.5 times that of AGS. The powder LIDT of (3AP)2(Sb4F13) is comparable with that of NaLu(SeO3)2, despite the substantial difference in their band gaps (3.05 vs. 5.30 eV).55
To gain further insights into the structure–property relationships of (3AP)2(Sb4F13) and (3PC)2(Sb4F14), theoretical calculations were carried out using density functional theory (DFT). The calculation results along the high symmetric points in the first Brillouin region revealed that (3AP)2(Sb4F13) is a direct bandgap compound with a bandgap of 2.28 eV (Fig. 8a), whereas (3PC)2(Sb4F14) is an indirect bandgap compound with a bandgap of 2.47 eV (Fig. S12a†). The calculated band gaps of (3AP)2(Sb4F13) and (3PC)2(Sb4F14) are smaller than the experimental values (3.05 and 3.71 eV, respectively), which can be attributed to the limitation of the GGA method. Accordingly, in order to accurately analyse the optical properties of (3AP)2(Sb4F13) and (3PC)2(Sb4F14), the scissor operators of 0.77 and 1.24 eV were adopted, respectively.
 | ||
| Fig. 8 Calculated band structure (a), total and partial densities of states (b), birefringence (c), electron density difference maps (d) and SHG density of d22 in the VB (e) and the CB (f) for (3AP)2(Sb4F13). | ||
The total and partial density of states of (3AP)2(Sb4F13) and (3PC)2(Sb4F14) are shown in Fig. 8b and S12b.† Since the optical properties of the compounds were mainly determined by the states near the Fermi level, we analyzed the top of the valence band (VB) and the bottom of the conduction band (CB). The VB near the Fermi level of (3AP)2(Sb4F13) is primarily composed of C-2p, and N-2p states, while the CB is mainly attributed to C-2p and N-2p states (Fig. 8b). Therefore, the band gap of (3AP)2(Sb4F13) is predominantly determined by C and N atoms. For (3PC)2(Sb4F14), the top of the VB is mainly contributed by O-2p orbitals, whereas the bottom of the CB is occupied by C-2p and N-2p orbitals (Fig. S12b†), indicating that the bandgap of (3PC)2(Sb4F14) is primarily dominated by O, C, and N atoms.
The refractive index dispersion curves depicted in Fig. 8c and S12c† demonstrate that (3AP)2(Sb4F13) and (3PC)2(Sb4F14) exhibit strong anisotropy. For (3AP)2(Sb4F13), the trend of optical anisotropy is n010 > n001 > n100, and the derived birefringence (Δncal.) is 0.267@546 nm (Fig. 8b), in good agreement with the experimental value (0.258@546 nm). The calculated birefringence of (3PC)2(Sb4F14) at 546 nm is 0.154, which also closely matches the experimental value (0.149@546 nm). The electron differential density (EDD) maps of (3AP)2(Sb4F13) and (3PC)2(Sb4F14) indicate the strong anisotropy of π-conjugated organic amine cations and the highly stereochemically active Sb3+ (Fig. 8d and S12d†), which is beneficial for generating the large birefringence. In addition, the π–π stacking of organic amine cations in these two compounds also facilitates the enhancement of their birefringence.
The second-order nonlinear optical susceptibilities of (3AP)2(Sb4F13) were also evaluated. With the constraints of Kleinman's symmetry and the space group (P1), its SHG tensor d22 gives the largest value (3.69 pm V−1), which agrees well with the experimental value. Furthermore, to precisely reveal the nonlinear origin of (3AP)2(S4F13), the SHG-weighted electron density (SHG density for short) of d22 was analyzed (Fig. 8e and f). Specifically, the SHG effect in the VB originates primarily from Sb-5s5p orbitals, the nonbonding N-2p orbitals, πp bonding orbitals between C–C bonds and a small number of F-2p orbitals, and in the CB, the dominating SHG sources are the antibonding orbitals of 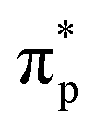 on 3AP+ groups, the unoccupied Sb-5p orbitals and some unoccupied F-2p orbitals. Numerically speaking, the SHG contribution percentages of helical [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cations were calculated to be 48.93% and 50.77%, respectively, indicating that the strong SHG effect of (3AP)2(S4F13) originates from the synergistic effect of the [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cations.
on 3AP+ groups, the unoccupied Sb-5p orbitals and some unoccupied F-2p orbitals. Numerically speaking, the SHG contribution percentages of helical [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cations were calculated to be 48.93% and 50.77%, respectively, indicating that the strong SHG effect of (3AP)2(S4F13) originates from the synergistic effect of the [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cations.
Conclusions
In summary, a universal synthesis method for polyfluoroantimonites has been developed for the first time, affording two novel polyfluoroantimonites, namely, (3PC)2(Sb4F14) and (3AP)2(Sb4F13). They exhibit totally different polymerization modes, which can be regulated by the organic amines. The 3AP+ cation with stronger hyperpolarizability results in a [Sb4F13]∞− helical polyfluoroantimonite chain, while the 3PC+ cation brings about a unique (Sb4F14)2− 4-MPR. The NCS and polar (3AP)2(Sb4F13) features a strong SHG effect of 8.1 × KDP, a large birefringence of 0.258@546 nm and a high LIDT value of 149.7 MW cm−2. Detailed structural analysis and theoretical calculations reveal that its strong SHG response originates from the synergistic effect of the helical [Sb4F13]∞− polyfluoroantimonite chain and π-conjugated 3AP+ cations. Our work demonstrates that the polyfluoroantimonite strategy is an effective approach for designing and synthesizing high-performance SHG materials. Further studies on NCS polyfluoroantimonites are in progress.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Jia-Hang Wu: conceptualization, investigation, data curation, and writing – original draft; Chun-Li Hu: software and formal analysis; Ya-Feng Li: data curation and visualization; Jiang-Gao Mao: project administration and funding acquisition; Fang Kong: methodology, writing – review & editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22031009, 22375201, 21921001 and 91963105) and the Natural Science Foundation of Fujian Province (Grant No. 2023J01216).Notes and references
- K. M. Ok, Toward the Rational Design of Novel Noncentrosymmetric Materials: Factors Influencing the Framework Structures, Acc. Chem. Res., 2016, 49(12), 2774–2785 CrossRef CAS PubMed.
- S. J. Han, A. Tudi, W. B. Zhang, X. L. Hou, Z. H. Yang and S. L. Pan, Recent Development of Sn(II), Sb(III)-based Birefringent Material: Crystal Chemistry and Investigation of Birefringence, Angew. Chem., Int. Ed., 2023, 62(26), e202302025 CrossRef CAS PubMed.
- X. Liu, Y. C. Yang, M. Y. Li, L. Chen and L. M. Wu, Anisotropic structure building unit involving diverse chemical bonds: a new opportunity for high-performance second-order NLO materials, Chem. Soc. Rev., 2023, 25, 8699–8720 RSC.
- X. M. Liu, P. F. Gong, Y. Yang, G. M. Song and Z. S. Lin, Nitrate nonlinear optical crystals: A survey on structure-performance relationships, Coord. Chem. Rev., 2019, 400, 213045 CrossRef CAS.
- P. S. Halasyamani, New nonlinear opportunities in the ultraviolet, Nat. Photonics, 2023, 17(8), 639–640 CrossRef CAS.
- Y. W. Kang and Q. Wu, A review of the relationship between the structure and nonlinear optical properties of organic-inorganic hybrid materials, Coord. Chem. Rev., 2024, 498, 215458 CrossRef CAS.
- P. F. Li, J. G. Mao and F. Kong, A survey of stereoactive oxysalts for linear and nonlinear optical applications, Mater. Today Phys., 2023, 37, 101197 CrossRef CAS.
- Z. X. Chen, W. Liu and S. P. Guo, A review of structures and physical properties of rare earth chalcophosphates, Coord. Chem. Rev., 2023, 474, 214870 CrossRef CAS.
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Achieving the full-wavelength phase-matching for efficient nonlinear optical frequency conversion in C(NH2)3BF4, Nat. Photonics, 2023, 17(8), 694–701 CrossRef CAS.
- Q. Lu, X. X. Jiang, K. N. Duanmu, C. Wu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Record Second-Harmonic Generation and Birefringence in an Ultraviolet Antimonate by Bond Engineering, J. Am. Chem. Soc., 2024, 146(14), 9975–9983 CrossRef PubMed.
- Y. L. Hu, C. Wu, X. X. Jiang, K. N. Duanmu, Z. P. Huang, Z. S. Lin, M. G. Humphrey and C. Zhang, Ultrashort Phase-Matching Wavelength and Strong Second-Harmonic Generation in Deep-UV-Transparent Oxyfluorides by Covalency Reduction, Angew. Chem., Int. Ed., 2023, e202315133 CAS.
- J. B. Wang, M. M. Zhu, Y. Q. Chu, J. D. Tian, L. L. Liu, B. B. Zhang and P. S. Halasyamani, Rational Design of the Alkali Metal Sn-Based Mixed Halides with Large Birefringence and Wide Transparent Range, Small, 2023, 2308884 Search PubMed.
- M. Yan, R. L. Tang, W. L. Liu and S. P. Guo, From Ba3Nb2O2F12(H2O)2 to Ba0.5NbO2F2(H2O): Achieving Balanced Nonlinear Optical Performance by O/F Ratio Regulation, Inorg. Chem., 2022, 61(50), 20709–20715 CrossRef CAS PubMed.
- P. F. Gong, Y. Yang, F. G. You, X. Y. Zhang, G. M. Song, S. Z. Zhang, Q. Huang and Z. S. Lin, ASbF3Cl (A = Rb, Cs): Structural Evolution from Centrosymmetry to Noncentrosymmetry, Cryst. Growth Des., 2019, 19(3), 1874–1879 CrossRef CAS.
- X. H. Dong, L. Huang, C. F. Hu, H. M. Zeng, Z. E. Lin, X. Wang, K. M. Ok and G. H. Zou, CsSbF2SO4: An Excellent Ultraviolet Nonlinear Optical Sulfate with a KTiOPO4 (KTP)-type Structure, Angew. Chem., Int. Ed., 2019, 58(20), 6528–6534 CrossRef CAS PubMed.
- X. H. Dong, H. B. Huang, L. Huang, Y. Q. Zhou, B. B. Zhang, H. M. Zeng, Z. E. Lin and G. H. Zou, Unearthing Superior Inorganic UV Second-Order Nonlinear Optical Materials: A Mineral-Inspired Method Integrating First-Principles High-Throughput Screening and Crystal Engineering, Angew. Chem., Int. Ed., 2024, e202318976 CAS.
- W. Y. Wang, X. Y. Wang, L. Xu, D. Zhang, J. L. Xue, S. Y. Wang, X. H. Dong, L. L. Cao, L. Huang and G. H. Zou, Centrosymmetric Rb2Sb(C2O4)2.5(H2O)3 and Noncentrosymmetric RbSb2(C2O4)F5: Two Antimony (III) Oxalates as UV Optical Materials, Inorg. Chem., 2023, 62(32), 13148–13155 CrossRef CAS PubMed.
- L. Wang, H. M. Wang, D. Zhang, D. J. Gao, J. Bi, L. Huang and G. H. Zou, Centrosymmetric RbSnF2NO3 vs. noncentrosymmetric Rb2SbF3(NO3)2, Inorg. Chem. Front., 2021, 8(13), 3317–3324 RSC.
- J. H. Wu, B. Zhang, T. K. Jiang, F. Kong and J. G. Mao, From Cs8Sb4Nb5O5F35 to Cs6Sb4Mo3O5F26: the first noncentrosymmetric fluoroantimonite with d0 transition metal, Chin. J. Struct. Chem., 2023, 42(1), 100016 CrossRef CAS.
- K. M. Ok and P. S. Halasyamani, The Lone-Pair Cation I5+ in a Hexagonal Tungsten Oxide-Like Framework: Synthesis, Structure, and Second-Harmonic Generating Properties of Cs2I4O11, Angew. Chem., Int. Ed., 2004, 116(41), 5605–5607 CrossRef.
- F. Ding, K. J. Griffith, W. Zhang, S. Cui, C. Zhang, Y. Wang, K. Kamp, H. Yu, P. S. Halasyamani, Z. Yang, S. Pan and K. R. Poeppelmeier, NaRb6(B4O5(OH)4)3(BO2) Featuring Noncentrosymmetry, Chirality, and the Linear Anionic Group BO2, J. Am. Chem. Soc., 2023, 145(9), 4928–4933 CrossRef CAS PubMed.
- W. J. Xie, R. L. Tang, S. N. Yan, N. Ma, C. L. Hu and J. G. Mao, Ba4B14O25: A Deep Ultraviolet Transparent Nonlinear Optical Crystal with Strong Second Harmonic Generation Response Achieved by a Boron-Rich Closed-Loop Strategy, Small, 2023, 2307072 Search PubMed.
- H. T. Qiu, F. M. Li, Z. Li, Z. H. Yang, S. L. Pan and M. Mutailipu, Breaking the Inherent Interarrangement of [B3O6] Clusters for Nonlinear Optics with Orbital Hybridization Enhancement, J. Am. Chem. Soc., 2023, 145(44), 24401–24407 CrossRef CAS PubMed.
- Y. C. Yan, J. H. Jiao, C. C. Tu, M. Zhang, Z. H. Yang and S. L. Pan, CsAB8O12F2·CsI (A = K+, NH4+): design of two fluorooxoborates with benign layered structures via a salt-inclusion strategy, J. Mater. Chem. C, 2022, 10(22), 8584–8588 RSC.
- H. N. Liu, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, Cs3[(BOP)3(B3O7)3]: A Deep-Ultraviolet Nonlinear Optical Crystal Designed by Optimizing Matching of Cation and Anion Groups, J. Am. Chem. Soc., 2023, 145(23), 12691–12700 CrossRef CAS PubMed.
- Q. Q. Chen, C. L. Hu, J. Chen, Y. L. Li, B. X. Li and J. G. Mao, [o-C5H4NHOH]2[I7O18(OH)·3H2O: An Organic-inorganic Hybrid SHG Material Featuring an [I7O18(OH)∞2- Branched Polyiodate Chain, Angew. Chem., Int. Ed., 2021, 60(32), 17426–17429 CrossRef CAS PubMed.
- J. Chen, C. L. Hu, F. F. Mao, B. P. Yang, X. H. Zhang and J. G. Mao, REI5O14 (RE=Y and Gd): Promising SHG Materials Featuring the Semicircle-Shaped I5O143- Polyiodate Anion, Angew. Chem., Int. Ed., 2019, 58(34), 11666–11669 CrossRef CAS PubMed.
- G. Zhang, J. G. Qin, T. Liu, Y. J. Li, Y. C. Wu and C. T. Chen, NaSb3F10: A new second-order nonlinear optical crystal to be used in the IR region with very high laser damage threshod, Appl. Phys. Lett., 2009, 95(26), 261104 CrossRef.
- L. A. Zemnukhova, A. A. Udovenko, N. V. Makarenko, S. I. Kuznetsov and T. A. Babushkina, Crystal structure and Sb NQR parameters of ammonium tridecafluorotetraantimonate(III) NH4Sb4F13, J. Struct. Chem., 2017, 58(4), 694–699 CrossRef CAS.
- H. T. Tian, C. S. Lin, X. Zhao, F. Xu, C. Wang, N. Ye and M. Luo, Ba(SO3CH3)2: A Deep-Ultraviolet Transparent Crystal with Excellent Optical Nonlinearity Based on a New Polar Non-π-Conjugated NLO Building Unit SO3CH3−, CCS Chem., 2023, 5, 2497–2505 CrossRef CAS.
- X. Y. Song, Z. P. Du, A. Belal, Y. Q. Li, Y. Zhou, Y. P. Song, W. Q. Huang, J. Y. Zheng, J. H. Luo and S. G. Zhao, A UV Solar-Blind Nonlinear Optical Crystal with Confined π-Conjugated Groups, Inorg. Chem. Front., 2023, 10(18), 5462–5467 RSC.
- X. H. Meng, X. Y. Zhang, Q. X. Liu, Z. Y. Zhou, X. X. Jiang, Y. G. Wang, Z. S. Lin and M. J. Xia, Perfectly Encoding π-Conjugated Anions in the RE5(C3N3O3)(OH)12(RE=Y, Yb, Lu) Family with Strong Second Harmonic Generation Response and Balanced Birefringence, Angew. Chem., Int. Ed., 2023, 62(1), e202214848 CrossRef CAS PubMed.
- J. Lu, X. Liu, M. Zhao, X. B. Deng, K. X. Shi, Q. R. Wu, L. Chen and L. M. Wu, Discovery of NLO Semiorganic (C5H6ON)+(H2PO4)−: Dipole Moment Modulation and Superior Synergy in Solar-Blind UV Region, J. Am. Chem. Soc., 2021, 143(9), 3647–3654 CrossRef CAS PubMed.
- Z. P. Zhang, X. Liu, X. M. Liu, Z. W. Lu, X. Sui, B. Y. Zhen, Z. S. Lin, L. Chen and L. M. Wu, Driving Nonlinear Optical Activity with Dipolar 2-Aminopyrimidinium Cations in (C4H6N3)+(H2PO3)−, Chem. Mater., 2022, 34(4), 1976–1984 CrossRef CAS.
- F. F. Mao, C. L. Hu, J. Chen, B. L. Wu and J. G. Mao, HBa2.5(IO3)6(I2O5) and HBa(IO3)(I4O11): Explorations of Second-Order Nonlinear Optical Materials in the Alkali-Earth Polyiodate System, Inorg. Chem., 2019, 58, 3982–3989 CrossRef CAS PubMed.
- H. T. Tian, N. Ye and M. Luo, Sulfamide: A Promising Deep-Ultraviolet Nonlinear Optical Crystal Assembled from Polar Covalent [SO2(NH2)2] Tetrahedra, Angew. Chem., Int. Ed., 2022, 61(17), e202200395 CrossRef CAS PubMed.
- Q. Q. Chen, C. L. Hu, M. Z. Zhang, B. X. Li and J. G. Mao, α- and β-(C4H5N2O)(IO3)·HIO3: Two SHG Materials Based on Organic–Inorganic Hybrid Iodates, Inorg. Chem., 2023, 62(32), 12613–12619 CrossRef CAS PubMed.
- F. F. Wu, Q. Y. Wei, X. Q. Li, Y. Liu, W. Q. Huang, Q. Chen, B. X. Li, J. H. Luo and X. T. Liu, Cooperative Enhancement of Second Harmonic Generation in an Organic-Inorganic Hybrid Antimony Halide, Cryst. Growth Des., 2022, 22(6), 3875–3881 CrossRef CAS.
- D. H. Lin, M. Luo, C. S. Lin, F. Xu and N. Ye, KLi(HC3N3O3).2H2O: Solvent-drop Grinding Method toward the Hydro-isocyanurate Nonlinear Optical Crystal, J. Am. Chem. Soc., 2019, 141(8), 3390–3394 CrossRef CAS PubMed.
- M. Zhang, B. B. Zhang, D. Q. Yang and Y. Wang, Synergistic Effect of π-Conjugated [C(NH2)3] Cation and Sb(III) Lone Pair Stereoactivity on Structural Transformation and Second Harmonic Generation, Inorg. Chem., 2021, 60(23), 18483–18489 CrossRef CAS PubMed.
- M. Yang, W. Liu and S. P. Guo, Sb5O7I: Exploration of Ternary Antimony-Based Oxyhalide as a Nonlinear-Optical Material, Inorg. Chem., 2022, 61(37), 14517–14522 CrossRef CAS PubMed.
- J. Kee and K. M. OK, Hydrogen-Bond-Driven Synergistically Enhanced Hyperpolarizability: Chiral Coordination Polymers with Nonpolar Structures Exhibiting Unusually Strong Second-Harmonic Generation, Angew. Chem., Int. Ed., 2021, 60(38), 20656–20660 CrossRef CAS PubMed.
- X. Hao, M. Luo, C. S. Lin, D. H. Lin, L. L. Cao and N. Ye, RE(H2C3N3O3)2·(OH).xH2O (RE = La, Y and Gd): potential UV birefringent materials with strong optical anisotropy originating from the (H2C3N3O3)- group, Dalton Trans., 2019, 48(32), 12296–12302 RSC.
- T. H. Wu, X. X. Jiang, C. Wu, Y. L. Hu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Ultrawide Bandgap and Outstanding Second-Harmonic Generation Response by a Fluorine-Enrichment Strategy at a Transition-Metal Oxyfluoride Nonlinear Optical Material, Angew. Chem., Int. Ed., 2022, 61(26), e202203104 CrossRef CAS PubMed.
- C. B. Jiang, X. X. Jiang, C. Wu, Z. P. Huang, Z. S. Lin, M. G. Humphrey and C. Zhang, Isoreticular Design of KTiOPO4-Like Deep-Ultraviolet Transparent Materials Exhibiting Strong Second-Harmonic Generation, J. Am. Chem. Soc., 2022, 144(44), 20394–20399 CrossRef CAS PubMed.
- J. J. Wu, J. L. Qi, Y. Guo, S. F. Yan, W. L. Liu and S. P. Guo, Reversible Tristate Structural Transition within Hybrid Copper (I) Bromides toward Tunable Multiple Emissions, Inorg. Chem. Front., 2023, 59, 16465–16469 Search PubMed.
- C. Sun, J. P. Zhang, Y. Q. Liu, Q. Q. Zhong, X. X. Xing, J. P. Li, C. Y. Yue and X. W. Lei, Lead-Free Hybrid Indium Perovskites with Highly Efficient and Stable Green Light Emissions, CCS Chem., 2022, 4, 3106–3121 CrossRef CAS.
- L. L. Cao, G. Peng, W. B. Liao, T. Yan, X. F. Long and N. Ye, A microcrystal method for the measurement of birefringence, CrystEngComm, 2020, 22(11), 1956–1961 RSC.
- J. H. Wu, C. L. Hu, T. K. Jiang, J. G. Mao and F. Kong, Highly Birefringent d0 Transition Metal Fluoroantimonite in the Mid Infrared Band: Order-Disorder Regulation by Cationic Size, J. Am. Chem. Soc., 2023, 145(44), 24416–24424 CrossRef CAS PubMed.
- Q. Wang, J. X. Ren, D. Wang, L. L. Cao, X. H. Dong, L. Huang, D. J. Gao and G. H. Zou, Low Temperature Molten Salt Synthesis of Noncentrosymmetric (NH4)3SbF3(NO3)3 and Centrosymmetric (NH4)3SbF4(NO3)2, Inorg. Chem. Front., 2023, 10(7), 2107–2114 RSC.
- Y. L. Deng, L. Huang, X. H. Dong, L. Wang, K. M. Ok, H. M. Zeng, Z. E. Lin and G. Zou, K2Sb(P2O7)F: Cairo Pentagonal Layer with Bifunctional Genes Reveal Optical Performance, Angew. Chem., Int. Ed., 2020, 59(47), 21151–21156 CrossRef CAS PubMed.
- X. M. Wen, J. Cheng, P. Q. Qian, Z. Z. Zhang, H. M. Zeng, L. Huang, G. H. Zou and Z. E. Lin, Synthesis and structure-dependent optical properties of two new organic–inorganic hybrid antimony(III) chlorides, Dalton Trans., 2024, 53(1), 260–266 RSC.
- J. Cheng, P. Q. Qian, M. Yang, L. Huang, H. M. Zeng, G. H. Zou and Z. E. Lin, Second-Harmonic Generation in Homochiral Antimony Halides Directed by l-Histidine, Inorg. Chem., 2023, 62(41), 16673–16676 CrossRef CAS PubMed.
- M. J. Zhang, B. X. Li, B. W. Liu, Y. H. Fan, X.-G. Li, H. Y. Zeng and G. C. Guo, Ln3GaS6 (Ln = Dy, Y): new infrared nonlinear optical materials with high laser induced damage thresholds, Dalton Trans., 2013, 42(39), 14223–14229 RSC.
- P. F. Li, C. L. Hu, J. G. Mao and F. Kong, A UV non-hydrogen pure selenite nonlinear optical material for achieving balanced properties through framework-optimized structural transformation, Mater. Horiz., 2024, 11, 1704–1709 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, theoretical calculations, crystallographic data, and measurements of physical properties. CCDC 2311987 and 2311988 for (3PC)2Sb4F14 and (3AP)2Sb4F13. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01716a |
| This journal is © The Royal Society of Chemistry 2024 |