
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePicking the tyrosine-lock: chemical synthesis of the tyrosyl-DNA phosphodiesterase I inhibitor recifin A and analogues†
Taylor B.
Smallwood
 a,
Lauren R. H.
Krumpe
b,
Colton D.
Payne
a,
Victoria G.
Klein‡
c,
Barry R.
O'Keefe
bd,
Richard J.
Clark
*a,
Christina I.
Schroeder
*ce and
K. Johan
Rosengren
*a
a,
Lauren R. H.
Krumpe
b,
Colton D.
Payne
a,
Victoria G.
Klein‡
c,
Barry R.
O'Keefe
bd,
Richard J.
Clark
*a,
Christina I.
Schroeder
*ce and
K. Johan
Rosengren
*a
aThe University of Queensland, School of Biomedical Sciences, Brisbane, QLD 4072, Australia. E-mail: richard.clark@uq.edu.au; j.rosengren@uq.edu.au
bMolecular Targets Program, Centre for Cancer Research, National Cancer Institute, National Institute of Health, Frederick, MD 21702, USA
cChemical Biology Laboratory, Centre for Cancer Research, National Cancer Institute, National Institute of Health, Frederick, MD 21702, USA
dNatural Products Branch, Centre for Cancer Research, National Cancer Institute, National Institute of Health, Frederick, MD 21702, USA
ePeptide Therapeutics, Genentech Inc, 1 DNA Way, South San Francisco, CA 94080, USA. E-mail: Schroeder.christina@gene.com
First published on 14th June 2024
Abstract
The peptide recifin A is the inaugural member of the structurally intriguing new fold referred to as a tyrosine-lock. Its central four stranded β-sheet is stabilized by a unique arrangement in which three disulfide bonds and their interconnecting backbone form a ring that wraps around one of the strands, resulting in a Tyr side chain being buried in the molecular core. Here we aimed to establish a synthetic route to this complex class of natural products. Full length recifin A was successfully generated through native chemical ligation chemistry joining two 21 amino acid residue fragments. Surprisingly, reduced linear recifin A readily adopts the correct, topologically-complex fold via random oxidation of the cysteines, suggesting it is highly energetically favored. Utilizing our synthetic strategy, we generated five recifin A analogues to investigate the structural role of the central Tyr residue and provide the first insights into the structure activity relationship of recifin A towards its cancer target tyrosyl-DNA phosphodiesterase I.
Introduction
Marine sponges are a diverse source of unique natural products, with many containing bioactive peptides and proteins. These molecules possess a wide range of pharmacological effects including cytotoxic and anticancer activities.1,2 Recently a novel peptide, recifin A, was isolated from the marine sponge Axinella sp. during a screening campaign to identify inhibitors of the enzyme tyrosyl-DNA phosphodiesterase 1 (TDP1).3 TDP1 is a DNA repair enzyme that recognizes stalled DNA-topoisomerase 1 (TOP1)/DNA covalent complexes and catalyzes the hydrolysis of the 3′-phosphodiester bond.4 Clinically, TDP1 can reduce the impact of chemotherapeutic TOP1 inhibiting drugs, such as the camptothecin derivates topotecan and irinotecan, resulting in diminished DNA damage and reduced efficacy of this class of anti-tumor agents.5 Therefore, chemotherapy using TOP1 inhibitors in combination with TDP1 inhibitors could be a promising new avenue for clinical cancer treatment.Recifin A is the first described peptidic and allosteric inhibitor of TDP1.3 Unlike previous small molecule TDP1 inhibitors that target the active site located in the C-terminal catalytic domain of TDP1, recifin A requires the presence of the unstructured N-terminal regulatory domain of TDP1 for its inhibitory activity. Recifin A comprises 42 amino acid residues, and solution Nuclear Magnetic Resonance (NMR) spectroscopy studies revealed that it is the first member of a new structural family of cysteine-rich peptides, named the Tyr-lock family of peptides.3 The recifin A secondary and tertiary structure is stabilized by three disulfide bonds, Cys5–Cys21, Cys11–Cys39 and Cys22–Cys42, a I–III, II–V, IV–VI arrangement (Fig. 1). Peptides with three disulfide bonds often form topologically complex arrangements referred to as inhibitory cystine knots, with a I–IV, II–V, III–VI disulfide bond arrangement, in which two disulfide bonds and their interconnecting backbone form a ring through which the third disulfide bond is threaded.6,7 However, what is unique about the recifin A structure is that all three disulfide bonds together with backbone segments form a ring that wraps around the third β-strand (residues 27–29). The fold of the peptide is further stabilized by Tyr6, which is deeply buried in the peptide core and locked in place by interactions with surrounding residues (Fig. 1).
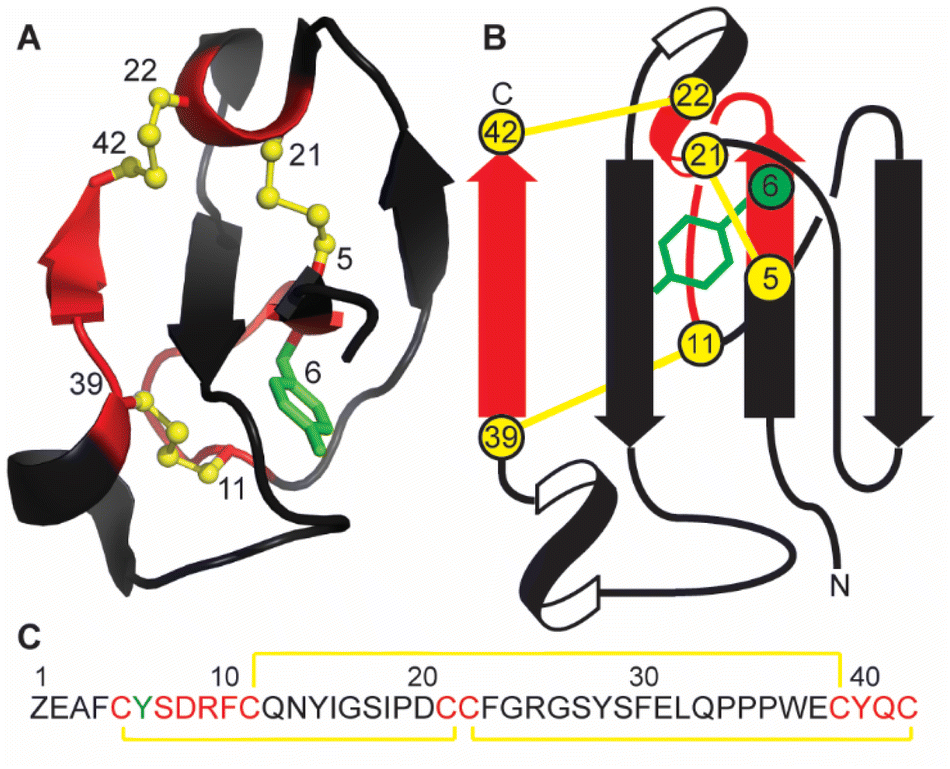 | ||
| Fig. 1 (A) Ribbon structure of recifin A showing the position of the buried Tyr6. (B) Schematic illustration showing threading of the third β-strand through the embedded ring formed by the three disulfide bonds. (C) The recifin A amino acid sequence. In all panels the disulfide bonds are highlighted in yellow, residues that form the ring in red, Tyr6 in green, and remaining residues in black. Z in the sequence represents pyroglutamate. | ||
For natural products, accessing sufficient native material for detailed activity and structural studies is often challenging. In the case of recifin A, to be able to further investigate the interaction between this peptide and TDP1, synthetic material is required. In this work we therefore focused on establishing a synthetic route for the production of recifin A, to allow for the generation of analogues, and to expand our knowledge of the key structural requirements of the Tyr-lock fold. We show that the linear recifin A can be assembled through ligation chemistry and efficiently oxidized to the correct disulfide isomer despite the complex fold. Functional data from analogues suggest that the TDP1 binding surface is extensive and that the buried tyrosine residue is critical for structural stability and consequently activity.
Results and discussion
Initial attempts at assembling the full-length recifin A via Fmoc-chemistry on 2-chlorotrityl resin were unsuccessful. Instead, it was necessary to employ a chemical ligation strategy. We chose to split the sequence between the central consecutive cysteines into two 21 amino acid fragments, and employ a native chemical ligation (NCL)8 strategy in which the C-terminal thioester was produced through hydrazide chemistry (Scheme 1).9,10 Individual N- and C-terminal recifin A fragments were successfully synthesized (Fig. S1 and Table S1†), and could be efficiently ligated into the full length peptide (Fig. S2†). The linear reduced peptide was subjected to thermodynamic oxidative folding using ammonium bicarbonate buffer in the presence of reduced and oxidized glutathione. Surprisingly, despite the expected complexity required for correct folding, a single dominant product appeared almost immediately under these conditions. This product was obtained in high purity after HPLC purification (Fig. S3†) and solution NMR spectroscopy revealed a well dispersed 1H NMR spectrum, implying that the peptide adopted an ordered structure in solution (Fig. S4†).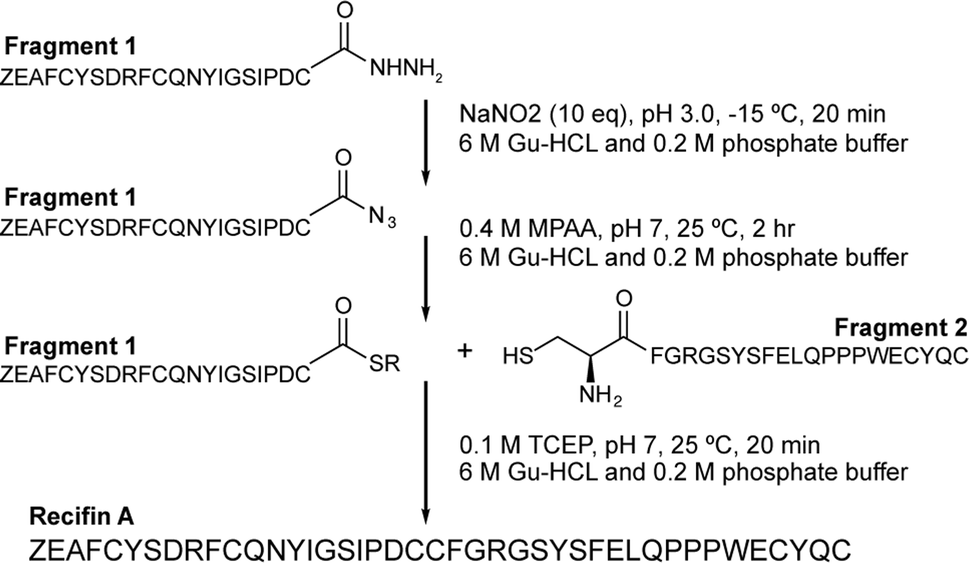 | ||
| Scheme 1 Synthetic strategy for recifin A using native chemical ligation of a C-terminal thioester produced by hydrazide chemistry and an N-terminal cysteine. Z – pyroglutamate. | ||
The native isolated recifin A and the synthetic version were compared using LC/MS analysis. Individual RP-HPLC analysis of the two peptides found they possessed the same retention time, and a co-elution experiment of the native and the synthetic recifin A showed no difference in retention time or peak shape (Fig. 2A). Comparing the two recifin A peptide's molecular charge envelope, identical ionization patterns and distribution of charge states were observed, with essentially identical isotopic distribution of [M + 3H]3+ (Fig. S5†). Furthermore, 2D NMR spectra including TOCSY and NOESY were recorded for synthetic recifin A and compared to the data used for structure determination of the native peptide, showing conserved peak patterns and positions (Fig. 2B). It should be noted that the NMR data of recifin A is highly sensitive to minute changes in pH conditions and concentration, making it difficult to replicate conditions perfectly. Consequently, some minor differences in chemical shifts are observed. A closer analysis of the chemical shifts and the recifin A structure revealed that any resonance with a notable perturbation is in close proximity to a titratable group, most prominently Glu31 and Glu38, thus sensing their respective protonation state. Altogether, these data verify that the synthetic recifin A possesses the same chemical properties as the isolated peptide.
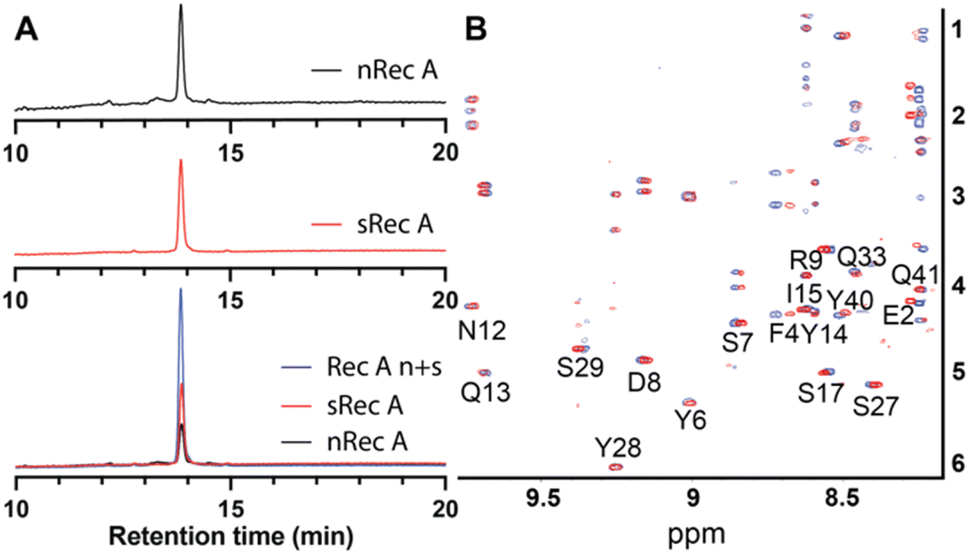 | ||
| Fig. 2 (A) Comparison of elution times of native (nRec A, red) and synthetic (sRec A, blue) recifin A and co-elution of native and synthetic recifin A (Rec A n + s, black). (B) Superposition of TOCSY spectra of native (red) and synthetic (blue) recifin A at 298 K. | ||
Based on the structure of the native recifin A peptide, we designed and synthesized five analogues (Fig. 3) to investigate the effects of the mutations on the peptide structure and activity. We first designed two peptides with mutations at the N-terminus. Recifin (3–42) is a truncated version of the native peptide, removing the first two N-terminal residues, pyroglutamic acid and glutamic acid. The [Pro1] recifin analogue replaces the N-terminal pyroglutamic acid residue with another five membered ring residue, proline. Next, we designed two analogues with modifications at Tyr6, the structurally important residue responsible for sterically stabilizing the native recifin A peptide core. This was conservatively replaced with another aromatic residue, phenylalanine ([Phe6] recifin), and with the smaller aliphatic residue alanine ([Ala6] recifin). The final recifin A analogue that was designed was [Ala10] recifin, where the surface-exposed Phe10 was replaced with Ala. Phenylalanine residues are rarely found on the surface of proteins, unless they are involved in intermolecular interactions.11,12 Therefore, it was hypothesized that Phe10 may be a key binding residue and involved in protein–protein interactions between recifin A and the regulatory domain of TDP1.
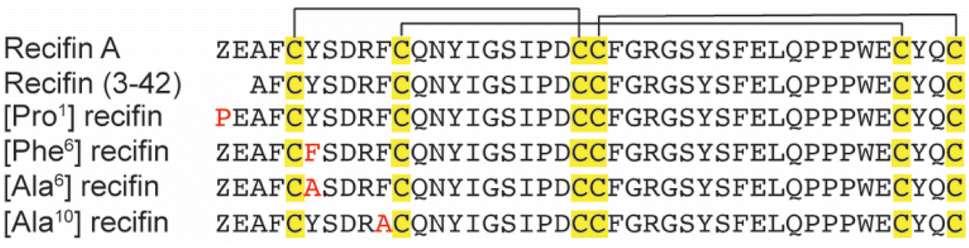 | ||
| Fig. 3 Aligned sequences of recifin A and analogues. Conserved cysteine residues are shown in yellow, with the black line highlighting the disulfide bond connection: Cys I–III, Cys II–V, and Cys IV–VI. Point mutations are shown in red. Z = pyroglutamic acid. | ||
Each recifin analogue was synthesized using the established hydrazide NCL methodology and folded under the same conditions as the synthetic recifin A (Fig. S1–S3 and Table S1†). All peptide analogues, apart from [Ala6] recifin, were found to oxidise into one isomer, which was confirmed by NMR spectroscopy to be correctly folded (Fig. S4†). Fig. 4 shows the comparison of the oxidative folding of native recifin A and [Phe6], highlighting that even with a change at this key position the correct fold is remarkably favoured. In contrast, folding of [Ala6] recifin resulted in multiple isomers and this analogue could not be purified to homogeneity (Fig. S3†). The 1D 1H NMR spectrum of partially purified peptide showed broad peaks lacking the wide dispersion characteristic of the recifin A fold, indicating that this variant was misfolded (Fig. S4†). The structures of all analogues, except for [Ala6] recifin, were further analyzed by 2D NMR spectroscopy. Secondary Hα chemical shifts revealed that the secondary structural features follow the same trend to that of the native recifin A peptide (Fig. S6†). All peptides were shown to possess a series of short β-strands and α-helices/turns, as indicated by significant stretches of positive and negative secondary chemical shifts, respectively.
 | ||
| Fig. 4 Oxidative folding of (A) recifin A and (B) [Phe6] recifin. HPLC traces of each time point taken for the oxidation of synthetic peptides. Oxidation was performed in 0.1 M ammonium bicarbonate (pH 8.0) with oxidized (0.5 mM) and reduced (2 mM) glutathione at a concentration of 0.125 mg mL−1 at room temperature. Aliquots were removed at time points 0, 8, and 48 h, quenched with 6 M Gu-HCl (pH 3.7). Samples were analyzed by analytical RP-HPLC on a C18 column using a gradient of 5% buffer B for the first 10 min followed by 5–65% B (buffer A: H2O/0.05% TFA; buffer B: 90% ACN/10% H2O/0.045% TFA) in 65 min. | ||
[Phe6] recifin was structurally investigated in more detail, given the peptide was able to fold despite a change to the class-defining Tyr6. Calculating a three-dimensional structure of [Phe6] recifin revealed a structure with essentially identical backbone conformation to native recifin A (Fig. 5). The key features in the Tyr-lock region observed in the native recifin A structure is maintained in the [Phe6] recifin analogue. Like in the native recifin A individual resonance signals are observed for the aromatic protons, resulting from the ring-flips being slowed down by the same steric packing. The removal of the Tyr6 hydroxyl group does however result in the loss of two hydrogen bonds. In recifin A it both donates a hydrogen bond to the backbone carbonyl of Glu31 and accepts a hydrogen bond from the backbone amide of Gln33. The lack of these interactions stabilizing this region results in increased dynamics, evident from significant line broadening of the backbone amides of residues 30–33. Interestingly two other side chain hydrogen bonds in the region, from Ser27 to Glu8 carbonyl and from Ser29 to Glu31 carbonyl are maintained, as these hydroxyl protons remain visible in the spectra, like in native recifin A. Thus, while the aromatic ring of the Phe residue is sufficient to maintain the so-called Tyr-lock, increased dynamics surrounding this region and minor differences in the backbone conformation of residues 31–33 are evident.
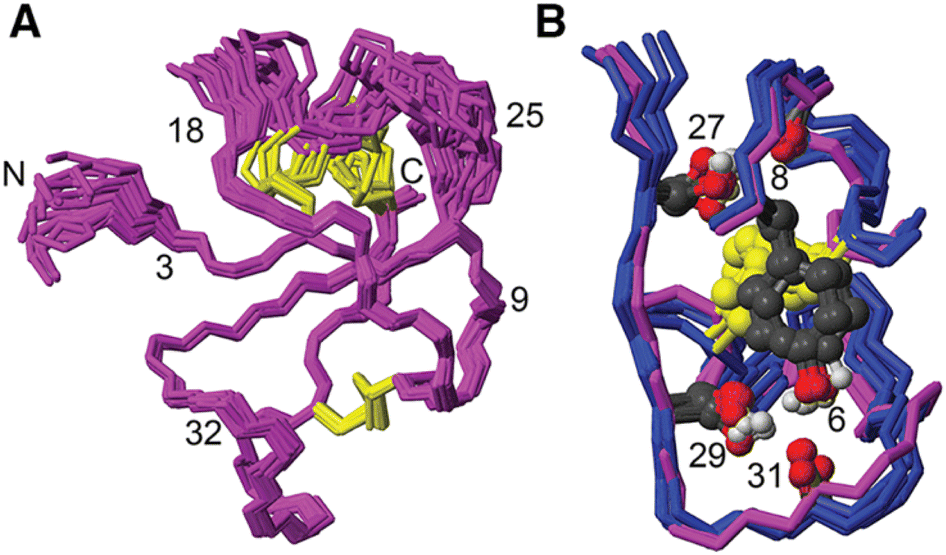 | ||
| Fig. 5 (A) Solution NMR structure of [Phe6] recifin showing disulfides in yellow. (B) Superposition of [Phe6] recifin (pink) and native recifin A (blue) (PDB ID: 6XN9), highlighting the similarities in the Tyr-lock region and hydrogen bonds. | ||
To evaluate the thermal stability for native and synthetic recifin A and analogues, we collected 1D 1H NMR spectra at increasing temperatures in 10 °C increments (20–60 °C) at 15 min intervals. Following collection of the final spectra at 60 °C, we allowed the sample to cool and collected an additional spectrum at 20 °C (Fig. S7†). Whilst we observed some expected chemical shift changes in the amide region with increased temperature, we did not observe any noticeable temperature dependent line broadening for any of the peptides, and the final spectra collected at 20 °C were identical to the initial spectra. This suggests that the β-sheets in combination with the complex disulfide bond framework renders these peptides highly stable towards thermal assault.
To investigate if our designed modifications had functional effects in terms of ability to inhibit TDP1, native recifin A and all synthetic analogs were tested side-by-side in a fluorescence resonance energy transfer (FRET) assay system. Native isolated recifin A was found to inhibit full-length TDP1 enzymatic activity in a concentration dependent manner with an IC50 of 0.80 μM, moderately higher than initially reported.2 The synthetic recifin A peptide and [Pro1] recifin were also found to inhibit TDP1 with equivalent potency (0.58 and 0.69 μM, respectively) in our assay, as shown in Table 1 and Fig. S8.† Surprisingly, neither [Phe6] recifin nor [Ala10] recifin showed any inhibition of TDP1 at the highest test dose of 10 μM, and the truncated analogue recifin 3–42, was also found to have no measurable TDP1 inhibitory activity. This suggests that recifin A engages TDP1 using an extended binding surface given that the N-terminal part of recifin A and Phe10 are spatially separated, yet both are important for TDP1 inhibitory activity. Since the replacement of the N-terminal pyroglutamic acid with Pro was tolerated the reduced binding of the truncated analogue is likely a result of the removal of the Glu2 negatively charged side chain. Thus, the interaction likely involves both charge attraction and hydrophobic packing. Importantly, recifin A requires the disordered N-terminal regulatory domain of TDP1 for activity,2 and these findings are consistent with an interaction in which this disordered region ‘wrap around’ recifin A, creating the extended binding surface. Alternatively, recifin A might be ‘sandwiched’ between the regulatory and catalytic domains, thus being involved in multiple interactions. Either arrangement could explain why reducing the rigidity and increasing structural dynamics by replacing the buried Tyr6 with a Phe is sufficient to reduce activity, despite this modification not directly affecting the peptide surface, as evident from our NMR structure. Further variants are required to be produced to explore the structure–activity relationship between recifin A and TDP1. Notable putative interaction surfaces include Arg9 and Arg25, which are adjacent to Phe10, and the aromatic cluster involving Tyr28, Phe30 and Tyr40 located proximal to the N-terminus.
| Peptide | IC50 μM | 95% CI | R 2 |
|---|---|---|---|
| Native recifin A | 0.80 | 0.08–7.93 | 0.90 |
| Synthetic recifin A | 0.58 | 0.20–1.77 | 0.96 |
| Recifin A (3–42) | >10 | ND | ND |
| [Pro1] recifin A | 0.69 | 0.26–1.88 | 0.97 |
| [Ala10] recifin A | >10 | ND | ND |
| [Phe6] recifin A | >10 | ND | ND |
Experimental
Materials
2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 9H-fluoren-9-ylmethoxycarbonyl (Fmoc) protected L-amino acids were obtained from Mimotopes, Australia. Sodium dihydrogen phosphate, disodium hydrogen phosphate, N,N-dimethylformamide (DMF) and HPLC-grade acetonitrile (ACN) were from Merck, USA. Peptide synthesis-grade trifluoroacetic acid (TFA) and diethyl ether were purchased from Novachem, Australia. Guanidine hydrochloride (Gu-HCl), sodium nitrite, tris(2-carboxyethyl)phosphine (TCEP), 2,2′-(ethylenedioxy)diethanethiol (DODT), triisopropylsilane (TIPS) and 4-mercaptophenylacetic acid (MPAA) were purchased from Sigma-Aldrich, Australia.Peptide synthesis
Native recifin A and analogues thereof could not successfully be assembled in one fragment using Fmoc solid phase peptide synthesis (SPPS) despite modifications to resin loading, coupling times and reagent equivalents used. Therefore, a native chemical ligation (NCL) approach using peptide hydrazides was applied to ligate the N- and C-terminal fragments between the 3rd and the 4th cysteine residues. This site was chosen as it is located at the center of the sequence, whereas all other cysteines are concentrated towards the N-terminus or C-terminus. An NCL could not be performed between Asp20 and Cys21 due to potential cyclisation between the sidechain carboxylic of Asp20 and the C-terminal hydrazide.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2.5:2.5) at room temperature for 2 h. TFA was removed under vacuum and peptide precipitated with ice-cold diethyl ether. The precipitate was filtered and dissolved in 50% ACN containing 0.05% TFA. The remaining diethyl ether was removed under vacuum and the peptide solution lyophilized. Crude peptides were purified by reverse phase-HPLC (RP-HPLC) on a C18 column (Phenomenex Jupiter 300 Å, 10 μm, 250 × 21.2 mm) using a gradient of 0–90% B (buffer A: 0.05% TFA; buffer B: 90% ACN/0.045% TFA) in 90 min at a flow rate of 8 mL min−1. Electrospray ionization-mass spectroscopy (ESI-MS) with declustering potential set to 40 was used to confirm the molecular mass of the synthesized peptide fragments using an ABSciex API2000 before lyophilization (Fig. S1†).
:5:2.5:2.5) at room temperature for 2 h. TFA was removed under vacuum and peptide precipitated with ice-cold diethyl ether. The precipitate was filtered and dissolved in 50% ACN containing 0.05% TFA. The remaining diethyl ether was removed under vacuum and the peptide solution lyophilized. Crude peptides were purified by reverse phase-HPLC (RP-HPLC) on a C18 column (Phenomenex Jupiter 300 Å, 10 μm, 250 × 21.2 mm) using a gradient of 0–90% B (buffer A: 0.05% TFA; buffer B: 90% ACN/0.045% TFA) in 90 min at a flow rate of 8 mL min−1. Electrospray ionization-mass spectroscopy (ESI-MS) with declustering potential set to 40 was used to confirm the molecular mass of the synthesized peptide fragments using an ABSciex API2000 before lyophilization (Fig. S1†).
LC-MS analysis of native and synthetic recifin A
Peptides were analyzed on an Agilent Q-TOF LC-MS system as previously described.3 Briefly, approximately 250 ng of peptide was injected onto an Poroshell 300SB-C18, 5 μm, 2.5 × 75 mm analytical HPLC column, heated to 40 °C and equilibrated in H2O/0.1% formic acid (A). A linear gradient from 100% A to 60% ACN + 0.1% formic was executed at a flow rate of 1 mL min−1 over 5 min to elute the peptides, followed by a 1 min gradient to 100% ACN + 0.1% formic acid. Spectra were collected within 100–3200 m/z range at 1 spectra per s, with internal reference mass correction at 121.05 and 1221.99 m/z. Peptide mass spectra were deconvoluted for mass determination using Agilent BioConfirm data analysis software (Fig. S5†).RP-HPLC co-elution of native and synthetic recifin A
Native, synthetic recifin A and a mixture of both peptides were analyzed for retention time using a Shimadzu Nexera RP-HPLC equipped with a LC-20AD pump and a SPD-M30A diod array detector on a Phenomenex Gemini column NX-C18 (5 μm, 110 Å) using a 0–60% B gradient over 20 min (buffer A: 0.05% TFA, buffer B: 90% ACN/0.045% TFA).NMR spectroscopy
Recifin analogues were dissolved in 90% H2O/10% D2O at a concentration of 1 mM, pH ∼4.9. Standard one- and two-dimensional data sets were recorded at 298 K on a Bruker Avance III 900 MHz spectrometer equipped with a cryoprobe and processed with Topspin 4.0.3 (Bruker Biospin), with data referenced to the residual water signal at 4.77 ppm. 1H–1H TOCSY13 (Total Correlation Spectroscopy) and NOESY14 (Nuclear Overhauser Spectroscopy) experiments were recorded with a sweep width of 12 ppm, 512 increments, and mixing times of 80 and 150 ms, respectively. TOCSY experiments were recorded with four scans and NOESY experiments with 32 scans. Additional 1H–13C and 1H–15N HSQC (Heteronuclear Single Quantum Coherence spectroscopy) spectra were recorded at natural abundance for [Phe6] recifin. 1H–13C HSQC data was recorded with 96 scans and 200 increments for and 15N data was recorded with 128 scans and 128 increments. The program CARA15 (Computer Aided Resonance Assignment) was used to assign the data using sequential assignment strategies16 and derive structural information.17 Analysis of the secondary structural features of the recifin analogues was achieved by direct comparison of each analogues secondary Hα shifts to that of native recifin. Secondary Hα shifts were determined by comparison of the observed chemical shift of each Hα proton to that of the equivalent random coil value.18 Additional TOCSY data were recorded at varying temperatures (288, 293, 298, 303 and 308 K) for [Phe6] recifin to monitor the temperature dependence of the backbone amide protons.21 and final structures were subjected to simulated annealing and refined in explicit water using CNS 1.21.22 Stereochemical analysis was conducted by MolProbity23 by comparing the generated [Phe6] recifin structures to that of structures published in the Protein Data Bank (PDB ID: 6XN9). The 20 best structures which contained no violations of distances or dihedral angles above 0.2 Å or 2°, and had low energy were chosen to represent the structure of [Phe6] recifin (Table S2†). MOLMOL24 was used to generate images of the secondary and tertiary structure of [Phe6] recifin. The structures, restraints, and chemical shifts have been submitted to the PDB and BRMB (Biological Magnetic Resonance Bank), the accessions codes are (8V2V) and (31130), respectively.
FRET-based TDP1 enzymatic activity inhibition assay
The FRET-based TDP1 enzymatic activity assay is based on the removal of an eosin-linked phospho-tryosyl fluorophore from the 3′ end of the DNA substrate (TAMRA-5′-GATCTAAAAGACTT-pY-3′-eosin) by TDP1 which results in an increase in fluorescence from the 5′ -linked TAMRA substrate. The assay has been described in detail previously but was slightly modified for this study.3 Briefly, 3-fold concentrated dilution series of peptides, TDP1 enzyme, and FRET substrate were prepared in FRET buffer (1X PBS pH 7.4, 80 mM KCl, 1 mM TCEP). TDP1, peptides, and substrate were then added sequentially to wells of a black, 384-well plate (Greiner 784900), mixed to combine, and analysed for one hour (520 nmex/550 nmem). Assay concentrations of TDP1 and FRET substrate were 1 nM and 0.25 μM, respectively. Results were normalized to assay controls (no TDP1 and un-inhibited TDP1), and dose–response curves and IC50 estimates were generated using GraphPad Prism software non-linear regression tools based on the 15 min timepoint (least squares fit with three parameters).Conclusions
We have here described the first chemical synthesis of the novel allosteric TDP1 inhibitor, recifin A, the inaugural member of the new Tyr-lock fold. We have shown that recifin A and analogues can readily be synthesized using hydrazide native chemical ligation methodology and that recifin A folds into primarily one species probably driven by the complex disulfide bond network folding around a buried aromatic residue. Evaluation of an initial series of recifin A analogues revealed that the central Tyr6 residue, giving the family its name, can be replaced with a Phe without significant changes in folding or 3D structure, but that it does increase structural dynamics and results in reduction of TDP1 inhibition potency. In contrast, replacing Tyr6 with an Ala is detrimental for folding. We also showed that replacing the N-terminal pyroglutamic acid with a Pro, [Pro1] recifin, did not alter activity, but that truncating the first two residues recifin (3–42), resulted in drastic reduction in inhibition of TDP1. Having established a synthetic route for recifin A opens the door for synthesis of further analogues enabling detailed studies of the allosteric mechanism by which recifin A inhibits TDP1. Understanding allosteric inhibition of TDP1 could lead to design of novel and selective TDP1 inhibitors sensitizing cells to TOP1 inhibitors for the treatment of cancer.Author contributions
TBS, LRHK, CDP, VGK – investigation, formal analysis, writing original draft. BRO, RJC, CIS, KJR – conceptualization, formal analysis, project administration, resources, writing original draft, writing – review and editing.Conflicts of interest
LRHK, BRO, CIS and KJR are co-inventors on a patent relating to tyrosyl-lock peptides. CIS is a Genentech Inc employee and a shareholder of Roche.Acknowledgements
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, Frederick National Lab, and the National Cancer Institute, Center for Cancer Research (Z01-BC 006150 and BC 006161).Notes and references
- Y. Hu, J. Chen, G. Hu, J. Yu, X. Zhu, Y. Lin, S. Chen and J. Yuan, Mar. Drugs, 2015, 13, 202–221 CrossRef PubMed.
- C. Jiménez, ACS Med. Chem. Lett., 2018, 9, 959–961 CrossRef PubMed.
- L. R. H. Krumpe, B. A. P. Wilson, C. Marchand, S. N. Sunassee, A. Bermingham, W. Wang, E. Price, T. Guszczynski, J. A. Kelley, K. R. Gustafson, Y. Pommier, K. J. Rosengren, C. I. Schroeder and B. R. O'Keefe, J. Am. Chem. Soc., 2020, 142, 21178–21188 CrossRef CAS PubMed.
- A. Zakharenko, N. Dyrkheeva and O. Lavrik, Med. Res. Rev., 2019, 39, 1427–1441 CrossRef CAS PubMed.
- T. M. Khomenko, A. L. Zakharenko, A. A. Chepanova, E. S. Ilina, O. D. Zakharova, V. I. Kaledin, V. P. Nikolin, N. A. Popova, D. V. Korchagina, J. Reynisson, R. Chand, D. M. Ayine-Tora, J. Patel, I. K. H. Leung, K. P. Volcho, N. F. Salakhutdinov and O. I. Lavrik, Int. J. Mol. Sci., 2019, 21, 126 CrossRef PubMed.
- E. Schwarz, Biol. Chem., 2017, 398, 1295–1308 CAS.
- N. L. Daly and D. J. Craik, Curr. Opin. Chem. Biol., 2011, 15, 362–368 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- J.-S. Zheng, S. Tang, Y.-K. Qi, Z.-P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- P. Chakrabarti and J. Janin, Proteins: Struct., Funct., Bioinf., 2002, 47, 334–343 CrossRef CAS PubMed.
- D. Talavera, D. L. Robertson and S. C. Lovell, PLoS One, 2011, 6, e21053 CrossRef CAS PubMed.
- L. Braunschweiler and R. R. Ernst, J. Magn. Reson., 1983, 53, 521–528 CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS.
- R. L. J. Keller, The computer aided resonance assignment tutorial, CANTINA Verlag, 1st edn, 2004 Search PubMed.
- K. Wüthrich, NMR of proteins and nucleic acids, John Wiley & Sons, New York, 1986 Search PubMed.
- C. I. Schroeder and K. J. Rosengren, Methods Mol. Biol., 2020, 2068, 129–162 CrossRef CAS PubMed.
- D. Wishart, C. Bigam, A. Holm, R. Hodges and B. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- Y. Shen and A. Bax, J. Biomol. NMR, 2013, 56, 227–241 CrossRef CAS PubMed.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249–261 CrossRef CAS PubMed.
- P. Güntert, Methods Mol. Biol., 2004, 278, 353–378 Search PubMed.
- A. T. Brünger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- R. Koradi, M. Billeter and K. Wüthrich, J. Mol. Graphics, 1996, 14, 51–55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC traces, MS data, NMR data. See DOI: https://doi.org/10.1039/d4sc01976h |
| ‡ Current address: Laboratory of Bioorganic Chemistry, National Institute for Diabetes and Digestive Kidney Diseases, National Institute of Health, Bethesda, MD, 20892, United States. |
| This journal is © The Royal Society of Chemistry 2024 |