
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing C2+ product selectivity in CO2 electroreduction by enriching intermediates over carbon-based nanoreactors†
Min
Wang
ab,
Chunjun
Chen
 ab,
Shuaiqiang
Jia
ab,
Shitao
Han
ab,
Xue
Dong
ab,
Dawei
Zhou
ab,
Ting
Yao
ab,
Minghui
Fang
ab,
Mingyuan
He
ab,
Wei
Xia
*ab,
Haihong
Wu
*ab and
Buxing
Han
*abc
ab,
Shuaiqiang
Jia
ab,
Shitao
Han
ab,
Xue
Dong
ab,
Dawei
Zhou
ab,
Ting
Yao
ab,
Minghui
Fang
ab,
Mingyuan
He
ab,
Wei
Xia
*ab,
Haihong
Wu
*ab and
Buxing
Han
*abc
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: wxia@chem.ecnu.edu.cn; hhwu@chem.ecnu.edu.cn; hanbx@iccas.ac.cn
bInstitute of Eco-Chongming, 20 Cuiniao Road, ChenjiaTown, Chongming District, Shanghai 202162, China
cBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 1st May 2024
Abstract
Electrochemical CO2 reduction reaction (CO2RR) to multicarbon (C2+) products faces challenges of unsatisfactory selectivity and stability. Guided by finite element method (FEM) simulation, a nanoreactor with cavity structure can facilitate C–C coupling by enriching *CO intermediates, thus enhancing the selectivity of C2+ products. We designed a stable carbon-based nanoreactor with cavity structure and Cu active sites. The unique geometric structure endows the carbon-based nanoreactor with a remarkable C2+ product faradaic efficiency (80.5%) and C2+-to-C1 selectivity (8.1) during the CO2 electroreduction. Furthermore, it shows that the carbon shell could efficiently stabilize and highly disperse the Cu active sites for above 20 hours of testing. A remarkable C2+ partial current density of−323 mA cm−2 was also achieved in a flow cell device. In situ Raman spectra and density functional theory (DFT) calculation studies validated that the *COatop intermediates are concentrated in the nanoreactor, which reduces the free energy of C–C coupling. This work unveiled a simple catalyst design strategy that would be applied to improve C2+ product selectivity and stability by rationalizing the geometric structures and components of catalysts.
Introduction
The escalating emission of greenhouse gases exacerbates climate change, posing a greater threat to the future of humanity. Carbon dioxide (CO2), one of the primary greenhouse gases, plays a crucial role in this process. Consequently, the electrocatalytic reduction of CO2 into valuable chemicals and fuels holds immense significance in mitigating climate change, promoting a closed carbon cycle, and facilitating the storage of renewable electricity.1–3 The primary outputs of the electrochemical CO2 reduction reaction (CO2RR) consist of C1 products and multicarbon (C2+) products. Within these products, C2+ products hold greater economic significance within the chemical industry due to their higher commercial value.4–6 Currently, extensive efforts have been devoted to developing catalysts to obtain high-productivity of C2+ products.7–10 Among them, copper (Cu) is recognized as the unique metal to efficaciously break the stable CO2 bonds and form C–C bonds, a key step towards C2+ products.11–14 However, the challenge of C2+ selectivity still limits its economic competitiveness, and more advanced strategies are needed to design high-performance catalysts.Based on previous studies, C–C coupling is considered a crucial route for the formation of C2+ products in the CO2RR, whereas the C–C coupling reaction relies heavily on the *CO intermediate dimerization.15 Achieving a local high concentration of *CO intermediates at the active sites is crucial for initiating the dimerization process between neighbouring *CO intermediates.16 But *CO species tended to diffuse from the catalyst surface to the electrolyte, and it may result in their premature discharge from the active sites.17 This premature release severely hampers the efficiency of C–C coupling, thus reducing the overall performance of the CO2RR process.18 A simple way to increase the local concentration of *CO species is to increase the diffusion resistance and extend the diffusion path length.
Previous studies have demonstrated that the confinement effect can alter the diffusion kinetics and effectively improve the local concentration of key intermediates.19 Sargent et al. applied finite element method (FEM) simulations and experimental analyses to explore the C–C coupling reactions by the confinement effect.20 They demonstrated that Cu2O with the cavity structure led to a high surface coverage of intermediates and improved the electrocatalytic conversion of CO-to-propanol. Yu et al. conducted a comprehensive investigation on the multicavities of Cu2O via intermediate confinement to enhance the selectivity of CO2 electroreduction to C2+ fuels.21 These findings revealed that this unique cavity structure effectively suppressed the loss of key intermediates, and enhanced their local concentration at active sites, thus increasing the chance of C–C coupling reactions. Studies on the steric confinement effect in the CO2RR have substantiated its capability to enhance the selectivity of multicarbon products.22,23 However, a persistent challenge in current studies about Cu-based nanoreactors is Cu surface reconstruction and compositional changes during electrolysis, which makes product selectivity and activity decline significantly.24 Also, the designability of pure Cu-based catalysts is very limited.
It is known that supported catalysts, in which active sites are anchored on supports, have some obvious advantages, such as the size and dispersion of the active sites can be tuned, and the synergy of the active sites and supports.25 Specifically, carbon materials exhibit advantages in electrocatalysis, including strong stability, ease of morphological adjustments and optimized reaction intermediate adsorption that make it an excellent candidate for supporting active sites.26 Xia et al. proposed a carbon protected indium oxide electrocatalyst, where the carbon layer not only prevents the reductive corrosion of indium oxide during electrolysis, but also optimizes the intermediate adsorption, thus improving the stability and activity of CO2 reduction.27 Therefore, it is important to introduce a carbon layer into catalyst design to achieve electrochemical stability while maintaining high activity.
Combining steric confined carbon supported nanoreactors with highly active Cu sites would establish a distinctive class of catalysts with both exceptional C2+ product selectivity and stability. Thus, we began using FEM simulations to explore the influence of cavity structures on the accumulation of *CO intermediates and C–C coupling. Guided by FEM results, we synthesized a Cu/C-cavity nanoreactor in which Cu species are supported in the carbon shell. The unique structure endows the Cu/C-cavity catalyst with high CO2 electrocatalytic performance. As a result, the nanoreactor achieved a remarkable multicarbon (C2+) product faradaic efficiency (FE) of 80.5% and a higher C2+-to-C1 ratio of 8.1 at −1.2 V vs. RHE. Based on the highly dispersed Cu active sites on the carbon support to prevent agglomeration, the nanoreactor performed continuous electrolysis for more than 20 hours. In situ Raman spectra and density functional theory (DFT) calculation results revealed that the cavity of the nanoreactor could concentrate *COatop intermediates and reduce their dimerization barriers. This work introduces a convenient and efficient synthesis strategy to enhance the selectivity of multicarbon products and stability for the electrochemical CO2RR. This strategy could applicable to other reactions, rationalizing the morphology and active components of the catalyst, and concentrating key intermediates through nanoconfinement to improve the selectivity of ideal products.
Results and discussion
It is assumed that the cavity structure nanoreactor could retard the diffusion kinetics through the confinement effect and concentrate *CO species, thus increasing the possibility for C–C coupling. To verify the hypothesis, we applied FEM simulations to explore the prospects of cavity structure in enhancing C2+ product selectivity. A hollow spherical shell model with circular openings (with an outer diameter of 39 nm and an inner diameter of 6.5 nm) was used to represent the nanoreactor immersed in an electrolyte (see details in the ESI†). The out flux of *CO and C2 products on the inner and outer surfaces of the nanoreactor was monitored. Results of the simulation indicated that CO2 molecules first diffused to the surface (Fig. 1a). Then, CO2 was adsorbed and reduced into *CO at the interior and outer surfaces of the spherical shell (Fig. 1b). Finally, the *CO species could desorb from the surface as C1 products (CO assumed) or be coupled with other *CO to form C2 products (C2H4 assumed). The surface C2 species could diffuse into the electrolyte or continue to react with another *CO to produce C3 products. Consistent with our hypothesis, the cavity structure could restrict the diffusion of internally generated *CO intermediates to the outside of the cavity (Fig. 1b, arrows), leading to the accumulation of *CO species inside the cavity and the concentration of the *CO species was significantly increased (Fig. 1b, colour map). The results lead to a high concentration of *CO intermediates required for the formation of C2 products, and facilitated the conversion to C2 products (Fig. 1c).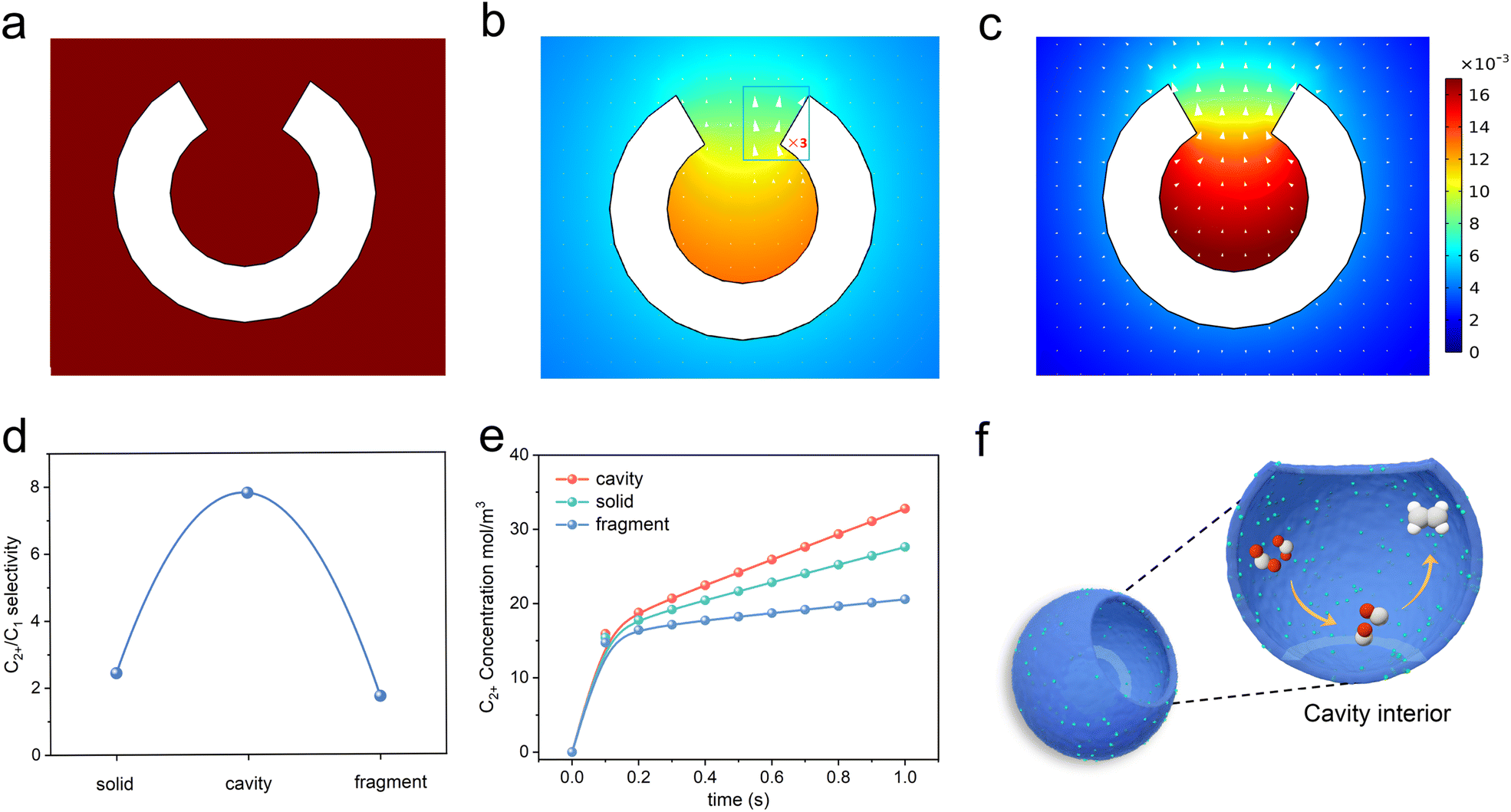 | ||
| Fig. 1 Computed concentration and flux distribution of species. (a) CO2, (b) *CO, and (c) C2+ concentrations (color scale, in mol L−1) and flux distributions (arrows) on the cavity structure. (d) Simulation results of the C2+/C1 product selectivity on the cavity, solid and fragment structure. (e) Simulation results of the time-dependent variation of C2+ concentration on cavity, solid and fragment structure. (f) The diagram displays how the cavity confinement effect promotes *CO intermediate dimerization and transformation to C2H4. Red, oxygen; grey, carbon; white, hydrogen. | ||
Furthermore, the microenvironments over the interface of the fully closed and fully open structures were also monitored by FEM. The solid and fragment models were used to represent the fully closed and fully open structure catalyst immersed in an electrolyte. As *CO species tended to escape from the catalyst surface to the electrolyte, both the solid and fragment structures cannot prevent the diffusion of *CO from its surface to the electrolyte (Fig. S1a–f†). Therefore, these two structures were ineffective in enhancing the coverage of *CO species at active sites, resulting in no enhancement in the yield of C2+ products. The solid and fragment structures exhibited limitations in promoting C–C coupling. Fig. 1d shows coincident simulating results of the C2+/C1 ratios of the three structures. The C2+-to-C1 simulated ratios of cavity, solid and fragment structures were 7.81, 2.44, 1.73, respectively.
We also simulated the time-dependent variation of C2+ concentration on the cavity, solid and fragment structure models, and the results are shown in Fig. 1e. The C2+ concentration on the three models kept increasing with time, and the cavity model was obviously larger than that of the solid and fragment structure models at the same time. This distinction arose from the cavity structure, which significantly slowed down the diffusion kinetics, and improved the local coverage of *CO, thus facilitating the formation of C2+ products. In contrast, the diffusion of the *CO on solid and fragment structures cannot be retarded. These simulation results showed that cavity structures could promote the formation of C2+ products through steric nanoconfinement effects (Fig. 1f).
Under the guidance of FEM simulation results, based on the synthesis mechanism of the self-template method,28 the carbon-based nanoreactor (Cu/C-cavity) was synthesized. The synthetic protocol of the Cu/C-cavity catalyst is illustrated in Fig. 2a. First, the template MET-5 was synthesized by a solvothermal method29 (Fig. S2†), and then coated with a polydopamine (PDA) outer layer to obtain MET-5@PDA (Fig. S3†). Thermogravimetric analysis of MET-5 and MET-5@PDA was carried out (Fig. S4†). The removal rate of template MET-5 could be regulated by controlling the calcination time and heating rate, which results in different structures,30 and the Cu/C-cavity and Cu/C-fragment (fully open structures) catalysts could be obtained. The Cu/C-solid (fully closed structures) was obtained by carbonizing MET-5.
 | ||
| Fig. 2 Morphology characterization of the Cu/C-cavity catalyst. (a) The synthesis process of the Cu/C-cavity catalyst. Cu/C-cavity catalyst imaged by (b) SEM, (c) TEM, and (d) HRTEM. (e) High-angle annular dark field (HAADF) and mapping images of the Cu/C-cavity catalyst, showing the homogeneous distribution of C (green), N (red), and Cu (blue). | ||
The scanning electron microscopy (SEM) images in Fig. 2b and S5† show the overall morphology of the Cu/C-cavity catalyst. Spherical particles with cavities were observed. In addition, transmission electron microscopy (TEM) images in Fig. 2c, S6 and S7† revealed the hollow morphology and open structures. The high-resolution TEM (HRTEM) image of the Cu/C-cavity catalyst displayed the lattice fringe of the crystal plane of Cu (111) (Fig. 2d). In addition, the SEM and TEM images confirmed the synthesis of Cu/C-solid and Cu/C-fragment catalysts (Fig. S8–S11†). The BET surface areas of Cu/C-cavity, Cu/C-solid, and Cu/C-fragment are shown in Fig. S12.† The high-angle annular dark field (HAADF) image in Fig. 2e further confirms the hollow spherical structure formation of the Cu/C-cavity catalyst. The energy-dispersive X-ray spectroscopy (EDX) elemental mapping images showed uniform distribution of C, N, and Cu in the Cu/C-cavity catalyst.
The chemical states and composition of the Cu/C-cavity, Cu/C-solid and Cu/C-fragment catalysts were confirmed by a series of techniques. The powder X-ray diffraction (XRD) patterns results are presented in Fig. 3a, and the diffraction patterns at 43.2, 50.4, and 74.1 could be indexed to the Cu (111), Cu (200), and Cu (220) planes, respectively (PDF #04-0836). The XPS survey spectra illustrated the existence of Cu, O, N, and C elements in the three materials (Fig. S13a†). The Cu 2p3/2 peak and Cu 2p1/2 peak in the XPS spectra were observed at 932.3 eV and 952.2 eV (Fig. 3b), respectively. The Cu LMM Auger verified that the Cu species in the Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts existed in the form of Cu0 (918.3 eV, predominant) and Cu+ (914.7 eV). The minor amount of Cu+ may result from the partial oxidation of Cu during characterization. In addition, the N 1s peak could be deconvolved into pyridinic-N (398.48 eV), graphitic-N (401.4 eV), and oxidic-N (402.35 eV) peaks (Fig. S13b†).31 The C 1s spectrum could be fitted into three peaks, corresponding to C–C (284.6 eV), C–N (285.9 eV), and C–O (287.2 eV) (Fig. S13c†).32 The O 1s spectrum could be deconvolved into O–C (530.8 eV), O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (532.1 eV), and OC–O (533.5 eV) (Fig. S13d†).33
C (532.1 eV), and OC–O (533.5 eV) (Fig. S13d†).33
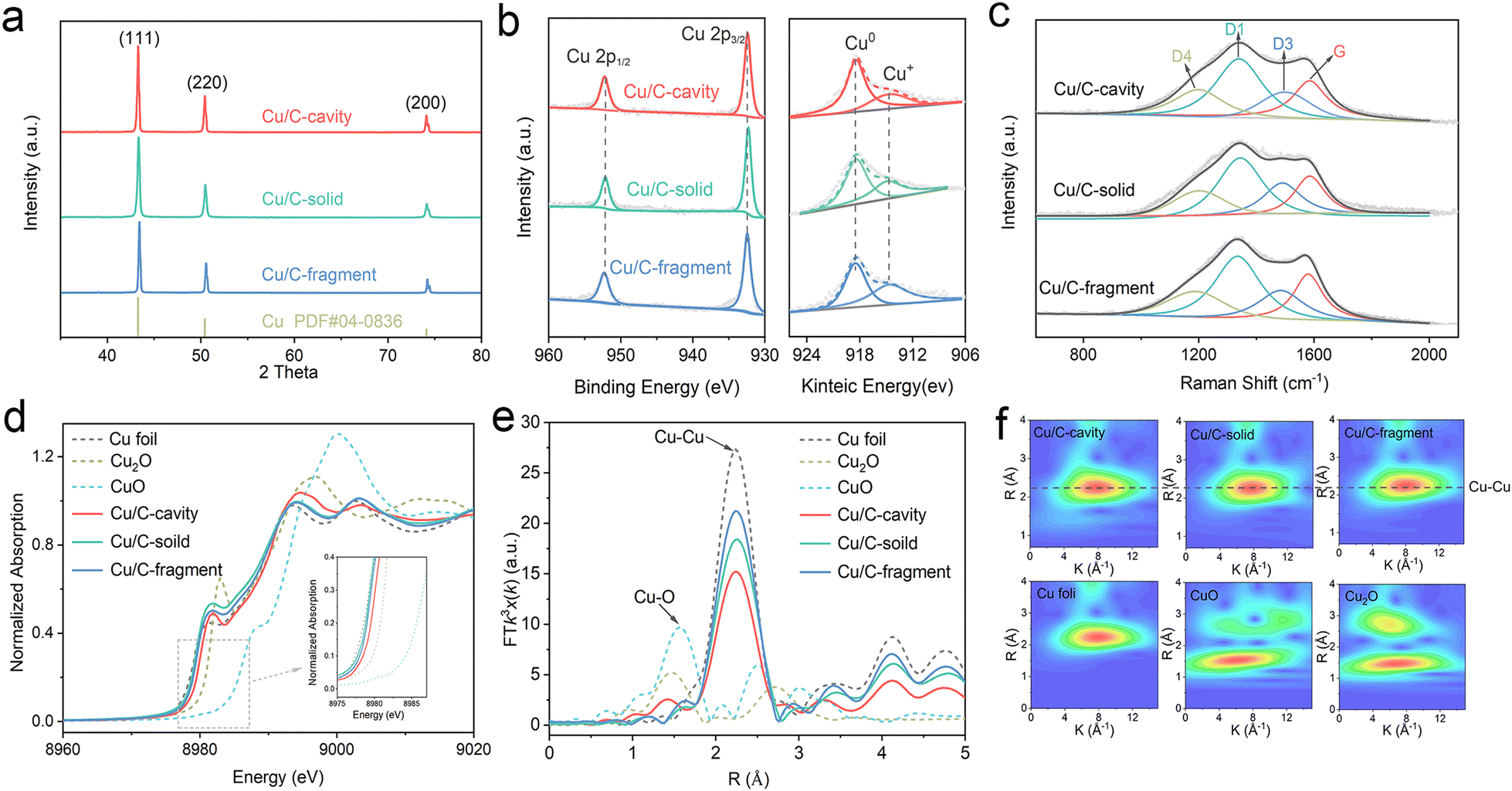 | ||
| Fig. 3 Chemical structural characterization of different catalysts. (a) XRD, (b) XPS spectra of Cu 2p and Cu LMM Auger, (c) Raman spectra, (d) Cu K-edge XANES, (e) Fourier transformed Cu K-edge EXAFS spectra and (f) Morlet WT of the k3-weighted EXAFS of Cu/C-cavity, Cu/C-solid and Cu/C-fragment with the references. | ||
The structures of Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts were further evaluated by Raman analysis (Fig. 3c). The Raman spectra could be deconvoluted into four peaks by Gaussian–Lorentzian numerical simulation,34 which were the graphene edges for the D1-band (ca. 1360 cm−1), topological defects for the D3-band (ca. 1500 cm−1), polyenes for the D4-band (ca. 1180 cm−1) and graphitic lattice for the G-band (ca. 1580 cm−1).35 The graphitization degree of carbon was inferred by the ratio of integrated areas of the D1 and G.36 The calculated ID1/IG ratios of the Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts were 1.65, 2.42, and 1.78, respectively, indicating that the Cu/C-cavity had a higher graphitization degree than Cu/C-solid and Cu/C-fragment catalysts.
The electronic information of the Cu/C-cavity, Cu/C-solid and Cu/C-fragment was further investigated by X-ray absorption spectroscopy (XAS) measurements. The Cu K-edge XANES spectra of the Cu/C-cavity, Cu/C-solid and Cu/C-fragment together with the references of Cu2O, CuO, and Cu foil are given in Fig. 3d. The X-ray absorption near edge structure (XANES) spectra (Fig. S14 and S15†) of Cu/C-cavity, Cu/C-solid and Cu/C-fragment were between the Cu foil and Cu2O, which proved that the Cu species was in the intermediate valence state between 0 and +1.37 The extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3e) of Cu/C-cavity, Cu/C-solid and Cu/C-fragment displayed a main peak at 2.24 Å, corresponding to the metal Cu–Cu bond.38 As shown in Fig. 3f, the Morlet Wavelet Transform (WT) of the k3-weighted extended X-ray absorption fine structure (EXAFS) further proved the existence of the Cu–Cu bond in the Cu/C-cavity, Cu/C-solid and Cu/C-fragment catalysts.39
The CO2 electroreduction activity over the Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts was first investigated in 0.1 M CsI aqueous solution using a typical H type cell (Fig. S16†). As shown in Fig. 4a, the linear sweep voltammetry (LSV) curves over the Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts exhibited a higher reduction current density in the CO2-saturated electrolyte than the N2-saturated electrolyte, demonstrating their CO2RR activity.40 Comparing these three catalysts, it was found that the Cu/C-cavity exhibited the highest reduction current density and the most positive onset potential compared to Cu/C-solid, and Cu/C-fragment catalysts. This indicated that the Cu/C-cavity achieved a higher CO2RR activity than the other two catalysts. The electrochemically active surface areas (ECSA) of the Cu/C-cavity, Cu/C-solid and Cu/C-fragment catalysts were obtained from the double-layer capacitance (Cdl). In Fig. S17,† the Cu/C-cavity catalyst has the largest ECSA, which demonstrates that the Cu/C-cavity catalyst has the highest CO2RR activity.41 Moreover, the Nyquist plots were recorded at the open-circuit potential to investigate the reaction kinetics of electrochemical processes. The electrochemical impedance spectroscopy (EIS) (Fig. S18†) shows that the Cu/C-cavity displayed the smallest Nyquist semicircle diameter compared to Cu/C-solid and Cu/C-fragment, suggesting a much faster interfacial charge-transfer kinetics.42 The local high ion concentration inside the cavity structure or the higher graphitization degree of Cu/C-cavity may improve the electrical conductivity, thus accelerating charge-transfer.43
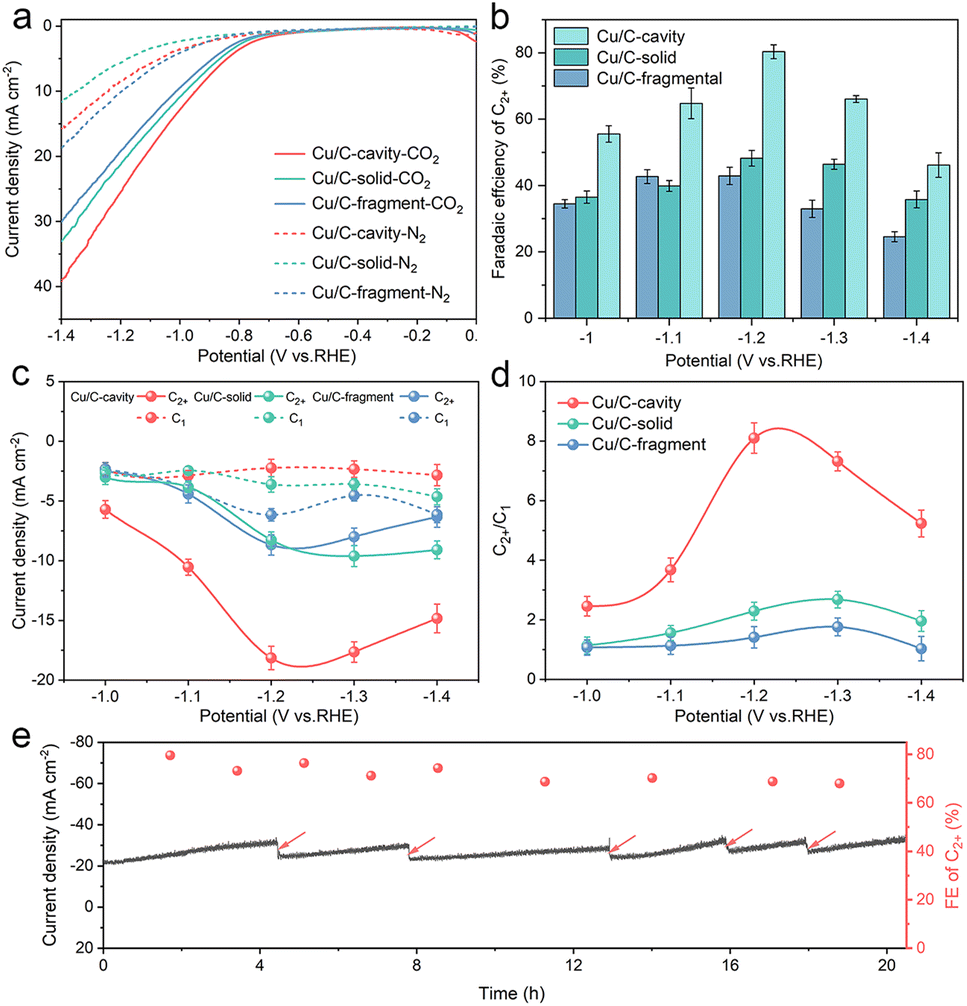 | ||
| Fig. 4 CO2 electrochemical reduction performance. (a) LSV curves on Cu/C-cavity, Cu/C-solid and Cu/C-fragment in CO2-saturated and N2-saturated 0.1 M CsI aqueous electrolyte. (b) C2+ FE from −1.0 V to −1.4 V vs. RHE. (c) C2+ and C1 partial current densities at −1.0 V to −1.4 V vs. RHE in CO2 saturated 0.1 M CsI aqueous electrolyte. (d) C2+/C1 products selectivity ratio from −1.0 V to −1.4 V vs. RHE in CO2 saturated 0.1 M CsI aqueous electrolyte. (e) Stability test of Cu/C-cavity at −1.2 V vs. RHE in CO2 saturated 0.1 M CsI aqueous electrolyte, the arrows indicate the renewal of the electrolyte. | ||
According to the results in Fig. S19a,† the Cu/C-cavity catalyst demonstrated the production of C2+ products within the potential range of −1.0 V to −1.4 V vs. RHE. The results showed that the FE of C2+ products over the Cu/C-cavity catalyst could reach up to 80.5% (1446.17 ppm) at −1.2 V vs. RHE (Fig. 4b). This efficiency comprised 52.2% C2H4, 18.8% C2H5OH, 5.4% CH3COOH, and 4.2% n-PrOH (Fig. S20†). Fig. S21† shows the liquid products in D2O (DMSO and phenol as an internal standard). Comparatively, the Cu/C-fragment and Cu/C-solid catalysts tended to produce CO-dominated C1 products (Fig. 4b, S19b, c and S20†). The FE over C2+ products on Cu/C-cavity was much higher than that over the Cu/C-solid and Cu/C-fragment catalysts. The above experimental results proved that the steric confinement effect of the cavity structure could promote C2+ product selectivity. The partial current densities of C2+ and C1 products of Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts were compared at different applied potentials. As depicted in Fig. 4c, it was evident that the Cu/C-cavity catalyst exhibited the highest C2+ partial current density, reaching 18.14 mA cm−2 at −1.2 V vs. RHE. In contrast, the Cu/C-solid and Cu/C-fragment showed lower maximum C2+ partial current densities of only 8.68 and 8.25 mA cm−2, respectively. The jC2+ normalised to ECSA and Cu content of Cu/C-cavity was larger than that of the Cu/C-solid and Cu/C-fragment catalysts (Tables S1 and S2†), indicating that the high C2+ products selectivity over the Cu/C-cavity resulted from its unique structure.
Fig. 4d discusses the ratios of C2+ to C1 products over the Cu/C-cavity, Cu/C-solid, and Cu/C-fragment catalysts. The Cu/C-cavity exhibited higher selectivity towards C2+ products across all tested potentials. Particularly at −1.2 V vs. RHE, the Cu/C-cavity catalyst achieved maximum selectivity for C2+ products, with a C2+/C1 ratio of approximately 8.1. The ratios over the Cu/C-solid and Cu/C-fragment catalysts were 2.7 and 1.8, respectively. To further confirm the universality of our Cu/C-cavity structure, we investigated the electrochemical CO2RR performance of Cu/C-cavity, Cu/C-solid, and Cu/C-fragment in 0.1 M KHCO3 solution using a typical H type cell (Fig. S22†), and the FE of C2+ products over the Cu/C-cavity catalyst could reach up to 61.2% at −1.2 V vs. RHE, with a C2+/C1 selectivity ratio of approximately 6.14. The ratios of the Cu/C-solid and Cu/C-fragment catalysts were 2.06 and 1.31, respectively. The Cu/C-cavity also exhibited higher C2+ selectivity than Cu/C-solid and Cu/C-fragment in 0.1 M KHCO3 solution. The experimental and simulated results showed excellent agreement, and it is also proved that the cavity structure is beneficial to improve the selectivity of C2+ products.
Furthermore, the electrochemical CO2RR performances of Cu/C-cavity, Cu/C-solid, and Cu/C-fragment were studied in 1 M KOH using a flow cell within the potential range of −0.8 V to −1.2 V vs. RHE (Fig. S23†). The Cu/C-cavity catalyst exhibited much higher selectivity for C2+ products (Fig. S24†), and the FE of C2+ could reach up to 75.2% (6990.76 ppm) with a C2+/C1 ratio of 3.57 at −1.0 V vs. RHE. The jC2+ could reach −323 mA cm−2, which is higher than those of most electrodes and reached industrial levels. In addition, we also conducted the stability test in a flow cell, which indicated that the stability of the electrode was satisfactory (Fig. S24e†). Fig. S25† shows the liquid products over Cu/C-cavity in the flow cell. However, the Cu/C-solid and Cu/C-fragment tended to produce CH4-dominated C1 products. The ratios of C2+/C1 over the Cu/C-solid and Cu/C-fragment catalysts were 1.15 and 1.34, respectively. The Cu/C-cavity also exhibited much higher C2+ selectivity than Cu/C-solid and Cu/C-fragment in the flow cell. Consequently, the above results proved that the confined geometric structure facilitates C–C coupling and the formation of multi-carbon compounds, which aligned with our previous hypothesis.
The stability of catalysts is a crucial parameter for the CO2RR. To assess the stability of the Cu/C-cavity catalyst, CO2 electrolysis was carried out at −1.2 V vs. RHE. As in Fig. 4e, the current density and FE did not change obviously over a period of 20 hours. The Cu/C-cavity nanoreactor was characterized by XRD and XPS techniques (Fig. S26†) after electrocatalysis showed no apparent change in the chemical state. The Cu K-edge spectra indicated that the Cu/C-cavity nanoreactor after electrocatalysis retained the characteristic feature of Cu(0), as can be further confirmed by the Cu–Cu coordination at 2.23 Å (Fig. S27†). From the TEM images (Fig. S28†) of the Cu/C-cavity after the CO2RR, the nanocavity structure basically remained intact, and no obvious agglomeration of particles was found. We conducted a comparative analysis of our carbon-based nanoreactor materials with other reported nanoconfinement reactors in the literature for CO2 electroreduction. The result proved that carbon-based nanoreactors have superior stability over other reported pure Cu-based nanoreactors (Table S3†). The superior stability is mainly from the Cu and C structure of the catalyst, and the carbon carrier effectively stabilizes the active site of Cu and protects it from the reaction environment.
To further explore the effect of nanoconfinement on the CO2RR, in situ Raman spectroscopy was conducted to monitor the key intermediates *CO of surface adsorption. Fig. 5a shows the in situ Raman spectra using the Cu/C-cavity catalyst during the CO2RR under different applied potentials in 0.5 M KHCO3 solution. The bands at 307 and 394 cm−1 were attributed to the CO frustrated rotation and Cu–CO vibration stretching, respectively, indicating the adsorption of *CO.44 The band at 524 cm−1 was ascribed to the adsorption of preliminary intermediates (such as CO2 ad) on the active sites.45 The band at 984 cm−1 was assigned to the *COO. Meanwhile, a much stronger band at 1067 cm−1, corresponding to carbonate could be observed. The adsorption bands in the range of 2007–2058 cm−1 were attributed to the atop-bound *CO (*COatop), which is a key intermediate of the CO–CO coupling.46 The *COatop peak could split into two bands. The low frequency band (LFB) at 2007 cm−1is ascribed to *COatop on the terrace (*COatop–L), and the high frequency band (HFB) at 2058 cm−1 is associated with *COatop on the low coordinated sites (*COatop–H). The *COatop peak intensity increased and then decreased with the potential scanned, indicating that *CO intermediates were accumulated and then consumed.
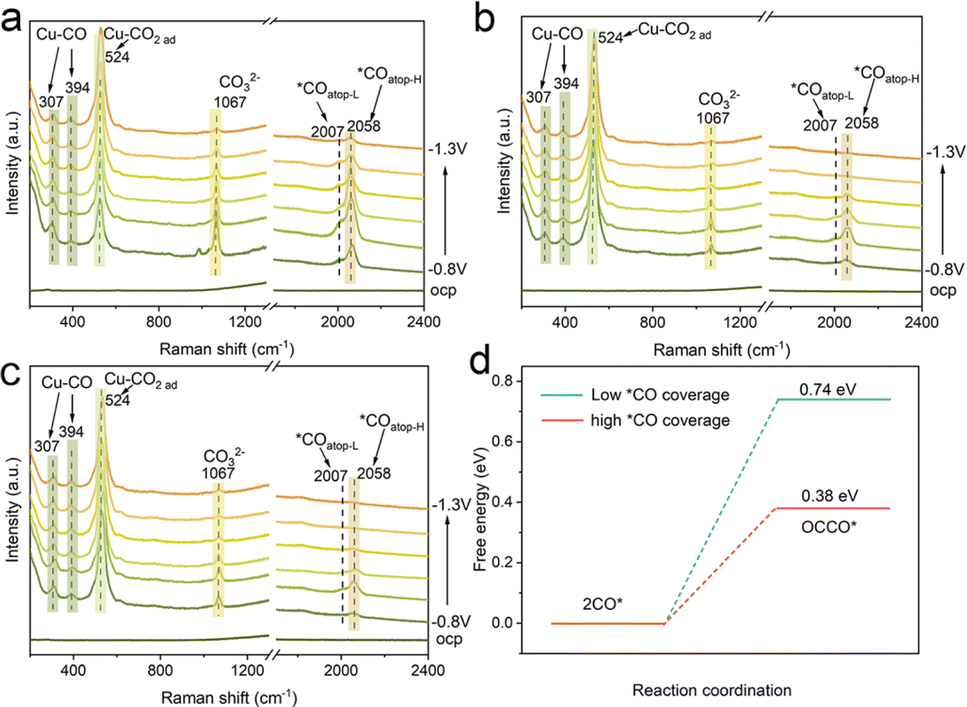 | ||
| Fig. 5 Mechanistic studies. In situ Raman spectra of the (a) Cu/C-cavity, (b) Cu/C-solid, and (c) Cu/C-fragment catalysts during the CO2RR under different applied potentials. (d) The free energy of the *CO dimerization step at low (blue) and high *CO coverage (red) on Cu(111). | ||
Furthermore, the in situ Raman spectra of Cu/C-solid and Cu/C-fragment were also recorded (Fig. 5b and c). The intensities of the peaks for linearly adsorbed *COatop on the Cu/C-solid and Cu/C-fragment were weak, implying less accumulation of *CO. In comparison, the peak intensity of *COatop (2058 cm−1) on the Cu/C-cavity was stronger than that over Cu/C-solid and Cu/C-fragment. The results suggested that *COatop intermediates were accumulated on the Cu/C-cavity, indicating that the cavity structure could enrich the local concentration of *CO intermediates.47 Overall, the in situ Raman results revealed that the cavity structure exhibited a higher coverage of *COatop compared to the solid and fragment structure, thus accelerating the process of *CO dimerization, resulting in superior CO2RR selectivity of C2+ products.
To further verify that the local high concentration of *CO could promote the rate of C–C coupling, the Gibbs free energy of the *CO dimerization step48 was studied using DFT (Fig. 5d, S29 and S30†). The Gibbs free energy value for the C–C coupling of *CO was found to be 0.74 eV at low *CO coverage, and decreased to 0.38 eV at high *CO coverage. The results suggested that a lower energy barrier for the C–C coupling reaction appeared at high *CO coverage. Thus, the probability of C–C coupling could be promoted by a high *CO coverage, which is consistent with our experimental results.
Conclusions
In summary, we discussed the correlation between the geometric structures of catalysts and the selectivity of C2+ products. We have demonstrated that cavity nanoreactors showed a significantly improved electrochemical CO2RR performance. The Cu/C-cavity exhibited a high C2+ FE of 80.5% and C2+/C1 selectivity ratio of 8.1, which is much higher than that over the Cu/C-solid and Cu/C-fragment. In addition, due to the highly dispersed Cu on the carbon support, the stability of Cu/C-cavity was better than that of most reported pure copper-based nanoreactor catalysts. The results of FEM simulation, in situ Raman and DFT supported that the cavity of nanoreactors enriched the local concentration of *CO intermediates, thus promoting the C–C coupling reactions. This research underscored the potential application of functionalized nanoreactors in the highly selective electrosynthesis of valuable fuels from CO2, while shedding light on the morphology and composition of catalysts having a significant effect on the performance of the CO2RR.Data availability
The authors declare that all data supporting the findings of this study are available within the paper [and its ESI†].Author contributions
M. W., W. X., H. H. W. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. M. W. performed the whole experiment. S. Q. J., T. Y., X. D., D. W. Z., and M. H. F. performed the analysis of experimental data. W. X., H. H. W., M. Y. H. and B. X. H. co-supervised the whole project. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2020YFA0710201; 2023YFA1507901), the China Postdoctoral Science Foundation Funded Project (2021M701211), and the National Natural Science Foundation of China (22293015, 21890761, and 22121002), and Fundamental Research Funds for the Central Universities. The X-ray absorption fine structure (XAFS) measurements were carried out at the 4B9A beamline of the Beijing Synchrotron Radiation Facility (BSRF).Notes and references
- S. I. Seneviratne, M. G. Donat, A. J. Pitman, R. Knutti and R. L. Wilby, Nature, 2016, 529, 477–483 CrossRef CAS PubMed.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chemistry, 2018, 4, 2571–2586 CrossRef CAS.
- X. She, Y. Wang, H. Xu, S. Chi Edman Tsang and S. Ping Lau, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- V. C. Hoang, V. G. Gomes and N. Kornienko, Nano Energy, 2020, 78, 105311 CrossRef CAS.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, e1804867 CrossRef PubMed.
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z. J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- Y. Wang, B. Li, B. Xue, N. Libretto, Z. Xie, H. Shen, C. Wang, D. Raciti, N. Marinkovic, H. Zong, W. Xie, Z. Li, G. Zhou, J. Vitek, J. G. Chen, J. Miller, G. Wang and C. Wang, Sci. Adv., 2023, 9, eade3557 CrossRef CAS PubMed.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- Y. Zang, T. Liu, P. Wei, H. Li, Q. Wang, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- Z. Ni, H. Liang, Z. Yi, R. Guo, C. Liu, Y. Liu, H. Sun and X. Liu, Coord. Chem. Rev., 2021, 441, 213983 CrossRef CAS.
- Z.-Z. Niu, L.-P. Chi, Z.-Z. Wu, P.-P. Yang, M.-H. Fan and M.-R. Gao, Natl. sci. open, 2023, 2, 20220044 CrossRef.
- F. Pan, X. Yang, T. O'Carroll, H. Li, K. J. Chen and G. Wu, Adv. Energy Mater., 2022, 2200586 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- P.-P. Yang and M.-R. Gao, Chem. Soc. Rev., 2023, 52, 4343–4380 RSC.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- P.-P. Yang, X.-L. Zhang, F.-Y. Gao, Y.-R. Zheng, Z.-Z. Niu, X. Yu, R. Liu, Z.-Z. Wu, S. Qin, L.-P. Chi, Y. Duan, T. Ma, X.-S. Zheng, J.-F. Zhu, H.-J. Wang, M.-R. Gao and S.-H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2016, 56, 796–800 CrossRef PubMed.
- L.-X. Liu, Y. Cai, H. Du, X. Lu, X. Li, F. Liu, J. Fu and J.-J. Zhu, ACS Appl. Mater. Interfaces, 2023, 15, 16673–16679 CrossRef CAS PubMed.
- X. Tan, K. Sun, Z. Zhuang, B. Hu, Y. Zhang, Q. Liu, C. He, Z. Xu, C. Chen, H. Xiao and C. Chen, J. Am. Chem. Soc., 2023, 145, 8656–8664 CAS.
- J. M. Yoo, H. Shin, D. Y. Chung and Y.-E. Sung, Acc. Chem. Res., 2022, 55, 1278–1289 CrossRef CAS PubMed.
- L. Yu, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 15294–15297 CrossRef CAS PubMed.
- Z. Wang, Y. Zhou, D. Liu, R. Qi, C. Xia, M. Li, B. You and B. Xia, Angew. Chem., Int. Ed., 2022, 61, e202200552 CrossRef CAS PubMed.
- L. Yu, H. B. Wu and X. W. D. Lou, Acc. Chem. Res., 2017, 50, 293–301 CrossRef CAS PubMed.
- H. Tong, S. Chen, P. Yang, C. Wang, J. Lu, X. Zeng, J. Tu, P. Wang, Z. Cheng and Q. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 52697–52705 CrossRef CAS PubMed.
- J. Wang, J. Wan and D. Wang, Acc. Chem. Res., 2019, 52, 2169–2178 CrossRef CAS PubMed.
- G. Wu, Y. Song, Q. Zheng, C. Long, T. Fan, Z. Yang, X. Huang, Q. Li, Y. Sun, L. Zuo, S. Lei and Z. Tang, Adv. Mater., 2022, 2202054 CAS.
- Z. Li, S. Ji, C. Xu, L. Leng, H. Liu, J. H. Horton, L. Du, J. Gao, C. He, X. Qi, Q. Xu and J. Zhu, Adv. Mater., 2022, 2209644 Search PubMed.
- Q. Wang, M. Dai, H. Li, Y. R. Lu, T. S. Chan, C. Ma, K. Liu, J. Fu, W. Liao, S. Chen, E. Pensa, Y. Wang, S. Zhang, Y. Sun, E. Cortés and M. Liu, Adv. Mater., 2023, 2300695 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- X. Yao, Y. Ke, W. Ren, X. Wang, F. Xiong, W. Yang, M. Qin, Q. Li and L. Mai, Adv. Energy Mater., 2018, 1803260 Search PubMed.
- C. Zhang, W. Shen, K. Guo, M. Xiong, J. Zhang and X. Lu, J. Am. Chem. Soc., 2023, 145, 11589–11598 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- T. Yang, X. Mao, Y. Zhang, X. Wu, L. Wang, M. Chu, C.-W. Pao, S. Yang, Y. Xu and X. Huang, Nat. Commun., 2021, 12, 6022 CrossRef CAS PubMed.
- Y. Rong, T. Liu, J. Sang, R. Li, P. Wei, H. Li, A. Dong, L. Che, Q. Fu, D. Gao and G. Wang, Angew. Chem., Int. Ed., 2023, e202309893 CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Y. Zhang, K. Li, M. Chen, J. Wang, J. Liu and Y. Zhang, ACS Appl. Nano Mater., 2019, 3, 257–263 CrossRef.
- Y. Sha, J. Zhang, X. Cheng, M. Xu, Z. Su, Y. Wang, J. Hu, B. Han and L. Zheng, Angew. Chem., Int. Ed., 2022, e202200039 CAS.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- X. Yan, M. Zhang, Y. Chen, Y. Wu, R. Wu, Q. Wan, C. Liu, T. Zheng, R. Feng, J. Zhang, C. Chen, C. Xia, Q. Zhu, X. Sun, Q. Qian and B. Han, Angew. Chem., Int. Ed., 2023, e202301507 CAS.
- K. Yao, J. Li, H. Wang, R. Lu, X. Yang, M. Luo, N. Wang, Z. Wang, C. Liu, T. Jing, S. Chen, E. Cortés, S. A. Maier, S. Zhang, T. Li, Y. Yu, Y. Liu, X. Kang and H. Liang, J. Am. Chem. Soc., 2022, 144, 14005–14011 CrossRef CAS PubMed.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2021, 61, e202113498 CrossRef PubMed.
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen, T.-K. Sham, J. Luo, Y. Yang, X. Xu and G. Zheng, Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01735h |
| This journal is © The Royal Society of Chemistry 2024 |