
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexible interactions of the rare-earth elements Y, La, and Lu with phosphorus in metallacyclohexane rings†
Yury
Minko
 ,
Taylor V.
Fetrow
,
Shikha
Sharma
,
Brenna K.
Cashman
and
Aaron M.
Tondreau
*
,
Taylor V.
Fetrow
,
Shikha
Sharma
,
Brenna K.
Cashman
and
Aaron M.
Tondreau
*
Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: tondreau_a@lanl.gov
First published on 8th July 2024
Abstract
A geometrically flexible bifunctional (bis)aminophosphine ligand was synthesized in a three-component, one-pot Kabachnik–Fields reaction using tertbutylphosphine, paraformaldehyde, and 3,5-dimethyl aniline. The product, bis((3,5-dimethylphenyl)aminomethyl)tertbutylphosphine (ArBiAMPtBu), containing two secondary amines and a tertiary phosphine, was isolated in good yields. Deprotonation of both N–H groups with (trimethylsilyl)methylpotassium (K-CH2SiMe3), followed by salt metathesis with LaI3, YI3, and LuI3 generated the corresponding MI(ArBiAMPtBu)(thf)3 complexes (M = Y (1), La (2), and Lu (3)) in good yields. A sterically encumbered indene, 1,3-diisopropyl-4,7-dimethyl-1H-indene, iPrMeInd, was deprotonated in situ and installed via salt-metathesis to generate the organometallic series of η5-indenide complexes, M(ArBiAMPtBu)(η5-iPrMeInd)(thf) (M = Y (4), La (5), and Lu (6)). 1H, 31P, 13C, and 89Y NMR experiments, IR spectroscopy, and single crystal X-ray diffraction (SC-XRD), were used to characterize these complexes. The Y–P coupling constant was found to be variable depending on the modifiable coordination environment of the metal center, indicating potential as both a spectroscopic handle as well as providing insight into the influence of additional ligands on the metal center.
Introduction
Hard, electropositive rare-earth elements (REEs) favorably coordinate harder oxygen-based ligands over softer ligands.1,2 Soft atom, such as phosphorus, coordination continues to draw interest as a means of observing covalency of REEs through overlap of the metal orbitals with the spatially extended p-orbitals.3,4 While bridging phosphinidene complexes of REEs have been known since 2008,5 recent studies have expanded the boundaries for REE–P interactions via a report of a terminal phosphinidene on Y, which exhibits a large JY–P coupling value indicating significant Y–P interaction.6 A range of anionic phosphide-bound Ln complexes has been established, showing wide diversity in their coordination modes.7–12 While neutral interactions with lanthanide metals dominate pnictogen–metal coordination, they remain difficult to control.13 A strategy for expanding this landscape is the use of bifunctional phosphines that may overcome unfavorable bonding properties by forcing the REE–P interaction via the use of a tether, a route that showed much promise in the nascent field.14–18 Continued development of new phosphorous-containing ligand systems featuring both neutral donor-to-metal (L-type) and anionic donor-to-metal (X-type) interactions deliver unconventional entryways into f-element-phosphorus coordination and provide insight into the nuances of these uncommon interactions.Bifunctional phosphines demonstrate promising versatility, spanning from metallodiphosphide complexes,19–22 (hydroxymethyl)-phosphines,23,24 to deprotonated alkyl phosphines.25,26 Bifunctional chelates such as PN27,28 and PNP29–34 have been employed on lanthanide complexes (Fig. 1A), where nitrogen acts as an anchor to force proximity and coordination between the lanthanide and the phosphorus atom. Phosphinoamide ligands were introduced by Nagashima,35,36 and subsequent works by Thomas37 explored heterobimetallic chemistry supported by this system. An additional subset of bifunctional (aminomethyl)phosphine ligands encompasses different P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratios, with either one amine group per phosphine38 or three amine groups per phosphine39 being the most commonly reported. Recent studies have employed (aminomethyl)phosphines in the synthesis of heterobimetallic complexes,31,40,41 including those that incorporate Ln–Ni heterobimetallic species (Fig. 1A).42 An alternative synthesis employs a 1:1 phosphine to amine ratio to produce cyclic ligands. Bullock, Ott, DuBois, and Helm,43–46 have shown that cyclic (aminomethyl)phosphine ligands coordinate phosphorus to a transition metal and the amine then works cooperatively with the transition metal for hydrogen splitting catalysis (Fig. 1A). Tris(aminomethyl)phosphine ligands have been reported, and their complexes with Y demonstrate through-space J-coupling between 31P and 89Y in the 31P{1H} NMR spectrum.47
:N ratios, with either one amine group per phosphine38 or three amine groups per phosphine39 being the most commonly reported. Recent studies have employed (aminomethyl)phosphines in the synthesis of heterobimetallic complexes,31,40,41 including those that incorporate Ln–Ni heterobimetallic species (Fig. 1A).42 An alternative synthesis employs a 1:1 phosphine to amine ratio to produce cyclic ligands. Bullock, Ott, DuBois, and Helm,43–46 have shown that cyclic (aminomethyl)phosphine ligands coordinate phosphorus to a transition metal and the amine then works cooperatively with the transition metal for hydrogen splitting catalysis (Fig. 1A). Tris(aminomethyl)phosphine ligands have been reported, and their complexes with Y demonstrate through-space J-coupling between 31P and 89Y in the 31P{1H} NMR spectrum.47
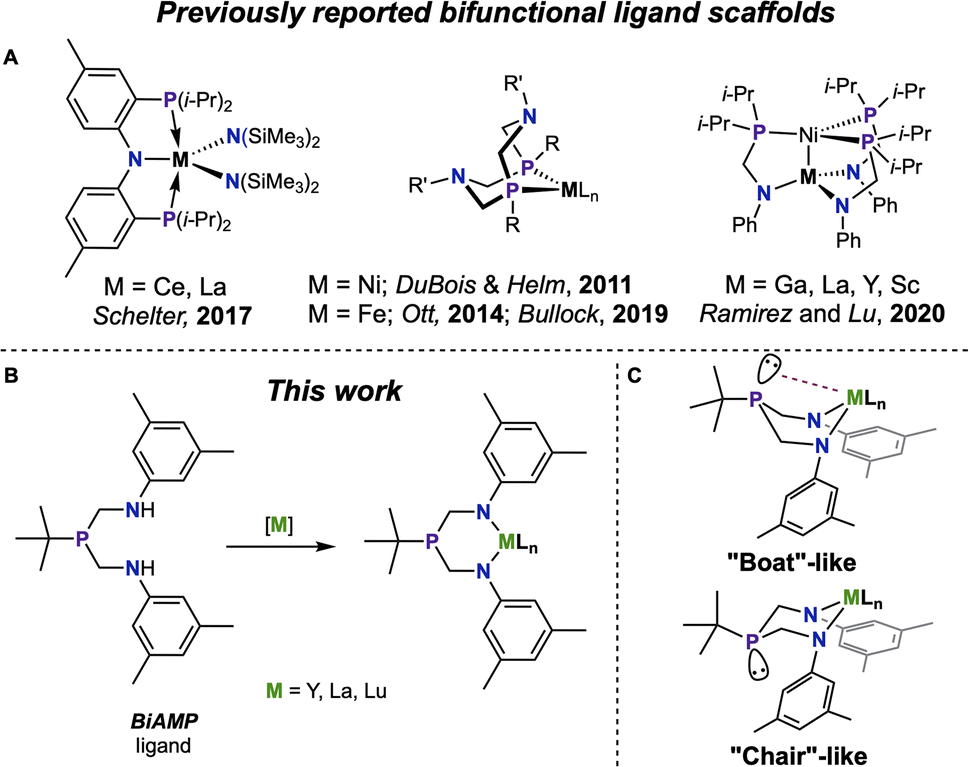 | ||
| Fig. 1 (A) Selected examples of previously disclosed bifunctional ligand scaffolds containing (aminomethyl)phosphine-based supporting ligands. (B) A simplified coordination scheme of bis(aminomethylene)phosphine ArBiAMPtBu ligand to form metal complexes. (C) The expected possible conformations defining the metal coordination sphere in Ln complexes with ArBiAMPtBu. | ||
The 2:1 bis(aminomethyl)phosphine motif can provide an additional entry point through which to investigate Ln–P interactions. Coordination of the deprotonated diamide to a Ln precursor is expected to form a six-membered metallacycle (Fig. 1B) with an inherent geometric flexibility allowing for both chair and boat conformations. The metallacycle can either adopt a chair conformation – and the distal phosphine will assume a non-interacting lone pair – or the metallocycle will adopt a boat-like conformation and the lone pair of the phosphine will be directed towards the central metal in a bonding interaction (Fig. 1C). We envisioned that this bonding would be affected by the coordination sphere of the metal, providing a spectroscopic handle in the 31P atom as a supplemental resource for investigation of the electronic structure of the metal center (Fig. S1†).
This report describes the coordination chemistry of the new phosphine-bearing diamine ligand ArBiAMPtBu. The series of diamagnetic rare earth element (REE) complexes MI(ArBiAMPtBu)(thf)3, where M = Y, La, and Lu, was isolated and characterized. Further derivatization with a bulky indenide-based ligand was performed in order to interrogate the influence of the large anionic donor on the metal center. NMR spectroscopy coupled to solid-state structural characterization was used to gain a better understanding of the bonding interactions between the metal center and the phosphorus atom of the BiAMP. Lanthanum and lutetium span the lanthanide series and present the extremes in ionic radius. Both La and Lu have high natural abundance spin active (I = 7/2) nuclei (99.91% (139La), 97.41% (175Lu)). The quadrupolar nuclei of 139La (quadrupole moment = 200 mb) (mb = millibarn; 1 barn = 10−28 m2) and 175Lu (quadrupole moment = 4970 mb) suggest that 31P NMR experiments will provide broadened resonances and the Ln–P coupling will remain unresolved, producing a single coalesced resonance. Yttrium was chosen for its comparably large ionic radius and its NMR-active nucleus (100% (89Y); I = 1/2). While 89Y NMR is notorious as a low-gamma nucleus with long T1 values (slow relaxation) requiring high concentrations,48 observed nuclear coupling, JY–P, has provided insight into the strength of Y–P bonding interactions.6,15,49–52
The bifunctional ArBiAMPtBu ligand scaffold provides a flexible phosphine moiety whose variable coordination strength can provide insight into the f-element–phosphine interactions. Despite this flexibility, the amide anchors strongly bind Ln centers and allow for further modification of the coordination sphere. Through coordination to diamagnetic, NMR active metal centers, 31P{1H} NMR coupling values can be used as a probe by which to gain a better understanding of f-element to phosphorus coordination and bonding.
Results and discussion
Ligand synthesis and characterization
The Kabachnik–Fields reaction,53,54 also known as the phospha-Mannich reaction,55 is an established entry point into (aminomethyl)phosphine compounds.56 Several substituted 1,5-diaza-3,7-diphosphacyclooctane scaffolds have been reported using this synthetic method.57,58 Non-cyclic bis(aminomethyl)phosphines have been sparsely reported, having been characterized as intermediates en route to substituted 1,5-diaza-3,7-diphosphacyclooctanes or 1,3-diaza-5-phosphacyclohexanes.59–61 Precise control of the stoichiometric ratio between RPH2, (CH2O)n, and aniline minimized the formation of undesired macrocycles and generated bis(aminomethyl)phosphine. For the synthesis of ArBiAMPtBu, tertbutylphosphine (tBuPH2), paraformaldehyde, and 3,5-dimethyl aniline were used in a 1:2:2 ratio (Fig. 2).
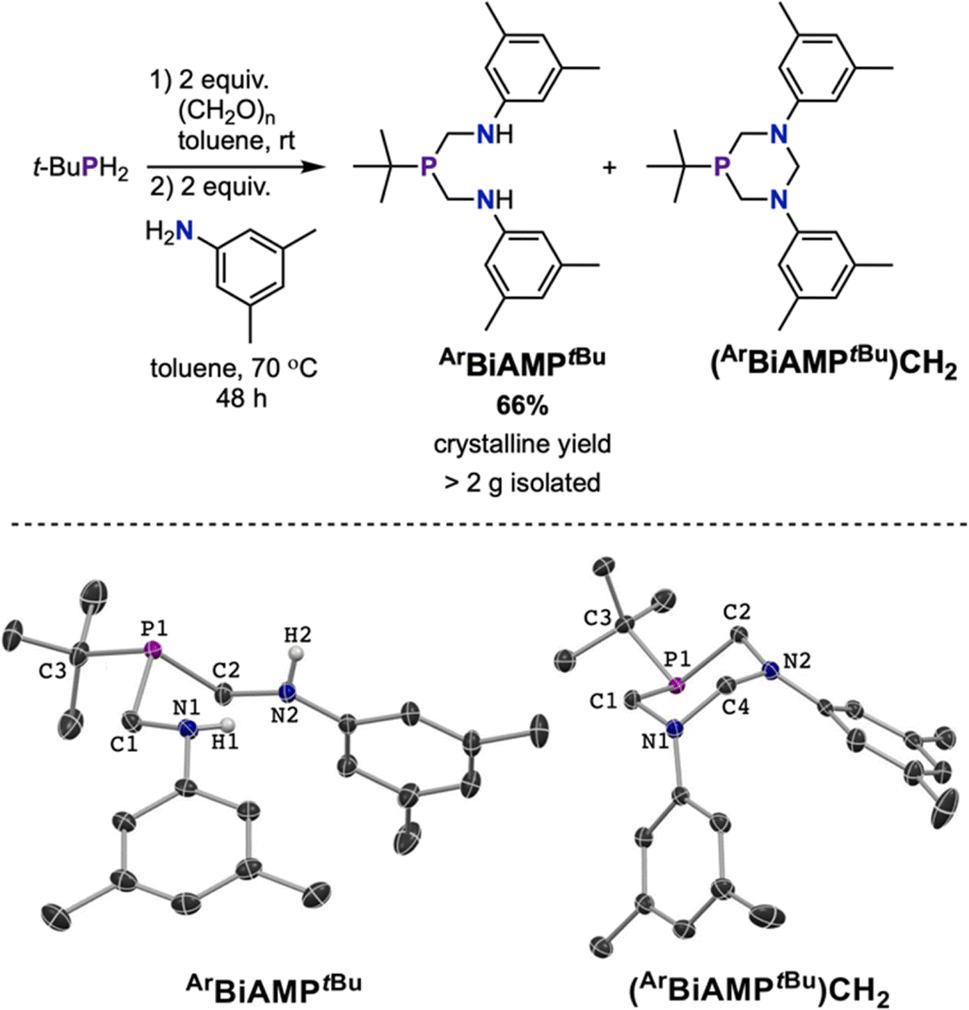 | ||
| Fig. 2 Top: optimized synthesis of the bis(aminomethyl) phosphine ArBiAMPtBu ligand under three-component, one-pot conditions. Bottom: solid state molecular structures of ArBiAMPtBu and cyclic side-product (ArBiAMPtBu)CH2 with hydrogen atoms shown where relevant or omitted for clarity. Thermal ellipsoids are presented at 50% probability. | ||
The initial reaction was performed using a commercially available 10 wt% solution of tBuPH2 in hexanes and two equivalents of paraformaldehyde, which were stirred until the visible paraformaldehyde suspension was consumed. At this time, 4 Å molecular sieves were added, followed by two equivalents of 3,5-dimethyl aniline. Work-up followed by NMR analysis of the tacky white solid in C6D6 suggested the presence of two major phosphorus containing products, with the 31P{1H} NMR spectrum containing resonances centered at δ −6.25 ppm and δ −41.24 ppm in an approximate ratio of >4:1, as well as those arising from minor additional impurities. Fractional crystallization of the product mixture from pentane furnished the desired ArBiAMPtBu in 45% yield (Fig. 2, bottom left). The major side-product was identified as 1,3-bis(3,5-dimethylphenyl)-5-tert-butyl-1,3-diaza-5-phosphacyclohexane ((ArBiAMPtBu)CH2), which was isolated in 6% crystalline yield (Fig. 2, bottom right).
NMR analysis of the isolated materials confirmed their assignments as the diamine (ArBiAMPtBu) and the heterocycle (Fig. S2–S7†). The 1H NMR spectrum of ArBiAMPtBu in C6D6 contained a broadened triplet resonance at δ 3.64 ppm that integrated 2:9 with a doublet located at δ 0.95 ppm, consistent with the presence of two N–H groups per tertbutyl-substituted phosphorus. Additionally, two resonances at δ 3.25 ppm and δ 3.12 ppm with characteristic splitting patterns (doublet of triplets and doublet of doublets, respectively) for two prochiral methylene groups were observed.
For (ArBiAMPtBu)CH2, no signals attributable to the N–H protons were observed in the 1H NMR spectrum. The methylene resonance signals adjacent to the phosphorus were observed as doublets of doublets at δ 3.83 ppm and δ 3.36 ppm. In addition, the two diastereotopic methylene protons of the CH2 group bridging the nitrogen atoms were observed at δ 5.08 ppm and δ 4.20 ppm. Further characterization of the ligand and cyclic byproduct was achieved by single-crystal X-ray diffraction (SC-XRD) (ESI S3.2 and S3.3†). These results confirmed that the targeted non-cyclic compound was formed as a major product in this three-component reaction. Our results contrast with prior reported examples of bis(hydroxymethylene)phosphines with primary amines or anilines, which have typically resulted in the formation of cyclic 1,3-diaza-5-phosphacyclohexane as the major product.56,60,61
Optimization of the reaction conditions were pursued to increase the yield of the desired (bis)amino ArBiAMPtBu ligand. Performing the reaction by combining the reactants simultaneously without the addition of molecular sieves improved the product yield and minimized the appearance of ArBiAMPtBuCH2. Analysis of the crude mixture after filtration and removal of volatiles revealed a significant improvement of the product ratio to 17:1 (ArBiAMPtBu:(ArBiAMPtBu)CH2) with minor impurities present. The target compound was purified by recrystallization from pentane, resulting in a colorless, crystalline product in 66% yield (Fig. 2). Importantly, this synthesis was performed with high reproducibility (five batches) on a multi-gram scale.
Rare-earth element coordination: MI(ArBiAMPtBu)(thf)3 synthesis and characterization
The ArBiAMPtBu ligand manifold was installed on Y, La, and Lu(III) iodides by salt metathesis with the deprotonated ligand. The deprotonation of ArBiAMPtBu was performed in THF using two equivalents of (trimethylsilyl)methylpotassium (K-CH2SiMe3) to generate [K2][ArBiAMPtBu], a pale yellow powder precipitated from THF. Analysis of the yellow material via1H and 13C NMR displayed resonances consistent with the expected (bis)potassium complex (Fig. S8–S10†). However, ingrowth of additional resonances over time indicated instability of [K2][ArBiAMPtBu]. Consistent results were found by employing a THF solution of [K2][ArBiAMPtBu] prepared in situ through deprotonation of ArBiAMPtBu in THF at −30 °C with two equivalents of K-CH2SiMe3. Subsequent metalation was accomplished by addition of the dipotassium salt solution to a cooled slurry of the THF adduct of the corresponding starting M(iii) iodide. After filtration of the formed KI salt, crystallization of the colorless products, MI(ArBiAMPtBu)(thf)3 (M = Y (1), La (2), Lu (3)), was accomplished in moderate to high yields (70–91%) out of cooled THF solutions either layered with pentane or using vapor diffusion techniques (Scheme 1). The yield was dependent on product crystallinity as the different metals each showed similar crude yields.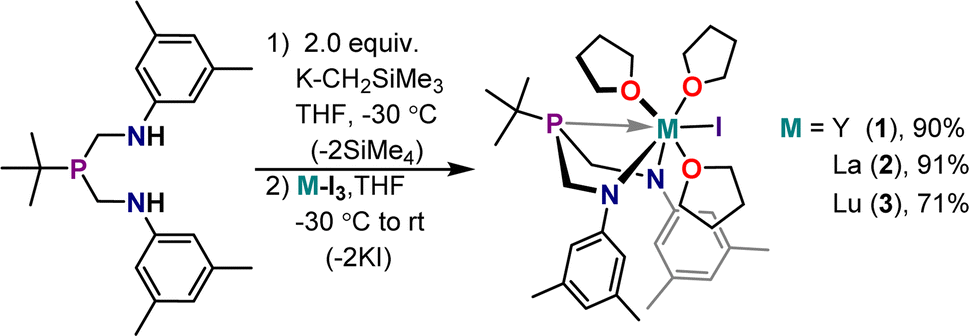 | ||
| Scheme 1 General synthetic route to diamagnetic complexes MI(ArBiAMPtBu)(thf)3, where M = Y (1), La (2), Lu (3). Yields for isolated crystalline products are reported. | ||
Crystals suitable for X-ray diffraction studies were formed from the reaction solutions, and SC-XRD experiments revealed that the isomorphous metal complexes 1–3 all crystallized in the orthorhombic space group P212121. For each compound of the series, X-ray data analysis revealed a coordination sphere around the metal that consisted of the ArBiAMPtBu ligand, three bound THF molecules, and the remaining iodide (Fig. 3). Additionally, the solid-state structures confirmed that the six-membered metallacycle adopts a distorted boat conformation, wherein the lone-pair of the phosphorus atom is directed towards the three different metal centers in 1–3. Including the phosphorus–metal interaction, the metal has a coordination number of seven. The REE geometry is best described as capped trigonal prismatic, which is distorted due to the constraints of the chelate.
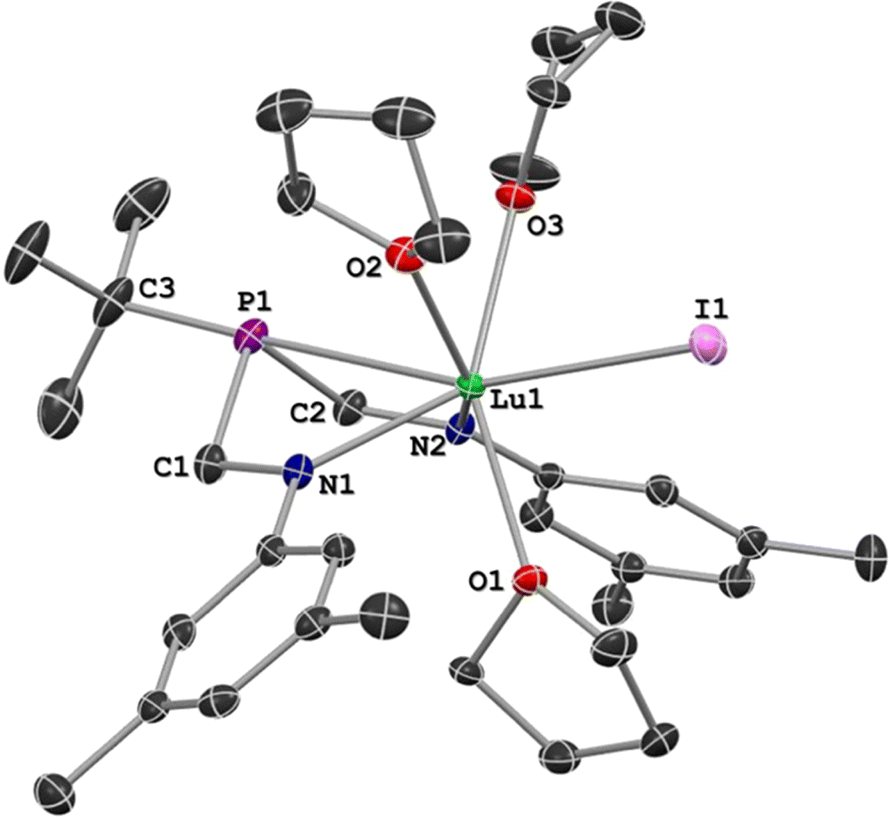 | ||
| Fig. 3 A representative solid state molecular structure of 3 shown with hydrogen atoms omitted for clarity and the thermal ellipsoids at 50% probability. | ||
The solid-state metrical parameters of 1, 2, and 3 generally display linear trends dependent on the ionic radius of the central metal atom. The distance between the metal and phosphorus linearly correlates to the corresponding ionic radius of the metal (Table 1),62,63 with Lu having the shortest distance (3.077 Å) and La having the longest (3.192 Å). The M–N distances are consistent with amide coordination to Y, La, and Lu with average distances of 2.296(6) Å, 2.404(5) Å, and 2.242(5) Å for 1, 2, and 3 respectively. For all three complexes, the C(3)–P–M angles are nearly linear (avg = 175.1°) with the angle becoming more linear as the ionic radius of the central metal decreases. In contrast, the observed P–M–I angles, which deviate significantly from linearity with an average angle of 160.9°, become more acute as the ionic radius of M decreases. Complete solid-state metrical parameters of 1, 2, and 3 are provided (Tables S1a and S1b, ESI 3.4–3.6†).
| D (Å) or ∠ (°) | 1 (Y) | 2 (La) | 3 (Lu) |
|---|---|---|---|
| a ESD (ESD = estimated standard deviation) values for the average were calculated using the root-mean-square method. | |||
| M–P | 3.124(2) | 3.192(1) | 3.077(1) |
| M–Navga | 2.296(6) | 2.404(5) | 2.242(5) |
| M–I | 3.119(2) | 3.226(6) | 3.046(4) |
| C3–P–M | 175.2(2) | 173.9(1) | 176.2(2) |
| P–M–I | 160.4(4) | 163.2(2) | 159.2(2) |
| N1–M–N2 | 91.7(1) | 88.8(1) | 92.2(1) |
NMR characterization of the complexes was performed in THF-d8 and structural integrity of the series MI(ArBiAMPtBu)(thf)3 in the solution phase was experimentally confirmed to be limited specifically to THF (Fig. S11–S20†). Upon dissolution of isolated crystalline 1 in solvents such as toluene, benzene, or methylene chloride, the colorless solution of the complex turns yellow and the 31P{1H} NMR spectrum shows resonances consistent with multiple species. Although our attempts to isolate individual products of decomposition were not successful, we propose decoordination of THF and potential formation of dimeric and/or oligomeric μ-iodo- and/or anilide-bridged structures to be responsible for the observed multiple signals in the 31P{1H} NMR spectra. This process was found to be reversible; after removal of solvent and redissolution of the solid residue in THF, the parent complexes were observed. Ligand exchange of coordinated THF of the (mono)iodide series was also observed in THF-d8 solutions. For example, integration of natural abundance THF in THF-d8 solutions gave variable and lower than expected values. This suggests that holding the isolated crystalline material under reduced pressure induces de-coordination of THF.
The expected resonance centered at δ −15.7 ppm in the 31P{1H} NMR spectrum was observed as a doublet with (JP–Y) = 2.7 Hz coupling, and the corresponding 89Y NMR resonance was observed as a doublet with (JY–P) = 2.6 Hz centered at δ 422.4 ppm (Fig. 4). Observation of coupling between the phosphorus and yttrium center indicates an interaction between the metal and phosphorus that contains orbital overlap impacting the nucleus. The coupling arises from nuclear interactions that are facilitated through orbital overlap, and various factors, such as bond geometry, bond type, and even through–bond interactions, contribute to the observed J-values. Including J-coupling in discussions of relative covalency of bonds within series of similar complexes provides additional tools to gauge subtle influences of bonding contributions. Observation of these values for Y suggests value in studying BiAMP complexes beyond REEs, specifically as a spectroscopic handle through which to interrogate the nature of phosphine-actinide bonding interactions. The phosphorus resonances for 2 and 3 appear as relatively sharp singlets located at δ −16.4 ppm (Δν1/2 = 2.8 Hz) and δ −18.2 ppm (Δν1/2 = 4.3 Hz), respectively. The lack of observed metal–phosphorus coupling likely arises from rapid relaxation owing to the quadrupolar nuclei, resulting in unresolved coupling of the expected octet that would arise from the abundant I = 7/2 139La and 175Lu centers.
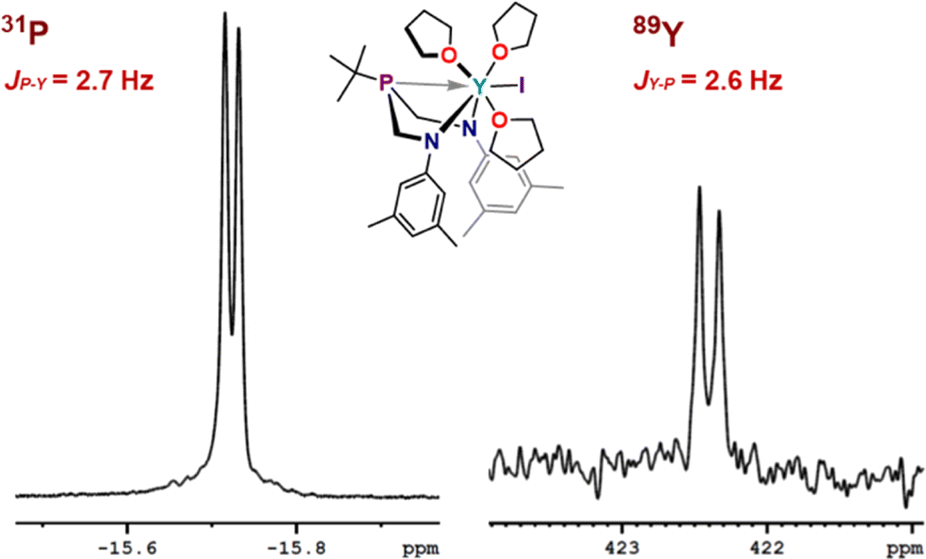 | ||
| Fig. 4 Expansions of the 31P{1H} and 89Y{1H} NMR spectra of (ArBiAMPtBu)YI(thf)3 (1) in THF-d8 (at 25 °C) showing metal-phosphorus coupling. | ||
Indenide complexes: M(ArBiAMPtBu)(η5-iPrMeInd)(thf)3 synthesis and characterization
The installation of the primary ligand ArBiAMPtBu and formation of diamagnetic complexes 1, 2, and 3 was successful. The presence of the iodide ligand allows for further chemistry at the rare-earth element center; one specific facet of interest is how substitution of the supporting anionic ligand affects the observed coupling between 89Y and 31P. The observed small coupling value provides a baseline from which we can start to investigate the effect of secondary supporting ligands on the observed M–P bond strength.A bulky, electron-rich indene ligand (1,3-diisopropyl-4,7-dimethyl-1H-indene, iPrMeInd)64 was chosen for addition to the coordination spheres of 1, 2, and 3. Indenyl moieties as ligands are known for being electronically flexible and capable of binding to a metal center through variable coordination modes (η1, η3, or η5). Additionally, adding steric protection by employing an indene with large substituents may offer additional stability that the iodide is unable to provide. The addition of [K][iPrMeInd] – made by in situ deprotonation of iPrMeInd using a stoichiometric amount of K-CH2SiMe3 in THF – to 1, 2, or 3 resulted in the formation of the corresponding indenyl species (Fig. 5a). The M(ArBiAMPtBu)(η5-iPrMeInd)(thf) complexes of Y (4) and Lu (6) were isolated as colorless to pale-yellow crystalline products in moderate yields (respectively 73.5% and 65%).
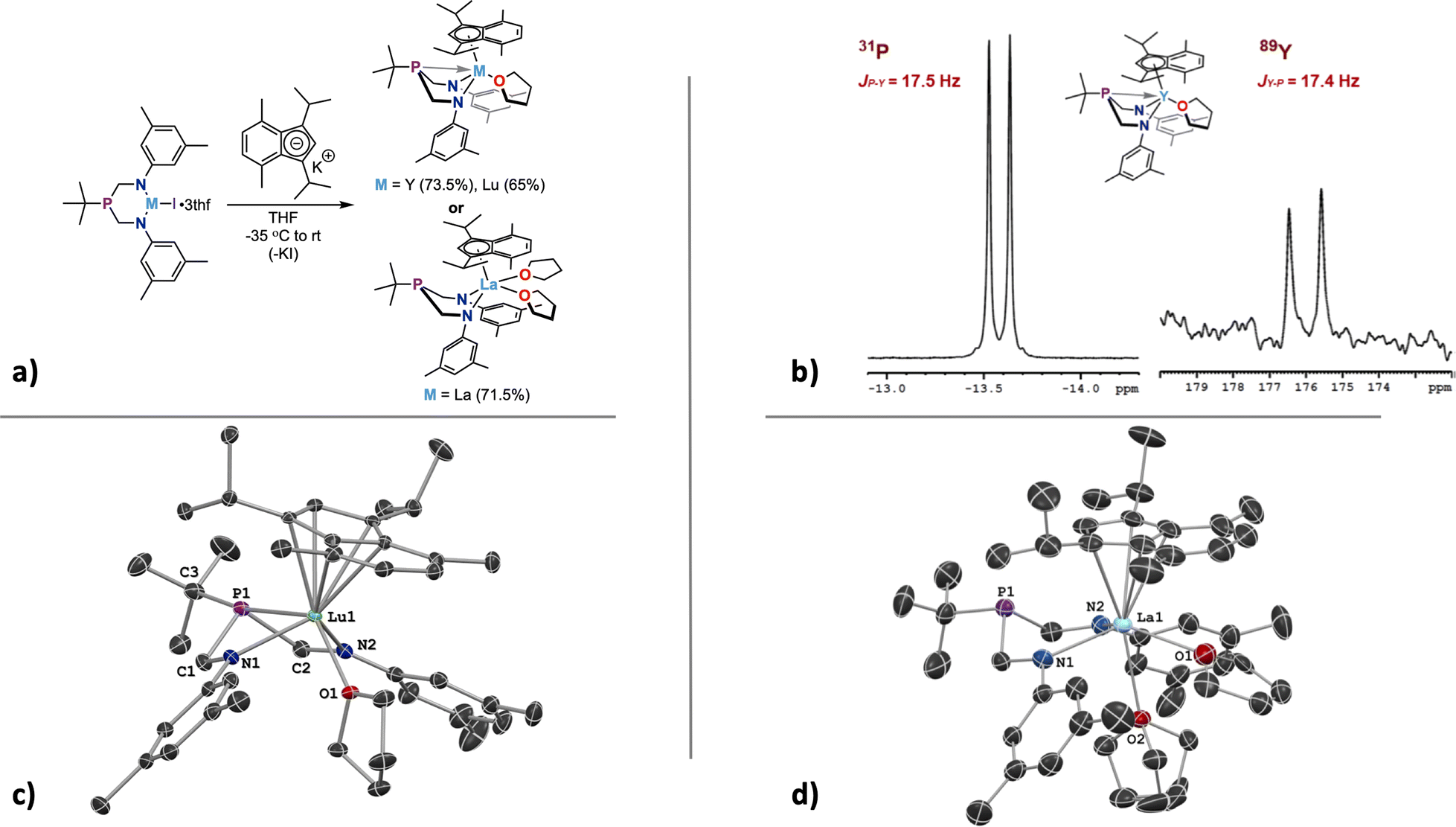 | ||
| Fig. 5 (a) General synthetic route to form diamagnetic M(ArBiAMPtBu)(η5-iPrMeInd)(thf)n, where M = Y (4, n = 1), La (5, n = 2), Lu (6, n = 1). Yields are reported for isolated crystalline products obtained by fractional crystallization. (b) Expanded section of both 31P{1H} and 89Y{1H} NMR spectra of 4 in THF-d8 (at 25 °C) showing metal-phosphorus coupling with increased J-coupling values shown. (c) The solid-state molecular structure of 6 is shown at 50% probability ellipsoids with hydrogen atoms omitted for clarity. (d) The solid-state molecular structure of 5 is shown at 50% probability ellipsoids with hydrogen atoms, disordered lattice solvent, and disorder of a bound THF arising from symmetry omitted for clarity. | ||
The substitution of η5-iPrMeInd onto 4 resulted in a much more pronounced M–P interaction as determined by the J-coupling. This effect was experimentally confirmed by 31P{1H} and 89Y NMR investigations of 4 (Fig. 5b). The corresponding 31P resonance signal (doublet) for this compound was shifted downfield to δ −13.6 ppm and exhibited a considerably increased J-coupling value ((JP–Y) = 17.5 Hz). The same increased value was observed for the doublet resonance in the 89Y NMR spectrum ((JY–P) = 17.4 Hz) that shifted to δ 176.0 ppm (Δδ = 246.4 ppm when compared to 1). NMR experiments performed on 5 and 6 showed no signs of M–P coupling, with broad singlet resonances observed in the 31P{1H} NMR spectrum at δ 18.1 and δ −14.5 ppm, respectively.
SC-XRD experiments were performed to confirm the identities of complexes 4 and 6, which are isostructural, and both were solved in the monoclinic space-group P21/c. The asymmetric unit cell contains one THF molecule in the interstitial space and a single THF molecule coordinated to the metal center (Fig. 5c). The metal center was found η5-coordinated to the indenyl ligand, and the ArBiAMPtBu ligand retained the boat conformation, maintaining the metal–phosphorus interaction. Introduction of the indenyl ligand resulted in a significant contraction of the P–M distance to 2.886(1) Å for 4 and 2.858(1) Å for 6 (compared to 3.124(2) Å for 1 and 3.077(1) Å for 3). A contraction of the metal-amide bond distances for 4, 5, and 6 is observed upon coordination of η5-iPrMeInd to the metal center.
In contrast to 4 and 6, our attempt to generate the isostructural La complex resulted exclusively in the formation of a bis-THF coordinated product La(ArBiAMPtBu)(η5-iPrMeInd)(thf)2 (5), which was confirmed by SC-XRD analysis (Fig. 5d). Complex 5 was isolated in 71% yield after crystallization from a THF–pentane mixture. The crystals formed as multi-crystalline clusters of colorless blocks, requiring careful separation prior to SC-XRD analysis. Data acquired for complex 5 was solved in the tetragonal space group ![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 421m. The ArBiAMPtBu ligand was found again in the boat conformation. Even with the sterically demanding indenide ligand, La was able to accommodate an additional THF ligand, inducing several structural changes as compared to 4 and 6 (Table 2).
421m. The ArBiAMPtBu ligand was found again in the boat conformation. Even with the sterically demanding indenide ligand, La was able to accommodate an additional THF ligand, inducing several structural changes as compared to 4 and 6 (Table 2).
| D (Å) or ∠ (°) | 4 (Y) | 5 (La) | 6 (Lu) |
|---|---|---|---|
| a ESD (ESD = estimated standard deviation) values for the average were calculated using the root-mean-square method; 5 has symmetry equivalent M–N bonds. | |||
| M–P | 2.886(1) | 3.658(1) | 2.858(1) |
| M–Navga | 2.252(3) | 2.388(3) | 2.214(3) |
| M–centroid | 2.389(1) | 2.645(2) | 2.343(1) |
| C3–P–M | 169.5 (1) | 167.6(1) | 170.5(1) |
| P–M–centroid | 111.0(3) | 92.3(2) | 111.3(4) |
| N1–M–N2 | 97.2(1) | 85.7(1) | 98.5 (1) |
A pronounced elongation was observed in the M–P distance, from 3.192(1) Å in 2 to 3.658(1) Å in 5, suggesting a significant weakening of the P–La interaction, with this distance outside of the sum of the covalent radii of La and P (3.15 Å) but remaining within the sum of van der Waals radii (4.78 Å). Additionally, the conformation of the ArBiAMPtBu is significantly changed, where the M–N–C atoms of the ligand are nearly planar, and the phosphorus atom protrudes above the formed plane. Overlays of the solid-state structures are provided to aid with the solid-state structural comparison (Fig. S51†) The centroid of the indene ligand is offset slightly with the oxygen atoms of the second THF with a centroid-La–O ∠ of 167.42(7)°. Further discussion of geometric data analysis is provided in ESI S.2.3.†
Complex 5 showed instability in diethyl ether, which was indicated by a color change to yellow and the observation of multiple phosphorus resonances in the 31P{1H} NMR spectrum. Attempts to crystallize the lanthanum complex from diethyl ether or toluene to generate a La complex with a single coordinated THF, analogous to 4 and 6, resulted in undesired reactivity and the formation of a dimeric product. The crystalline material that formed out of diethyl ether was collected as thin, polycrystalline plates, and the SC-XRD data was adequate to provide a connectivity structure only (Fig. S60†). The data confirmed the loss of THF ligands and formation of a dimer derived from 5. The connectivity structure obtained showed that the phosphorus atom is indeed hemilabile, and the six-membered metallacycle will rearrange into the chair conformation in appropriate conditions. A similar experiment was performed with 4, which crystallized out of diethyl ether in a different space-group (1) with a molecule of ether in the asymmetric unit cell (Fig. S57a†), otherwise, the metrical parameters were not notably different. The coordination differences and instability observed with the lanthanum congeners can be attributed to the metal's propensity for a higher coordination number due to its larger ionic radius as compared to Y and Lu.
Triethylphosphine oxide substitution
The observed Ln–P bond shortening – and correlated increase in the JY–P-value – upon installation of η5-iPrMeInd was significant. The products 4 and 6 showed Ln-P contraction of nearly 0.2 Å, while 5 exhibited an La–P bond expansion to a length outside of the sum of covalent radii. These results suggest that the M–P bond can be tailored by substitution of secondary ligands surrounding the lanthanide centers. Substitution of the bound THF with a stronger phosphine-oxide was then pursued to observe the effect the addition of this hard, strongly donating oxygen atom has on the M–P coupling value. By choosing triethylphosphine oxide (TEPO), we can also compare the relative Lewis acidities of the metal centers through an established technique.Gutmann–Beckett analysis offers one of the most commonly applied and convenient experiments to measure relative Lewis-acidities via a calculated acceptor number (AN).65–67 The experiment employs TEPO as a hard Lewis-base that undergoes a distinctive 31P{1H} NMR chemical shift change (Δδ) induced by coordination to a Lewis acidic center.67 This method was originally applied to assess the effective Lewis acidity68 of boranes,69 arylboronic catechol esters,70 and silylium ions.71 Guttmann–Beckett analysis has also found use in complexes of the transition metals,72 alkaline-earth metals,73 and with rare-earth elements.74 A prior study also employed simple rare-earth salts to correlate Lewis acidity to observed Lewis-acid catalyzed rates of reactivity.75
Complexes 4, 5, and 6 present an opportunity to study molecular systems that presumably can coordinate a single molecule of TEPO. Employing NMR-tube reactions, the addition of a single equivalent of triethylphosphine oxide to solutions of 4, 5, and 6 resulted in ligand exchange with the bound THF molecule, as determined by NMR spectroscopy, to generate M(ArBiAMPtBu)(η5-iPrMeInd)(TEPO) complexes (M = Y (7), La (8), Lu (9)). The 31P{1H} NMR spectra of these complexes comprised two resonances with varying coupling, giving rise to different splitting patterns at room temperature (Fig. 6, Top).
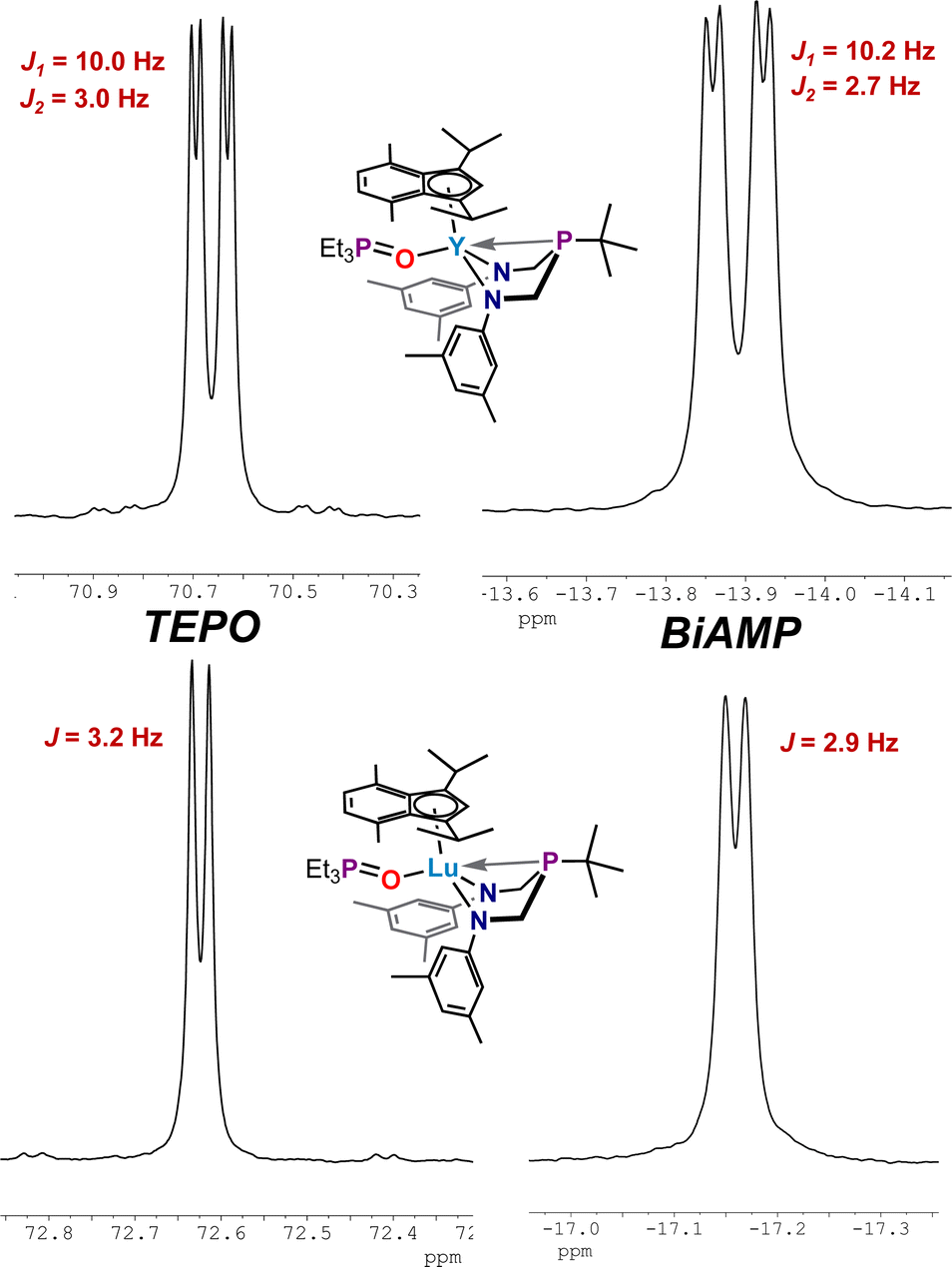 | ||
| Fig. 6 Resonances from the 31P{1H} NMR spectra of (ArBiAMPtBu)(η5-iPrMeInd)M(TEPO) adducts in THF-d8 (at 25 °C); 7 (Y) (top), 9 (Lu) (Bottom), with expansions of the observed resonances attributed to TEPO (left) and BiAMP (right). | ||
For 7, coupling to the 89Y nucleus induced additional splitting; the 31P{1H} NMR spectrum comprised two doublets of doublets with TEPO resonance shifted downfield to δ 70.66 ppm (JP–Y = 10.0 Hz, JP–P = 2.9 Hz) and ArBiAMPtBu phosphorus resonance centered at δ −13.89 ppm (JP–Y = 10.2 Hz, JP–P = 2.8 Hz). A resonance of low signal strength appearing as pseudo-triplet at δ 170.5 ppm was observed in the 89Y NMR spectrum, with an observed coupling of JY–P = 10 Hz. We attribute this splitting pattern to near-coincident coupling observed to the 1JP–Y of the BiAMP phosphine and 2JP–Y from the coupling of the TEPO phosphine through the oxygen atom. The 31P{1H} NMR spectrum of 9 showed two doublets (Fig. 6, Bottom): one centered at δ 72.62 ppm (JP–P = 3.2 Hz) corresponding to the downfield shifted phosphine oxide of the coordinated TEPO and the second resonance centered at δ −17.21 ppm (JP–P = 2.9 Hz) attributed to the phosphorus resonance of ArBiAMPtBu. Complex 8 shows two broadened singlets at 25 °C centered at δ 68.78 ppm (Δν1/2 = 10.8 Hz) (TEPO) and δ −13.77 ppm (Δν1/2 = 26.7 Hz) (BiAMP) by 31P{1H} NMR spectroscopy. Variable temperature 31P{1H} NMR experiments allowed for the observation of J-coupling in 8 at −50 °C. In this case, the respective resonances for TEPO and BiAMP ligands were observed as doublets at δ 70.29 (JP–P = 3.2 Hz) and δ −14.97 (JP–P = 2.8 Hz).
Gutmann–Beckett acceptor numbers (AN) for each (ArBiAMPtBu)(η5-iPrMeInd)M(TEPO) adduct (7–9) were calculated using the Δδ values of the metal-coordinated TEPO resonance measured at 25 °C (298 K) according to the equation AN = 2.21 × (δsample − 45.05) that takes into the account the “free” TEPO phosphorus resonance observed at δ 45.05 ppm in THF-d8 with 31P{1H} NMR spectroscopy. As predicted, the observed change in the resonance frequency arising from the TEPO group follows a roughly linear trend (AN(7) = 56.6; AN(8) = 52.4; AN(9) = 60.9 based on established Shannon radii of the metals.62,63 However, the observed trend is imperfect; recent reports describe the variables which prevent Gutmann–Beckett analysis from being a universal indicator of Lewis-acidity.67 In our case the trend is consistent but falls far outside of linearity (Graph S1†) and we can hypothesize several reasons for this. Complex 7 bears a metal (Y) with a lower principal quantum number, 4d, than the lanthanide metals La and Lu (4f/5d). The 4f electrons are considered core-like and non-participatory in bonding, while d-orbitals tend to dominate bonding. Additionally, the La analogue 8 may be able to reversibly accommodate an additional THF as a ligand from the THF-d8 solvent, which would produce broadened resonances through dynamic averaging. Evidence for an unknown dynamic process was found by performing VT NMR experiments, during which a doublet (JP–P) was observed at −50 °C (Fig. 7).
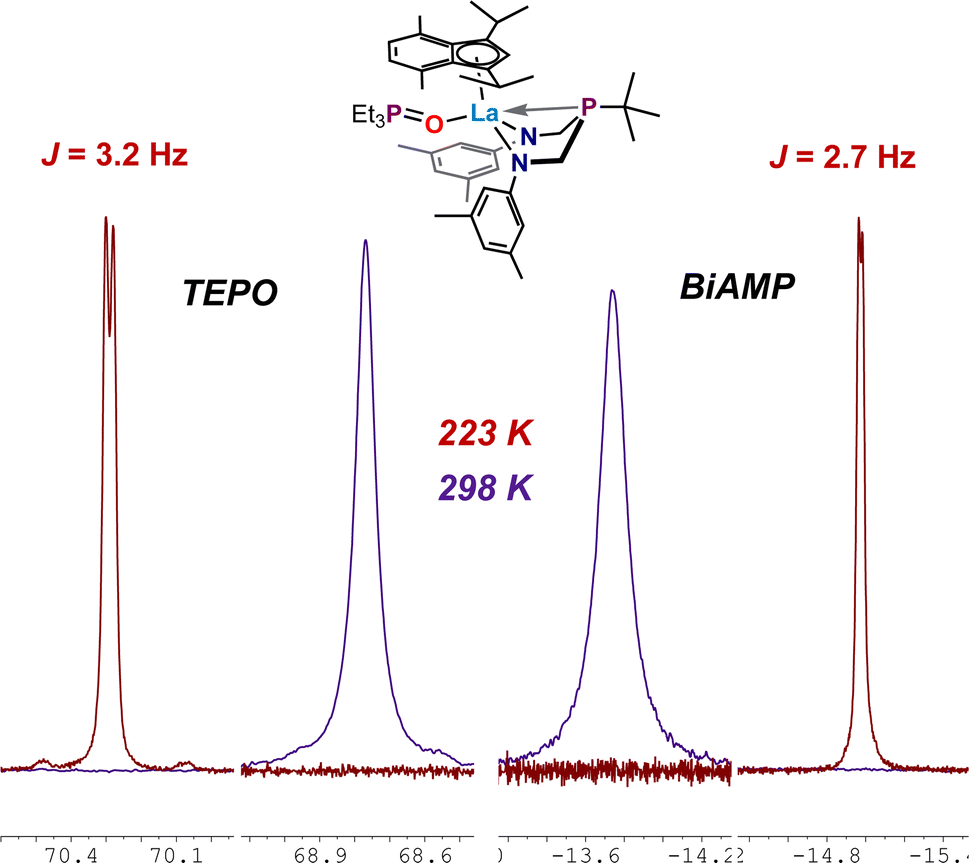 | ||
| Fig. 7 Resonances from the 31P{1H} NMR spectra of 8, (ArBiAMPtBu)(η5-iPrMeInd)La(TEPO), in THF-d8 recorded at 25 °C (298 K) and −50 °C (223 K), with the expansions of the resonances attributed to TEPO (left) and BiAMP (right). | ||
An unfortunate physical consequence arising from the addition of TEPO was the decreased crystallinity of the resultant complexes that necessitated investigating these molecules solely from material generated and analyzed in situ. Complexes 7–9 were observed as tacky solids, and attempted crystallization using various solvents and methods were non-productive. Thus, solid-state analysis and the related metrical parameters were not obtained. While these data would have allowed for further correlation between structural and spectroscopic parameters, we were nevertheless able to confirm the substitution of TEPO onto the metal center. The obtained NMR parameters from these complexes proved useful; specifically, the 89Y–31P coupling were determined to increase along the series 1 < 7 < 4. We rationalize this observed trend based on our original hypothesis on the electrostatic nature of the M–P interaction, where the electron rich, tris(thf) iodide environment of 1 is observed with the smallest coupling. The largest coupling was observed in 4, where η5-iPrMeInd substitutes the iodide and two THF ligands. A substantial decrease in coupling was observed upon substituting the coordinated THF with TEPO in 7, as the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O donor would be the stronger donor in the spectrochemical series. Future efforts will be dedicated to understanding the effects of additional ligands at the metal center, either to increase or decrease the observed coupling values in order to aid future predictive ability. Further exploration of REE–P bonding and covalency, using J coupling as a tool, may further aid our understanding of fundamental bonding descriptions.
O donor would be the stronger donor in the spectrochemical series. Future efforts will be dedicated to understanding the effects of additional ligands at the metal center, either to increase or decrease the observed coupling values in order to aid future predictive ability. Further exploration of REE–P bonding and covalency, using J coupling as a tool, may further aid our understanding of fundamental bonding descriptions.
Conclusions
A new bifunctional ligand, ArBiAMPtBu, was prepared in good yields in a one-pot reaction using the Kabachnik–Fields condensation. The synthesis of the new ligand was optimized to reduce the amount of observed cyclic byproduct, and the reaction consistently produced a 66% yield of highly pure crystalline ArBiAMPtBu. The coordination chemistry of three diamagnetic rare-earth elements with the deprotonated ArBiAMPtBu ligand was investigated. Installation of the doubly deprotonated, dipotassium salt of the ligand onto the THF-adducts of MI3 salts was achieved to generate the mono(iodide) complexes of Y (1), La (2), and Lu (3) that crystallize as THF adducts MI(ArBiAMPtBu)(thf)3. These compounds proved unstable in non-coordinating solvents as observed by 31P{1H} NMR spectroscopy. Subsequent organometallic derivatization of 1, 2, and 3 to form η5-indenyl products 4, 5, and 6 was accomplished by performing a salt metathesis with [K][η5-iPrMeInd]. And while the products proved non-isolable, the TEPO derivatives 7–9 were characterized using NMR techniques to determine the effect of a stronger donor ligand in the coordination sphere.For complexes 1–6, SC-XRD analysis was used to evaluate potential bonding between the Ln and phosphorus, and a linear trend was observed correlating the ionic radius of the metal to the M–P distances found in the solid-state data. NMR spectroscopy confirmed direct Y–P bonding as determined through both 31P and 89Y NMR spectroscopy. The strength of the JP–Y-coupling increased from 2.7 Hz to 17.5 Hz, following installation of the η5-iPrMeInd. The increase in bond strength also manifested as an observable contraction of the Y–P bond. Lanthanum complex 5 exhibited an elongation of the La–P bond to that of a non-bonding interaction owing to an additional ligand coordination, which supports our hypothesis that increasing the electron density at the metal center will result in a weaker electrostatic interaction between the two moieties. For Lu, the substitution also led to a significant contraction of the M–P bond, but the 31P{1H} NMR spectra did not show resolved coupling but rather a broadened resonance, which is likely attributable to the I = 7/2 nucleus with inherently small J-values. Complexes 7–9 were characterized using NMR spectroscopy (Fig. S31–S41†) as the crystallization proved difficult, which precluded solid-state characterization.
This preliminary exploration of the chemistry of ArBiAMPtBu as a supporting ligand for f-block metals produced encouraging results. Additional reactions and further chemical derivatization may begin to elucidate the necessary conditions for M–P bond expansion and contraction, with the goal of predicting and controlling the observed nuclear coupling. We hope to further explore the extent to which the flexible phosphorous moiety enables the ligand to act as a spectroscopic handle. The M–P bond was sensitive to the coordination environment around the metal center and attempts to understand and control that sensitivity will support future work with the BiAMP system, including efforts targeting separations, frustrated Lewis-acid base pair reactivity, and actinide coordination and bonding studies.
Data availability
The data supporting this manuscript, including experimental descriptions, spectra for all complexes, CCDC deposition numbers and crystallography refinement tables, have been uploaded as part of the ESI.†Author contributions
This work was conceived by AMT and executed at Los Alamos National Laboratory under his guidance. YM performed the majority of the synthesis and characterization presented herein. SS provided the supporting organic ligand chemistry necessary to perform this study. TVF supported aspects of the spectroscopy. BKC aided with the final organization and edits.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of Los Alamos National Laboratory, which is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001). This work, from concept to implementation, was funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372, DE-AC52-06NA25396) and AMT and YM are grateful for their support to conduct this research. We would like to thank Sarah K. Tondreau for assistance with copyediting and organization. SS was supported by Laboratory Directed Research and Development program of Los Alamos National Laboratory under project number 2019057ECR. TVF was supported by the Glenn T. Seaborg Institute with a postdoctoral fellowship.Notes and references
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- C. Comuzzi, P. Di Bernardo, R. Portanova, M. Tolazzi and P. Zanonato, Polyhedron, 2002, 21, 1385–1391 CrossRef CAS.
- J. Du, P. J. Cobb, J. Ding, D. P. Mills and S. T. Liddle, Chem. Sci., 2024, 15, 13–45 RSC.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- J. D. Masuda, K. C. Jantunen, O. V. Ozerov, K. J. T. Noonan, D. P. Gates, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 2408–2409 CrossRef CAS PubMed.
- T. E. Rieser, P. Wetzel, C. Maichle-Mössmer, P. Sirsch and R. Anwander, J. Am. Chem. Soc., 2023, 145, 17720–17733 CrossRef CAS PubMed.
- M. Berardini, T. J. Emge and J. G. Brennan, Inorg. Chem., 1995, 34, 5327–5334 CrossRef CAS.
- G. P. A. Y. Rabe and A. L. Rheingold, Inorg. Chem., 1995, 34, 4521–4522 CrossRef CAS.
- G. W. Rabe, J. Riede and A. Schier, Inorg. Chem., 1996, 35, 40–45 CrossRef CAS PubMed.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1997, 36, 3212–3215 CrossRef CAS PubMed.
- F. A. Watt, N. Dickmann, R. Schoch and S. Hohloch, Inorg. Chem., 2020, 59, 13621–13631 CrossRef CAS PubMed.
- F. A. Watt, K. N. McCabe, R. Schoch, L. Maron and S. Hohloch, Chem. Commun., 2020, 56, 15410–15413 RSC.
- M. D. Fryzuk, T. S. Haddad and D. J. Berg, Coord. Chem. Rev., 1990, 99, 137–212 CrossRef CAS.
- T. D. Tilley, R. A. Andersen and A. Zalkin, J. Am. Chem. Soc., 1982, 104, 3725–3727 CrossRef CAS.
- M. D. Fryzuk and T. S. Haddad, J. Am. Chem. Soc., 1988, 110, 8263–8265 CrossRef CAS.
- M. D. Fryzuk, T. S. Haddad and S. J. Rettig, Organometallics, 1991, 10, 2026–2036 CrossRef CAS.
- M. Visseaux, A. Dormond, M. M. Kubicki, C. Moïse, D. Baudry and M. Ephritikhine, J. Organomet. Chem., 1992, 433, 95–106 CrossRef CAS.
- H. H. Karsch, V. Graf, M. Reisky and E. Witt, Eur. J. Inorg. Chem., 1998, 10, 1403–1406 Search PubMed.
- T. S. Targos, R. P. Rosen, R. R. Whittle and G. L. Geoffroy, Inorg. Chem., 1985, 24, 1375–1379 CrossRef CAS.
- R. T. Baker, T. H. Tulip and S. S. Wreford, Inorg. Chem., 1985, 24, 1379–1383 CrossRef CAS.
- L. Gelmini, L. C. Matassa and D. W. Stephan, Inorg. Chem., 1985, 24, 2585–2588 CrossRef CAS.
- F. Lindenberg, T. Gelbrich and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 1995, 621, 771–778 CrossRef.
- G. S. Ferguson and P. T. Wolczanski, Organometallics, 1982, 4, 1601–1605 CrossRef.
- G. S. Ferguson, P. T. Wolczanski, L. Parkanyi and M. C. Zonnevylle, Organometallics, 1985, 7, 1967–1979 CrossRef.
- N. E. Schore, S. J. Young, M. M. Olmstead and P. Hofmann, Organometallics, 1983, 2, 1769–1780 CrossRef CAS.
- D. R. Tueting, M. M. Olmstead and N. E. Schore, Organometallics, 1992, 11, 2235–2241 CrossRef CAS.
- B. Liu, D. Cui, J. Ma, X. Chen and X. Jing, Chem.–Eur. J., 2007, 13, 834–845 CrossRef CAS PubMed.
- S. Li, W. Miao, T. Tang, D. Cui, X. Chen and X. Jing, J. Organomet. Chem., 2007, 692, 4943–4952 CrossRef CAS.
- A. V. Zabula, Y. Qiao, A. J. Kosanovich, T. Cheisson, B. C. Manor, P. J. Carroll, O. V. Ozerov and E. J. Schelter, Chem.–Eur. J., 2017, 23, 17923–17934 CrossRef CAS PubMed.
- L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909–1913 CrossRef CAS PubMed.
- W. W. N. O, X. Kang, Y. Luo and Z. Hou, Organometallics, 2014, 33, 1030–1043 CrossRef CAS.
- L. Wang, D. Cui, Z. Hou, W. Li and Y. Li, Organometallics, 2011, 30, 760–767 CrossRef CAS.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Organometallics, 2015, 34, 4647–4655 CrossRef CAS.
- T. Cantat, B. L. Scott, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2009, 48, 2114–2127 CrossRef CAS PubMed.
- Y. Sunada, T. Sue, T. Matsumoto and H. Nagashima, J. Organomet. Chem., 2006, 691, 3176–3182 CrossRef CAS.
- H. Nagashima, T. Sue, T. Oda, A. Kanemitsu, T. Matsumoto, Y. Motoyama and Y. Sunada, Organometallics, 2006, 25, 1987–1994 CrossRef CAS.
- J. W. Napoline, S. J. Kraft, E. M. Matson, P. E. Fanwick, S. C. Bart and C. M. Thomas, Inorg. Chem., 2013, 52, 12170–12177 CrossRef CAS PubMed.
- P. Cui, X. Huang, J. Du and Z. Huang, Inorg. Chem., 2021, 60, 3249–3258 CrossRef CAS PubMed.
- H. Han, M. Elsmaili and S. A. Johnson, Inorg. Chem., 2006, 45, 7435–7445 CrossRef CAS PubMed.
- P. Cui, C. Wu, J. Du, G. Luo, Z. Huang and S. Zhou, Inorg. Chem., 2021, 60, 9688–9699 CrossRef CAS PubMed.
- B. L. Ramirez, P. Sharma, R. J. Eisenhart, L. Gagliardi and C. C. Lu, Chem. Sci., 2019, 10, 3375–3384 RSC.
- B. L. Ramirez and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 5396–5407 CrossRef CAS PubMed.
- Q. Liao, T. Liu, S. I. Johnson, C. M. Klug, E. S. Wiedner, R. Morris Bullock and D. L. DuBois, Dalton Trans., 2019, 48, 4867–4878 RSC.
- A. Orthaber, M. Karnahl, S. Tschierlei, D. Streich, M. Stein and S. Ott, Dalton Trans., 2014, 43, 4537–4549 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- P. Das, R. M. Stolley, E. F. van der Eide and M. L. Helm, Eur. J. Inorg. Chem., 2014, 4611–4618 CrossRef CAS.
- R. Raturi, J. Lefebvre, D. B. Leznoff, B. R. McGarvey and S. A. Johnson, Chem.–Eur. J., 2008, 14, 721–730 CrossRef CAS PubMed.
- E. Kolehmainen, in Encyclopedia of Spectroscopy and Spectrometry, ed. J. C. Lindon, D. W. Koppenaal and G. E. Tranter, Elsevier, Amsterdam, 3rd edn, 2017, pp. 366–374 Search PubMed.
- A. A. Pinkerton and W. L. Earl, J. Chem. Soc., Dalton Trans., 1978, 3, 267–272 RSC.
- A. A. Pinkerton and W. L. Earl, J. Chem. Soc., Dalton Trans., 1979, 9, 1347–1349 RSC.
- K. W. Feindel and R. E. Wasylishen, Can. J. Chem., 2004, 82, 27–44 CrossRef CAS.
- S. O. Grim and S. A. Sangokoya, J. Chem. Soc., Chem. Commun., 1984, 1599–1600 RSC.
- M. I. Kabachnik and T. Y. Medved, Dokl. Akad. Nauk SSSR, 1952, 83, 689–692 CAS.
- E. K. Fields, J. Am. Chem. Soc., 1952, 74, 1528–1531 CrossRef CAS.
- G. Keglevich and E. Balint, Molecules, 2012, 17, 12821–12835 CrossRef CAS PubMed.
- E. I. Musina, V. V. Khrizanforova, I. D. Strelnik, M. I. Valitov, Y. S. Spiridonova, D. B. Krivolapov, I. A. Litvinov, M. K. Kadirov, P. Lonnecke, E. Hey-Hawkins, Y. H. Budnikova, A. A. Karasik and O. G. Sinyashin, Chem.–Eur. J., 2014, 20, 3169–3182 CrossRef CAS PubMed.
- V. G. Märkl, G. Y. Jin and C. Schoerner, Tetrahedron Lett., 1980, 21, 1409–1412 CrossRef.
- A. A. Karasik, I. O. Georgiev, E. I. Musina, O. G. Sinyashin and J. Heinicke, Polyhedron, 2001, 20, 3321–3331 CrossRef CAS.
- R. M. Kuznetsov, A. S. Balueva, T. M. Serova and G. N. Nikonov, Russ. J. Gen. Chem., 2001, 71, 899–902 CrossRef CAS.
- A. A. Karasik, I. O. Georgiev, O. G. Sinyashin and E. Hey-Hawkins, Polyhedron, 2000, 19, 1455–1459 CrossRef CAS.
- A. A. Karasik, R. N. Naumov, R. Sommer, O. G. Sinyashin and E. Hey-Hawkins, Polyhedron, 2002, 21, 2251–2256 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- K. Mikami, M. Terada and H. Matsuzawa, Angew. Chem., Int. Ed., 2002, 41, 3554–3572 CrossRef CAS PubMed.
- G. P. McGovern, F. Hung-Low, J. W. Tye and C. A. Bradley, Organometallics, 2012, 31, 3865–3879 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS.
- L. Greb, Chem.–Eur. J., 2018, 24, 17852–18129 CrossRef PubMed.
- M. Mewald, R. Frohlich and M. Oestreich, Chem.–Eur. J., 2011, 17, 9406–9414 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, M. Jakubczyk, A. Sporzyński and G. Żukowska, Inorg. Chem. Commun., 2011, 14, 1753–1755 CrossRef.
- H. Großekappenberg, M. Reißmann, M. Schmidtmann and T. Müller, Organometallics, 2015, 34, 4952–4958 CrossRef.
- M. Fischer, M. C. Wolff, E. del Horno, M. Schmidtmann and R. Beckhaus, Organometallics, 2020, 39, 3232–3239 CrossRef CAS.
- C. A. Fischer, A. Rosch, H. Elsen, G. Ballmann, M. Wiesinger, J. Langer, C. Farber and S. Harder, Dalton Trans., 2019, 48, 6757–6766 RSC.
- R. R. Golwankar, T. D. Curry II, C. J. Paranjothi and J. D. Blakemore, Inorg. Chem., 2023, 62, 9765–9780 CrossRef CAS PubMed.
- J. J. Jennings, B. W. Wigman, B. M. Armstrong and A. K. Franz, J. Org. Chem., 2019, 84, 15845–15853 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2328028–2328036. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02077d |
| This journal is © The Royal Society of Chemistry 2024 |