
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering charge transfer dynamics of a lead halide perovskite–nickel(II) complex for visible light photoredox C–N coupling†
Vishesh
Kumar
a,
Sunil Kumar
Patel
 b,
Ved
Vyas
a,
Deepak
Kumar
a,
E. Siva
Subramaniam Iyer
*b and
Arindam
Indra
*a
b,
Ved
Vyas
a,
Deepak
Kumar
a,
E. Siva
Subramaniam Iyer
*b and
Arindam
Indra
*a
aDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, 221005, UP, India. E-mail: arindam.chy@iitbhu.ac.in
bSchool of Chemical and Materials Sciences, Indian Institute of Technology Goa, Ponda, Goa, India. E-mail: essiyer@iitgoa.ac.in
First published on 25th July 2024
Abstract
Photoredox catalysis involving perovskite quantum dots (QDs) has gained enormous attention because of their high efficiency and selectivity. In this study, we have demonstrated CsPbBr3 QDs as photocatalysts for the C–N bond formation reaction. The introduction of Ni(dmgH)2 (dmgH = dimethyl glyoximato) as a cocatalyst with CsPbBr3 QDs facilitates photocatalytic C–N coupling to form a wide variety of amides. The optimized interaction between the cocatalyst and photocatalyst enhances charge transfer and mitigates charge recombination, ultimately boosting photocatalytic performance. The photocatalytic activity is notably influenced by the variation in the amount of cocatalyst and 7 wt% Ni(dmgH)2 produces the best yield (92%) of amide. Femtosecond transient absorption spectroscopy reveals that the dynamics of the trap states of QDs are affected by cocatalyst. Further, Ni(dmgH)2 facilitates molecular oxygen activation to form superoxide radicals, which further initiates the radical pathway for the C–N coupling.
Introduction
Visible light-driven photoredox catalysis has been widely explored as a versatile and effective platform for the synthesis of different organic compounds.1–3 When exposed to visible light, semiconductor photocatalysts undergo a redistribution of surface electron clouds, triggered by the excitation of electrons from the valence band (VB) to the conduction band (CB).4–8 This photophysical process leads to the concurrent generation of both reducing and oxidizing centers on the photocatalyst surface.9,10 The photogenerated electrons and holes with the appropriate lifetimes are efficiently transported to the catalytic sites to initiate the redox processes.11–13In this context, CsPbBr3 QDs have been explored as the photoredox catalyst for various organic reactions.14,15 The kinetic energy associated with the CB electrons of QDs and the favorable reduction potential enable them to overcome the energy barriers and facilitate charge transfer, ultimately favoring photocatalytic activity. High light-absorption efficiency, low exciton binding energy, high carrier mobility, and optimized electron–hole diffusion length make them ideal for photoredox catalysis.16,17 Further, the long-lived active carriers of CsPbBr3 QDs promote photocatalytic organic reactions.18
For example, CsPbBr3 QDs have been employed to selectively oxidize benzyl alcohols to benzaldehydes under visible light irradiation.19 Similarly, photoredox catalysis of CsPbBr3 QDs has been explored for the oxidative coupling of thiols to disulfides, cross-coupling of phosphonates with tertiary amines, α-alkylation of aldehydes, etc.20 Recently, Yan groups explored the photoredox properties of CsPbBr3 QDs for organic transformation reactions such as C–C, C–O, and C–N coupling reactions.21 CsPbBr3 QDs have been also explored by other groups for facilitating coupling reactions including C–C,22,23 N–N,24 C–S,25 or C–P25 bond formation processes.
On the other hand, the photocatalytic production of amide by the reaction of aldehyde and amine has been explored by different groups.26,27 Amides serve as the foundational components of natural peptides and crucial intermediates in the production of polymers, agrochemicals, and pharmaceuticals.27–30 While simple, the traditional thermal condensation of carboxylic acids and amines is limited to substrate scope because of the harsh reaction conditions.31 Similarly, traditional approaches involving derivatives of activated carboxylic acids and amines, such as the Beckmann rearrangement, Schmidt and Staudinger reaction often result in a substantial waste production.32 This ongoing challenge of achieving efficient amide synthesis while minimizing waste has prompted the exploration of novel photocatalytic strategies from both industrial and academic standpoints.
However, most of the photochemical methods for amide synthesis in the homogeneous medium suffer from drawbacks like costly catalysts, use of non-oxygen oxidizing agents, and limited tolerance towards secondary amines. Simpler methodologies using photocatalysts like phenazine salt, Rose Bengal, and aminoanthraquinone derivatives were found to be promising.26 Further, heterogeneous photoredox catalysts (Ag/g-C3N4, Ni/g-C3N4, Ag2O/P–C3N4, TiO2, and Fe3O4/PDA/CdS) have been demonstrated for oxidative amination of aldehydes.26
In this context, we have explored the photoredox properties of CsPbBr3 QDs for the formation of a series of structurally versatile amides by the reaction of aldehydes and amines. To improve the charge transfer dynamics and access catalytic active sites, [Ni(dmgH)2] was used as the cocatalyst with CsPbBr3 QDs. The combination of [Ni(dmgH)2] and CsPbBr3 produced high efficiency for the oxidative C–N bond formation by the reaction of secondary amines and aldehydes to form structurally diverse amides (highest yield = 92%). The catalyst can be recycled four times with a minimum loss of initial activity. Further, the transient absorption spectroscopic studies have revealed that [Ni(dmgH)2] acts as an electron funnel, enabling fast electron transfer from QDs to the catalytic centers.
Results and discussion
Syntheses and characterizations of the catalysts
Metal halide perovskite CsPbBr3 QDs were synthesized by hot-injection method following a literature report (Experimental†).33 The powder X-ray diffraction (PXRD) revealed the monoclinic crystal structure of CsPbBr3 QDs (JCPDS card no. 18-0364) (Fig. S1a and b†).33,34 The transmission electron microscopy (TEM) study revealed the average particle size as 5.5 ± 1 nm (Fig. S1c†). HRTEM detected the lattice spacing corresponding to (200), (210), (202), (211), and (002) planes of monoclinic CsPbBr3 (Fig. S1c–e†).35,36 The energy-dispersive X-ray (EDX) spectroscopy confirmed the presence of the elements Cs, Pb, and Br (Fig. S2†). For the photoredox reaction, [Ni(dmgH)2] was used as the cocatalyst to improve the efficiency and selectivity of the reaction. The addition of different weight% of [Ni(dmgH)2] (3, 5, 7, and 9 wt%) into the reaction mixture showed a significant variation in the photocatalytic activity (Table S1†) (see later). Therefore, we have carried out a detailed optoelectronic characterization of the catalyst systems having different amounts of [Ni(dmgH)2] with CsPbBr3 QDs. The UV-visible diffuse reflectance spectroscopy (DRS) revealed the bandgap of CsPbBr3 QDs as 2.28 eV and the addition of [Ni(dmgH)2] did not alter the bandgap of the photocatalyst (Fig. S3a and b†).37 However, a broad peak for ligand-to-metal charge transfer (LMCT) in [Ni(dmgH)2] was detected after the introduction of the cocatalyst to CsPbBr3 (Fig. S3a†).38 The absorption maxima was shifted to lower wavelength (blue shift) due to the electronic interaction of [Ni(dmgH)2] with CsPbBr3 QDs.In Mott–Schottky (MS) plot, both CsPbBr3 QDs and [Ni(dmgH)2] exhibited positive slopes within the frequency range of 0.5–1.5 kHz, signifying the formation of an n–n-type heterojunction. Based on MS plots, we have determined the flat band potentials as −0.99 eV and −0.59 eV vs. NHE (normal hydrogen electrode) for CsPbBr3 and [Ni(dmgH)2], respectively (Fig. 1b and S4†).38–40 As the conduction band minimum (CBM) of an n-type semiconductor is about 0.1 or 0.2 eV higher than the flat band potential, the CBMs of CsPbBr3 QDs and [Ni(dmgH)2] are determined to be −1.09 eV and −0.69 eV, respectively. The valence band maximum (VBM) of CsPbBr3 QDs is calculated to be 1.19 eV vs. NHE. Therefore, the Fermi level energy (Ef) of CsPbBr3 QDs is high enough to transfer the photogenerated electrons from the CB of CsPbBr3 to the LUMO of [Ni(dmgH)2] (Fig. 1c).41
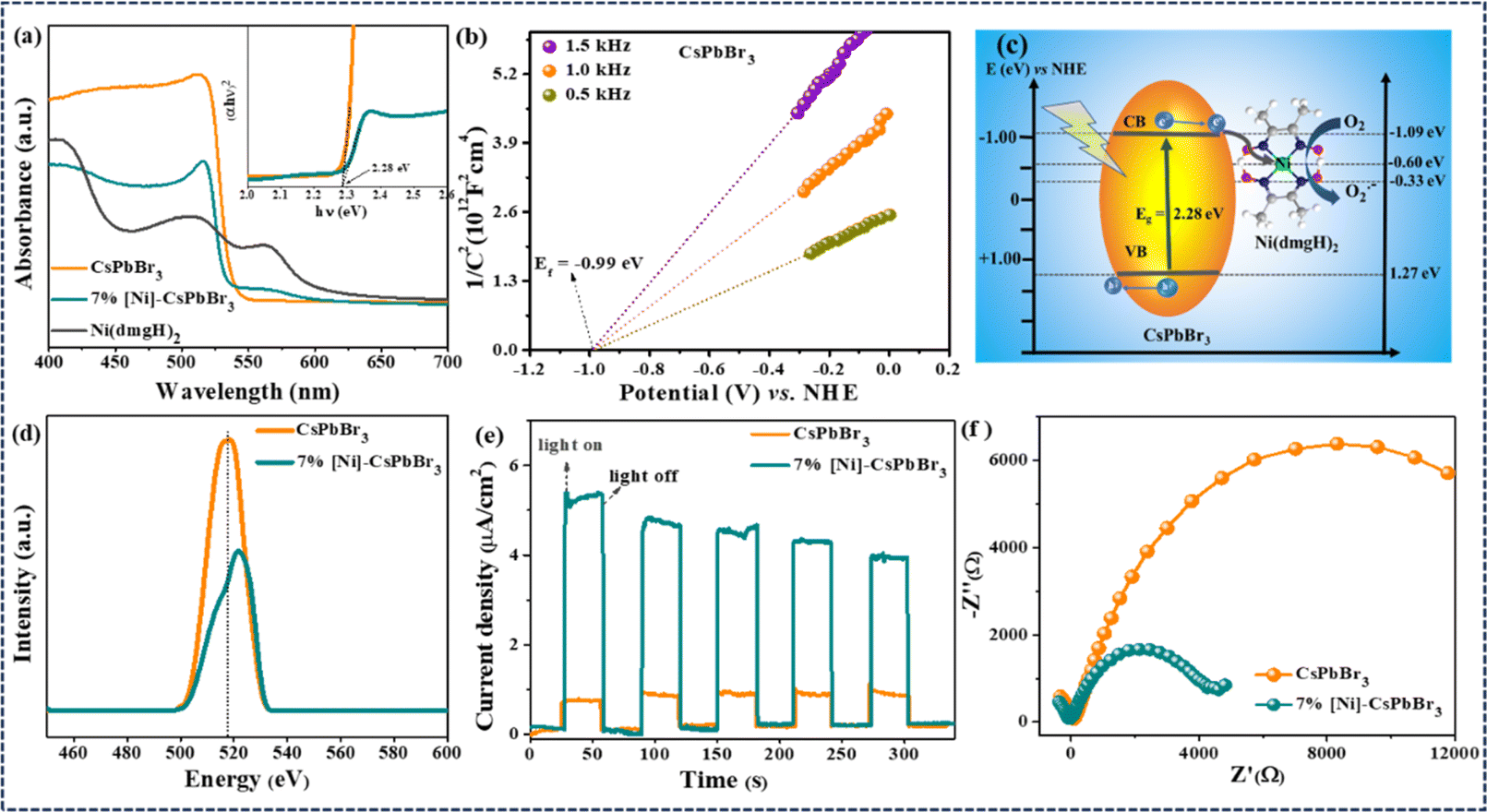 | ||
| Fig. 1 (a) The UV-vis diffuse reflectance spectra of CsPbBr3 QDs, 7% [Ni]-CsPbBr3 and [Ni(dmgH)2], inset showing corresponding Tauc plot, demonstrating no significant change in the bandgap after the introduction of the cocatalyst to CsPbBr3 QDs. (b) Mott–Schottky plot of CsPbBr3 QDs. (c) Depiction of the conduction band minima and valence band maxima for CsPbBr3 QDs and [Ni(dmgH)2], derived from Tauc plot and Mott–Schottky studies. (d) Photoluminescence spectra of CsPbBr3 QDs and 7% [Ni]-CsPbBr3 showing a reduction in the charge recombination process after the introduction of cocatalyst to CsPbBr3 QDs. (e) Photocurrent measurements of CsPbBr3 QDs and 7% [Ni]-CsPbBr3 under light/dark conditions. (f) Electrochemical impedance spectroscopic studies of CsPbBr3 QDs and 7% [Ni]-CsPbBr3, revealing the low charge transfer resistance for 7% [Ni]-CsPbBr3 (condition for recording photocurrent and EIS: electrolyte: 0.1 M TBAPF6 in ethyl acetate (20 mL), photocatalyst@FTO: working electrode, Ag/AgCl: reference electrode, and Pt-wire: counter electrode; 0.0 V vs. Ag/AgCl was applied for photocurrent measurements. Light source: 15 W blue LED). | ||
Photoluminescence (PL) spectrum of CsPbBr3 QDs showed the emission peak at 517 nm (Fig. 1d).42 The emission peak intensity is gradually decreased with the increasing amount of cocatalyst as the introduction of [Ni(dmgH)2] into CsPbBr3 solution significantly reduced the charge recombination process (Fig. 1d and S5†). In [Ni(dmgH)2], the strong π-acceptor conjugation in the ligand backbone facilitates the electron transfer from CsPbBr3 QDs to the cocatalyst, minimizing the charge recombination process.43 The trend in the PL intensity is also reflected in the photocatalytic C–N coupling reaction, where the best activity is observed with 7% [Ni]-CsPbBr3 (Table S4†).
For a more comprehensive understanding, we recorded the PL of [Ni(dmgH)2] as well (Fig. S5†). PL reveals that [Ni(dmgH)2] has emission peaks at 507 nm and 562 nm.38 A slight shift in the two fluorescence peaks was observed in 7% [Ni]-CsPbBr3 compared to pure [Ni(dmgH)2], which can be attributed to the charge transfer from CsPbBr3 QDs to the cocatalyst (Fig. S5†).
Further, the photocurrent response of the catalyst systems was recorded under on/off cycling of the light (Fig. 1e and S6†). Compared to CsPbBr3 QDs, [Ni]-CsPbBr3 photocatalysts showed a substantial improvement in the photocurrent density, and the best photocurrent response was achieved with 7% [Ni]-CsPbBr3, indicating efficient separation of charges after cocatalyst addition. The [Ni(dmgH)2] captures the photogenerated electrons from CsPbBr3 QDs, effectively minimizing charge recombination and enhancing the charge transfer.42 The effective electron transfer was revealed through electrochemical impedance spectroscopy (EIS) measurements (Fig. S7†). The Nyquist plots of [Ni]-CsPbBr3 exhibited noticeably smaller diameters in comparison to that of CsPbBr3 QDs and 7% [Ni]-CsPbBr3 showed the lowest charge-transfer resistance (Rct) value (Fig. 1f).44,45
The HOMO–LUMO energy gap of the cocatalyst was determined using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in the presence of light and in the dark (Fig. 2a and S8†). In ground state, the energy gap between CB of CsPbBr3 QDs and LUMO of [Ni(dmgH)2] is high. In the presence of light, the HOMO of [Ni(dmgH)2] shifts to a more positive potential value while the LUMO moves to a further negative potential. This leads to a reduction in the energy gap between the conduction band potential of CsPbBr3 and the LUMO of [Ni(dmgH)2], facilitating the transfer of the photogenerated CB electrons to the cocatalyst. As a result, O2 molecules are faster reduced to superoxide, improving the rate of photocatalytic C–N coupling reaction (see later).
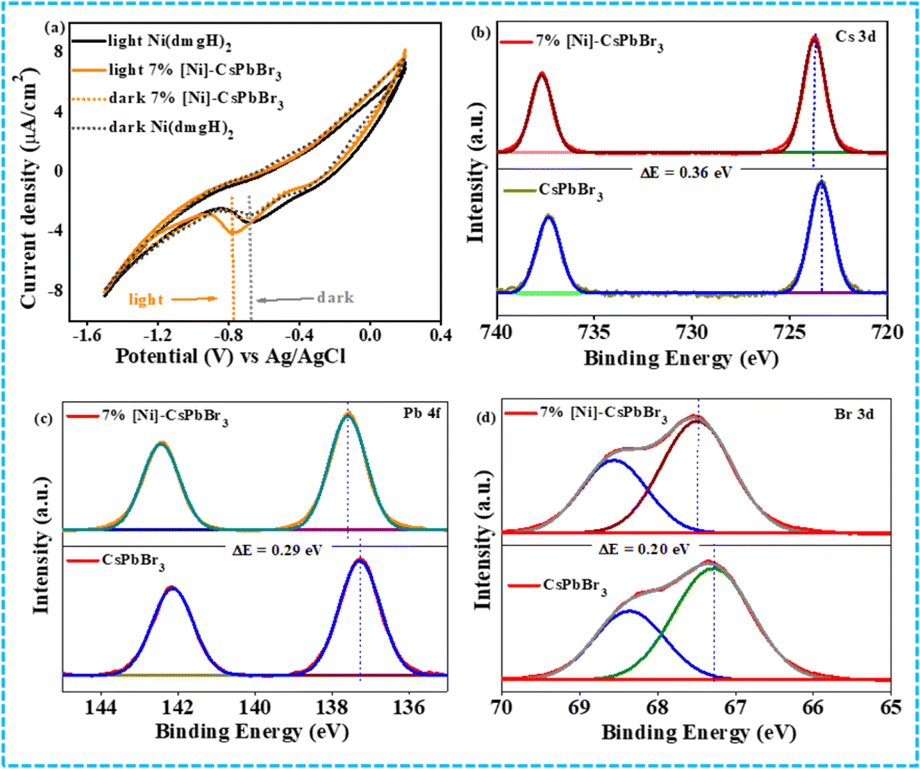 | ||
| Fig. 2 (a) Cyclic voltammogram of CsPbBr3 QDs and 7% [Ni]-CsPbBr3 in the dark and light. XPS spectra of CsPbBr3 and 7% [Ni]-CsPbBr3 (b) Cs 3d, (c) Pb 4f, (d) Br 3d. | ||
The X-ray photoelectron spectroscopy (XPS) study has also shown strong electronic interaction between CsPbBr3 QDs and cocatalyst (Fig. 2b–d). The comparison of Cs 3d XPS of CsPbBr3 QDs and 7% [Ni]-CsPbBr3 showed a positive shift (0.33 eV) in the binding energies of Cs 3d5/2 (723.82 eV) and Cs 3d3/2 (737.79 eV) peaks after the cocatalyst incorporation, indicating electron transfer from the CB of photocatalyst to cocatalyst.
Similarly, Pb 4f and Br 3d XPS exhibited positive shifts in the binding energies in 7% [Ni]-CsPbBr3 compared to that of CsPbBr3. In contrast, a negative shift of Ni 2p3/2 and 2p1/2 peaks was detected in 7% [Ni]-CsPbBr3 compared to that in [Ni(dmgH)2] due to the electron accumulation in the LUMO of the cocatalyst (Fig. S9†).38 The C 1s, N 1s, and O 1s XPS spectra of 7% [Ni]-CsPbBr3 are presented in Fig. S9.† Consequently, these findings provide strong evidence of electron transfer from CsPbBr3 QDs to [Ni(dmgH)2].
Role of cocatalyst on photoredox activity
The optimization of the photoredox C–N coupling reaction was performed using 4-nitrobenzaldehyde (1) and piperidine (2) as the model substrates (Table S2†). CsPbBr3 QDs produced amide by the photocatalytic reaction with a reduced yield (see later). However, the integration of the Ni-complex with CsPbBr3 QDs significantly improved the photocatalytic activity (Table S2†). A series of Ni(II)-complexes and salts were explored as the cocatalyst {[Ni(dmgH)2], [Ni(salen)2], [Ni(acac)2], and Ni(OAc)2·4H2O} with CsPbBr3 QDs (Table S2†). Among different cocatalysts, [Ni(dmgH)2] was found to be the most effective for the photoredox C–N coupling reaction. The amount of cocatalyst has also a significant effect on the photoredox activity of [Ni]-CsPbBr3 (Table S2†). With the increasing wt% of [Ni(dmgH)2], the photoredox activity was increased to achieve a higher yield of amide. Interestingly, the best yield of amide was obtained with 7% [Ni]-CsPbBr3, and further increase in the amount of [Ni(dmgH)2] decreased the yield of the product (Table S2†).Based on the above studies, it is clear that a cocatalyst should possess an optimized reduction potential and improve the photogenerated charge separation and transport process of CsPbBr3 QDs, enabling it to accept electrons from the CsPbBr3 CB and relay the electron to atmospheric molecular oxygen (from air) to form superoxide radicals. It should be mentioned here that CsPbBr3 QDs can also activate molecular oxygen to form superoxide radicals. However, the cocatalyst [Ni(dmgH)2] helps in the relay of the photogenerated electrons from the CB of CsPbBr3 to O2 because of the suitable energy levels and redox potentials.
To understand the process, we have conducted a comparison of the energy levels and reduction potentials of the three components: (i) CB position of CsPbBr3 QDs, (ii) single electron reduction potential of [Ni(dmgH)2], and (iii) reduction potential of O2 to O2˙−. The MS study confirms that the reduction potential of [Ni(dmgH)2] (−0.59 V vs. NHE) is higher enough to reduce O2 to O2˙− (−0.33 V vs. NHE) (Fig. 1c and S4†). As the superoxide radical is responsible for the activation of the aldehyde, the rate-determining step is not the initial photoexcitation or charge injection but the subsequent conversion of O2 to O2˙−. Therefore, the optimized energy levels of the photocatalyst CB and cocatalyst LUMO can enhance the overall photoredox performance.
Optimization of the photoredox reaction conditions
Further optimization of the photoredox reaction conditions has been carried out in different aprotic solvents (tetrahydrofuran (THF), acetonitrile, toluene, and 1,4 di-oxane). Among these, THF was found to be the suitable solvent for the photoredox C–N bond formation. To understand the role of photocatalyst, cocatalyst, light, and O2, photoredox reactions with all the necessary combinations have been performed (Table S2†). In the absence of photocatalyst or light, the reaction did not take place. In the N2 environment (absence of O2), a trace amount (<5%) of product formation was detected. This result clearly shows the importance of the activation of molecular oxygen to superoxide radicals for the photoredox reaction. However, in the absence of the cocatalyst, CsPbBr3 QDs can also activate molecular O2 and produce 48% yield of amide.Substrate scope for the C–N bond formation
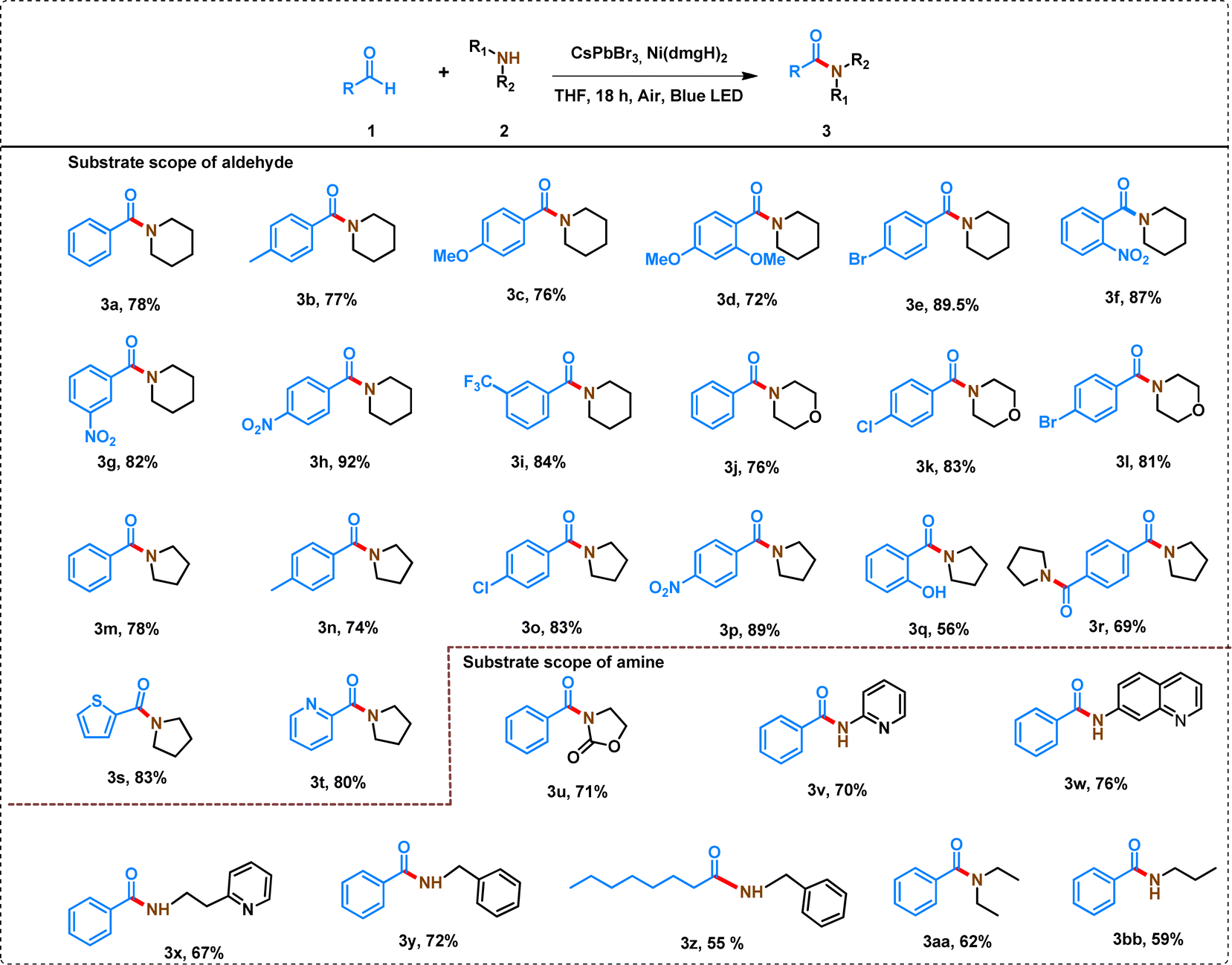
The C–N coupling strategy involving derivatives of benzaldehyde and piperidine demonstrated a wide substrate scope and remarkable tolerance to diverse functional groups, leading to the formation of amides in good to excellent yields (Table 1). The reactivity of aldehyde derivatives depends on the presence of electron-donating and electron-withdrawing groups in the phenyl ring of benzaldehydes. Substrates with the electron-donating group in the phenyl ring of benzaldehydes (para-methyl-benzaldehyde and para-methoxy benzaldehyde) showed comparatively low yields (Table 1, 3a–3c). The steric crowding in the ortho-position of benzaldehyde further decreased the yield of the amide (Table 1, 3d).| a Reaction conditions: aldehyde (0.5 mmol), amine (1 mmol), 7% [Ni]-CsPbBr3 (10 mg), THF (2.5 mL), 15 W blue LED, air, temperature: 35 ± 3 °C, time: 18 h. *In the case of 3r, aldehyde and pyrrolidine were taken 0.5 mmol and 2 mmol, respectively. In all the cases, isolated yield of the product was reported and the products were characterized by 1H and 13C NMR (Table S5). |
|---|
|
In contrast, benzaldehydes having electron-withdrawing groups (–NO2, and –CF3) in the phenyl ring produced high yields, ranging from 82% to 92% (Table 1, 3f–3i). Further, the position of the substituent (ortho-, meta-, para-nitro) in the phenyl ring of benzaldehydes has shown a significant effect on the yield of the product (Table 1, 3f–3h). Even, 4-bromobenzaldehyde produced a good yield of amide (89.5%) under the similar reaction conditions (Table 1, 3e). The reaction of morpholine with different benzaldehyde derivatives produced slightly lower yield of amide than the corresponding piperidine derivative (Table 1, 3j–3l).
The reaction of pyrrolidine with different substituted benzaldehydes produced a wide variety of amides (Table 1, 3m–3p). A similar trend for the substitution in the phenyl ring of benzaldehyde was observed when 5-membered pyrrolidine was used instead of 6-membered piperidine. However, in all the cases, a slightly lower yield (74–89%) was attained with pyrrolidine compared to piperidine because of increased ring strain in the cationic amine radical of pyrrolidine (see later). Interestingly, the process can even tolerate hydroxyl group at the ortho-position of benzaldehyde to produce a yield of 56% of amide (Table 1, 3q). The reaction of terephthaldehyde with pyrrolidine produced 69% yield of 3q (Table 1, 3r). The heterocyclic aldehydes (thiophene-2-carbaldehyde and pyridine 2-carbaldehyde) also produced >80% yield of amides (Table 1, 3s–3t).
A wide variety of amines have also been studied to form amides with moderate to high yield. The effect of the variation of amine (with a similar structure) is not so pronounced in the yield of amides. For example, when 6-membered piperidine was replaced by morpholine, the yield was not significantly affected (Table 1, 3a, and 3k). Similarly, 6-membered piperidine and 5-membered pyrrolidine produced similar yields (Table 1, 3a, and 3m). However, the reaction of 2-oxolidone with benzaldehyde lowered the yield of the amide (Table 1, 3u).
Interestingly, aniline derivatives of pyridine and quinoline (2-aminopyridine, 2-(2-pyridyl)ethylamine, and 7-aminoquinoline) also produced a high yield of amides when reacted with benzaldehyde (Table 1, 3v–x). However, the reaction of aniline with benzaldehyde led to the formation of a mixture of products, which cannot be separated by column chromatography. Even benzylamine produced 72% yield of amide reacting with benzaldehyde (Table 1, 3y). However, the yield of the amide was decreased when aliphatic aldehyde was reacted with benzylamine (Table 1, 3z). Octanal showed only 61% conversion under similar reaction conditions with a 55% yield of amide and 6% yield of N-octylidene-1-phenylmethanamine as a byproduct following the hydrogen atom transfer (HAT) mechanism (Table S5,†3z′).46 A decrease in the yield of amides was also observed when benzaldehyde reacted with open-chain aliphatic amines (Table 1, 3aa, and 3bb).
Further, the catalytic recyclability test was performed four times with a minimum loss of activity (Fig. S10†). The recovered catalyst did not show any change in the UV-visible spectrum (Fig. S11†).
Effect of cocatalyst on the excited state dynamics of CsPbBr3 QDs
The femtosecond transient absorption spectroscopy (TAS) was used to investigate the impact of [Ni(dmgH)2] on the dynamics of carrier relaxation in CsPbBr3 QDs (Fig. S12†). The samples were excited above the band edge of CsPbBr3 (370 nm) to generate charge carriers, which subsequently formed trap states.34,35 The TAS of CsPbBr3 reveals specific features (Fig. S12a–g†): (i) a prominent photobleach band at 518 nm (PB1). (ii) A less pronounced negative band (390–450 nm: PB2). (iii) A broad absorption band (410–510 nm: ESA1). (iv) Another excited state absorption band (530–560 nm: ESA2). This second absorption band evolves and decays rapidly within 800 fs. A concomitant broadening and a red shift of ESA1 are observed within this time scale. The ESA2 is ascribed to the intra-band relaxation of the hot electron in the vibrational levels of the conduction bands of CsPbBr3.35 As the relaxation takes place, the energy gap between the relaxed states and higher energy states increases which leads to a blue shift of the ESA2. This overlaps with the wavelengths of ESA1 and appears to broaden and enhance the intensity of ESA1.The TA of 7% [Ni]-CsPbBr3 showed similar spectral features for the photobleach bands and the lower wavelength ESA band (PB3, PB4, and ESA3) (Fig. S12h–n†). The excited absorption band, akin to ESA2 in CsPbBr3, was not detected in 7% [Ni]-CsPbBr3. Photo-induced absorption ESA3 appeared at 510 nm while the photo bleach band PB3 was red-shifted (526 nm) compared to that of CsPbBr3 (Fig. S12h–n†). Although several spectral features were observed to be similar in QDs and 7% [Ni]-CsPbBr3, the rates at which the spectra evolve are significantly different. This suggests that the same electronic states are involved but their formation rates are different.
The decay dynamics at different wavelengths are monitored by fitting the kinetic traces to a sum of exponentials. The kinetic trace at 400 nm corresponds to the bleach of the ground state to the higher energy states (Fig. 3a(1)). The 550 nm kinetics belongs to the recovery of bleach of ground state as this wavelength matches the lowest energy band in the steady state spectrum. A negative signal in the TA can be assigned to bleach as well as stimulated emission. The hot electrons undergo intra-band relaxation processes. Had the signal at 390 nm to 430 nm been exclusively from the stimulated emission of hot electrons, then this band would have evolved along with ESA2. However, as noted from TA spectra (Fig. S12†) this blue band exists well beyond ESA2 absorption. ESA2 rises and falls within 800 fs, while the PB2 signatures exist even in the 2 ns spectra. The 7% [Ni]-CsPbBr3 spectra also showed the negative band in the same region, however, the ESA2-type band was not observed. Based on the electrochemical spectroscopic studies, an ultrafast electron transfer from perovskite to cocatalyst can be proposed. If the negative band is originated from the stimulated emission of hot electrons, it should not be detected in 7% [Ni]-CsPbBr3. As the higher energy negative band was detected even in the presence of cocatalyst, we assigned it as a photobleach band.
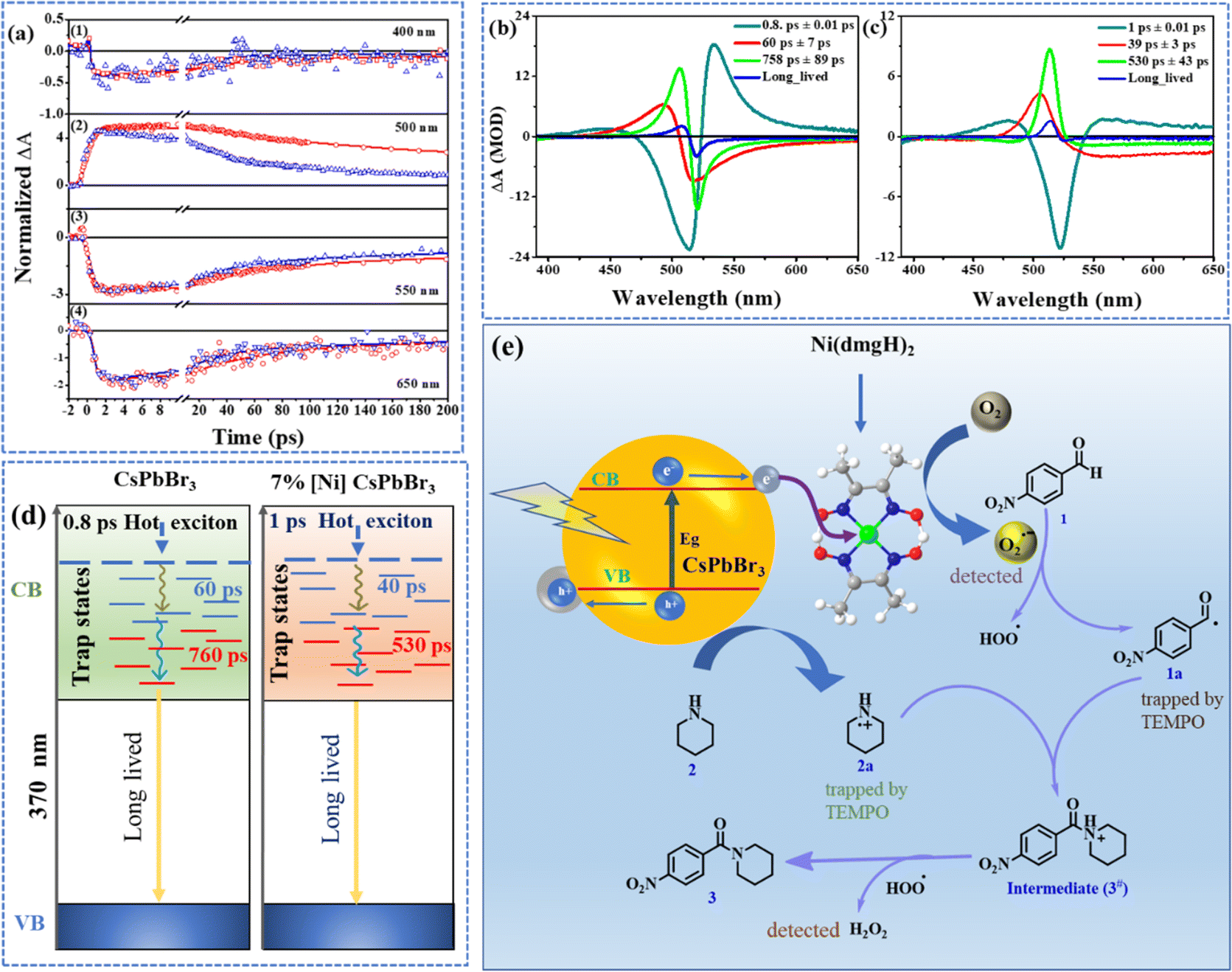 | ||
| Fig. 3 (a) Kinetic traces at (1) 400 nm (2) 500 nm (3) 550 nm and (4) 650 nm for CsPbBr3 (red) and 7% [Ni]-CsPbBr3 (blue). The solid lines are fitted lines. The decay-associated spectra (DAS) of (b) CsPbBr3 and (c) 7% [Ni]-CsPbBr3. (d) Schematic illustration of the formation and relaxation of charge carriers of CsPbBr3 and 7% [Ni]-CsPbBr3. (e) Proposed mechanism of C–N bond formation. | ||
The comparison of the kinetic traces at 500 nm has shown that the rise and decay of the signal of CsPbBr3 is faster than 7% [Ni]-CsPbBr3 (Fig. 3a(2)). The rise times were calculated to be 98 ± 5 ps for CsPbBr3 and 41 ± 2 ps for 7% [Ni]-CsPbBr3. The recovery of bleach (550 nm) was found to be faster for 7% [Ni]-CsPbBr3 than pristine QDs (Fig. 3a(3)). The same inference was drawn from 400 nm traces as well. The lifetimes at 550 nm decreased from 60 ± 8 ps and 817 ± 165 ps for CsPbBr3 to 31 ± 3 ps and 293 ± 32 ps for 7% [Ni]-CsPbBr3. Similar observations were obtained for kinetic traces at 650 nm (Fig. 3a(4)) and the time constants are listed in Table S3.† The decay-associated spectra (DAS) revealed three ultrafast components for both systems (Fig. 3b and c).47 The global lifetimes are obtained as 0.8 ± 0.01 ps, 60 ± 7 ps, and 758 ± 89 ps for CsPbBr3 and 1 ± 0.01 ps, 39 ± 3 ps, and 530 ± 43 ps for 7% [Ni]-CsPbBr3.
In addition to the above three components, a long-lived component, associated with the charge recombination process, that did not decay within the timescale of our measurement, was also observed (Fig. S13†). The DAS at 0.8 ± 0.01 ps showed a strong positive band (533 nm) corresponding to ESA2. The strong positive band in the DAS at 60 ± 7 ps matched with ESA1. The amplitude at the peak of ESA1 of the 760 ps DAS is stronger than that observed in the 60 ps DAS. This is in line with the observation that the ESA1 becomes stronger over the range of 500 ps and then decays. The DAS has a longer lifetime having a positive band at the same wavelength 506 nm associated with the ESA1 and PB1 bands (Fig. S12†). PB1 is attributed to the depopulation of the ground state, as its peak position aligns with the lowest energy steady-state absorption of the CsPbBr3 (Fig. S12a†).48,49 The 0.8 ps rise in PB1 signal resulting from 370 nm excitation is attributed to the gradual transition of the high energy excitons towards the band edge excitonic state through intra-band relaxation.
The loading of [Ni(dmgH)2] on CsPbBr3 results in faster kinetics in the TAS (Fig. 3d). A faster decay arises from the increased rate of depopulation of the involved states by an excited state process due to the migration of the charge from CsPbBr3 to [Ni(dmgH)2]. This is manifested as a decrease in lifetime in 7% [Ni]-CsPbBr3 and is a reflection of the formation of a trap state from CsPbBr3. Earlier works on CsPbBr3 have also indicated that the exciton bleach in the picosecond to nanosecond time scales serves as a reliable indicator of electron trapping.48,50,51
The electrochemical studies showed that light-induced charge transfer was facilitated from CsPbBr3 to [Ni(dmgH)2] (Fig. 2a and S8†). We take the aid of TA measurements to estimate the rate of this migration. The lifetimes of exciton formation and cooling are affected by cocatalyst. The inverse of lifetime corresponds to the rate constant of the associated process. Hence the difference in rate constants of [Ni(dmgH)2] loaded and pristine CsPbBr3 will serve as a measure of the electron transfer process (eqn S1†). The formation time of the trap state decreases from 60 ± 8 ps for CsPbBr3 to 31 ± 3 ps for 7% [Ni]-CsPbBr3. Substituting these lifetimes yields a rate constant of ket = 1.6 × 1010 s−1. The observed electron transfer rate agrees with the rate constant typically observed in other semiconductor metal systems where electrons have relaxed to the bottom of the conduction band.51,52
Reaction mechanism
The reaction of 4-nitrobezaldehyde and piperidine in the inert atmosphere resulted <5% product formation, highlighting the role of oxygen. Furthermore, when we introduced electron and hole scavengers (AgNO3 and TEA or ethanol) into the reaction mixture, the yield of the product decreased significantly, suggesting the involvement of free-radical pathways (Fig. S14†). When superoxide radical scavenger para-benzoquinone was introduced into the reaction mixture, the yield of the product dropped to only 10% (Fig. S14†).16In the presence of [Ni(dmgH)2], faster electron transfer from the CB of CsPbBr3 to cocatalyst takes place, followed by the reduction of surface-bound O2, producing superoxide radicals. Considering these findings, we have proposed a mechanism for the photo-induced amide formation reaction (Fig. 3e). Under visible light irradiation, [Ni]-CsPbBr3 is initially photoexcited, promoting an electron from the VB to CB. This photogenerated electron in the CB is relayed to atmospheric oxygen by cocatalyst to form a superoxide radical (O2˙−). Concurrently, the holes (h+) in the VB are transferred to amine (2), resulting in the formation of amine radical cation (2a). The superoxide radical abstract proton from 4-nitrobenzaldehyde forms acyl radical (1a) along with a hydroperoxyl radical (˙OOH).26,53–58 Interestingly, intermediate 1a and 2a were trapped by TEMPO and identified using NMR spectroscopy (Fig. S15a†) and mass spectrometry (Fig. S15b†).59 The radical coupling of intermediates 1a and 2a produces cationic species 3#, which is further oxidized by ˙OOH to final product 3.53,54 As a result, H2O2 was formed as the reduction product of O2. Interestingly, we were able to detect the formation of H2O2 (Fig. S16†).60,61
The higher efficiency of O2 reduction by 7% [Ni]-CsPbBr3 compared to CsPbBr3 was also confirmed (Fig. S16a†). Further, an increase in H2O2 concentration was observed with increasing reaction time (Fig. S16b†). Therefore, it is clear that 7% [Ni]-CsPbBr3 is more effective in the activation of molecular oxygen compared to CsPbBr3. In addition, superoxide radical was also detected by UV-visible spectroscopy (Fig. S17†).62,63 A higher amount of O2˙− formation was also observed for 7% [Ni]-CsPbBr3 compared to the pristine QDs.
Therefore, in this study, we are able to detect the reaction intermediates (1a, 2a) by mass-spectroscopy and NMR, active radical spices (O2˙−) and H2O2 by UV-visible spectroscopy (Fig. S15–S17†). Based on the previous literature, the formation of ˙OOH radicals and 3# has been proposed to complete the catalytic cycle.60–63
Conclusion
In summary, we have demonstrated the potential of CsPbBr3 QDs for the photoredox C–N coupling reaction. The introduction of cocatalyst improved the photoredox process and the best catalytic activity was obtained with [Ni(dmgH)2] because of the suitable band alignment. [Ni(dmgH)2] enhances photogenerated charge transfer dynamics of CsPbBr3 QDs. 7% [Ni]-CsPbBr3 produced the best photoredox activity forming amide with 92% yield. Ultrafast TA measurement showed that electron transfer to [Ni(dmgH)2] originated from CsPbBr3, exhibiting a rate constant of ket = 1.6 × 1010 s−1. The trap states decayed significantly fast, thereby enhancing the photocatalytic activities. Importantly, the optimal reaction conditions for these transformations avoid non-oxygen oxidants, harsh reaction conditions, ligands, additives, and environmentally hazardous solvents and bases. This study has shown the potential of photoredox reactions using semiconductor QDs for the synthesis of valuable compounds.Data availability
All the data are available in the ESI.†Author contributions
VK involved in the synthesis of the catalysts, their characterization, photocatalytic studies, data interpretation, and manuscript writing. VV involved in product separation (column chromatography) and NMR. DK carried out (photo)electrochemical characterization and data interpretation. SKP and ESSI conducted transient absorption measurements and wrote their part. The project design, conceptualization, supervision, data interpretation, manuscript writing, and editing were performed by AI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AI would like to thank DST-SERB, India for the core research grant (Grant No.: CRG/2023/002395). VK is thankful to CSIR [09/1217/(0084)/2020-EMR-II] for providing the senior research fellowship.Notes and references
- C. Russo, F. Brunelli, G. C. Tron and M. Giustiniano, J. Org. Chem., 2023, 88, 6284–6293 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- D. Sun, Y. Chen, X. Yu, Y. Yin and G. Tian, Chem. Eng. J., 2023, 462, 142084 CrossRef CAS.
- P. Sha, L. Huang, J. Zhao, Z. Wu, Q. Wang, L. Li, D. Bu and S. Huang, ACS Catal., 2023, 13, 10474–10486 CrossRef CAS.
- A. Wang, M. Du, J. Ni, D. Liu, Y. Pan, X. Liang, D. Liu, J. Ma, J. Wang and W. Wang, Nat. Commun., 2023, 14, 6733 CrossRef CAS PubMed.
- Y. Zheng, Y. Chen, B. Gao, B. Lin and X. Wang, Adv. Funct. Mater., 2020, 30, 2002021 CrossRef CAS.
- A. Indra, R. Beltrán-Suito, M. Müller, R. P. Sivasankaran, M. Schwarze, A. Acharjya, B. Pradhan, J. Hofkens, A. Brückner, A. Thomas, P. W. Menezes and M. Driess, ChemSusChem, 2021, 14, 306–312 CrossRef CAS PubMed.
- A. Indra, P. W. Menezes, M. Schwarze and M. Driess, New J. Chem., 2014, 38, 1942–1945 RSC.
- A. Indra, P. W. Menezes, K. Kailasam, D. Hollmann, M. Schröder, A. Thomas, A. Brückner and M. Driess, Chem. Commun., 2016, 52, 104–107 RSC.
- S. Alemdar, A. Basak and O. Metin, ACS Appl. Mater. Interfaces, 2023, 15, 48096–48109 CrossRef CAS PubMed.
- A. P. Deshmukh, K. Patil, S. Ogale and T. Bhave, ACS Appl. Electron. Mater., 2023, 5, 1536–1545 CrossRef CAS.
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W. H. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- H. Jiang, M. Liu, X. Lian, M. Zhu and F. Zhang, Angew. Chem., Int. Ed., 2024, 136, e202318850 CrossRef.
- I. Rosa-Pardo, C. Casadevall, L. Schmidt, M. Claros, R. E. Galian, J. Lloret-Fillol and J. P. Prieto, Chem. Commun., 2020, 56, 5026–5029 RSC.
- Q. Fan, H. Zhang, D. Liu, C. Yan, H. Zhu, Z. Xie and Z. Le, J. Org. Chem., 2023, 88, 7391–7400 CrossRef CAS PubMed.
- J. Lee, A. Kumar and H. Tuysuz, Angew. Chem., Int. Ed., 2024, 202404496 Search PubMed.
- K. Mishra, D. Guyon, J. San Martin and Y. Yan, J. Am. Chem. Soc., 2023, 145, 17242–17252 CrossRef CAS PubMed.
- Y. Dong, Y. Feng, Z. Li, H. Zhou, H. Lv and G. Y. Yang, ACS Catal., 2023, 13, 14346–14355 CrossRef CAS.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- K. Chen, X. Deng and G. Dodekatos, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- Y. Yuan, H. Zhu, K. H. Kimball, T. Cai, W. Shi, Z. Wei, H. Yang, Y. Candler, P. Wang, J. He and O. Chen, Angew. Chem., Int. Ed., 2020, 59, 22563–22569 CrossRef CAS PubMed.
- J. S. Martin, X. Zeng, X. Chen, C. Miller, C. Han, Y. Lin, N. Yamamoto, X. Wang, S. Yazdi, Y. Yan, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2021, 143, 11361–11369 CrossRef CAS PubMed.
- W. Wu, Y. C. Wong, Z. K. Tan and J. Wu, Catal. Sci. Technol., 2018, 8, 4257–4263 RSC.
- L. Xu, S. Z. Zhang, W. Li and Z. H. Zhang, Chem.–Eur. J., 2021, 27, 5483–5491 CrossRef CAS PubMed.
- L. Deng, L. Chen, L. Zhu, Y. Li, J. Ou-Yang, S. Wu, P. Chen, S. Shen, J. Guo, Y. Zhou, C. T. Au and S. F. Yin, Chem. Eng. Sci., 2022, 261, 117960 CrossRef CAS.
- B. Goel, V. Vyas, N. Tripathi, A. K. Singh, P. W. Menezes, A. Indra and S. K. Jain, ChemCatChem, 2020, 12, 5743–5749 CrossRef CAS.
- V. Vyas, V. Kumar and A. Indra, Chem. Commun., 2024, 60, 2544–2547 RSC.
- O. G. Mountanea, D. Psathopoulou, C. Mantzourani, M. G. Kokotou, E. A. Routsi, D. Tzeli, C. G. Kokotos and G. Kokotos, Chem.–Eur. J., 2023, 29, 2300556 CrossRef PubMed.
- N. Martín and F. G. Cirujano, Catal. Commun., 2022, 164, 106420 CrossRef.
- A. Hassan Tolba, M. Krupička, J. Chudoba and R. Cibulka, Org. Lett., 2021, 23, 6825–6830 CrossRef CAS PubMed.
- Y. C. Pu, Y. H. Chuang, M. W. Zheng, Y. J. Chang and S. H. Liu, J. Environ. Chem. Eng., 2023, 11, 109103 CrossRef CAS.
- H. Wang, W. Lu, P. Xu, J. Luo, K. Yao, J. Zhang, X. Wei, S. Peng, H. Cheng, H. Hu and K. Sun, ACS Sustain. Chem. Eng., 2023, 11, 5963–5972 CrossRef CAS.
- J. Ding, X. Deng, J. Fan, Y. Wang, Z. Li and Q. Liang, Inorg. Chem., 2023, 62, 16493–16502 CrossRef CAS PubMed.
- X. Deng, Q. Liang, J. Fan, X. Yan, H. Si, H. Huang, Z. Li and Z. Kang, Chem. Eng. J., 2023, 475, 146385 CrossRef CAS.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 14613 Search PubMed.
- S. Liu, F. Li, T. Li and W. Cao, J. Colloid Interface Sci., 2023, 642, 100–111 CrossRef CAS PubMed.
- Z. Zhang, L. Li, Y. Jiang and J. Xu, Inorg. Chem., 2022, 61, 3351–3360 CrossRef CAS PubMed.
- J. Ma, W. Meng, L. Zhang, F. Li and T. Li, RSC Adv., 2021, 11, 5035–5043 RSC.
- Y. Zhang, L. Shi, H. Yuan, X. Sun, X. Li, L. Duan, Q. Li, Z. Huang, X. Ban and D. Zhang, Chem. Eng. J., 2022, 430, 132820 CrossRef CAS.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- S. Y. Ali, K. D. Reddy and A. K. Manna, J. Phys. Chem. A, 2019, 123, 9166–9174 CrossRef CAS PubMed.
- Y. F. Xu, M. Z. Yang, H. Y. Chen, J. F. Liao, X. D. Wang and D. Bin Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- W. Gong, Y. Li, Y. Yang, H. Guo and X. Niu, J. Mater. Chem. C, 2023, 11, 6963–6970 RSC.
- A. J. McMillan, M. Sieńkowska, P. Di Lorenzo, G. K. Gransbury, N. F. Chilton, M. Salamone, A. Ruffoni, M. Bietti and D. Leonori, Angew. Chem., Int. Ed., 2021, 133, 7208–7215 CrossRef.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D. D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atatüre, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS PubMed.
- C. Harris and P. V. Kamat, ACS Nano, 2010, 4, 7321–7330 CrossRef CAS PubMed.
- K. Wu, H. Zhu, Z. Liu, W. Rodr and T. Lian, J. Am. Chem. Soc., 2012, 10337–10340 CrossRef CAS PubMed.
- G. Nanoparticles, W. Virginia and U. States, J. Phys. Chem. Lett., 2011, 2, 2125–2129 CrossRef.
- Z. Akrami and M. Hosseini-Sarvari, Eur. J. Org. Chem., 2022, 202200429 CrossRef.
- A. Dey, S. Chakraborty, A. Singh, F. A. Rahimi, S. Biswas, T. Mandal and T. K. Maji, Angew. Chem., Int. Ed., 2024, e202403093 CAS.
- F. K. C. Leung, J. F. Cui, T. W. Hui, K. K. Y. Kung and M. K. Wong, Asian J. Org. Chem., 2015, 4, 533–536 CrossRef CAS.
- L. Deng, L. Chen, L. Zhu, Y. Li, J. Ou-Yang, S. Wu, P. Chen, S. Shen, J. Guo, Y. Zhou and C. T. Au, Chem. Eng. Sci., 2022, 261, 117960 CrossRef CAS.
- D. Leow, Org. Lett., 2014, 16, 5812–5815 CrossRef CAS PubMed.
- A. Banerjee, Z. Ley and M. Y. Ngai, Synthesis, 2019, 51, 303–333 CrossRef CAS PubMed.
- L. Wang, M. Yu, C. Wu, N. Deng, C. Wang, X. Yao and A. Syn, Catal., 2016, 358, 2631–2641 CAS.
- L. Magerusan, C. Socaci, F. Pogacean, M. C. Rosu, A. R. Biris, M. Coros, A. Turza, V. Floare-Avram, G. Katona and S. Pruneanu, RSC Adv., 2016, 6, 79497–79506 RSC.
- A. K. Singh, D. Hollmann, M. Schwarze, C. Panda, B. Singh, P. W. Menezes and A. Indra, Adv. Sustainable Syst., 2021, 5, 2000288 CrossRef CAS.
- J. Luo, X. Wei, Y. Qiao, C. Wu, L. Li, L. Chen and Ji. Shi, Adv. Mater., 2023, 35, 2210110 CrossRef CAS PubMed.
- A. K. Singh, K. Bijalwan, N. Kaushal, A. Kumari, A. Saha and A. Indra, ACS Appl. Nano Mater., 2023, 6, 8036–8045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including synthesis, characterization, spectroscopic and photoelectrochemical measurement, characterization, catalytic studies, 1H NMR, and 13C NMR spectroscopy. See DOI: https://doi.org/10.1039/d4sc03023k |
| This journal is © The Royal Society of Chemistry 2024 |